Embed Size (px)
DESCRIPTION
real time pcr
Citation preview

T A B L E O F C O N T E N T ST A B L E O F C O N T E N T ST A B L E O F C O N T E N T ST A B L E O F C O N T E N T ST A B L E O F C O N T E N T S
Table of Contents
The Basics of PCR and RT-PCR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2The Basics of PCR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2The Basics of RT-PCR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
Increasing RT-PCR Sensitivity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3Isolating High-Quality RNA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3Using RNase H– Reverse Transcriptases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4Higher RT Incubation Temperatures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5Additives to Enhance RT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5RNase H Treatment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5Improving Detection of Small Amounts of RNA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6One-Step vs. Two-Step RT-PCR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
Improving RT-PCR Specificity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7Priming cDNA Synthesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7Higher RT Incubation Temperatures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7Minimizing Contaminating Genomic DNA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
RT-PCR Applications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85´ and 3´ RACE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8Quantifying mRNA Expression . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
Improving PCR Specificity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10Primer Design . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10Primer Annealing Temperature . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10Touchdown PCR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11Primer Concentration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11Primer Purity and Stability . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11Hot Start . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12Magnesium Concentration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13Additives to Enhance PCR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13Nested PCR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
Increasing PCR Sensitivity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14Template Quality . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14Template Concentration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14Enzyme Choice . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
Improving Fidelity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15Enzymes with Proofreading . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15Enzyme Mixes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15Other Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
Other Things to Consider . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16Amplifying Long Targets . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16Prevention of Carry-Over Contamination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16Purification of PCR Products . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
PCR Applications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17Multiplex PCR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17Genotyping with Dinucleotide Repeat Markers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17Tools for Detecting Polymorphisms . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
Troubleshooting Guide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
Related Products . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
Selected Primer Sequences . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
CHAPTER 1
CHAPTER 2
CHAPTER 3
CHAPTER 4
CHAPTER 5
CHAPTER 6
CHAPTER 7
CHAPTER 8
CHAPTER 9
CHAPTER 10
CHAPTER 11
CHAPTER 12
APPENDIX A

N6
N6
N6 N6 N6
N6 N6
N6N6 N6
The Basics of PCR
The Polymerase Chain Reaction (PCR)process uses multiple cycles of tem-plate denaturation, primer annealing,
and primer elongation to amplify DNA sequences (1). It is an exponential processsince amplified products from the previouscycle serve as templates for the next cycle of
amplification, making it a highly sensitivetechnique for the detection of nucleic acids.Typically, enough amplified product is generated after 20 to 30 cycles of PCR so that it can be visualized on an ethidium bromide-stained gel. The reaction is composedof several components (table 1). The templatecan include purified genomic or plasmidDNA; RNA converted to cDNA; or unpuri-fied, crude biological samples such as bacte-rial colonies or phage plaques. The primersdetermine the sequence and the length of theamplified product. The most frequently used thermostable polymerase is Taq DNA polymerase. This enzyme is appropriate forroutine amplifications, but the use of otherthermostable polymerases can enhance results.Amplification reactions also contain buffer,deoxynucleotide triphosphates, and magne-sium. The magnesium ion concentrationaffects enzyme activity, primer annealing,melting temperature of the template and thePCR product, fidelity, and primer-dimer formation (2). How the interaction of thesecomponents, cycling parameters, as well asother factors contribute to successful PCRwill be discussed in the following chapters.
The Basics of RT-PCRRT-PCR combines cDNA synthesis from
RNA templates with PCR (figure 1) to providea rapid, sensitive method for analyzing geneexpression. RT-PCR is used to detect or quantify the expression of messages, oftenfrom small amounts of RNA (3,4). In addition,the technique is used to analyze differentialgene expression or clone cDNAs withoutconstructing a cDNA library (5,6). RT-PCR ismore sensitive and easier to perform thanother RNA analysis techniques, includingNorthern blots, RNase protection assays,in situ hybridization, and S1 nuclease assays(7,8).
The template for RT-PCR can be totalRNA or poly(A)+ selected RNA. RT reac-tions can be primed with random primers,oligo(dT), or a gene-specific primer (GSP)using a reverse transcriptase. RT-PCR canbe carried out either in two-step or one-step
formats. In two-step RT-PCR, each step isperformed under optimal conditions. cDNAsynthesis is performed first in RT buffer and one tenth of the reaction is removed for PCR. In one-step RT-PCR, reverse tran-scription and PCR take place sequentially in a single tube under conditions optimizedfor both RT and PCR.
This Guide describes the keys to suc-cessful RT-PCR and PCR. High sensitivity(getting enough product from small samples)and high specificity (selective amplificationof only the desired product) are the hallmarks of successful PCR. Optimal RT-PCR andPCR can be obtained through careful exper-imental design, including selecting the appro-priate enzymes, designing optimal primers,using different buffers and additives, estab-lishing cycling parameters, and preparinghigh quality templates.
2 T H E B A S I C S O F P C R A N D R T - P C R
C H A P T E R 1C H A P T E R 1C H A P T E R 1C H A P T E R 1C H A P T E R 1
The Basics of PCR and RT-PCR
FIGURE 1. RT-PCR overview.
Cells or Tissue
AAAAAAA(A)n
AAAAAAA(A)n
AAAAAAA(A)n AAAAAAA(A)n
AAAAAAA(A)n
AAAAAAA(A)n
TTTTTTTT
AAAAAAA(A)nTTTTTTTT
AAAAAAA(A)nTTTTTTTT
AAAAAAA(A)n
random hexamer primingoligo(dT) priming
RNA Isolation
cDNA Synthesis
PCR Amplification
gene specific priming
AAAAAAA(A)n
AAAAAAA(A)n
AAAAAAA(A)n
GSP
Table 1
Component Final Concentration
Template 104–106 copies of DNA template
Primer 1 0.1–0.5 µM
Primer 2 0.1–0.5 µM
10X Reaction buffer 1X
Magnesium 1.0–3.0 mM
dNTP mix 200 µM each dNTP
Thermostable DNA polymerase 1–4 units/100 µl reaction
TABLE 1. Reaction components.

Isolating High-Quality RNA
Successful cDNA synthesis starts withhigh-quality RNA. High-quality RNAis substantially full length, and does
not contain inhibitors of reverse transcrip-tases such as EDTA or SDS (9). The qualityof the RNA dictates the maximum amount ofsequence information that can be convertedinto cDNA. One popular RNA isolation protocol is the single-step method whichuses guanidine isothiocyanate/acidic phenol(10). The TRIZOL® Reagent method (see figure
below) is an improvement on this single-stepmethod and can be used to isolate high qual-ity, undegraded RNA from various cells andtissues (11,12). The TRIZOL Reagent methodcan be used to isolate RNA from as little as100 cells or 1 mg of tissue (13).
Oligo(dT) selection for poly(A)+ RNA is typically not necessary. Amplified productis detectable when either total RNA orpoly(A)+ RNA is used as the starting tem-plate (figure 2) (14). In addition, isolatingpoly(A)+ RNA may lead to variability ofmRNA enrichment between samples, whichcan bias the detection or quantification of message. However, poly(A)+ RNA mayincrease the sensitivity of detection whenanalyzing rare messages.
RNA isolated from RNase-rich samples,such as the pancreas, may need to be storedin formamide to maintain high quality RNA,due to the carry-over of trace amounts ofRNase (15). This is especially true for longterm storage. RNA isolated from rat pancreasand stored in water showed substantial degra-dation after just 1week, whereas RNA isolatedfrom rat liver, which contains lower amountsof RNases, was stable in water for 3 years(15). In addition, transcripts >4 kb are more
susceptible to degradation by trace amountsof RNases than shorter transcripts (16). Toincrease the stability of stored RNA samples,dissolve the RNA in deionized formamideand store at –70°C. The formamide used forstorage of RNA must not contain impuritiesthat cause RNA degradation (16). RNA frompancreas was stable for at least 1 year in form-amide. When ready to use the RNA, simplyprecipitate it by adding NaCl to 0.2 M followed by 4 volumes of ethanol. Incubate 3 to 5 min at room temperature and centrifugeat 10,000 × g for 5 min.
RNase inhibitors are often added to RTreactions to help improve cDNA length andyield. Add RNase inhibitors to the first-strand reaction in the presence of buffer and a reducing agent, such as DTT, since procedures prior to cDNA synthesis thatdenature the inhibitor will release boundRNases that can degrade RNA. Protein RNaseinhibitors protect RNA against degradation by RNases A, B, and C, but do not protectagainst RNases found on the skin. Thus, evenwhen using these inhibitors be careful not tointroduce RNases from your fingertips.
Outline of Procedure for TRIzOL® Reagent
C H A P T E R 2C H A P T E R 2C H A P T E R 2C H A P T E R 2C H A P T E R 2
I N C R E A S I N G R T - P C R S E N S I T I V I T Y 3
Increasing RT-PCR Sensitivity
FIGURE 2. Comparison of total RNA and poly(A)+ RNA in RT-PCR.cDNA was synthesized in duplicate from 5 or 1 µg total HeLa RNA(lanes 1 and 2, respectively) or from 500 ng or 50 ng of poly(A)+
HeLa RNA (lanes 3 and 4, respectively) with oligo(dT) primers andSUPERSCRIPT™ II RT. The amplified targets were a 377-bp fragmentnear the 5´ end of the DNA polymerase ε mRNA and a 643-bp frag-ment of the replication protein A mRNA, which are both moderatelyabundant targets. One tenth of the cDNA reaction was amplified for30 cycles using Taq DNA polymerase.
–377 bp
–643 bp
100
bp
DNA
Ladd
er
100
bp
DNA
Ladd
er
Total RNA Poly(A)+ RNA1 2 3 4
Panel A
Panel B
TRIZOL Reagent can isolate high-quality RNA from as little as 100 cells or 1 mg of tissue.
Elapsed Time <1 h
Homogenize sample in TRIZOL Reagent
Separate Phases(add chloroform)
Precipitate RNA
Wash and Solubilize
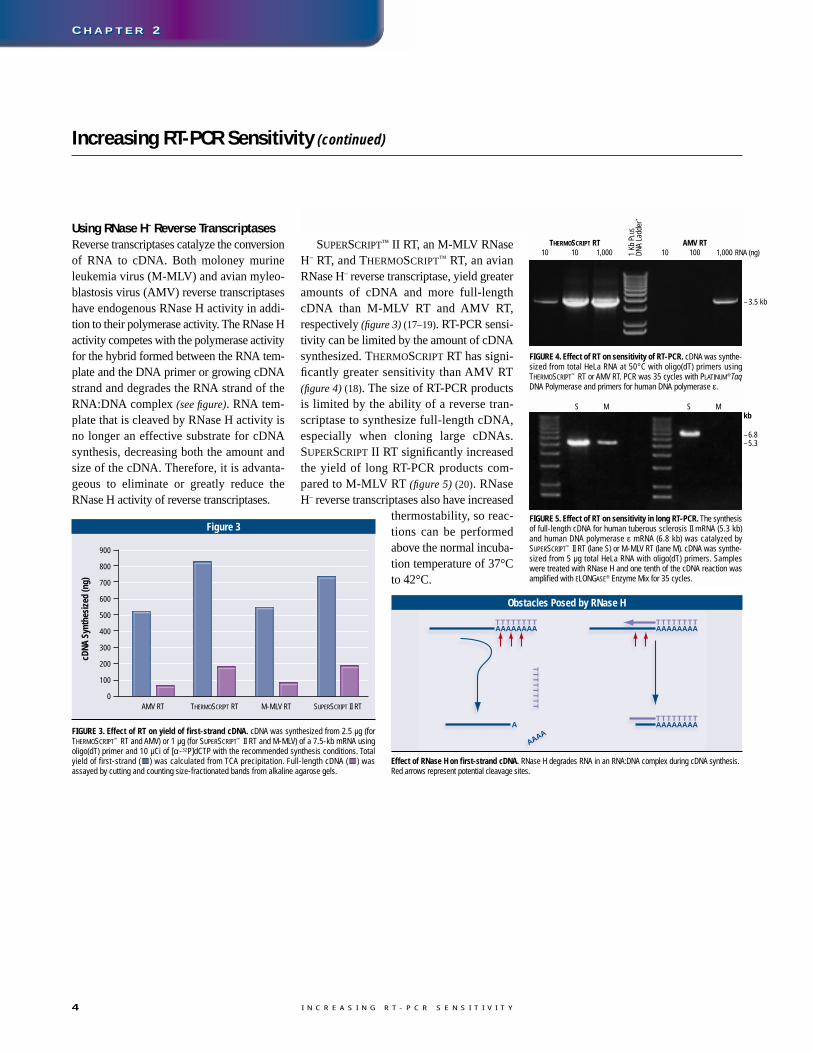
Using RNase H– Reverse TranscriptasesReverse transcriptases catalyze the conversionof RNA to cDNA. Both moloney murineleukemia virus (M-MLV) and avian myleo-blastosis virus (AMV) reverse transcriptaseshave endogenous RNase H activity in addi-tion to their polymerase activity. The RNase Hactivity competes with the polymerase activityfor the hybrid formed between the RNA tem-plate and the DNA primer or growing cDNAstrand and degrades the RNA strand of theRNA:DNA complex (see figure). RNA tem-plate that is cleaved by RNase H activity is no longer an effective substrate for cDNAsynthesis, decreasing both the amount andsize of the cDNA. Therefore, it is advanta-geous to eliminate or greatly reduce theRNase H activity of reverse transcriptases.
SUPERSCRIPT™ II RT, an M-MLV RNaseH– RT, and THERMOSCRIPT™ RT, an avianRNase H– reverse transcriptase, yield greateramounts of cDNA and more full-lengthcDNA than M-MLV RT and AMV RT,respectively (figure 3) (17–19). RT-PCR sensi-tivity can be limited by the amount of cDNAsynthesized. THERMOSCRIPT RT has signi-ficantly greater sensitivity than AMV RT(figure 4) (18). The size of RT-PCR products is limited by the ability of a reverse tran-scriptase to synthesize full-length cDNA,especially when cloning large cDNAs.SUPERSCRIPT II RT significantly increasedthe yield of long RT-PCR products com-pared to M-MLV RT (figure 5) (20). RNaseH– reverse transcriptases also have increased
thermostability, so reac-tions can be performedabove the normal incuba-tion temperature of 37°Cto 42°C.
10 10 1,000THERMOSCRIPT RT
10 100 1,000AMV RT
FIGURE 4. Effect of RT on sensitivity of RT-PCR. cDNA was synthe-sized from total HeLa RNA at 50°C with oligo(dT) primers usingTHERMOSCRIPT™ RT or AMV RT. PCR was 35 cycles with PLATINUM®TaqDNA Polymerase and primers for human DNA polymerase ε.
1 Kb
PLU
SDN
A La
dder
™
Figure 3
FIGURE 3. Effect of RT on yield of first-strand cDNA. cDNA was synthesized from 2.5 µg (forTHERMOSCRIPT™ RT and AMV) or 1 µg (for SUPERSCRIPT™ II RT and M-MLV) of a 7.5-kb mRNA usingoligo(dT) primer and 10 µCi of [α-32P]dCTP with the recommended synthesis conditions. Totalyield of first-strand ( ) was calculated from TCA precipitation. Full-length cDNA ( ) wasassayed by cutting and counting size-fractionated bands from alkaline agarose gels.
cDNA
Syn
thes
ized
(ng)
900
800
700
600
500
400
300
200
100
0THERMOSCRIPT RT M-MLV RTAMV RT SUPERSCRIPT II RT
Effect of RNase H on first-strand cDNA. RNase H degrades RNA in an RNA:DNA complex during cDNA synthesis.Red arrows represent potential cleavage sites.
Obstacles Posed by RNase H
FIGURE 5. Effect of RT on sensitivity in long RT-PCR. The synthesisof full-length cDNA for human tuberous sclerosis II mRNA (5.3 kb) and human DNA polymerase ε mRNA (6.8 kb) was catalyzed bySUPERSCRIPT™ II RT (lane S) or M-MLV RT (lane M). cDNA was synthe-sized from 5 µg total HeLa RNA with oligo(dT) primers. Samples were treated with RNase H and one tenth of the cDNA reaction was amplified with ELONGASE® Enzyme Mix for 35 cycles.
S M S Mkb
–6.8–5.3
RNA (ng)
–3.5 kb
4 I N C R E A S I N G R T - P C R S E N S I T I V I T Y
Increasing RT-PCR Sensitivity (continued)
C H A P T E R 2C H A P T E R 2C H A P T E R 2C H A P T E R 2C H A P T E R 2
TTTTTTTTAAAAAAAA
TTTTTTTTAAAAAAAA
TTTTTTTTAAAAAAAA
AAAA
TT
TT
TT
TT
A

Higher RT Incubation TemperaturesHigher RT incubation temperatures (table 2)
can help disrupt RNA secondary structureand increase the yield of product (18). Formany RNA templates, incubating the RNAand primer at 65°C in the absence of buffersor salts and quickly chilling on ice eliminatesmost secondary structure and allows primerbinding. However, secondary structure maystill exist in some templates even after heatdenaturation. The amplification of productsfrom these difficult templates can beimproved by using THERMOSCRIPT RT andperforming the RT reactions at higher tem-perature (figure 6) (18). Higher temperaturescan also improve specificity, especially whenusing a gene-specific primer (GSP) for cDNAsynthesis (see page 7). If using a GSP, makesure the Tm of the primer is as high as the
planned incubation temperature. Do not useoligo(dT) and random primers above 60°C;random primers require an initial 10 minincubation at 25°C before increasing the temperature to 60°C. In addition to using
elevated RT temperatures, increase specificityby shifting the RNA/primer mix directlyfrom the 65°C denaturation temperature tothe RT incubation temperature and add pre-warmed 2X reaction mix (cDNA synthesishot start). This helps prevent intramolecularbase pairing that can occur at lower temper-atures. Using a thermal cycler can simplify
the multiple temperatureshifts required for RT-PCR.
Tth thermostable poly-merase, which functions as aDNA polymerase in the pres-ence of Mg++ and a reversetranscriptase in the presenceof Mn++, can also be incu-bated up to 65°C. However,the presence of Mn++
during PCR reduces fidelity.This makes Tth polymeraseless suitable for applicationsthat require highly accurateamplification, such as cloningcDNAs. In addition, Tthpolymerase is a less efficientreverse transcriptase, whichreduces sensitivity and, since
a single enzyme is used for both reverse transcription and PCR, a control reactionwithout RT cannot be used to distinguishamplified cDNA from amplified contami-nating genomic DNA.
Additives to Enhance RTAdditives including glycerol and DMSO canbe added to first-strand synthesis reactions to help destabilize nucleic acid duplexes andmelt RNA secondary structure. Up to 20%glycerol or up to 10% DMSO can be usedwithout effecting SUPERSCRIPT II RT or M-MLV RT activity (9). AMV RT will alsotolerate up to 20% glycerol without loss ofactivity. For maximum RT-PCR sensitivity in SUPERSCRIPT II RT reactions, add 10%glycerol and incubate at 45°C. If one tenth ofthe RT reaction is added to the PCR, the finalconcentration of glycerol in the amplificationreaction is 0.4%, which will not inhibit PCR.
RNase H TreatmentTreating cDNA reactions with RNase H priorto PCR can improve sensitivity. For some targets, it is thought that the RNA in thecDNA reaction may prevent binding of theamplification primers. In these cases, RNaseH treatment can improve sensitivity. RNaseH treatment is often necessary when ampli-fying longer, full-length cDNA targets, such
as the low copy target tuberous sclerosis II(figure 7) (20). For this difficult target, RNase Htreatment improved the signal from cDNAgenerated with either SUPERSCRIPT II RT orM-MLV RT. For many RT-PCR applications,RNase H treatment is optional since incu-bation at 95°C for the PCR denaturation stepoften hydrolyzes the RNA in the RNA:cDNAcomplex.
C H A P T E R 2C H A P T E R 2C H A P T E R 2C H A P T E R 2C H A P T E R 2
Table 2
Reverse Transcriptase Incubation Temperature
AMV 37°C–45°C
M-MLV 37°C
SUPERSCRIPT™ II RT 37°C–50°C
THERMOSCRIPT™ RT 42°C–65°C*
*RNA begins to hydrolyze at temperatures above 65°C. For targets≤1 kb, 70°C first-stand synthesis temperatures can be used. Fortargets >1 kb, use first-strand synthesis temperatures ≤65°C.
TABLE 2. RT incubation temperatures.
FIGURE 7. Effect of RNase H treatment on RT-PCR. The synthesisof full-length cDNA for human tuberous sclerosis II mRNA (5.3 kb)from 5 µg of total HeLa RNA was catalyzed by SUPERSCRIPT™ II RT (S),M-MLV RT (M), or AMV RT (A). One half of the reaction was treatedwith RNase H for 30 min. An amount of the treated or untreated RTreaction equivalent to 0.5% of the starting RNA was amplified for 35cycles with ELONGASE® Enzyme Mix.
–5.3 kb
S M A+ RNase H
S M A– RNase H
1 Kb PLUSDNA Ladder™50°C 55°C 60°C 50°C 55°C 60°C
THERMOSCRIPT RT AMV RT
FIGURE 6. Effect of temperature on a difficult template. cDNA was synthesized at the indi-cated temperatures from 10 ng of soybean total RNA with THERMOSCRIPT™ RT or AMV RT and agene-specific primer for 18S rRNA. 40 cycles of PCR was performed with one tenth of thecDNA reaction and PLATINUM® Taq DNA Polymerase High Fidelity.
1.5 kb–
I N C R E A S I N G R T - P C R S E N S I T I V I T Y 5

Improving Detection of Small Amounts of RNART-PCR is particularly challenging when onlysmall amounts of RNA are available. Theaddition of glycogen as a carrier during RNAisolation helps improve the yield from smallsamples (13). RNase-free glycogen is added atthe same time TRIZOL Reagent is added.Glycogen is water soluble and remains in theaqueous layer with the RNA to aid subsequentprecipitation. The recommended concen-
tration of RNase-free glycogen is 250 µg/mlfor <50 mg of tissue or <106 cultured cells.
The addition of acetylated BSA (figure 8) toRT reactions performed with SUPERSCRIPT IIRT can increase sensitivity (21). Also, for smallamounts of RNA, reducing the amount ofSUPERSCRIPT II RT and adding 40 units of RNASEOUT™ Ribonuclease Inhibitor canenhance the level of detection. If glycogen is
used during the RNA isolation, the addition ofBSA or RNase inhibitor to SUPERSCRIPT II RTreactions is still recommended.
One-Step vs. Two-Step RT-PCRTwo-step RT-PCR is popular and useful fordetecting multiple messages from a singleRNA sample. However, a one-step RT-PCRmethod offers other benefits (table 3) (22).One-step RT-PCR is easier to use when pro-cessing large numbers of samples, and helpsminimize carry-over contamination sincetubes do not need to be opened betweencDNA synthesis and amplification. Since theentire cDNA sample is amplified, one-stepRT-PCR can provide greater sensitivity,down to 0.1 pg total RNA (figure 9) (22). Forsuccessful one-step RT-PCR, use an antisensegene-specific primer (GSP) to prime cDNAsynthesis.
6 I N C R E A S I N G R T - P C R S E N S I T I V I T Y
Increasing RT-PCR Sensitivity (continued)
1 2 3 4 5 6 71 2 3 4 5 6 7A B
FIGURE 8. Effect of BSA on RT-PCR sensitivity. 50 pg, 5 pg, 500 fg, 50 fg, 5 fg, and 0.5 fg (lanes 1 to 6, respectively) of CAT mRNA transcriptwere reverse transcribed without (Panel A) or with (Panel B) 0.5 µg of acetylated BSA using SUPERSCRIPT™ II RT. The “no-RT” control contained 50 pg of CAT transcript (lane 7). 30 cycles of PCR was performed with Taq DNA polymerase.
Table 3
TWO-STEP PROCEDURE ONE-STEP PROCEDURE
Prime first-strand cDNA with: Prime first-strand cDNA with:Oligo(dT) GSP primersRandom hexamersGSP primers
Provides: Provides:• Flexibility • Convenience
Choice of primers. Amplification enzymes premixed with Choice of amplification enzyme. reverse transcriptase.
• Ability to optimize for difficult RT-PCR. Fewer pipetting steps and reduced chances Combine with PLATINUM® enzymes for higher specificity. of contamination.Combine with PLATINUM Pfx DNA Polymerase • High sensitivity.for greater fidelity. • Ideal for analysis of large numbers of samples.
• Ideal for detecting or quantifying several • Ideal for quantitative PCR.messages from a single sample.
Notes on amplification enzymes and product size:• Use Taq DNA Polymerase or PLATINUM Taq DNA Polymerase for products <4 kb.• Use PLATINUM Pfx DNA Polymerase or PLATINUM Taq DNA Polymerase High Fidelity for products <12 kb.• Use ELONGASE® Enzyme Mix for products >12 kb.• For one-step RT-PCR, use Taq DNA Polymerase or PLATINUM Taq DNA Polymerase for products <3.5 kb; and use PLATINUM Taq
DNA Polymerase High Fidelity for products <9 kb.
TABLE 3. Comparison of two-step and one-step RT-PCR.
100
bp
DNA
Ladd
er
1 2 3 4 5 6
–353 bp
FIGURE 9. Sensitivity of one-step RT-PCR. A β-actin fragment was amplified from 0, 0.1, 1, 10, 102, and 103 pg of total HeLa RNA(lanes 1 to 6, respectively) using the SUPERSCRIPT™ One-Step RT-PCRSystem. Reactions were incubated at 50°C for 30 min; 94°C for 2 min; then 40 cycles of 94°C for 15 s and 55°C for 30 s and 68°Cfor 90 s; followed by 68°C for 5 min. Reactions contained 200 nM ofsense and antisense primer.
C H A P T E R 2C H A P T E R 2C H A P T E R 2C H A P T E R 2C H A P T E R 2
500 bp–

Priming cDNA Synthesis
First-strand cDNA synthesis reactionsmay be primed using three differentmethods. The relative specificity of
each method influences the amount and thevariety of the cDNA synthesized.
Random primers are the least specific ofthe three methods. The primers anneal tomultiple sites along the entire transcript to generate short, partial-length cDNAs. This method is often used to capture 5´ endsequences and make cDNA from RNA templates with regions of secondary structureor pause sites that reverse transcriptases can-not copy (2,3). To maximize the size of thecDNA, the ratio of primers to RNA may needto be determined empirically for each RNApreparation. The starting concentration rangeis 50 to 250 ng of random primers per 20 µlreaction. Since the majority of the cDNAsynthesized from total RNA with randomprimers is ribosomal, poly(A)+ selected RNAis often used as the template.
Oligo(dT) priming is more specific thanrandom primers. It hybridizes to the poly(A)tails found at the 3´ ends of most eukaryoticmRNAs (23). Since poly(A)+ RNA constitutesapproximately 1% to 2% of a total RNA pop-ulation, the amount and complexity of cDNAis considerably less than when randomprimers are used. Because of its higher speci-ficity, oligo(dT) priming generally does notrequire optimization of the primer-to-RNAratio or poly(A)+ selection. The use of 0.5 µgof oligo(dT) primer per 20 µl reaction is recommended. Oligo(dT)12-18 is suitable formany RT-PCR applications. Oligo(dT)20 isprovided with the THERMOSCRIPT RT-PCRSystem, since it has greater thermostabilityfor use at higher RT incubation temperatures.
A gene-specific primer (GSP) is the mostspecific primer for the RT step. GSPs areantisense oligonucleotides that hybridize to
specific RNA target sequences, instead ofannealing to entire RNA populations as withrandom primers or oligo(dT). The samerules for designing PCR primers are appliedto the design of the GSP for the RT reaction(see chapter 5). A GSP can be the samesequence as the amplification primer whichanneals closest to the 3´ end of the message;or a GSP can be designed to anneal down-stream of the reverse amplification primer.Some targets require the design of morethan one antisense GSP for successful RT-PCR, since secondary structure of the RNAtarget may prevent primer binding. The useof 1 pmole antisense GSP in a 20-µl first-strand reaction is recommended.
Higher RT Incubation TemperaturesTo take full advantage of the specificity provided by GSPs, use an RT with greaterthermostability. Thermostable RTs can beincubated at higher temperatures to increasereaction stringency (17,18). For example, if aGSP has an annealing temperature of 55°C,the specificity conferred by the GSP is notfully utilized if the RT reaction is performedwith AMV RT or M-MLV RT under lowstringency at 37°C. However, SUPERSCRIPT
II RT and THERMOSCRIPT RT allow for reaction temperatures of 50°C and greater(table 2, page 5), which can eliminate the nonspecific products generated at lower temperatures (figure 10). When maximizingspecificity, add the RNA/primer mix directlyfrom the 65°C denaturation temperature tothe RT incubation temperature and add pre-warmed 2X reaction mix (cDNA synthesishot start). This helps prevent intramolecularbase pairing that can occur at lower temper-atures. Using a thermal cycler can simplifythe multiple temperature shifts required forRT-PCR.
Minimizing Contaminating Genomic DNAOne potential difficulty encountered with RT-PCR is genomic DNA contamination ofRNA. Using a good RNA isolation tech-nique, such as TRIZOL Reagent, minimizesthe amount of contaminating genomic DNAin the RNA preparation. To avoid generatingproducts from genomic DNA, remove the contaminating DNA by treating RNAwith DNase I, Amplification Grade beforereverse transcription. To terminate theDNase I digestion, incubate the sample at65°C for 10 min in the presence of 2.0 mMEDTA. The EDTA chelates the magnesiumand prevents magnesium dependent hydrol-ysis of the RNA that can occur at highertemperatures.
To distinguish amplified cDNA fromamplified contaminating genomic DNA,design primers that anneal in separateexons. The PCR products derived fromcDNA will be shorter than products ampli-fied from contaminating genomic DNA. Inaddition, perform a control experiment with-out RT for each RNA template to determinewhether a given fragment is of genomicDNA or cDNA origin. Products generated inthe absence of RT are of genomic origin.
I M P R O V I N G R T - P C R S P E C I F I C I T Y 7
Improving RT-PCR Specificity
C H A P T E R 3C H A P T E R 3C H A P T E R 3C H A P T E R 3C H A P T E R 3
50°C 55°C 60°C1 Kb
PLU
SDN
A LA
DDER
™
– 2 kb
FIGURE 10. Effect of RT reac-tion temperature on RT-PCRspecificity. cDNA was synthe-sized from 1 µg of HeLa RNAwith THERMOSCRIPT™ RT and aGSP designed to anneal to the human DNA polymerase εmRNA. THERMOSCRIPT RT wasadded to a prewarmed reactionmixture and 35 cycles of PCRwas performed using one tenthof the cDNA with PLATINUM®TaqDNA Polymerase.

5´ and 3´ RACE
Rapid amplification of cDNA Ends(RACE) is a procedure to captureunknown sequences at either the 5´
or 3´ end of a transcript. Unlike conventionalRT-PCR, which employs two sequence-specific primers, RACE uses one sequence-specific primer and either the poly(A) tail ofmRNAs (3´ RACE) or a homopolymeric tailadded to cDNA ends (5´ RACE) (figure 11).RACE has been used for the amplificationand cloning of rare mRNAs (23-25). RACEproducts can be cloned, directly sequenced,used to prepare probes, or combined to gener-ate full-length cDNA (25-27). One method forjoining 5´ and 3´ RACE products is to use the sequence information generated by 5´ and 3´ RACE to design new primers whichwill amplify the entire cDNA sequence (26).The use of RNase H– RT and high-fidelitythermostable polymerases allows higher fidelity amplifications of longer sequences to generate full-length cDNA clones.
5´ RACE is more challenging and less specific than RT-PCR applications, since only
one of the amplification primers is gene-specific. 5´ RACE products may be a singleproduct, multiple products, or even a smearwith no distinguishable products (figure 12)
(28). The quality of the result depends on thespecificity of the GSPs used in first-strandsynthesis and amplification, the specificity ofthe anchor primer during amplification, thecomplexity and abundance of the target, andthe length of the product. Amplification withnested primers (up to three rounds of nestedamplification) and using size-selected ampli-fied product as the target in successive roundsof amplification increases the specificity of 5´RACE (see nested PCR discussion on page13).Increasing the RT incubation temperature andPCR annealing temperature, as well as de-creasing the magnesium concentration in theamplification reaction, can enhance specificity.
5´ RACE sensitivity is affected by the RTused and secondary structure at the 3´-end ofthe cDNA that may inhibit cDNA tailing.Incomplete cDNA synthesis lowers the yieldof full-length product and contributes to the
smearing pattern observedwith some targets since shortercDNAs are tailed and ampli-fied. Since SUPERSCRIPT II RTgenerates more full-lengthcDNA, it can increase thelevel of detection of 5´ endsequence, especially for tran-scripts >1 kb (28). Double-stranded 3´ termini and hairpinstructures impair tailing ofcDNA by decreasing the availability of the 3´-OH fortailing. The initial incubationof the cDNA at 94°C followedby chilling on ice helps to disrupt secondary structure.Some difficult targets mayrequire the addition of DMSOfor effective tailing (figure 12)
(28). The tailing enzyme, ter-
minal deoxynucleotidyl transferase is tolerantof up to 20% DMSO.
If no products are visible or if only asmear is visible after nested amplification, aSouthern blot can be used to detect products.This requires internal sequence informationto use as a probe.
Quantifying mRNA ExpressionQuantifying mRNA expression is challengingRT-PCR, but offers advantages over tradi-tional RNA analysis methods. RT-PCR ismore sensitive than Northern blot or ribo-nuclease protection analysis and requires lessRNA and sequence information. However,RT-PCR involves two enzymatic steps thatcan contribute to variability in the amount ofRT-PCR product. The amount of RNA con-verted to cDNA affects yield, but the majordifficulty in quantitative PCR is the expo-nential nature of PCR, in which small vari-ations between samples translates to largedifferences in product yield(29). Two popular
8 R T - P C R A P P L I C A T I O N S
C H A P T E R 4C H A P T E R 4C H A P T E R 4C H A P T E R 4C H A P T E R 4
RT-PCR Applications
FIGURE 11. Summary of the 5´ RACE procedure.
Figure 11
5´ mRNA
GSP1
(A)n
(A)n5´3´
3´
3´CC...CC
3´CC...CC5´ GI ... IG
Anneal first strandprimer, GSP1,to mRNA
GSP2
Copy mRNA into cDNA withSUPERSCRIPTTM II RT
Degrade RNA with RNase Mix
Purify cDNA withGLASSMAX® SpinCartridge
Tail purified cDNA with dCTP and TdT
PCR amplify dC-tailed cDNA using theAbridged Anchor Primer and nested GSP2.
Reamplify primaryPCR product using AUAP, or UAP, and nested GSP
5´
5´
5´
Abridged Anchor Primer
nestedGSP
5´3´
UAP
AUAP
5´ GI ... IG3´ CC...CC
FIGURE 12. 5´ RACE. cDNA wassynthesized from 5 µg HeLa totalRNA with SUPERSCRIPT™ II RT at45°C. 10 µl of purified cDNA wastailed in the presence of 10%DMSO. 1 µl of the tailing reactionwas directly amplified for 40 cycleswith primers for human tuberoussclerosis and ELONGASE® EnzymeMix (primary PCR 2.8 kb; lane 1). A10 µl gel plug was removed aroundthe 2.8-kb band. The DNA waseluted in 50 µl of TE. Nested PCR (30cycles) was performed using 1 µl ofsize-selected PCR product (nestedPCR 2.7 kb; lane 2). The gel isstained with SYBR® Green I.
–kb
––––2.8–2.7
1 2 1 Kb
DN
A La
dder
C H A P T E R 4C H A P T E R 4C H A P T E R 4C H A P T E R 4C H A P T E R 4
R T - P C R A P P L I C A T I O N S 9
methods for quantifying mRNA abundanceare competitive, quantitative PCR and real-time PCR (4,30).
In competitive PCR, an exogenous RNAtranscript (internal standard RNA) is addedbefore the RT step to control for sample-to-sample variation. The internal standard RNAis reverse transcribed and amplified with the target to control for differences in the efficiency of cDNA conversion and ampli-fication. Quantitation is performed by co-amplification of the specific target sequencetogether with known concentrations of theinternal standard RNA. The abundance of thetarget is determined by comparing the signalobtained for the internal standard to the signalfor the target.
The internal standard RNA shares thesame primer recognition sites with the tar-get, but is a different size. Several methodsexist to generate internal standards (31). Oneof the simplest methods involves installingthe primer recognition sites on nonspecificspacer DNA by PCR (31). This product canbe cloned into a vector carrying the T7 orSP6 RNA polymerase promoter, or the RNApromoter sequences can be included on the forward primer in order to install thesequences by PCR (32). Then, RNA standardscan be generated by in vitro transcription.Competitive, quantitative PCR can be per-formed in one-step RT-PCR using a GSP orperformed in a two-step reaction using aGSP or oligo(dT). Oligo(dT) sequence canbe installed by adding poly(dT) sequencesto the reverse primer (32).
Competitive RT-PCR is an end-pointanalysis that requires the amplification efficiency of the target and the internal stan-dard to be equal. Some prior knowledge of the amount of starting target is necessary to establish a concentration range for the internal standard. For accurate quantitation,reactions containing more standard than tar-get and less standard than target are required.
Real-time RT-PCR is non-competitive and detects the product as it is being formed.Several probes can be used to assay the accu-mulation of product including fluorogenic 5´ nuclease probes and Molecular Beacons(33,34). The first method is based on the 5´nuclease activity of Taq DNA polymerase,which is used to cleave a hybridization probeat the branch point during extension (33). Theassay uses probes with two fluorescent labels,one dye serves as a reporter and the otherquenches its signal until the probe is cleaved.
Molecular Beacons contain a 5´ fluoro-phore and 3´ quencher. These probes aredesigned with a hairpin structure bringingthe fluorophore and quencher in close prox-imity to quench fluorescence (34). The stemstructure is composed of 5 to 7 nucleotidesand is GC rich. The outer loop structure iscomposed of 15 to 30 nucleotides and iscomplementary to the target sequence. Whenthe target sequence is present, themolecular beacon hybridizes tothe target, relaxing the hairpinstructure and allowing fluores-cence.
For both probes, fluorescentmeasurements are made in realtime during each cycle of PCRusing a thermal cycler coupled toan optical excitation and detectiondevice. The amount of fluores-cence released is proportional tothe amount of PCR product. Thecycle at which the amount ofreleased fluorescence crosses anestablished baseline fluorescenceis known as the threshold cycle or CT. The CT value is inverselyproportional to the amount ofstarting target. Thus, high-copy-number samples will have low CT
values and low-copy-numbersamples will have high CT values(figure 13).
For absolute quantification of messages, astandard curve is generated using an in vitrotranscribed RNA. The CT value is determinedfor different concentrations of the in vitrotranscribed RNA. Then the CT values areplotted against the RNA concentrations togenerate a standard curve for quantifying thelevel of expression in unknown samples(figure 13, panel B).
Real-time RT-PCR offers advantages over competitive RT-PCR. Fluorescencemonitoring is highly sensitive and highlyaccurate with a linear-dose response over awide range of target concentrations (figure 13)
(30). Samples do not require post-amplifi-cation analysis and little prior knowledge oftarget abundance is required. Use a two-stepRT-PCR strategy for quantifying multiplemessages in a single RNA sample and use aone-step strategy for added conveniencewhen processing large numbers of samples.
-400
-200
0
200
400
600
800
1,000
1,200
1,400
1,600
1,800
2,000
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40 42 44
0
5
10
15
20
25
30
35
40
45
50
105104103102101 106
Slope: -3.581Y-Intercept: 41.56CorrelationCoefficient: 0.993
500 ng50 ng5 ng500 pg
50 pg5 pg0
Total HeLa RNA
Fluorescence Threshold
FIGURE 13. Real-time PCR quantitation of retinoblastoma mRNA. RT-PCR ofduplicate samples of total HeLa RNA was performed with the PLATINUM® QuantitativeRT-PCR THERMOSCRIPT™ One-Step System (cDNA synthesis was performed at 60°C)and detected using a FAM-labeled Molecular Beacon probe (400 nM) on an ABIPrism® 7700. Panel A. Relative fluorescence intensity of duplicate samples duringPCR. Panel B. Standard curve.
Rela
tive
Fluo
resc
ence
Thre
shol
d Cy
cle
(CT)
Cycle Number
Starting Quantity (pg total HeLa RNA)
Panel A
Panel B

Primer Design
Careful primer design is one of themost important aspects of PCR. Theideal primer pair anneals to unique
sequences that flank the target and not to other sequences in the sample. Poorlydesigned primers may amplify other, non-target sequences. The following guidelinesdescribe the desirable characteristics of aprimer sequence that increase specificity:• Typical primers are 18 to 24 nucleotides in
length. The primer needs to be long enoughfor the sequence to be unique and to reducethe probability of the sequence being foundat non-target sites. However, primers greaterthan 24 nucleotides do not confer greaterspecificity. Longer sequences can hybridizewith some mismatching, which decreasesspecificity, and hybridize slower than shortersequences, which may decrease yield (2).
• Select primers that are 40% to 60% GC ormirror the GC content of the template.
• Design primers with G or C residues in the5´ and central regions. This increases theprimer’s stability and confers hybridizationstability with the target sequence.
• Avoid complementary sequences at the 3´ end of primer pairs. This prevents ampli-fication from the primers themselves toform primer-dimers.
• Avoid a GC-rich 3´ end. Design primers to contain 3 As or Ts within the last 5nucleotides (35).
• Avoid mismatches at the 3´ end. The last 3´nucleotide needs to anneal to the templatefor the polymerase to catalyze extension(36).
• Avoid sequences with the potential to forminternal secondary structure. This desta-bilizes primer annealing.Additional sequences that are not present
on the target, such as restriction sites and promoter sequences, can be added to the 5´end of a primer without affecting specificity.These sequences are not included when esti-mating the Tm of a primer. However, check
these regions for complementarity and internalsecondary structures.
Sometimes only limited sequence infor-mation is available for primer design. Forexample, if only the amino acid sequence is known, degenerate primers are designed.Degenerate primers are a mix of differentsequences representing all possible bases coding for a single aminoacid. To improve specificity,minimize the degeneracy con-sulting codon usage tables forbase preference in differentorganisms (2). In addition,deoxyinosine may be used inplaces where more than onebase is possible. Inosine pairswith all bases and will lower the annealingtemperature of the primer. Do not includedegenerate bases at the 3´ end of the primer,since annealing of the last three bases on the3´ end can be enough to initiate PCR at thewrong sites. Use higher primer concentrations(1 µM to 3 µM) because many of the primersin the degenerate mixture are not specific forthe target (37).
Primer Annealing TemperatureAnother important parameter for primers isthe melting temperature (Tm). This is thetemperature at which 50% of the primer andits complementary sequence are present in aduplex DNA molecule. The Tm is necessaryto establish an annealing temperature forPCR. Ideally, the annealing temperature is
low enough to guarantee efficient annealingof the primer to the target, but high enoughto minimize nonspecific binding. Reason-able annealing temperatures range from55°C to 70°C. Annealing temperatures aregenerally set about 5°C below the Tm of theprimers.
There are several formulas for estimatingthe Tm (38-40). Table 4 lists two of the com-monly used formulas for determining aprimer’s Tm. The first formula was derived forhybridization in high salt (1 M) and is validfor primers <18 bases. The second formulaestimates Tm based on GC content and saltconcentration. The most reliable method fordetermining the Tm of a primer is the nearest-neighbor analysis (38). This method predictsthe hybridization stability of a primer from
the primary sequence and the identity of theneighboring bases. Most computer programsuse nearest-neighbor analysis.
The Tm can vary significantly dependingon the formula used and the primer sequence.Since most formulas provide an estimatedTm value, the annealing temperature is only astarting point. Specificity can be increasedby analyzing several reactions with increas-ingly higher annealing temperatures. Beginat 5°C below the estimated Tm and increasethe annealing temperature in 2°C incre-ments. Higher annealing temperatures canreduce the formation of both primer-dimerand nonspecific products. For best results,both primers need to have a similar Tm.Primer pairs whose Tm varies by more than5°C exhibit greater mispriming due to the
10 I M P R O V I N G P C R S P E C I F I C I T Y
C H A P T E R 5C H A P T E R 5C H A P T E R 5C H A P T E R 5C H A P T E R 5
Improving PCR Specificity
Table 4
Simple Formula (39) (valid for primers <18 bases)
Tm = 4°C × (G + C) + 2°C × (A + T)
Tm for Oligonucleotides (40) (dependent on salt concentration)
Tm = 81.5 + 16.6 × (log10[Na+]) + 0.41 × (%G + C) – 675/n
Where n = number of bases and [Na+] = monovalent (Na+ or K+) cations
TABLE 4. Formulas to estimate Tm.
Careful primer design is one of the most important
aspects of PCR. Poorly designedprimers may amplify other,
non-target sequences.

use of lower annealing temperatures duringcycling. If two primers have a different Tms,set the annealing temperature 5°C below the lowest Tm. Or to increase specificity,perform 5 cycles at the annealing temper-ature established by the primer with thehigher Tm and then the remaining cycles atthe annealing temperature established bylower Tm. This allows some copies of the target to be generated under more stringentconditions.
Touchdown PCRTouchdown PCR increases sensitivity byusing stringent annealing conditions duringthe early cycles of PCR (41). Cycling beginswith annealing temperature approximately5°C above the estimated Tm. It is incremen-tally decreased by 1°C to 2°C every cycleuntil the annealing temperature is about 5°C below the Tm. Only targets with thegreatest homology will be amplified. Theseproducts will continue to be amplified andwill compete out nonspecific productsamplified in later cycles. Touchdown PCR is useful in applications where the degree ofhomology between the target sequences andthe primer are unknown, such as AFLP®
DNA fingerprinting.
Primer ConcentrationPrimer concentration can affect specificity.Optimal primer concentration typically fallsbetween 0.1 to 0.5 µM. Higher concen-trations of primer may result in the amplifi-cation of nonspecific products.
To determine primer concentration, readthe optical density at 260 nm (OD260). Then,using Beers Law (formula 1) calculate theconcentration by using the absorbance andthe reciprocal of the millimolar extinctioncoefficient (nmol/OD). The millimolarextinction coefficient can be calculatedusing formula 2 (42). Unlike large double-stranded DNA molecules where an averaged
extinction coefficient can be used, the use ofthe extinction coefficient calculated for the primer is essential to accurately deter-mine the concentration (42). This is becauseprimers are short and the base compositioncan vary greatly. Both the extinction coeffi-cient (OD/µmol) and the reciprocal of theextinction coefficient (µmol/OD) are pro-vided on the GIBCO BRL® Custom Primercertificate of analysis. In addition, do notestimate primer concentration on ethidiumbromide-stained gels using oligonucleotidesas standards, since the staining capability ofthe standard and the primer can vary greatlydepending on their sequence (43).
Primer Purity and StabilityStandard purity Custom Primers are suffi-cient for most PCR applications (table 5).With Life Technologies® PARALLEL ARRAY
SYNTHESIS™ technology, desalting is not nec-essary. The benzoyl and isobutyryl groupsremoved by desalting are found in loweramounts with PARALLEL ARRAY SYNTHESIS
technology, compared to other methods, andthus do not interfere with PCR. Some appli-cations require purification to remove anyless than full-length sequences generatedduring synthesis. These truncated sequencesare generated because DNA synthesis chem-istry is not 100% efficient. It is a cyclicalprocess in which DNA is synthesized 3´→5´using chemical reactions that are repeatedfor each base added. Failures can occur duringany cycle. Longer primers, especially primers>50 bases, have a greater portion of truncatedsequences and may require purification.
Primer yield is affected by the efficiencyof the synthesis chemistry and by the methodof purification. Life Technologies guaranteesthe total oligonucleotide yield as a minimumnumber of OD units (table 6, page 12). CustomPrimers are shipped lyophilized. It is best toreconstitute primers in TE [10 mM Tris-HCl(pH 8.0), 1 mM EDTA] to bring the finalconcentration to 100 µM. TE is better thandeionized water since the pH of the water isoften slightly acidic and can cause hydrolysisof the oligonucleotide.
The stability of the primerdepends on storage conditions.Store lyophilized and reconsti-tuted primers at –20°C. Primersreconstituted in TE at ≥10 µMare stable for >6 months at–20°C, but only <1 week whenstored at room temperature(15°C to 30°C). Lyophilizedprimers are stable >1 year at–20°C, and up to 2 monthswhen stored at room tempera-ture (15°C to 30°C).
I M P R O V I N G P C R S P E C I F I C I T Y 11
C H A P T E R 5C H A P T E R 5C H A P T E R 5C H A P T E R 5C H A P T E R 5
Table 5
Application Minimum Suggested Purity
PCR Standard
First-strand cDNA synthesis for RT-PCR Standard
AFLP® technology Standard
PCR using primers with 5´ sequences Cartridge(restriction endonuclease sites, RNA polymerase promoter sites, etc)
PCR primers >50 bases PAGE
Cycle sequencing Standard
Isothermal sequencing Desalted
Site-directed mutagenesis Cartridge
CFLP™ technology Desalted
TABLE 5. Minimum recommended primer purity for PCR applications.
Formula 1
Concentration= A260 × dilution factor × the reciprocal of theextinction coefficient × conversion factors
Example: To calculate the concentration of an oligonucleotidein 1 ml, measure the A260 of 10 µl of the oligonucleotide in 990 µl of water (1:100 dilution). If the A260 = 0.14 OD and the oligonucleotide has a reciprocal extinction coefficient of 4.9 nmol/OD, the concentration would be calculated as follows:
Concentration = 0.14 OD × 100 × 4.9 nmol × 1 µmol × 103 ml ml OD 103 nmol L
= 69 µM
Formula 2
Millimolar Extinction Coefficient of Oligonucleotide =
A(15.2) + C(7.05) + G(12.01) + T(8.4) at pH 8.0
Where A, C, G, and T are the number of dAs, dCs, dGs,and dTs (35).

12 I M P R O V I N G P C R S P E C I F I C I T Y
C H A P T E R 5C H A P T E R 5C H A P T E R 5C H A P T E R 5C H A P T E R 5
Hot StartHot-start PCR is one of the most importantmethods, in addition to good primer design,for increasing PCR specificity. Even thoughthe optimal extension temperature for TaqDNA polymerase is approximately 72°C, thepolymerase has activity at room temperature(44). Thus, nonspecific products are oftengenerated during PCR set-up and at the startof thermal cycling when reactions are brieflyincubated at temperatures well below theannealing temperature (45,46). Once thesenonspecific products are formed they can beefficiently amplified. Hot-start PCR is partic-ularly effective when the sites available fordesigning primers are limited due to thelocation of genetic elements, such as site-directed mutagenesis, expression cloning, orthe construction and manipulation of geneticelements for DNA engineering.
A popular method that limits Taq DNApolymerase activity is to set-up amplificationreactions on ice and place them into a pre-heated thermal cycler. This method is simpleand inexpensive, but does not completely
inactivate enzyme activity and therefore doesnot completely eliminate the amplification ofnonspecific products.
Hot start delays DNA synthesis by with-holding one of the essential componentsuntil the thermal cycler reaches the denatur-
ation temperature. Most manual hot-startmethods involve the delayed addition of TaqDNA polymerase, which can be cumber-some especially for high-throughput applica-tions (46). Other hot-start methods use waxbarriers to encapsulate an essential compo-nent, such as magnesium or enzyme, or tophysically separate reaction components,such as template and buffer, from each other.Melting of the wax during thermal cyclingreleases and mixes all of the componentstogether. Like manual hot-start methods,wax barrier methods can be cumbersome,prone to contamination and unreliable inhigh-throughput applications.
PLATINUM DNA Polymerases are con-venient and highly-effective for automatic hot-start PCR (figure 14). PLATINUM TaqDNA Polymerase is comprised of recombi-nant Taq DNA polymerase complexed withmonoclonal antibodies to Taq DNA poly-merase (47). The antibodies prevent enzymeactivity during PCR set-up and even duringprolonged incubations at room temperature.
Taq DNA polymerase is released into thereaction after incubation at 94°C during thedenaturation step, restoring full polymeraseactivity. In comparison to chemically modi-fied Taq DNA polymerase for hot-start PCR,PLATINUM enzymes do not require pro-
longed incubation at 94°C (10 to 15 min) toactivate the polymerase. Greater than 90%of Taq DNA polymerase activity is restoredin 2 min at 94°C with PLATINUM Taq DNAPolymerase (48).
Magnesium ConcentrationMagnesium affects several aspects of PCRincluding DNA polymerase activity, whichcan affect yield; and primer annealing, whichcan affect specificity. The dNTPs and template bind magnesium and reduce theamount of free magnesium available forenzyme activity. The optimal magnesiumconcentration varies for each primer pair andtemplate, but the starting concentration intypical PCR containing 200 µM dNTPs is1.5 mM (Note: for real-time quantitativePCR use 3 to 5 mM magnesium with fluo-rescent probes) (2). Higher concentrations offree magnesium can result in greater yield,but can also increase nonspecific amplifi-cation and reduce fidelity (49,50). To deter-mine the best concentration, perform amagnesium titration using from 1 mM to 3 mM in 0.5 mM increments. To reduce the
Improving PCR Specificity (continued)
4.1 kb–
–114 bp
1 2 31 2 3100
bp
DNA
Ladd
er
1 Kb
DN
A La
dder
FIGURE 14. Improved specificity with PLATINUM® Taq DNA Poly-merase. Panel A. Detection of cloned HIV DNA in human genomicDNA. 1,000 copies of plasmid DNA with the HIV gag region wasmixed with 100 ng of genomic DNA and amplified with primers to thegag region. Lane 1. Taq DNA polymerase with room temperatureassembly. Lane 2. Taq DNA polymerase with manual hot start byaddition of enzyme at 94°C. Lane 3. PLATINUM Taq DNA Polymerasewith room temperature assembly. Panel B. Amplification of 4.1 kb ofhuman β-globin from 100 ng of human genomic DNA. Lane 1. TaqDNA polymerase with room temperature assembly. Lane 2.Taq DNApolymerase with assembly on ice and placed in a preheated (80°C)thermal cycler. Lane 3. PLATINUMTaq DNA Polymerase with room tem-perature assembly.
Panel A Panel B
Table 6
Minimum Yield (OD) for Different Primer Purities*Standard/
Number of Bases Synthesis Scale Desalted Cartridge HPLC PAGE
<20 50 nmole 2 2 NA NA200 nmole 8 8 3 1
1 µmole 20 20 10 310 µmole 200 NA 100 30
≥20 50 nmole 5 2 NA NA200 nmole 20 10 3 1
1 µmole 50 25 15 510 µmole 500 NA 150 50
Note: For primers >50 bases, PAGE purification is recommended and HPLC is not an option.NA is not available.
*These yields are seen with the following 5´ modifications: biotin, fluorescein (FITC), rhodamine, primary amines (NH2), phosphate (PO4),HEX, TET, FAM, and phosphorothioates (S-Oligos). Other modifications may have slightly lower yields.
TABLE 6. Minimum oligonucleotide yield for the various purification methods.

need for magnesium optimization, usePLATINUM Taq DNA Polymerase. PLATINUM
Taq DNA Polymerase functions over abroader range of magnesium concentrationthan Taq DNA polymerase so less optimi-zation is required (figure 15) (47).
Additives to Enhance PCROptimization of the annealing temperature,primer design, and magnesium concentra-tions is adequate to achieve high specificityamplification of many targets. However,some targets, including those with a high-GC content, require additional measures.Additives that affect DNA melting temper-ature provide another method for improvingproduct specificity and yield (figure 16).Complete denaturation of the template is
required to obtain the best results. Addi-tionally, secondary structure can preventprimer binding and enzymatic elongation.PCR additives, including formamide,DMSO, glycerol, betaine, and PCRX
Enhancer Solution can enhance amplifi-cation (51-53). Their proposed mechanism isto lower the melting temperature, therebyaiding primer annealing and helping theDNA polymerase extend through regions of secondary structure (53). PCRX Solutionhas additional benefits. It requires less
magnesium optimization and works withPLATINUM Taq DNA Polymerase (figure 16)
and PLATINUM Pfx DNA Polymerase. Thus,combining PLATINUM technology and addi-tives enhances specificity while minimizingthe need for the third approach-magnesiumoptimization. For best results, optimize theconcentration of the additives (figure 17),especially DMSO, formamide, and glycerolthat inhibit Taq DNA polymerase (46,52).
Nested PCRSequential rounds of amplification usingnested primers can improve specificity andsensitivity (3). The first round is a standardamplification of 15 to 20 cycles. A smallaliquot of the initial amplification is diluted1:100 to 1:1,000 and added to the second
round of amplifica-tion with 15 to 20cycles. Alternatively,the initial amplifi-cation product can be size selected bygel purification. Two different primer setsare used for the tworounds of amplifi-cation. The secondamplification uses anested set of primersthat bind to the targetjust inside the first
set of primers. The chance of amplifyingmultiple targets is reduced with nested PCR since fewer targets will be comple-mentary to both sets of primers; whereasperforming the same total number of cycles(30 to 40) with the same set of primersoften amplifies nonspecific targets. NestedPCR can increase the sensitivity from limitedamounts of target, such as amplifying a raremessage, and increase the specificity ofmore challenging PCR applications such as 5´ RACE.
C H A P T E R 5C H A P T E R 5C H A P T E R 5C H A P T E R 5C H A P T E R 5
FIGURE 16. Improved specificity with PCRx Enhancer Solution.A 156-bp fragment (62% GC content), was amplified from 100 ng ofhuman genomic DNA using PLATINUM® Taq DNA Polymerase. MgCl2was titrated (1.0, 1.5, 2.0, 2.5 mM, lanes 1 to 4 respectively) using:standard PCR buffer (panel A) or PCRx Amplification Buffer with 1XPCRx Enhancer Solution (panel B). Cycling was 35 cycles of 94°C for30 s; 60°C for 30 s, and 68°C for 1 min.
–156 bp62% GC
Panel A Panel B
0X 1X 2X 2.5X 3X 3.5X 4X
–149 bp78% GC
FIGURE 17. Titration of PCRx Enhancer Solution. A 149-bp (78%GC) trinucleotide repeat-containing sequence was amplified from100 ng of human genomic DNA with PLATINUM® Taq DNA Polymerasein 1X PCRx Amplification Buffer with varying amounts of PCRxEnhancer Solution (0 to 4X).
1 2 3 4 1 2 3 4
FIGURE 15. Broader magnesium range with PLATINUM® Taq DNA Polymerase. A 2.8-kb region of thehuman β-globin gene was amplified from 100 ng of human genomic DNA. Panel A.Taq DNA polymerasewith assembly on ice and placed in a preheated (80°C) thermal cycler. Panel B. PLATINUM Taq DNA Poly-merase with room temperature assembly.
100
bpDN
A La
dder
I M P R O V I N G P C R S P E C I F I C I T Y 13
1.0 1.4 1.8 2.2 2.6 3.0
(mM MgCl2)
1.0 1.4 1.8 2.2 2.6 3.0
(mM MgCl2)
Panel A Panel B
2.8 kb–

14 I N C R E A S I N G P C R S E N S I T I V I T Y
C H A P T E R 6C H A P T E R 6C H A P T E R 6C H A P T E R 6C H A P T E R 6
Increasing PCR Sensitivity
Template Quality
Template quality affects product yield. Anumber of contaminants found in DNApreparations can inhibit PCR (46).
Reagents such as SDS, which are used in stan-dard genomic DNA preparations, can inhibitamplification reactions at concentrations as low as 0.01%. Newer methods for isolation of high-quality genomic DNA include DNAZOL®
Reagents (53), which are guanidine-detergentlysing solutions, and the FTA® Products,
which is a matrix-bound method for the storage and purification of DNA fromblood and other biological samples (table 7) (55).
Template ConcentrationThe amount of starting template is important for obtaining good product yields. For mostamplifications, 104 to 106 starting target molecules allows sufficient amplification to visualize the product on an ethidium bro-mide-stained gel (2). The optimal amount of template required depends on the size of thegenome (table 8) (56). For example, 100 ng to1 µg of human genomic DNA, correlating to 3 × 104 to 3 × 105 molecules, is sufficient todetect a PCR product from a single-copy gene.For plasmid DNA, which is much smaller,the amount of DNA added to PCR is in thepicogram range.
Enzyme ChoiceIn addition to using high-quality templateDNA, the choice of polymerase also affectsyield (see table on page 22). PLATINUM poly-merases provide better yield than other poly-merases because they prevent amplificationof nonspecific product during PCR set-up(figure 18). For high-sensitivity PCR of longproducts (up to 12 kb), choose an enzymemix, preferably in a PLATINUM format, suchas PLATINUM Taq DNA Polymerase HighFidelity. This enzyme combines the benefits ofPLATINUM technology with those of enzymemixes (Taq DNA Polymerase mixed with aproofreading polymerase).
Table 8
Target Molecules/µg Amount of DNA (µg) Genomic DNA Size (bp)* of Genomic DNA for ~105 Molecules
E. coli 4.7 × 106 1.8 × 108 0.001
Saccharomyces cerevisiae 2.0 × 107 4.5 × 107 0.01
Arabidopsis thaliana 7.0 × 107 1.3 × 107 0.01
Drosophila melanogaster 1.6 × 108 6.6 × 105 0.5
Homo sapiens 2.8 × 109 3.2 × 105 1.0
Xenopus laevis 2.9 × 109 3.1 × 105 1.0
Mus musculus 3.3 × 109 2.7 × 105 1.0
Zea mays 1.5 × 1010 6.0 × 104 2.0
pUC 18 plasmid DNA 2.69 × 103 3.4 × 1011 1 × 10 –6
* Haploid genome size
TABLE 8. Correlation of genome size and number of molecules.
1 Kb
PLU
SDN
A LA
DDER
™
1 2 3 4 5 1 2 3 4 5 1 2 3 4 5
FIGURE 18. Amplification with different thermostable polymerases. Human blood was spotted onFTA® Cards. DNA was amplified directly from 1 to 3 mm punches of washed FTA Cards in 50 µl using TaqDNA polymerase (panel A), PLATINUM® Taq DNA Polymerase (panel B), and PLATINUM Taq DNA PolymeraseHigh Fidelity (panel C). Amplified products were 4.1, 5.2, 7.5, 8.0, and 8.4 kb in lanes 1 to 5, respectively.
Taq DNA Polymerase PLATINUMTaq DNA Polymerase PLATINUMTaq DNA Polymerase High Fidelity
TABLE 7. Methods for genomic DNA isolation.
Table 7
Method Description
Proteinase K/phenol extraction Classic method
DNAZOL® Reagents Fast, phenol-free DNA isolation
FTA® Products Matrix-bound purification and storage
–kb
–8.4
–4.1

C H A P T E R 7C H A P T E R 7C H A P T E R 7C H A P T E R 7C H A P T E R 7
Enzymes with Proofreading
PCR applications involving cloningand sequence analysis, expression of PCR products, or site-directed
mutagenesis require high-fidelity PCR. Taq DNA polymerase is considered a low-fidelity polymerase since it lacks 3´ to 5´exonuclease (proofreading) activity. The useof thermostable polymerases with 3´ exo-nuclease activity improve fidelity (57,58).However, these polymerases can give loweryields than Taq DNA polymerase. PLATINUM
Pfx DNA Polymerase has significantly betterfidelity than Taq DNA polymerase andoffers the advantage of high yield and speci-ficity of PLATINUM products (figure 19) (59).
Enzyme MixesMixing Taq DNA polymerase with a secondpolymerase with 3´ exonuclease activity provides greater fidelity than Taq DNA polymerase alone and allows for higher yield and amplification of longer templates.The GIBCO BRL high fidelity enzyme mix,PLATINUM Taq DNA Polymerase HighFidelity has 6-times greater fidelity than TaqDNA polymerase alone and can amplify upto 12 kb.
Other ParametersBesides enzyme, high dNTP or magnesiumconcentrations can reduce fidelity. Decreasingthe concentration of dNTPs from 200 µM to 25-50 µM can increase accuracy. If the concentration is not the same for all fournucleotides, the fidelity will be effected. Performing fewer cycles of PCR also can help increase fidelity, since the probability ofa mutation increases with increasing cyclenumber and product length.
Improving Fidelity
1 Kb
PLU
SDN
A LA
DDER
™
1 Kb
PLU
SDN
A LA
DDER
1 Kb
PLU
SDN
A LA
DDER
FIGURE 19. High sensitivity and specificity with PLATINUM® Pfx DNA Polymerase. Genomic DNA (100, 50, 10, 5, 1, and 0 ng, lanes 1 to 6)was amplified with primers for a 1.1-kb fragment of human thrombospondin for 35 cycles. Panel A. 1 unit of PLATINUM Pfx DNA Polymerasewith room temperature set-up. Panel B. 2.5 units of PfuTurbo™ DNA Polymerase with set-up on ice. Panel C. 1 unit of Taq DNA Polymerasewith set-up on ice.
Panel A Panel B Panel C
–1.1 kb
1 2 3 4 5 6 1 2 3 4 5 6 1 2 3 4 5 6
I M P R O V I N G F I D E L I T Y 15

Amplifying Long Targets
When amplifying targets >5 kb, usean enzyme or enzyme mix forlong PCR to obtain the best yield
(see table on page 22 and figure 18 on page 14).Taq DNA polymerase does not efficientlyamplify longer targets (>5 kb), presumablybecause it lacks 3´→5´exonuclease activityand cannot correct dNTP misincorporations(60). The elongation rate from a mismatch isgreatly reduced, which decreases the yield oflonger products. Mixing Taq DNA polymerasewith a thermostable DNA polymerase con-taining 3´→5´ exonuclease activity can allow
amplification of targets up to 30 kb (61).PLATINUM Pfx DNA Polymerase (has 3´→5´exonuclease activity) can also amplify longerproducts (≤12 kb).
In addition to choosing the correct ther-mostable polymerase, the amplification oflonger products requires changes to theextension times, denaturation times, andbuffer pH of the standard protocol. Increaseextension times to 1 min/kb to allow thepolymerase to complete synthesis. Typicallythe extension temperature is lowered to68°C for more effective long PCR (60,61).Since the elongation times are long, up to 20 min for a 20-kb target, a buffer with ahigher initial pH is used. If the pH fallsbelow pH 7.0, the DNA can depurinate. To protect the template against damage,minimize the denaturation time at 94°C to30 s or less during each cycle and limit thepreamplification denaturation time to 94°Cfor ≤1 min. Primers are designed in the samemanner as those used in a standard protocol,with primers 18 to 24 bases giving goodproduct yields (60,61).
Prevention of Carry-Over ContaminationPCR is susceptible to contamination prob-lems because it is a sensitive amplificationtechnique. Small amounts of contaminatingDNA from an exogenous source can beamplified along with the desired template. A common source of contamination occurswhen previously amplified products areintroduced into new amplification reactions.This is called carry-over contamination.Purified DNA from other samples and clonedDNA can also be sources of contamination.
Carry-over contamination can be mini-mized by using good laboratory proceduresduring PCR. Use aerosol-resistant tips to prevent aerosols from reaching the pipette barrel. Designate separate areas for PCRsample set-up and post-amplification analysisand change gloves before preparing new reactions. Always perform a negative control,without template, to check for contami-nation. Use premixed reaction componentsinstead of adding each reagent to individualreactions.
One method to prevent carry-over con-tamination uses uracil DNA glycosylase(UDG) (62). This enzyme (also known asuracil-N-glycosylase or UNG) removesuracil residues from DNA. Substitutingdeoxyuracil for thymine in amplificationreactions allows previously amplified prod-ucts to be distinguished from template DNA.Since the previously amplified products are susceptible to UDG, newly assembled reactions are treated with UDG before PCRto destroy carry-over products.
Purification of PCR Products Residual reaction components includingprimers, nucleotides, and thermostable poly-merases can inhibit cloning, sequencing, andlabeling reactions. Thus, it is often necessaryto purify PCR products before further manip-ulation. Clean-up processes involving multiplephenol:chloroform extractions followed byPEG precipitation (63) or isopropanol pre-
cipitation (2) are effective but time con-suming and can result in product losses. TheCONCERT™ Rapid PCR Purification Systempurifies PCR products from reaction com-ponents in 10 min. It is effective for a widerange of PCR fragments, from 80 bp to 10 kb, and provides efficient recovery of upto 95% of the original sample (figure 20) (64).
Some PCR products may require purifi-cation, from nonspecific PCR products thatcan interfere with cloning or sequencing,by gel electrophoresis. The desired productis purified from the agarose using theCONCERT Gel Extraction System, a silica-based technology for rapid isolation of DNAfragments from gels.
When cloning PCR products by restrictionendonucleases, TAQUENCH™ PCR CloningEnhancer provides an alternative to the clean-up of PCR products. If 3´-recessed termini are generated by digestion, residualTaq DNA polymerase and dNTPs from thePCR can fill in the 3´-recessed termini (65).This lowers the cloning efficiency and gen-erates clones ligated in the improper readingframe. However, the addition of TAQUENCH
Enhancer before restriction digestion mini-mizes Taq DNA polymerase activity for effi-cient cloning (65).
16 O T H E R T H I N G S T O C O N S I D E R
C H A P T E R 8C H A P T E R 8C H A P T E R 8C H A P T E R 8C H A P T E R 8
Other Things to Consider
The Concert Rapid PCR PurificationSystem purifies PCR fragments
from 80 bp to 10 kb.Hi
gh D
NAM
ASS™
Ladd
er
–kb
–3
–0.7
–primers
A B A B1 2
FIGURE 20. Purification of PCR fragments following amplifi-cation. Completed PCRs containing ~1 µg of amplified product werespiked with 1.2 µg of primer (lane 1) or 3.5 µg of primer (lane 2).Primers were 36 to 40 bases. Spiked reactions were purified, andaliquots of the eluate before (lane A) and after (lane B) purificationwere analyzed by agarose electrophoresis.

Multiplex PCR
In multiplex PCR, multiple primer setsare used simultaneously to amplify several different loci. This complex
PCR often results in low product yield andrequires higher concentrations of magnesium(67). A false negative result may be obtained
if reactions are not optimized correctly. Theincreased robustness of PLATINUM Taq DNAPolymerase over a broad range of magne-sium concentrations increases success inmultiplex reactions (figure 21).
Genotyping with Dinucleotide Repeat Markers Determining the size of the amplified dinucle-otide repeat region can be challenging sinceTaq DNA polymerase often adds a nontem-plated nucleotide to PCR products. The frac-tion of products in an amplification reactioncontaining an extra nucleotide is sequenceand primer dependent and can vary greatly.However, PLATINUM GENOTYPE Tsp DNAPolymerase exhibits minimal nontemplatednucleotide addition to help increase the accu-racy of allele size determination (figure 22)
(66). Simply substitute PLATINUM GENOTYPETsp DNA Polymerase for Taq DNA poly-merase in these amplifications.
Tools for Detecting Polymorphisms Amplified Restriction Fragment Polymor-phism (AFLP) technology is a technique forfingerprinting genomic DNA from plantsand microorganisms (figure 23) (68,69). RFLP,AP-PCR, and RAPD are also commonDNA fingerprinting techniques. RFLP is a
hybridization-based methodwhich is time consuming andrequires a large amount ofgenomic DNA. AP-PCR andRAPD are faster, PCR-basedmethods, but both are sensi-tive to reaction conditionswhich can affect the repro-ducibility of the techniques.
AFLP technology com-bines the reliability of RFLPand the convenience of aPCR-based fingerprintingmethod for a more robustand informative method.DNA fingerprints generatedwith AFLP technology canbe used as a tool to identify a
specific DNA sample or to assess the simi-larity between samples (figure 24) (69).
A number of parameters influence AFLPresults, including the number of selectivenucleotides in the primers (69). The numberof amplified bands decreases when the number of selective nucleotides increases.Larger genomes require more selectivenucleotides to amplify an appropriate number of DNAs, since more restrictionfragments are generated from digesting alarger genome.
The template quality can also affectAFLP results. Poor quality template mayinhibit restriction endonucleases, resultingin incomplete digestion and fewer amplifiedbands visible on the gel (69). However, theAFLP technique is tolerant of variation intemplate concentration, with no differenceobserved with amounts of DNA between100 ng to 5 µg.
Figure 21
C H A P T E R 9C H A P T E R 9C H A P T E R 9
P C R A P P L I C A T I O N S 17
C H A P T E R 9C H A P T E R 9
PCR Applications
n
n+1
n+1n
FIGURE 22. Comparison of DNA polymerases in genotyping of dinucleotide repeat markers. Amplification of nga106 locus ofArabidopsis thaliana generated withTaq DNA polymerase (66% n + 1)or PLATINUM® GENOTYPE Tsp DNA Polymerase (0% n + 1).
FIGURE 24. A typical AFLP®
fingerprint. The polymorphism ofsoybean ecotypes was performedusing the AFLP System I and theselective primers EcoR I+ACCand Mse I+CAA. Ecotypes wereHolladay; Morgan; Hytest; Essex;Bass; T135; P88; PI88788; P83;PI83495; P90; PI90763; lanes 1to 9, respectively.
2,500–2,000–1,500–1,000–
500–0–
2,500–2,000–1,500–1,000–
500–0–
150 155 160
Taq DNA Polymerase(66% n + 1)
PLATINUM GENOTYPE TspDNA Polymerase
(0% n + 1)M
FIGURE 21. PLATINUM® Taq DNA Polymerase provides greater sensitivity for multiplexPCR. Multiplex amplification of the dystrophin gene was performed with 5 different primersets from human genomic DNA withTaq DNA polymerase (panel A) or PLATINUM Taq DNAPolymerase (panel B). Using the indicated primer and template concentration, productsranging from 268 bp to 547 bp were amplified from human genomic DNA.
Figure 23
5´3´
TTAAAATT
3´5´
GAATTCCTTAAG
+EcoR I Mse I
+EcoR I adapter Mse I adapter
TAATTCG AAT
Mse I adapter
TTAAEcoR I adapter
AATTCNTTAAGN
NTTANAAT
preselective amplification withEcoR I primer +AMse I primer +C
denaturing polyacrylamide gel electrophoresis
selective amplificationwith primers +3
AATTCATTAAGT CAAT
GTTA
primer +1
primer +3
AATTCAACTTAAGTTG
TA
AACAATTTGTTA
5´ A
5´ AAC
AAC 5´
C 5´
Mse I adapter sequences EcoR I adapter sequences
FIGURE 23. Schematic of the AFLP® Analysis System.
Nucleotides
Fluo
resc
ence
Inte
nsity
1 2 3 4 5 6 7 8 9
Panel A Panel B
bp
547–459——
416–360——
268–
250 200 200 100100 250 500 500
M
250 200 200 100100 250 500 500
primer (nM)template (ng)
18 T R O U B L E S H O O T I N G G U I D E
C H A P T E R 1 0C H A P T E R 1 0C H A P T E R 1 0C H A P T E R 1 0C H A P T E R 1 0
Troubleshooting Guide
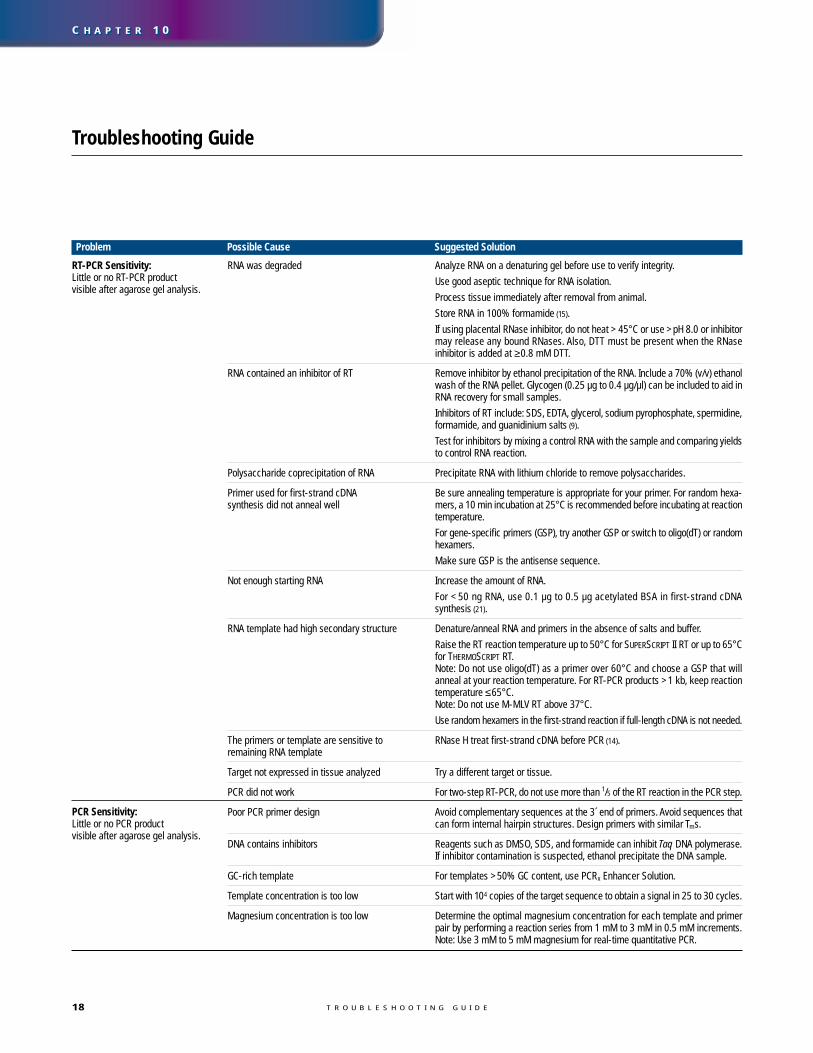
Problem Possible Cause Suggested Solution
RT-PCR Sensitivity: Little or no RT-PCR product visible after agarose gel analysis.
PCR Sensitivity: Little or no PCR product visible after agarose gel analysis.
RNA was degraded
RNA contained an inhibitor of RT
Polysaccharide coprecipitation of RNA
Primer used for first-strand cDNA synthesis did not anneal well
Not enough starting RNA
RNA template had high secondary structure
The primers or template are sensitive to remaining RNA template
Target not expressed in tissue analyzed
PCR did not work
Poor PCR primer design
DNA contains inhibitors
GC-rich template
Template concentration is too low
Magnesium concentration is too low
Analyze RNA on a denaturing gel before use to verify integrity.
Use good aseptic technique for RNA isolation.
Process tissue immediately after removal from animal.
Store RNA in 100% formamide (15).
If using placental RNase inhibitor, do not heat >45°C or use >pH 8.0 or inhibitormay release any bound RNases. Also, DTT must be present when the RNaseinhibitor is added at ≥0.8 mM DTT.
Remove inhibitor by ethanol precipitation of the RNA. Include a 70% (v/v) ethanolwash of the RNA pellet. Glycogen (0.25 µg to 0.4 µg/µl) can be included to aid inRNA recovery for small samples.
Inhibitors of RT include: SDS, EDTA, glycerol, sodium pyrophosphate, spermidine,formamide, and guanidinium salts (9).
Test for inhibitors by mixing a control RNA with the sample and comparing yieldsto control RNA reaction.
Precipitate RNA with lithium chloride to remove polysaccharides.
Be sure annealing temperature is appropriate for your primer. For random hexa-mers, a 10 min incubation at 25°C is recommended before incubating at reactiontemperature.
For gene-specific primers (GSP), try another GSP or switch to oligo(dT) or randomhexamers.
Make sure GSP is the antisense sequence.
Increase the amount of RNA.
For <50 ng RNA, use 0.1 µg to 0.5 µg acetylated BSA in first-strand cDNA synthesis (21).
Denature/anneal RNA and primers in the absence of salts and buffer.
Raise the RT reaction temperature up to 50°C for SUPERSCRIPT II RT or up to 65°Cfor THERMOSCRIPT RT.Note: Do not use oligo(dT) as a primer over 60°C and choose a GSP that willanneal at your reaction temperature. For RT-PCR products >1 kb, keep reactiontemperature ≤65°C.Note: Do not use M-MLV RT above 37°C.
Use random hexamers in the first-strand reaction if full-length cDNA is not needed.
RNase H treat first-strand cDNA before PCR (14).
Try a different target or tissue.
For two-step RT-PCR, do not use more than 1/5 of the RT reaction in the PCR step.
Avoid complementary sequences at the 3´ end of primers. Avoid sequences thatcan form internal hairpin structures. Design primers with similar Tms.
Reagents such as DMSO, SDS, and formamide can inhibit Taq DNA polymerase.If inhibitor contamination is suspected, ethanol precipitate the DNA sample.
For templates >50% GC content, use PCRx Enhancer Solution.
Start with 104 copies of the target sequence to obtain a signal in 25 to 30 cycles.
Determine the optimal magnesium concentration for each template and primerpair by performing a reaction series from 1 mM to 3 mM in 0.5 mM increments.Note: Use 3 mM to 5 mM magnesium for real-time quantitative PCR.
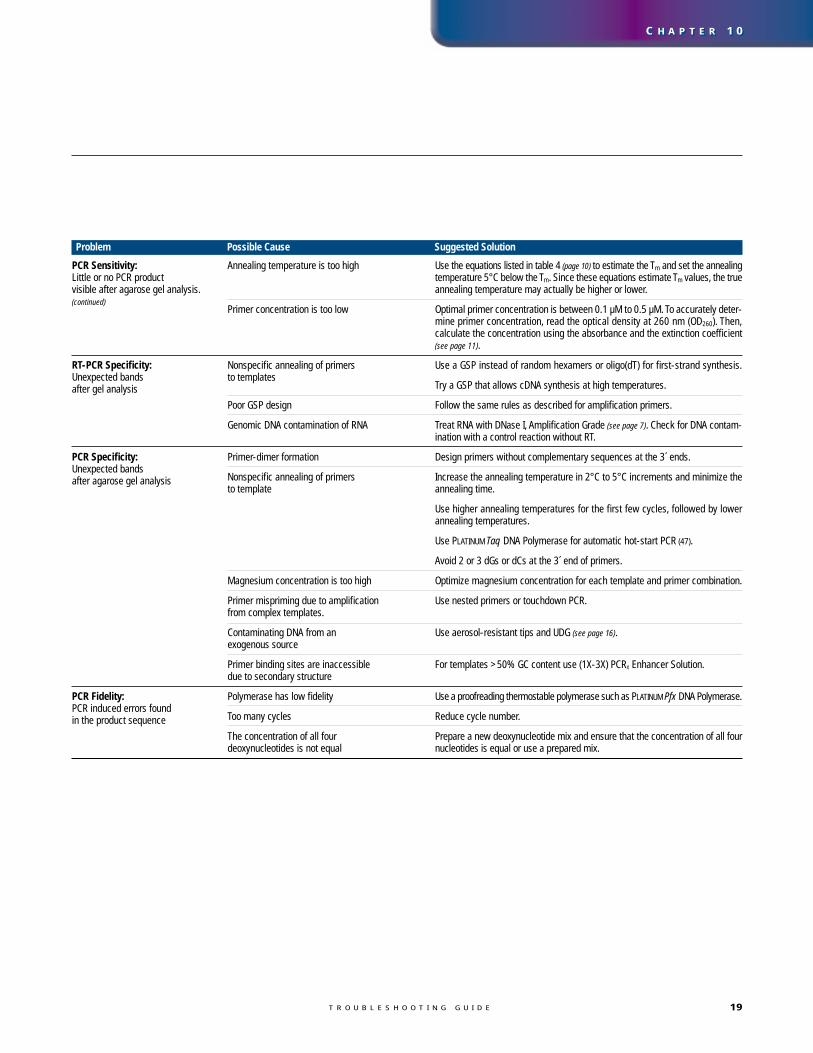
T R O U B L E S H O O T I N G G U I D E 19
C H A P T E R 1 0C H A P T E R 1 0C H A P T E R 1 0C H A P T E R 1 0C H A P T E R 1 0
Problem Possible Cause Suggested Solution
PCR Sensitivity: Little or no PCR product visible after agarose gel analysis.(continued)
RT-PCR Specificity: Unexpected bands after gel analysis
PCR Specificity: Unexpected bands after agarose gel analysis
PCR Fidelity: PCR induced errors found in the product sequence
Annealing temperature is too high
Primer concentration is too low
Nonspecific annealing of primers to templates
Poor GSP design
Genomic DNA contamination of RNA
Primer-dimer formation
Nonspecific annealing of primers to template
Magnesium concentration is too high
Primer mispriming due to amplification from complex templates.
Contaminating DNA from an exogenous source
Primer binding sites are inaccessible due to secondary structure
Polymerase has low fidelity
Too many cycles
The concentration of all four deoxynucleotides is not equal
Use the equations listed in table 4 (page 10) to estimate the Tm and set the annealingtemperature 5°C below the Tm. Since these equations estimate Tm values, the trueannealing temperature may actually be higher or lower.
Optimal primer concentration is between 0.1 µM to 0.5 µM. To accurately deter-mine primer concentration, read the optical density at 260 nm (OD260). Then,calculate the concentration using the absorbance and the extinction coefficient(see page 11).
Use a GSP instead of random hexamers or oligo(dT) for first-strand synthesis.
Try a GSP that allows cDNA synthesis at high temperatures.
Follow the same rules as described for amplification primers.
Treat RNA with DNase I, Amplification Grade (see page 7). Check for DNA contam-ination with a control reaction without RT.
Design primers without complementary sequences at the 3´ ends.
Increase the annealing temperature in 2°C to 5°C increments and minimize theannealing time.
Use higher annealing temperatures for the first few cycles, followed by lowerannealing temperatures.
Use PLATINUM Taq DNA Polymerase for automatic hot-start PCR (47).
Avoid 2 or 3 dGs or dCs at the 3´ end of primers.
Optimize magnesium concentration for each template and primer combination.
Use nested primers or touchdown PCR.
Use aerosol-resistant tips and UDG (see page 16).
For templates >50% GC content use (1X-3X) PCRX Enhancer Solution.
Use a proofreading thermostable polymerase such as PLATINUM Pfx DNA Polymerase.
Reduce cycle number.
Prepare a new deoxynucleotide mix and ensure that the concentration of all fournucleotides is equal or use a prepared mix.

C H A P T E R 1 1C H A P T E R 1 1C H A P T E R 1 1C H A P T E R 1 1C H A P T E R 1 1
20 R E F E R E N C E S
References
1. Saiki, R.K., Scharf, S., Faloona, F., Mullis, K.B., Horn, G.T.,Erlich, H.A., and Arnheim, N. (1985) Science 230, 1350.
2. Innis, M.A., Gelfand, D.H., Sinsky, J.J., and White, T.J.(1990) PCR Protocols, A Guide to Methods and Applications. Academic Press, San Diego, California.
3. Dieffenbach, C.W. and Dveksler, G.S. (1995) PCR Primer:A Laboratory Manual. Cold Spring Harbor LaboratoryPress, Cold Spring Harbor, New York.
4. Siebert, P.D. and Larrick, J.W. (1991) Nature 359, 557.
5. Liang, P. and Pardee, A.B. (1992) Science 257, 967.
6. Frohman, M.A. (1993) Methods Enzymol. 218, 340.
7. Foley, K.P., Leonard, M.W., and Engel, J.D. (1993) Trends in Genetics 9, 380.
8. Mocharla, H., Mocharla, R., and Hodes, M.E. (1990) Gene 93, 271.
9. Gerard, G.F. (1995) FOCUS ® 16, 102.
10. Chomczynski, P. and Sacchi, N. (1987) Anal. Biochem.162, 156.
11. Chomczynski, P. (1993) BioTechniques 15, 532.
12. Simms, D., Cizdziel, P.E., and Chomczynski, P. (1994)FOCUS 15, 99.
13. Bracete, A.M. and Fox, D.K. (1999) FOCUS 21, 38.
14. Fox, D.K. and Nathan, M. (1997) FOCUS 19, 48.
15. Bracete, A.M., Fox, D.K., and Simms, D. (1998) FOCUS 20, 82.
16. Nakajima, D., Nakayama, M., and Ohara, O. (1998) FOCUS 20, 80.
17. Gerard, G.F., D’Alessio, J.M., and Kotewicz, M.L. (1989)FOCUS 11, 66.
18. Schwabe, W., Lee, J.E., Nathan, M., Xu, R.H., Sitaraman,K., Smith, M., Potter, R.J., Rosenthal, K., Rashtchian, A.,and Gerard, G.F. (1998) FOCUS 20, 30.
19. Schuster, D.M., Darfler, M., Lee, J.E., and Rashtchian, A.(1998) FOCUS 20, 34.
20. Nathan, M., Mertz, L., and Fox, D.K. (1995) FOCUS 17, 78.
21. Nathan, M. and Fox, D.K. (1997) FOCUS 19, 50.
22. Lee, H.E., Sitaraman, K., Schuster, D., and Rashtchian, A.(1997) FOCUS 19, 39.
23. Frohman, M.A., Dush, M.K., and Martin, G.R. (1988) Proc. Nat. Acad. Sci. USA 85, 8998.
24. Schuster, D.M., Buchman, G.W., and Rashtchian, A. (1992)FOCUS 14, 46.
25. Loh, Y., Elliott, J.F., Cwirla, S., Lanier, L.L., and Davis, M.M.(1989) Science 243, 217.
26. Cowell, I. and Austin, C.A. (1997) Methods in MolecularBiology: cDNA Library Protocols. Humana Press, Totawa,New Jersey.
27. Fitscher, B.A., Riedel, H.D., Young, K.C., and Stremmel, W.(1998) Biochem. Biophys. Acta 1443, 381.
28. Fox, D.K., Westfall, B., Nathan, M., Hughes, A.J.,Rashtchian, A., and Schuster, D.M. (1996) FOCUS 18, 33.
29. Freeman, W.M., Walker, S.J., and Vrana, K.E. (1999)BioTechniques 26, 112.
30. Heid, C.A., Stevens, J., Livak, K.J., and Williams, P.M.(1996) Genome Res. 6, 986.
31. Zimmerman, K. and Mannhalter, J.W. (1996) BioTechniques 21, 268.
32. Vanden Heuvel, J.P., Tyson, F.L., and Bell, D.A. (1993)BioTechniques 14, 395.
33. Holland, P.M., Abramson, R.D., Watson, R., and Gelfand,D.H. (1991) Proc. Natl. Acad. Sci. USA 88, 7276.
34. Tyagi, S., Bratu, D.P., and Kramer, F.R. (1998) Nature Biotechnology 16, 49.
35. Lear, W., McDonel, M., Kashyap, S., and Boer, P. (1995)BioTechniques 18, 84.
36. Kwok, S., Kellogg, D.E., McKinney, N., Spasic, D.,Goda, L., Levenson, C., and Sinsky, J. (1990)Nucleic Acids Res. 18, 999.
37. White, B.A. (1993) PCR Protocols, Current Methods andApplications. Humana Press, Totowa, New Jersey.
38. Breslauer, K.J., Frank, R., Blocker, H., and Marky, L.A.(1986) Proc. Natl. Acad. Sci. USA 83, 3746.
39. Nelson, T. and Brutlag, D. (1979) Methods Enzymol. 68, 41.
40. Rychlik, W., Spencer, W.J., and Rhoads, R.E. (1990)Nucleic Acids Res. 18, 6409.
41. Don, R.H., Cox, P.T., Wainwright, B.J., Baker, K., andMattick, J.S. (1991) Nucleic Acids Res. 19, 4008.
42. Fox, D.K. (1998) FOCUS 20, 84.
43. Sewall, A., Natarajan, P., and Fox, D.K. (1999) FOCUS 21, 2.
44. Li, H., Cui, X., and Arnheim, N. (1990) Proc. Natl. Acad.Sci. USA 87, 4580.
45. Chou, Q., Russell, M., Birch, D., Raymond, J., and Bloch,W. (1992) Nucleic Acids Res. 20, 1717.
46. Erlich, H.A. (1989) PCR Technology, Principles and Applications for DNA Amplification. Stockton Press,New York, New York.
47. Westfall, B., Sitaraman, K., Solus, J., Hughes, J., andRashtchian, A. (1997) FOCUS 19, 46.
48. Westfall, B., Darfler, M., Xu, R., and Rashtchian, A. (1998)FOCUS 20, 17.
49. Eckert, K.A. and Kunkel, T.A. (1990) Nucleic Acids Res. 18, 3739.
50. Williams, J.F. (1989) BioTechniques 7, 762.
51. Pomp, D. and Medrano, J.F. (1991) BioTechniques 10, 58.
52. Varadaraj, K. and Skinner, D.M. (1994) Gene 140, 1.
53. Baskaran, N., Kandpal, R., Bhargava, A., Glynn, M.,Bale, A., and Weissman, S. (1996) Genome Res. 6, 633.
54. Ally, A.H. and Chomczynski, P. (1995) FOCUS 17, 70.
55. Hansen, P. and Blakesley, R. (1998) FOCUS 20, 72.
56. Brown, T.A. (1991) Molecular Biology Labfax. AcademicPress, Inc., San Diego, California.
57. Cline, J., Braman, J.C., and Hogrefe, H.H. (1996) Nucleic Acids Res. 24, 3546.
58. Tindall, K.R. and Kunkel, T.A. (1988) Biochemistry 27, 6008.
59. Westfall, B., Sitaraman, K., Lee, J.E., Borman, J., andRashtchian, A. (1999) FOCUS 21, 46.
60. Westfall, B., Sitaraman, K., Berninger, M., and Mertz, L.(1993) FOCUS 17, 62.
61. Cheng, S., Fockler, C., Barnes, W.M., and Higuchi, R.(1994) Proc. Natl. Acad. Sci. USA 91, 5695.
62. Longo, M.C., Berninger, M.S., and Hartley, J.L. (1990)Gene 93, 125.
63. Paithankar, K. R. and Prasand, K.S. (1991) Nucleic Acids Res. 19, 1346.
64. Young, A., Xu, L., Goldsborough, M., and Blakesley, R.(1999) FOCUS 21, 11.
65. Fox, D.K., Nathan, M., Natarajan, P., Bracete, A.M., and Mertz, L. (1998) FOCUS 20, 15.
66. Jordan, H., Darfler, M., and Solus, J. (1999) FOCUS 21, 4.
67. Chamberlain, J.S., Gibbs, R.A., Ranier, J.E., Nguyen, P.N.,and Caskey, C.T. (1988) Nucleic Acids Res. 16, 11141.
68. Lin, J.J., Kuo, J., and Ma, J. (1996) Nucleic Acids Res. 24, 3649.
69. Lin, J.J. and Kuo, J. (1997) FOCUS 17, 66.
70. Raff, T., van der Giet, M., Endemann, D., Wiederholt, T.,and Paul, M. (1997) BioTechniques 23, 456.
71. Khiri, H., Reyneir, P., Peyrol, N., Lerique, B., Torresani, J.,and Planells, R. (1996) Mol. Cell Probes 10, 201.
72. Tokunaga, K., Taniguchi, H., Shimizu, M., and Sakiyama, S.(1986) Nucleic Acids Res. 14, 2829.
73. Strehlau, J., Pavlakis, M., Lipman, M., Shapiro, M.,Vasconcellos, L., Harmon, W., and Strom, T. (1997) Proc. Natl. Acad. Sci. USA 94, 695.
74. Arcari, P., Martinelli, R., and Salvatore, F. (1984) Nucleic Acids Res. 12, 9179.
75. Zhao, J., Araki, N., and Nishimoto, S.K. (1995) Gene 155, 159.
76. Fort, P.H., Marty, L., Piechaczyk, M., el Sabrouty, S.,Dani, C.H., Jeanteur, P.H., and Blanchard, J.M. (1985)Nucleic Acids Res. 13, 1431.
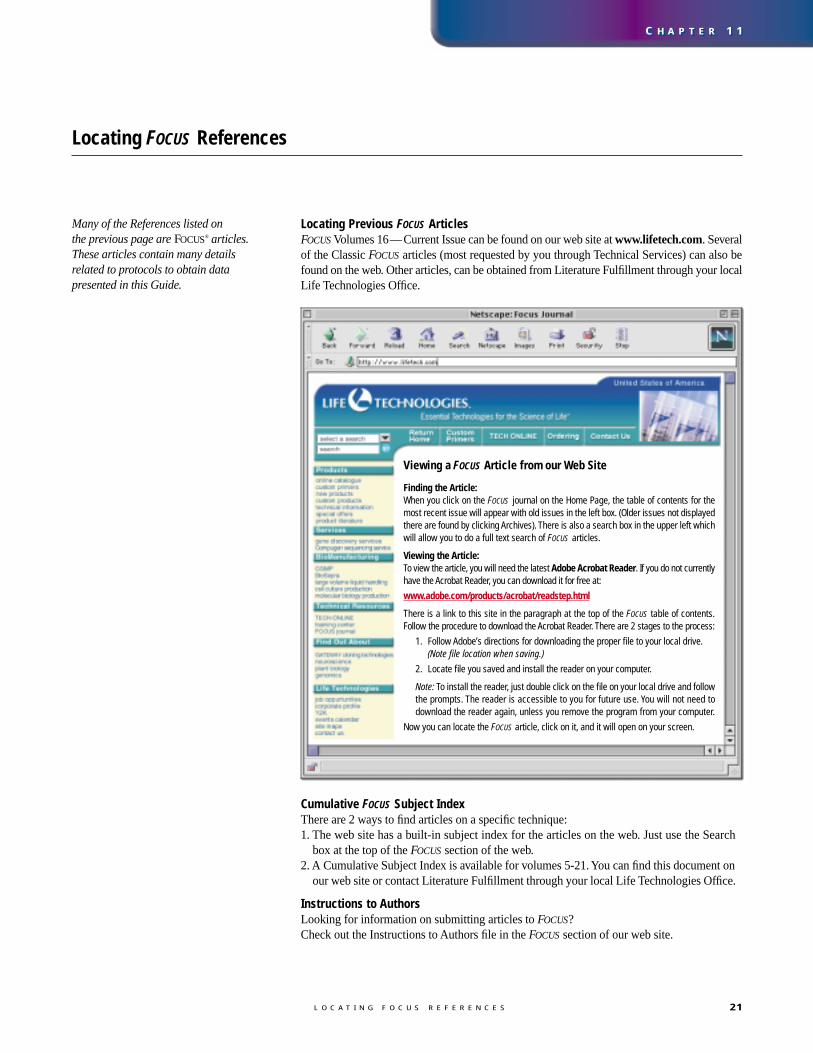
C H A P T E R 1 1C H A P T E R 1 1C H A P T E R 1 1C H A P T E R 1 1C H A P T E R 1 1
L O C A T I N G F O C U S R E F E R E N C E S 21
Locating FOCUS References
Locating Previous FOCUS ArticlesFOCUS Volumes 16— Current Issue can be found on our web site at www.lifetech.com. Severalof the Classic FOCUS articles (most requested by you through Technical Services) can also befound on the web. Other articles, can be obtained from Literature Fulfillment through your localLife Technologies Office.
Cumulative FOCUS Subject IndexThere are 2 ways to find articles on a specific technique:1. The web site has a built-in subject index for the articles on the web. Just use the Search
box at the top of the FOCUS section of the web.2. A Cumulative Subject Index is available for volumes 5-21. You can find this document on
our web site or contact Literature Fulfillment through your local Life Technologies Office.
Instructions to AuthorsLooking for information on submitting articles to FOCUS? Check out the Instructions to Authors file in the FOCUS section of our web site.
Viewing a FOCUS Article from our Web Site
Finding the Article:When you click on the FOCUS journal on the Home Page, the table of contents for themost recent issue will appear with old issues in the left box. (Older issues not displayedthere are found by clicking Archives). There is also a search box in the upper left whichwill allow you to do a full text search of FOCUS articles.
Viewing the Article:To view the article, you will need the latest Adobe Acrobat Reader. If you do not currentlyhave the Acrobat Reader, you can download it for free at:
www.adobe.com/products/acrobat/readstep.html
There is a link to this site in the paragraph at the top of the FOCUS table of contents.Follow the procedure to download the Acrobat Reader. There are 2 stages to the process:
1. Follow Adobe’s directions for downloading the proper file to your local drive.(Note file location when saving.)
2. Locate file you saved and install the reader on your computer.
Note: To install the reader, just double click on the file on your local drive and followthe prompts. The reader is accessible to you for future use. You will not need todownload the reader again, unless you remove the program from your computer.
Now you can locate the FOCUS article, click on it, and it will open on your screen.
Many of the References listed on the previous page are FOCUS® articles. These articles contain many details related to protocols to obtain data presented in this Guide.
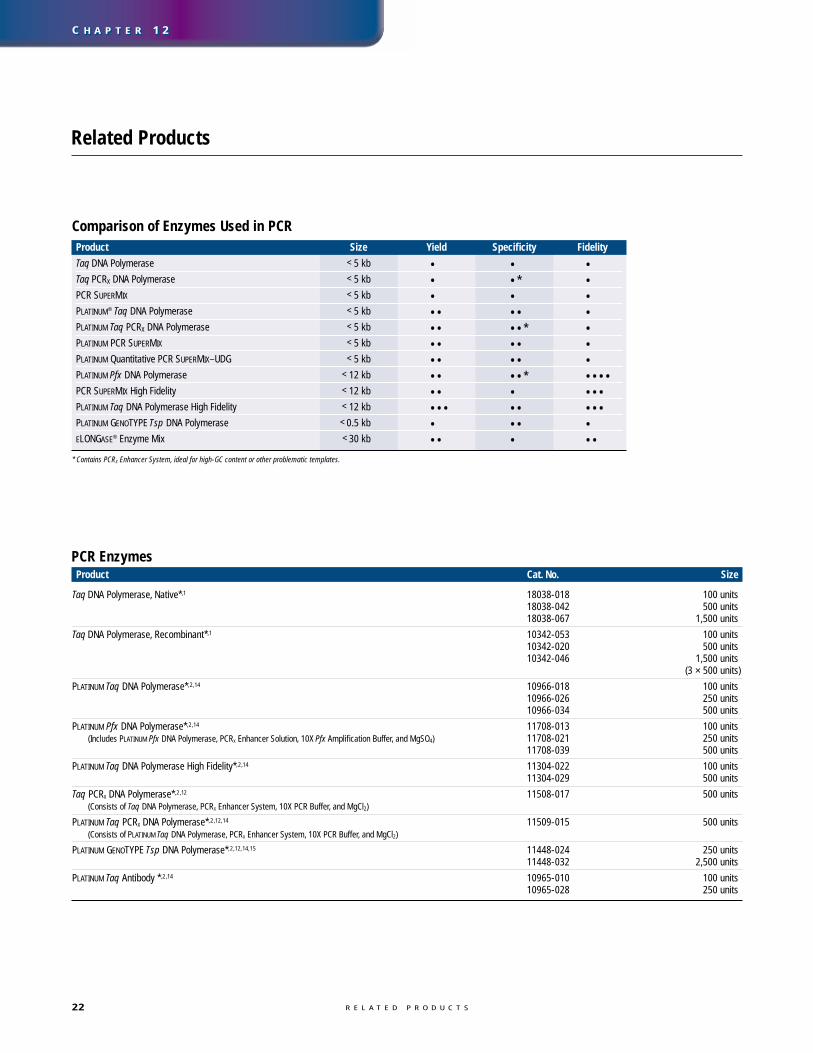
22 R E L A T E D P R O D U C T S
C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2
Related Products
Product Cat. No. Size
Taq DNA Polymerase, Native*,1 18038-018 100 units18038-042 500 units18038-067 1,500 units
Taq DNA Polymerase, Recombinant*,1 10342-053 100 units10342-020 500 units10342-046 1,500 units
(3 × 500 units)
PLATINUM Taq DNA Polymerase*,2,14 10966-018 100 units10966-026 250 units10966-034 500 units
PLATINUM Pfx DNA Polymerase*,2,14 11708-013 100 units(Includes PLATINUM Pfx DNA Polymerase, PCRX Enhancer Solution, 10X Pfx Amplification Buffer, and MgSO4) 11708-021 250 units
11708-039 500 units
PLATINUM Taq DNA Polymerase High Fidelity*,2,14 11304-022 100 units11304-029 500 units
Taq PCRX DNA Polymerase*,2,12 11508-017 500 units(Consists of Taq DNA Polymerase, PCRX Enhancer System, 10X PCR Buffer, and MgCl2)
PLATINUM Taq PCRX DNA Polymerase*,2,12,14 11509-015 500 units(Consists of PLATINUM Taq DNA Polymerase, PCRX Enhancer System, 10X PCR Buffer, and MgCl2)
PLATINUM GENOTYPE Tsp DNA Polymerase*,2,12,14,15 11448-024 250 units11448-032 2,500 units
PLATINUM Taq Antibody *,2,14 10965-010 100 units10965-028 250 units
PCR Enzymes
Comparison of Enzymes Used in PCRProduct Size Yield Specificity Fidelity
Taq DNA Polymerase <5 kb • • •Taq PCRX DNA Polymerase <5 kb • •* •PCR SUPERMIX <5 kb • • •PLATINUM® Taq DNA Polymerase <5 kb •• •• •PLATINUM Taq PCRX DNA Polymerase <5 kb •• ••* •PLATINUM PCR SUPERMIX <5 kb •• •• •PLATINUM Quantitative PCR SUPERMIX–UDG <5 kb •• •• •PLATINUM Pfx DNA Polymerase <12 kb •• ••* ••••PCR SUPERMIX High Fidelity <12 kb •• • •••PLATINUM Taq DNA Polymerase High Fidelity <12 kb ••• •• •••PLATINUM GENOTYPE Tsp DNA Polymerase <0.5 kb • •• •ELONGASE® Enzyme Mix <30 kb •• • ••
* Contains PCRX Enhancer System, ideal for high-GC content or other problematic templates.

C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2
R E L A T E D P R O D U C T S 23
Product Cat. No. Size
PCR SUPERMIX*,2,12 10572-014 100 reactions
PLATINUM PCR SUPERMIX*,2,12,14 11306-016 100 reactions
PCR SUPERMIX High Fidelity*,2,12 10790-020 100 reactions
PLATINUM Quantitative PCR SUPERMIX-UDG*,2,9,12,14, 24 11730-017 100 reactions11730-025 500 reactions
PCR Reagent System*,2 10198-018 100 reactions
ELONGASE Amplification System*,1,2 10481-018 100 reactions
ELONGASE Enzyme Mix*,1,2 10480-010 100 reactions
Product Cat. No. Size
SUPERSCRIPT™ II RNase H– Reverse Transcriptase5 18064-014 2,000 units18064-022 10,000 units18064-071 4 × 10,000 units
SUPERSCRIPT RNase H– Reverse Transcriptase5 18053-017 10,000 units
SUPERSCRIPT First-Strand Synthesis System for RT-PCR5 11904-018 50 reactions(Includes oligo(dT)12-18, random hexamers, SUPERSCRIPT II RT, RNASEOUT™ RNase Inhibitor 10X RT buffer,Mg solution, 10 mM dNTP mix, E. coli RNase H, control RNA and primers, and DEPC treated water)
SUPERSCRIPT One-Step RT-PCR System*,2,5 10928-018 25 reactions(Includes SUPERSCRIPT II RT/Taq DNA Polymerase enzyme mix, 2X reaction buffer, Mg solution) 10928-026 100 reactions
SUPERSCRIPT One-Step RT-PCR System with PLATINUM Taq DNA Polymerase2,5,14 10928-034 25 reactions(Includes SUPERSCRIPT II RT/PLATINUM Taq DNA Polymerase Mix, 2X Reaction Mix, and MgSO4) 10928-042 100 reactions
SUPERSCRIPT One-Step RT-PCR for Long Templates2,5,14 11922-010 25 reactions(Includes SUPERSCRIPT II RT/PLATINUMTaq DNA Polymerase High Fidelity Mix, 2X Reaction Mix, and MgSO4) 11922-028 100 reactions
THERMOSCRIPT™ RT-PCR System*,2,13 11146-024 25 reactions(Includes THERMOSCRIPT RT, 5X cDNA Synthesis Buffer, oligo(dT)20, random hexamers, 0.1 M DTT, 11146-016 100 reactions10 mM dNTP Mix, RNASEOUT RNase Inhibitor, DEPC-treated water, E. coli RNase H)
THERMOSCRIPT RT-PCR System and PLATINUMTaq DNA Polymerase*,2,13,14 11146-057 25 reactions11146-032 100 reactions
THERMOSCRIPT RT-PCR System and PLATINUMTaq DNA Polymerase High Fidelity*,2,13,14 11146-040 100 reactions
PLATINUM Quantitative RT-PCR THERMOSCRIPT One-Step System*,2,13,14,24 11731-015 100 reactions(Includes THERMOSCRIPT PLUS/PLATINUM Taq Mix, 2X THERMOSCRIPT Reaction Mix, and MgSO4) 11731-023 500 reactions
M-MLV Reverse Transcriptase 28025-013 40,000 units28025-021 200,000 units
RT-PCR Primer and Control Set 10929-016 20 reactions(Includes HeLa RNA and β-actin primers)
Ribonuclease H 18021-014 30 units18021-071 120 units
DNase I, Amplification Grade 18068-015 100 units
RNASEOUT Recombinant Ribonuclease Inhibitor 10777-019 5,000 units
DEPC-treated Water 10813-012 4 × 1.25 ml
5´ RACE System for Rapid Amplification of cDNA Ends, Version 2.0 4,5 18374-058 10 reactions
3´ RACE System for Rapid Amplification of cDNA End 4,5 18373-019 20 reactions
PCR Enzymes (continued)
RT-PCR
24 R E L A T E D P R O D U C T S
Related Products (continued)
C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2
Product Cat. No. Size
AFLP® Analysis System I (Genome size of 5 × 108 to 6 × 109)17 10544-013 each(Contains AFLP Core Reagent Kit and AFLP Starter Primer Kit)
AFLP Analysis System II (Genome size of 1 × 108 to 5 × 108)17 10717-015 each(Contains AFLP Core Reagent Kit and AFLP Small Genome Primer Kit)
AFLP System for Microorganisms (Designed for bacteria and fungi)17 11352-010 each(Contains AFLP Core Reagent Kit and AFLP Microorganism Primer Kit)
AFLP Non-radioactive Probe 10822-013 125 µl
Other Applications
Product Cat. No. Size
TRIZOL® Reagent 15596-026 100 ml15596-018 200 ml
TRIZOL LS Reagent 10296-010 100 ml10296-028 200 ml
DNAZOL® Reagent 10503-027 100 ml10503-035 200 ml
DNAZOL BD Reagent 10974-020 100 ml10974-038 200 ml
Plant DNAZOL Reagent 10978-021 100 ml
Buffered-Saturated Phenol 15513-039 100 ml
Proteinase K 25530-015 100 mg
FTA® Cards 4,16 10786-010 100/pkg
FTA GeneCard4,16 10786-036 100/pkg
FTA Purification Reagent 4,16 10876-019 250 ml
CONCERT™ Rapid PCR Purification System 11458-015 50 reactions11458-023 250 reactions
CONCERT Rapid Gel Extraction System 11456-019 50 reactions11456-027 250 reactions
TAQUENCH™ PCR Cloning Enhancer 4,12,14 11265-014 100 units11265-022 250 units
Nucleic Acid Purification
Product Cat. No. Size
GIBCO BRL® Custom Primers Please Inquire
10 mM dNTP Nucleotide Mix4 18427-013 100 µl
100 mM dNTP Nucleotide Set4 10297-018 4 × 250 µl
Reaction Components
Product Cat. No. Size
100 bp DNA Ladder 15628-019 50 µg
1 Kb PLUS DNA LADDER™ 10787-018 250 µg
0.2-ml PCR Tubes 10795-011 1,000 tubes/bag
0.5-ml PCR Tubes 10795-037 1,000 tubes/bag
0.2-ml PCR Tubes with Flat Caps 10795-151 1,000 tubes/bag
0.2-ml PCR Tubes with Attached Strip Caps 10795-144 125 strips/bag
Related PCR Products
Product Cat. No. Size
HOTSTART 50® Storage and Reaction Tubes (20 to 50 µl reactions) 10331-015 96 tubes
HOTSTART 100® Storage and Reaction Tubes (50 to 100 µl reactions) 10332-013 96 tubes
HOTSTART Micro 20® Storage and Reaction Tubes (15 to 25 µl reactions) 10467-017 96 tubes
HOTSTART Micro 50® Storage and Reaction Tubes (25 to 50 µl reactions) 10468-015 96 tubes
Silicone Oil 10890-010 4 ml
Uracil DNA Glycosylase9 18054-015 100 units
Thin-wall Reaction Tubes with Wax Beads
Product Cat. No. Size
ART REACH™ 10670-016 960 tips/pkg
ART 20P 14862-015 1,000 tips/pkg
ART 20E 14862-023 960 tips/pkg
ART 200 14867-014 960 tips/pkg
ART 1000 14868-012 800 tips/pkg
ART ®, Aerosol Resistant Tips™, Brand Pipet Tips
C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2
R E L A T E D P R O D U C T S 25
These products are for laboratory research use only and are not intended for human oranimal diagnostic, therapeutic, or other clinical uses, unless otherwise stated.
ABI PRISM® is a registered trademark of Perkin Elmer Corporation.AFLP® is a registered trademark of Keygene n.v.ART®, Aerosol Resistant Tips™, and REACH™ are marks of Molecular Bio-Products, Inc. CFLP™ is a trademark of Third Wave Technologies, Inc.,DNAZOL® and TRIZOL® are registered trademarks of Molecular Research Center, Inc.,FTA® is a registered trademark of Flinders Technologies PTY Ltd. HOTSTART 50®, HOTSTART 100®, HOTSTART Micro 20®, and HOTSTART Micro 50® areregistered trademarks of Molecular Bio-Products, Inc.PfuTurbo™ is a trademark of Stratagene.SYBR® is a registered trademark of Molecular Probes, Inc. TaqMan® is a registered trademark of Roche Molecular Systems, Inc.
* Purchase of this product is accompanied by a limited license to use it in the Poly-merase Chain Reaction (PCR) process for research and development in conjunctionwith a thermal cycler whose use in the automated performance of the PCR process is covered by the up-front license fee, either by payment to Perkin-Elmer or aspurchased, i.e., an authorized thermal cycler. This product is sold under licensingarrangements with F. Hoffmann-La Roche Ltd, Roche Molecular Systems, Inc., andThe Perkin-Elmer Corporation.
U.S. Academic/TECH-LINE™: (800) 828-6686U.S. Government Orders/TECH-LINE: (888) 584-8929 U.S. Industrial Orders/TECH-LINE: (800) 874-4226Internet: www.lifetech.com

Limited Label Licenses1 A license under U.S. Patents 4,683,202, 4,683,195, 4,965,188, and 5,075,216 ortheir foreign counterparts, owned by Hoffmann-La Roche Inc. and F. Hoffmann-La Roche Ltd (“Roche”), has an up-front fee component and a running-loyaltycomponent. The purchase price of this product includes limited, nontransferablerights under the running-royalty component to us only this amount of the productto practice the polymerase chain reaction (PCR) and related processes describedin said patents solely for the research and development activities of the purchaserwhen this product is used in conjunction with a thermal cycler whose use iscovered by the up-front fee component. Rights to the up-front fee componentmust be obtained by the end user in order to have a complete license. These rightsunder the up-front fee component may be purchased from Perkin-Elmer orobtained by purchasing an Authorized Thermal Cycler. No right to perform ofoffer commercial services of any kind using PCR including, without limitation,reporting the results of the purchaser’s activities for a fee or other commercialconsideration, is hereby granted by implication or estoppel. Further onpurchasing licenses to practice the PCR Process may be obtained by contactingthe Director of Licensing at the Perkin-Elmer Corporation, 850 Lincoln CentreDrive, Foster City, California 94404 or at Roche Molecular Systems, Inc., 1145Atlantic Avenue, Alameda, California 94501.
2 A license under U.S. Patent 4,683,202, 4,683,195, and 4,965,188 or their foreigncounterparts, owned by Hoffman-La Roche Inc. and F. Hoffmann-La Roche Ltd(“Roche”), has an up-front component and a running-royalty component. Thepurchase price of this product includes limited, nontransferable rights under therunning-royalty component to use only this amount of the product to practice thePolymerase Chain Reaction (“PCR”) and related processes described in saidpatents solely for the research and development activities of the purchaser whenthis product is used in conjunction with a thermal cycler whose use is covered bythe up-front fee component. Rights to the up-front fee component must beobtained by the end user in order to have a complete license. These rights underthe up-front fee component may be purchased from Perkin-Elmer or obtained bypurchasing an Authorized Thermal Cycler. No right to perform of offercommercial services of any kind using PCR, including without limitationreporting the results of the purchaser’s activities for a fee or other commercialconsiderations, is hereby granted by implication or estoppel. Further informationon purchasing licenses to practice the PCR Process may be obtained bycontacting the Director of Licensing at the Perkin-Elmer Corporation, 850Lincoln Centre Drive, Foster City, California 94404 or at Roche MolecularSystems, Inc., 1145 Atlantic Avenue, Alameda, California 94501.
4 This product optimized for use in the Polymerase Chain Reaction (PCR)covered by patents owned by Hoffman-La Roche Inc. and F. Hoffman- La RocheLtd (“Roche”). No license under these patents to use the PCR process is conveyedexpressly or by implication to the purchaser by the purchase of this product. Alicense to use the PCR Process for certain research and development activitiesaccompanies the purchase of certain reagents from licensed supplies such as LifeTechnologies, Inc. when used in conjunction with an Authorized Thermal Cycler,or is available form The Perkin-Elmer Corporation. Further information onpurchasing licenses to practice the PCR Process may be obtained by contacting theDirector of Licensing at the Perkin-Elmer Corporation, 850 Lincoln Centre Drive,Foster City, California 94404 or at Roche Molecular Systems, Inc., 1145 AtlanticAvenue, Alameda, California 94501.
5 Purchase of this product conveys to the buyer only the non-transferable right touse the product in research conducted by the buyer. The buyer cannot sell orotherwise transfer this product to a third party. Life Technologies, Inc. reserves allother rights, and this product may not be used in any manner other than providedherein. In particular, without prior written agreement between the buyer and LifeTechnologies, Inc., no rights are conveyed to the buyer to use the product inmanufacturing, or to resell the product or components of the product as a stand-alone reagent or in a kit, whether or not such stand-alone reagent is resold or suchkit is resold for use in research. If the purchaser is not willing to accept thelimitations of this Label License, Life Technologies, Inc. is willing to accept returnof the product with a full refund. For information on purchasing a license to thisproduct for purposes other than research, contact the Director of Licensing, 9800Medical Center Drive, Rockville, MD 20850. Phone (301) 610-8000. Fax (301)610-8383.
9 The use of uracil-N-glycosylase for carryover prevention in amplificationreactions is the subject of claims in U.S. Patents 5,035,996, 5,638,896, and foreignequivalents owned by Life Technologies, Inc. Purchase of this product conveys the right to use it for research purposes only in processes claimed in theaforementioned patents. No rights to use in other applications including thediagnosis of disease in humans, animals, or plants, under any patents owned byLife Technologies, Inc. are conveyed by the purchase of this product.
12 The purchase of this product conveys to the buyer the non-transferable right touse the product and components of the product in research conducted by thebuyer. The buyer cannot sell or otherwise transfer this product or its componentsto a third party and in particular, no rights are conveyed to the buyer to use theproduct or its components for commercial purposes. Commercial purposes meansany activity for which a party receives consideration and may include, but is notlimited to, (1) use of the product or its components in manufacturing, (2) use ofthe product or its components to provide a service, information or data, (3) use ofthe product or its components for diagnostic purposes, or (4) resell the product orits components, whether or not such product or its components are resold for usein research. If purchaser is not willing to accept the limitations of this limited usestatement, Life Technologies, Inc. is willing to accept return of the products witha full refund. For information on purchasing a license to this product for purposesother than research, contact the Director of Licensing, 9800 Medical CenterDrive, Rockville, MD 20850. Phone (301) 610-8000. Fax (301) 610-8383.
26 R E L A T E D P R O D U C T S
C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2
Related Products (continued)

C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2C H A P T E R 1 2
R E L A T E D P R O D U C T S 27
13 Purchase of this product conveys to the buyer the non-transferable right to usethe product and components of the product in research conducted by the buyer.The buyer cannot sell or otherwise transfer this product or its components forcommercial purposes. Commercial purposes means any activity for which a partyreceives consideration and may include, but is not limited to, (1) use of theproduct or its components in manufacturing, (2) use of the product or itscomponents to provide a service, information, or data, (3) use of the product or itscomponents for diagnostic purposes, or (4) resell the product or its components,whether or not such product or its components are resold for use in research. Ifpurchaser is not willing to accept the limitations of this limited use statement,Life Technologies, Inc. is willing to accept return of the products with a fullrefund. For information on purchasing a license to this product for purposes otherthan research, contact the Director of Licensing, 9800 Medical Center Drive,Rockville, MD 20850. Phone (301) 610-8000. Fax (301) 610-8383.
14 Licensed to Life Technologies, Inc. under U.S. Patent 5,338,671, 5,587,287,and foreign equivalents for use in research only.
15 Unless indicated otherwise, purchase of this product does not convey to thepurchaser a license to practice any claim of any patent, including but not limitedto U.S. Patents 5,075,217 and 5,766,847, German Patent 3,834,636, and foreignequivalents. Users of this product should determine if any license is requiredunder these or any other patents.
16 Licensed to Life Technologies, Inc. under U.S. Patents 5,496,562, 5,807,527,5,756,126, and foreign equivalents for laboratory research and human identitytesting. Purchase of this product conveys to the buyer only the non-transferableright to use the product for laboratory research and non-commercial humanidentity testing. Use of this product for any commercial purposes or alteration ofFTA Cards and related products in any manner is prohibited without the priorwritten consent of Life Technologies, Inc. Material supplied by Life Technologies,Inc. is not intended for human or animal diagnostic or therapeutic uses. Forinformation concerning the availability of product and licenses to practice thepatented technology for commercial purposes in human identity testing, contactLife Technologies, Inc., Rockville, MD. For information concerning theavailability of product and licenses to practice the patented technology for clinicaldiagnosis or therapeutic uses contact FITZCO, Inc., Maple Plain, MN.
17 The AFLP technique is covered by patents or patented applications owned byKeygene n.v. This product is sold under license from Keygene n.v. This kit maybe used for research purposes only. For use of this kit in plant breeding, contactthe Director of Licensing, Life Technologies, Inc., 9800 Medical Center Drive,Rockville, MD 20850. Phone (301) 610-8000. Fax (301) 610-8383. The use ofthis kit for any other purpose, whether commercial or noncommercial, includingbut not limited to the use for clinical, diagnostic, and/or therapeutic purposes; orfor providing services to third parties, requires a license from Keygene n.v., P.O.Box 216, 6700 AE Wageningen, The Netherlands.
24 The use of TaqMan® fluorogenic probes in 5´ nuclease assays is covered by U.S.Patent Nos. 5,210,015 and 5,487,972, owned by Roche Molecular Systems, Inc.,and by U.S. Patent No. 5,538,848, owned by PE Corporation. Purchase of theproduct does not provide a license to use this patented technology. A license topractice this technology must be obtained from PE Biosystems, 850 LincolnCenter Drive, Forest City, CA 94404, or from Roche Molecular Systems, Inc.,1145 Atlantic Avenue, Alameda, CA 94501.
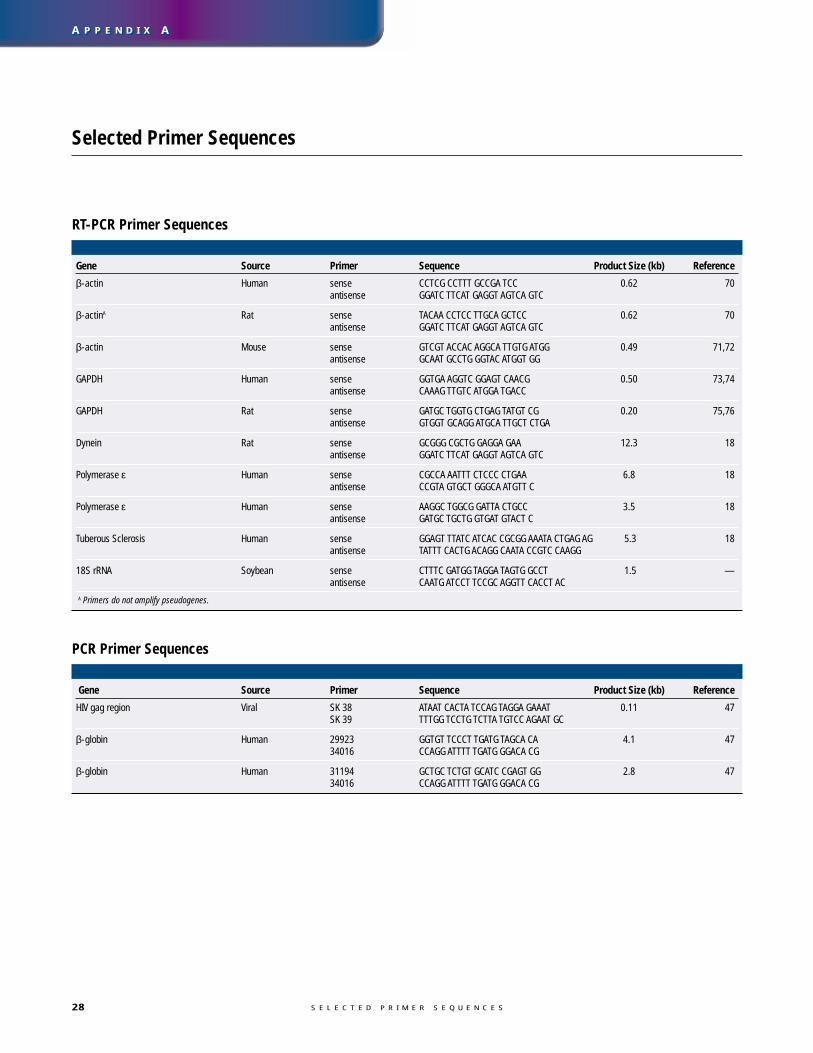
28 S E L E C T E D P R I M E R S E Q U E N C E S
Selected Primer Sequences
RT-PCR Primer Sequences
Gene Source Primer Sequence Product Size (kb) Reference
β-actin Human sense CCTCG CCTTT GCCGA TCC 0.62 70antisense GGATC TTCAT GAGGT AGTCA GTC
β-actinA Rat sense TACAA CCTCC TTGCA GCTCC 0.62 70antisense GGATC TTCAT GAGGT AGTCA GTC
β-actin Mouse sense GTCGT ACCAC AGGCA TTGTG ATGG 0.49 71,72antisense GCAAT GCCTG GGTAC ATGGT GG
GAPDH Human sense GGTGA AGGTC GGAGT CAACG 0.50 73,74antisense CAAAG TTGTC ATGGA TGACC
GAPDH Rat sense GATGC TGGTG CTGAG TATGT CG 0.20 75,76antisense GTGGT GCAGG ATGCA TTGCT CTGA
Dynein Rat sense GCGGG CGCTG GAGGA GAA 12.3 18antisense GGATC TTCAT GAGGT AGTCA GTC
Polymerase ε Human sense CGCCA AATTT CTCCC CTGAA 6.8 18antisense CCGTA GTGCT GGGCA ATGTT C
Polymerase ε Human sense AAGGC TGGCG GATTA CTGCC 3.5 18antisense GATGC TGCTG GTGAT GTACT C
Tuberous Sclerosis Human sense GGAGT TTATC ATCAC CGCGG AAATA CTGAG AG 5.3 18antisense TATTT CACTG ACAGG CAATA CCGTC CAAGG
18S rRNA Soybean sense CTTTC GATGG TAGGA TAGTG GCCT 1.5 —antisense CAATG ATCCT TCCGC AGGTT CACCT AC
A Primers do not amplify pseudogenes.
PCR Primer Sequences
Gene Source Primer Sequence Product Size (kb) Reference
HIV gag region Viral SK 38 ATAAT CACTA TCCAG TAGGA GAAAT 0.11 47SK 39 TTTGG TCCTG TCTTA TGTCC AGAAT GC
β-globin Human 29923 GGTGT TCCCT TGATG TAGCA CA 4.1 4734016 CCAGG ATTTT TGATG GGACA CG
β-globin Human 31194 GCTGC TCTGT GCATC CGAGT GG 2.8 4734016 CCAGG ATTTT TGATG GGACA CG
A P P E N D I X AA P P E N D I X AA P P E N D I X AA P P E N D I X AA P P E N D I X A

PCR
Enzymes including:Taq DNA PolymerasePLATINUM Enzymes (high specificity)PLATINUM Pfx DNA Polymerase (high fidelity)PLATINUM GENOTYPE Tsp DNA PolymeraseHigh Fidelity Enzyme MixesPCRx DNA Polymerases (high GC content)SUPERMIXESPLATINUM Quantitative PCR SUPERMIX-UDGELONGASE Reagents
PCR Reagent SystemCustom PrimersdNTPsUracil DNA GlycosylaseDistilled Water, DNase-RNase-FreeART TipsPCR TubesHOTSTART Storage and Reaction TubesSilicone OilMICROMATE Labels
Electrophoretic Analysis of PCR Products
25, 50, 250, and 500 bp DNA Ladders10, 100, and 123 bp DNA Ladders1 Kb PLUS DNA LadderHORIZON Electrophoresis ApparatusSUNRISE Electrophoresis ApparatusModels 250, 250 EX, and 125 Power SupplyElectrophoresis Reagents
AgaroseAgarose 100010X BLUEJUICE BufferTBE BufferTAE BufferEthidium Bromide
Kodak Digital Science EDAS Systems
Cloning PCR Products
CONCERT Rapid PCR Purification SystemTAQUENCH Cloning EnhancerRestriction EndonucleasesT4 DNA LigaseCompetent CellsCLONEAMP SystemsPCR Cloning System with GATEWAY Technology
First-Strand cDNA Synthesis (RT-PCR)
SUPERSCRIPT First-Strand Synthesis RT-PCR SystemSUPERSCRIPT One-Step RT-PCR SystemsTHERMOSCRIPT RT-PCR SystemsPLATINUM Quantitative RT-PCR THERMOSCRIPT System3´ RACE System5´ RACE SystemCustom PrimersDNAse I, Amplification GradedNTPsRibonuclease HM-MLV RTSUPERSCRIPT II Reverse TranscriptaseRT-PCR Primer and Control Set
Plasmid DNA Isolation
CONCERT Rapid Plasmid Purification SystemCONCERT High Purity Plasmid Purification System
Genomic DNA Isolation
DNAZOL ReagentsFTA Cards and ReagentPhenol ProductsPRETAQ Thermophilic ProteaseProteinase K
RNA Isolation
TRIZOL ReagentsMESSAGEMAKER SystemGLASSMAX RNA SystemRNASEOUT Ribonuclease InhibitorProteinase KRNA Molecular Size StandardsDEPC-treated Water
PCR and RT-PCR Product Groupings
P C R A N D R T - P C R P R O D U C T G R O U P I N G S 29





























