Embed Size (px)
Citation preview

38 Bertrand ME, Simoons ML, Fox KA, Wallentin LC, Hamm CW,McFadden E, et al. Management of acute coronary syndromes:acute coronary syndromes without persistent ST segment elevation.Recommendations of the Task Force of the European Society ofCardiology. Eur Heart J 2000;21:1406-32.
39 Braunwald E, Antman EM, Beasley JW, Califf RM, Cheitlin MD,Hochman JS, et al. ACC/AHA guidelines for the managementof patients with unstable angina and non-ST-segment elevationmyocardial infarction. A report of the American College ofCardiology/American Heart Association Task Force on PracticeGuidelines (Committee on the Management of Patients WithUnstable Angina). J Am Coll Cardiol 2000;36:970-1062.
40 Canto JG, Shlipak MG, Rogers WJ, Malmgren JA, Frederick PD,Lambrew CT, et al. Prevalence, clinical characteristics, and mor-tality among patients with myocardial infarction presenting withoutchest pain. JAMA 2000;283:3223-9.
41 Turi ZG, Rutherford JD, Roberts R, Muller JE, Jaffe AS, Rude RE,et al. Electrocardiographic, enzymatic and scintigraphic criteria ofacute myocardial infarction as determined from study of 726 patients(a MILIS study). Am J Cardiol 1985;55(13 Pt 1):1463-8.
42 Pope JH, Aufderheide TP, Ruthazer R, Woolard RH, Feldman JA,Beshansky JR, et al. Missed diagnoses of acute cardiac ischemia inthe emergency department. N Engl J Med 2000;342:1163-70.
43 Lee TH, Rouan GW, Weisberg MC, Brand DA, Acampora D,Stasiulewicz C, et al. Clinical characteristics and natural history ofpatients with acute myocardial infarction sent home from the emer-gency room. Am J Cardiol 1987;60:219-24.
44 Hamm CW, Goldmann BU, Heeschen C, Kreymann G, Berger J,Meinertz T. Emergency room triage of patients with acute chest painby means of rapid testing for cardiac troponin T or troponin I. NEngl J Med 1997;337:1648-53.
45 Newby LK, Kaplan AL, Granger BB, Sedor F, Califf RM, OhmanEM. Comparison of cardiac troponin T versus creatine kinase-MBfor risk stratification in a chest pain evaluation unit. Am J Cardiol2000;85:801-5.
46 Adams 3rd JE, Davila-Roman VG, Bessey PQ, Blake DP, LadensonJH, Jaffe AS. Improved detection of cardiac contusion with cardiactroponin I. Am Heart J 1996;131:308-12.
47 Swaanenburg JC, Klaase JM, DeJongste MJ, Zimmerman KW, DuisHJ ten. Troponin I, troponin T, CKMB-activity and CKMB-mass asmarkers for the detection of myocardial contusion in patients whoexperienced blunt trauma. Clin Chim Acta 1998;272:171-81.
Aanvaard op 1 november 2000
466 Ned Tijdschr Geneeskd 2001 10 maart;145(10)
Neuronale migratiestoornissen van de hersenschors zijnontwikkelingsstoornissen die gepaard kunnen gaan meteen trias van klinische manifestaties: mentale retardatie,epilepsie en hypotonie.1 2 Neuronale migratiestoornis-sen komen bij meer dan 1% van de bevolking voor enbij 20-40% van de onbehandelbare vormen van epilep-sie.3 Ze worden gekenmerkt door een aberrante archi-tectuur en een verkeerde differentiatie van de hersen-schors.4-8 Hun vóórkomen is wisselend en complex; hetgaat om meer dan 25 syndromen. Classificaties zijn ge-maakt op grond van ontstaanswijze,1 morfologischbeeld4 9 en vooral beeldvormend neurologisch onder-zoek.10 11 Globaal kan de volgende indeling worden ge-maakt: (a) agyrie/pachygyrie met de verschillende vor-men van lissencefalie (hersenen met een ‘glad’ opper-vlak, dat wil zeggen geen of weinig windingen); (b) he-terotopieën, zoals de subcorticale-bandheterotopie; (c)polymicrogyrie, en (d) corticale dysplasieën (figuur 1).Veel onderzoek naar de mechanismen van migratie-stoornissen werd de laatste jaren verricht aan de ‘reeler’-
muis (‘to reel’ = ‘waggelen’), waarbij door de afwezig-heid van het eiwit ‘reeline’ vroeg in de ontwikkeling demigratie van neuronen naar de hersenschors stopt.12 13
Inmiddels is hiervan het coderende gen gekloneerd.14
Ook van enkele humane neuronale migratiestoornissenis inmiddels het gen bekend.
In dit derde overzicht over ontwikkeling en ontwik-kelingsstoornissen van het humane brein worden deneuronale migratiestoornissen van de grote hersenenbehandeld.15 16 Na een kort overzicht van het dierexpe-
Capita selectaOntwikkeling en ontwikkelingsstoornissen van het humane brein. III.Neuronale migratiestoornissen van de grote hersenen
h.j.ten donkelaar, m.lammens, p.wesseling, h.o.m.thijssen, w.o.renier en f.j.m.gabreëls
samenvatting– Neuronale migratiestoornissen van de hersenschors vormeneen heterogene groep van afwijkingen die worden gekenmerktdoor mentale retardatie, epilepsie en hypotonie.– Ze komen voor bij 1% van de bevolking en bij 20-40% vande onbehandelbare vormen van epilepsie.– Stoornissen aan het begin van de migratie leiden tot nodulai-re heterotopieën. Bilaterale periventriculaire nodulaire hete-rotopieën komen familiair voor en zijn X-gebonden. Hierbijzijn corticale neuronen door de afwezigheid van het eiwit fila-mine 1 niet in staat hun positie aan het ventrikeloppervlak teverlaten.– De grote groep lissencefalieën kan in een aantal syndromenworden verdeeld, waarbij mutaties in verschillende genen(LIS1, DCX, RELN) leiden tot de voor deze groep kenmer-kende agyrie en pachygyrie.– Een aantal van deze afwijkingen, vooral de kleinere nodulai-re heterotopieën en focale corticale dysplasie, komt in aan-merking voor neurochirurgische excisie.
Universitair Medisch Centrum St Radboud, Postbus 9101, 6500 HBNijmegen.Instituut voor Neurologie: dr.H.J.ten Donkelaar, arts-anatoom; dr.M.Lammens en dr.P.Wesseling, neuropathologen (beiden tevens: Insti-tuut voor Pathologie); prof.dr.W.O.Renier, kinderneuroloog (tevens:Interdisciplinair Kinderneurologisch Centrum).Instituut voor Radiologie: prof.dr.H.O.M.Thijssen, neuroradioloog.Interdisciplinair Kinderneurologisch Centrum: prof.dr.F.J.M.Gabreëls,kinderneuroloog.Correspondentieadres: dr.H.J.ten Donkelaar ([email protected]).

rimentele werk aan deze stoornissen, bespreken wijaspecten van het symptomenbeeld, beeldvormend on-derzoek en neuropathologie van neuronale migratie-stoornissen bij de mens. Hierbij is gekozen voor een be-nadering vanuit het perspectief van de normale neuro-nale migratie van de hersenschors.15 16
diermodelonderzoekenDe reeler-muis werd in 1951 min of meer bij toeval ont-dekt door Falconer.17 18 Dit is een autosomaal recessievemuizenmutant met afwijkingen in zowel het cerebellumals in de hersenschors. De zogenaamde preplaat, de eer-ste schorslaag die wordt aangelegd,13 16 19 wordt wel ge-vormd, maar bij de vorming van de corticale plaat nietgesplitst in de marginale zone en de subplaat (figuur 2).In plaats hiervan blijft de preplaat bestaan als een ‘su-perplaat’. Dit resulteert in een ‘omgekeerde’ hersen-
schors, waarbij de vroege populatie neuronen opper-vlakkig komt te liggen en de late populatie neuronen indiepere delen van de hersenschors. Ondanks de aber-rante lokalisatie van de subplaatneuronen, die een be-langrijke rol spelen bij de ingroei van thalamocorticalevezels in de hersenschors, bereiken deze vezels wel dejuiste schorsgebieden en de neuronen die overeenko-men met laag IV.20 Ook de corticobulbaire en cortico-spinale vezelsystemen zijn vergelijkbaar met die in denormale situatie.21 De oorsprongcellen van de piramide-baan zijn echter verspreid door de gehele schors en nietbeperkt tot laag V.
Het eiwit reeline wordt voornamelijk door de Cajal-Retzius-cellen geproduceerd. De mogelijke werking er-van is samengevat in figuur 3. Reeline remt het enzym-complex p35/Cdk5-kinase in migrerende neuronen. Ditcomplex remt op zijn beurt de activiteit van het adhe-siemolecuul N-cadherine. Remming van het p35/Cdk5-kinase activeert dan ook via het N-cadherine de adhesietussen neuronen, waardoor de migratie van neuronenwordt gestopt. De reeler-muis vertoont, evenmin als devergelijkbare ‘scrambler’-mutaties,18 convulsies, een fe-nomeen dat wel bij de p35–/–-mutatie (Cdk5-knock-out-muis) wordt gevonden.7 22 In de hersenschors van zowelp35–/–- als Cdk5–/–-muizen kunnen migrerende neuro-nen de door N-cadherine gemedieerde adhesie niet on-derdrukken, waardoor ze niet in staat zijn voorbij desubplaatneuronen te migreren. Het humane reeline-gen(RELN) is gelokaliseerd op chromosoom 7q22 en is na-genoeg identiek aan dat van muizen.23 Onlangs werd ereen humane pendant van de reeler-muis gevonden bijeen vorm van lissencefalie die gepaard gaat met cere-bellaire hypoplasie.24
neuronale migratiestoornissen bij de mensVoor een aantal neuronale migratiestoornissen beginthet duidelijk te worden waar het normale ontwikke-lingsproces is verstoord (tabel). Dit geldt voor de liss-encefalieën en voor veel heterotopieën. Voor polymi-
Ned Tijdschr Geneeskd 2001 10 maart;145(10) 467
figuur 1. Schematisch overzicht van de verschillende typenneuronale migratiestoornissen.7 ccal = corpus callosum; Cd =nucleus caudatus; ci = capsula interna; hip = hippocampus; lv =laterale ventrikel; thal = thalamus.
periventriculaire nodulaireheterotopie
poly-micro-gyrie
subcorticale-bandheterotopie
leptomeningealeheterotopie
focale corticale dysplasie
agyrie
Cdci
ccal
thal
pons
lv
hip
figuur 2. Schema van de normale (a) en abnormale (b; de ‘reeler’-muis) vorming van de corticale plaat.19 Migrerende postmito-tische neuronen (mgn) bereiken langs radiaire gliacellen (rgc) de corticale plaat. Het eiwit reeline (zwarte stippen in de margi-nale zone) speelt een belangrijke rol bij het beëindigen van deze migratie. Bij de reeler-muis vindt geen splitsing plaats van depreplaat in de marginale zone en de subplaat. Cajal-Retzius-cellen (CR) en subplaatneuronen (spn) vormen de zogenaamdesuperplaat; cpn = corticale-plaatneuron.
marginale zone
corticale plaat
corticaleplaat
subplaat
super-plaat
intermediaire zone
ventriculaire zone
a b
crogyrie en andere corticale afwijkingen is dit echterminder duidelijk. De grote groep lissencefalieën en he-terotopieën kan pathogenetisch worden verdeeld in 4hoofdgroepen:8 (a) stoornissen aan het begin van de mi-gratie (de periventriculaire heterotopieën); (b) stoornis-sen in het verdere migratieproces (het lissencefalie type1 en het ‘dubbele-cortex’-syndroom); (c) problemen bijde passage van de subplaat, zoals bij de reeler-muis, enwaarschijnlijk ook bij de humane lissencefalievorm diegepaard gaat met cerebellaire hypoplasie; (d) verstorin-gen van de corticale architectuur, waarbij de hersen-schors op meerdere plaatsen wordt onderbroken en eenzeer onregelmatig aspect krijgt (‘cobble-stone’- of type-2-lissencefalie). Kenmerkend voor deze heterogenegroep van afwijkingen is de trias ernstige mentale re-tardatie, epilepsie en hypotonie. Een aantal van dezeneuronale migratiestoornissen, vooral de kleinere nodu-laire heterotopieën en focale corticale dysplasie, komt inaanmerking voor neurochirurgische excisie.25
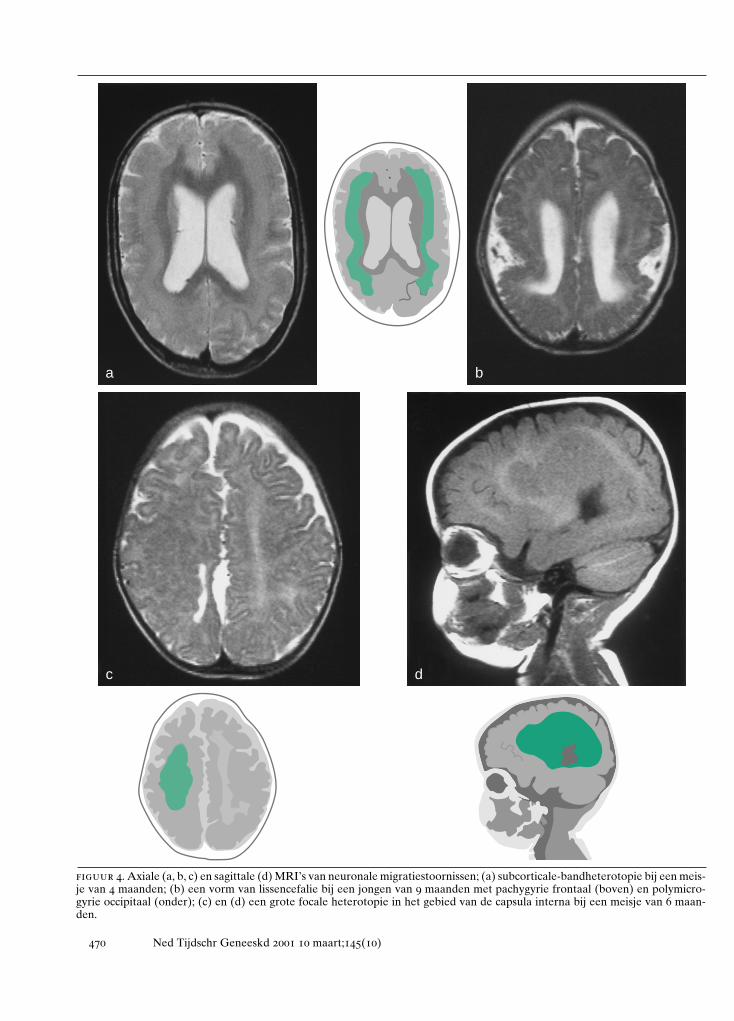
lissencefalieën en heterotopieënPeriventriculaire heterotopieën. Neuronen van de her-senschors ontstaan in de ventriculaire zone van hettelencephalon.16 Hierna treedt migratie op naar de cor-ticale plaat. Bij periventriculaire heterotopieën is eenbepaalde populatie neuronen hiertoe niet in staat enworden nodulaire heterotopieën gevormd (zie figuur 1,4c, 4d en 5d). Deze clusters van neuronen puilen dikwijlsuit in de ventrikelholte en zijn met CT- en MRI-onder-zoek op te sporen. Bilaterale periventriculaire nodulai-re heterotopieën (BPNH) komen familiair voor en zijnX-gebonden.26 Jongens met BPNH-mutaties overlijdenmeestal al vóór de geboorte.27 Meisjes met BPNH heb-ben een normale intelligentie. De belangrijkste klinischemanifestatie is het optreden van epilepsie, die waar-schijnlijk wordt veroorzaakt door de nodulaire hetero-topieën. Het syndroom werd gelokaliseerd op Xq28 enhet betreffende gen werd geïdentificeerd als filamine 1(FLN1). FLN1 codeert een actinebindend fosfoproteïne(filamine 1), dat onder andere betrokken is bij de re-gulatie van de celmigratie.28 Epilepsie bij unilateraleof bilaterale periventriculaire nodulaire heterotopieënkan therapieresistent zijn ten gevolge van de intrinsiekeepileptogeniciteit van de periventriculaire heteroto-pieën.29
Klassieke lissencefalie (type-1-lissencefalie) en het‘dubbele-cortex-syndroom’. Bij de zogenaamde klassie-ke lissencefalie hebben de hersenen een overwegendglad oppervlak en een dikke, meestal vierlagige schors.Wanneer karakteristieke craniofaciale afwijkingen, zo-als microcefalie, een hoog voorhoofd, een prominentachterhoofd, een brede neusbrug, micrognathie, eenlang filtrum en een prominente bovenlip aanwezig zijn,wordt van het Miller-Dieker-syndroom gesproken; zon-der deze afwijkingen, vooral aan het aangezicht, spreektmen van een ‘geïsoleerde lissencefalie’.30 De klassiekelissencefalie wordt gekenmerkt door diepe mentale re-tardatie, epilepsie, voedingsproblemen en een korte le-vensduur. Bij de klassieke lissencefalie zijn de hersenenmeestal kleiner dan normaal. Bij ernstige microcefaliewordt meestal de term ‘microlissencefalie’ gebruikt.9 31
Bij de klassieke lissencefalie vertonen de hersenen eengyrering die varieert van volledige afwezigheid van gyri(‘agyrie’) tot aanwezigheid van een tiental sulci over hetgehele brein (‘pachygyrie’). Kenmerkend is de dikke,vierlagige hersenschors (figuur 6a), die van buiten naarbinnen de volgende lagen heeft: (a) een moleculairelaag; (b) een dunne oppervlakkige laag van neuronen;(c) een celarme laag, en (d) een opvallend brede laagvan neuronen die aan een smalle laag periventriculairewitte stof grenst. Bij het Miller-Dieker-syndroom wordter meestal een gedeeltelijke deletie van de korte armvan chromosoom 17 (band 17p13.3) gevonden.9 Pa-tiënten met geïsoleerde lissencefalie hebben meestaleen minder ernstige graad van lissencefalie dan bij hetMiller-Dieker-syndroom.9 10 Deleties of mutaties vantenminste twee genen leiden tot deze klassieke vorm vanlissencefalie. Het Miller-Dieker-syndroom en de geïso-leerde vorm van lissencefalie worden veroorzaakt doordeleties van het LIS1- of PAFAH1B1-gen.32 Prenatale
468 Ned Tijdschr Geneeskd 2001 10 maart;145(10)
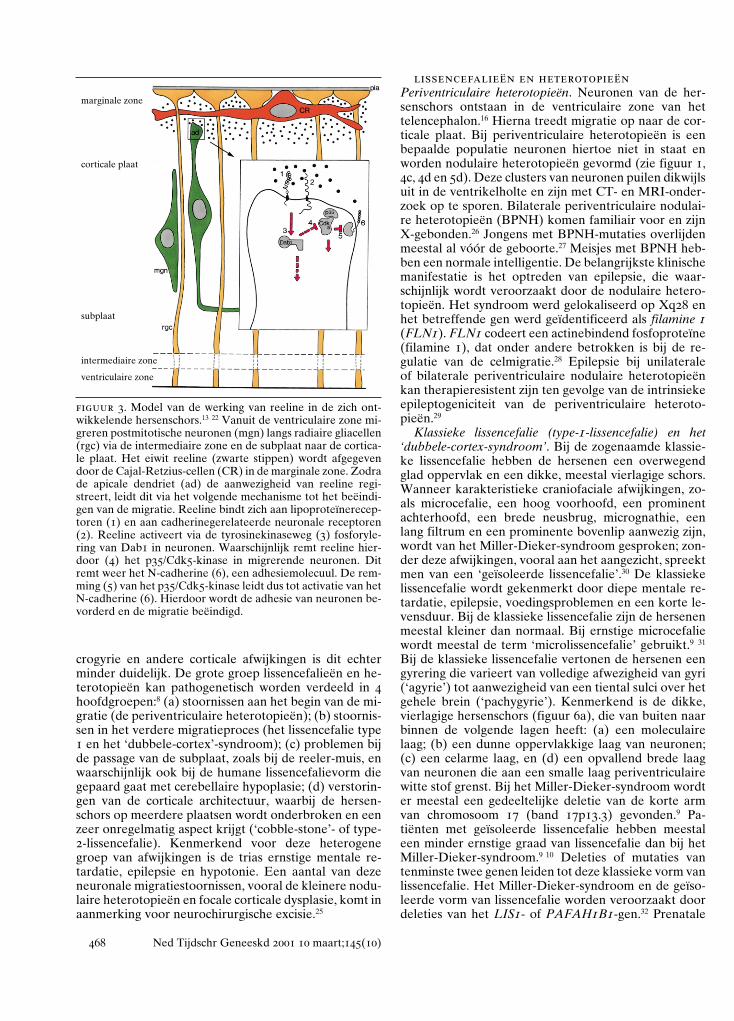
figuur 3. Model van de werking van reeline in de zich ont-wikkelende hersenschors.13 22 Vanuit de ventriculaire zone mi-greren postmitotische neuronen (mgn) langs radiaire gliacellen(rgc) via de intermediaire zone en de subplaat naar de cortica-le plaat. Het eiwit reeline (zwarte stippen) wordt afgegevendoor de Cajal-Retzius-cellen (CR) in de marginale zone. Zodrade apicale dendriet (ad) de aanwezigheid van reeline regi-streert, leidt dit via het volgende mechanisme tot het beëindi-gen van de migratie. Reeline bindt zich aan lipoproteïnerecep-toren (1) en aan cadherinegerelateerde neuronale receptoren(2). Reeline activeert via de tyrosinekinaseweg (3) fosforyle-ring van Dab1 in neuronen. Waarschijnlijk remt reeline hier-door (4) het p35/Cdk5-kinase in migrerende neuronen. Ditremt weer het N-cadherine (6), een adhesiemolecuul. De rem-ming (5) van het p35/Cdk5-kinase leidt dus tot activatie van hetN-cadherine (6). Hierdoor wordt de adhesie van neuronen be-vorderd en de migratie beëindigd.
marginale zone
corticale plaat
subplaat
intermediaire zone
ventriculaire zone

diagnose van een 17p13.3-deletie is mogelijk aan dehand van een biopsie van chorionvilli met behulp vanfluorescentie-in-situhybridisatie en de restrictiefrag-mentlengte-polymorfismetechniek.
Een tweede, X-gebonden vorm van lissencefalie(XLIS) komt op verschillende wijze bij jongens en meis-jes tot expressie.33 34 Bij jongens is het fenotype verge-lijkbaar met dat van de klassieke lissencefalie. Wel is ereen verschil in lokalisatie: de afwijkingen ten gevolgevan LIS1-mutaties komen vooral in de pariëtale en oc-cipitale schorsgebieden tot expressie, terwijl de XLIS-vormen vooral in de frontaalkwab tot uitdrukkingkomen.34 Meisjes met dit syndroom daarentegen zijnminder ernstig geretardeerd en hebben ook minder ern-stige vormen van epilepsie. Zij vertonen een subcortica-le-bandheterotopie, waarbij er onder de hersenschorsnog een tweede schors aanwezig is (zie figuur 1 en 4a).Er wordt dan ook wel van het ‘dubbele-cortexsyndroom’gesproken.9 35 Opmerkelijk is dat bij subcorticale-band-heterotopie, ondanks de epileptische activiteit, de sub-corticale laminaire heterotopieën wel degelijk ooknormale activiteit vertonen, zoals met behulp van func-tionele MRI werd aangetoond.36 Het gen voor deze X-gebonden vorm van lissencefalie, doublecortine (DCX),dat codeert voor het eiwit doublecortine, is gelokali-seerd op de lange arm van het X-chromosoom op posi-
tie Xq22.3.35 Een interessant diermodel voor de subcor-ticale-bandheterotopie is de tish-mutant (‘telencephalicinternal structural heterotopia’) bij de rat.37 Bij dezemalformatie bij de rat, die gepaard gaat met convulsies,wordt onder de gewone hersenschors een tweede hete-rotypische cortex gevormd met vergelijkbare verbindin-gen als bij de gewone schors.
Lissencefalie met cerebellaire hypoplasie. Bij de ree-ler-muis zijn, zoals eerder toegelicht, corticale neuronenniet in staat de subplaat te passeren. Bij deze muis wor-den ook ernstige cerebellaire afwijkingen gevonden. Bijeen humaan type lissencefalie, dat gepaard gaat metcerebellaire hypoplasie (LCH), werd onlangs een muta-tie van het reeline-gen gevonden op chromosoom 7q22(LCHRELN).24 Bij MRI-onderzoek bleek de lissencefalieveel minder uitgesproken te zijn dan bij de klassiekevorm van lissencefalie en werd een ernstige hypoplasievan het cerebellum gevonden. Bij de aangedane kinde-ren is de ontwikkeling van zowel de cognitie als de mo-toriek ernstig gestoord.
‘Cobble-stone’-lissencefalie (type-2-lissencefalie). Dezogenaamde cobble-stone-lissencefalie of type-2-lissen-cefalie wordt gekenmerkt door een hobbelig aspect vande hersenschors, een abnormale witte stof, verwijdeventrikels en afwijkingen aan de hersenstam en het ce-rebellum, waaronder cerebellaire polymicrogyrie.9 De
Ned Tijdschr Geneeskd 2001 10 maart;145(10) 469
Neuronale migratiestoornissen
aandoening over- chromosoom- genproduct klinische manifestatieserving locatie
neuronale heterotopie in witte stofbilaterale periventriculaire nodulaire heterotopie XL Xq28 filamine 1 epilepsie
(bij meisjes; bij jongens letaal)
klassieke lissencefalieMiller-Dieker-syndroom AD 17p13.3 LIS1 ernstige mentale retardatie, epilepsie en spasticiteit geïsoleerde lissencefalie AD 17p13.3 LIS1 mentale retardatie, epilepsieX-gebonden lissencefalie (XLIS, bij jongens),
respectievelijk subcorticale-bandheterotopie (XLIS of DCX, bij meisjes) XL Xq22.3 doublecortine mentale retardatie met geringere expressie bij
meisjes, epilepsie
lissencefalie met cerebellaire hypoplasielissencefalie met cerebellaire hypoplasie (RELN) AR 7q22 LCHRELN ernstige mentale retardatie, epilepsie en hypotonie
‘cobble-stone’-lissencefalieWalker-Warburg-syndroom AR onbekend onbekend ernstige retardatie, hypotonie, hydrocefalus, oogafwij-
kingen en cerebellaire afwijkingen‘spier-oog-hersenziekte’ AR 1p32-p34 onbekend meestal ernstige retardatie, epilepsie, hypotonie en
glaucoomcongenitale Fukuyama-spieratrofie AR 9q31-33 fukutine ernstige retardatie, in 20% van de gevallen epilepsie,
hypotonie en geringe spasticiteit
polymicrogyrieAicardi-syndroom XL Xp22 onbekend ernstige retardatie, chorioretinopathie, agenese van
corpus callosum, infantiele spasmenZellweger-syndroom (heterogeen) AR zeker 10 peroxisomale cerebrohepatorenaal syndroom: mentale retardatie,
verschillende enzymen epilepsie, hypotonie, hepatomegalie, hartafwijkin-genen gen, polycystische nieren
andere afwijkingenKallmann-syndroom XL Xp22.3 KAL1 anosmie, hypothalamisch of hypogonadotroop
AD, AR onbekend onbekend hypogonadisme, secundaire retardatie
AD = autosomaal dominant; AR = autosomaal recessief; XL = X-gebonden.
470 Ned Tijdschr Geneeskd 2001 10 maart;145(10)
figuur 4. Axiale (a, b, c) en sagittale (d) MRI’s van neuronale migratiestoornissen; (a) subcorticale-bandheterotopie bij een meis-je van 4 maanden; (b) een vorm van lissencefalie bij een jongen van 9 maanden met pachygyrie frontaal (boven) en polymicro-gyrie occipitaal (onder); (c) en (d) een grote focale heterotopie in het gebied van de capsula interna bij een meisje van 6 maan-den.
a b
c d

hersenschors is chaotisch gestructureerd door een com-binatie van agyrie, pachygyrie en polymicrogyrie meteen onregelmatig oppervlak. Van buiten naar binnenkunnen de volgende lagen worden onderscheiden: (a)een brede, buitenste meningeale zone met proliferatiesvan neuronen en gliacellen in de leptomeningen; (b) eenbuitenste cellulaire zone met brede onregelmatige op-hopingen van neuronen; (c) een diepere cellulaire zonevan onregelmatig gerangschikte neuronen; (d) heteroto-pieën die gedeeltelijk in de witte stof liggen en (e) eenbinnenste gliazone. Het hersenoppervlak is zeer onre-gelmatig en lijkt sterk op een straat geplaveid met kas-seien. De leptomeningeale heterotopieën worden overi-
gens ook bij andere aangeboren afwijkingen van hetcentrale zenuwstelsel gevonden, zoals bij het syndroomvan Zellweger (zie figuur 6b), bij verkregen destructie-ve aandoeningen, zoals porencefalie ten gevolge vanvasculaire stoornissen of infecties, en bij het foetale al-coholsyndroom.6
De cobble-stone-lissencefalie gaat meestal gepaardmet oogmisvormingen en spierdystrofie en komt in ver-schillende varianten voor.9 Het Walker-Warburg-syn-droom wordt gekenmerkt door diffuse agyrie, hydroce-falus, oogafwijkingen en cerebellaire afwijkingen (figuur5a en 5b). Het is autosomaal recessief en komt over degehele wereld voor. De precieze lokalisatie van het be-
Ned Tijdschr Geneeskd 2001 10 maart;145(10) 471
figuur 5. Neuropathologische bevindingen bij neuronale migratiestoornissen; (a) en (b) een casus met het Walker-Warburg-syn-droom met diffuse agyrie (a) en ernstige hydrocefalus (b); (c) polymicrogyrie; (d) een kleine periventriculaire heterotopie naasthet laterale ventrikel.
a b
c d
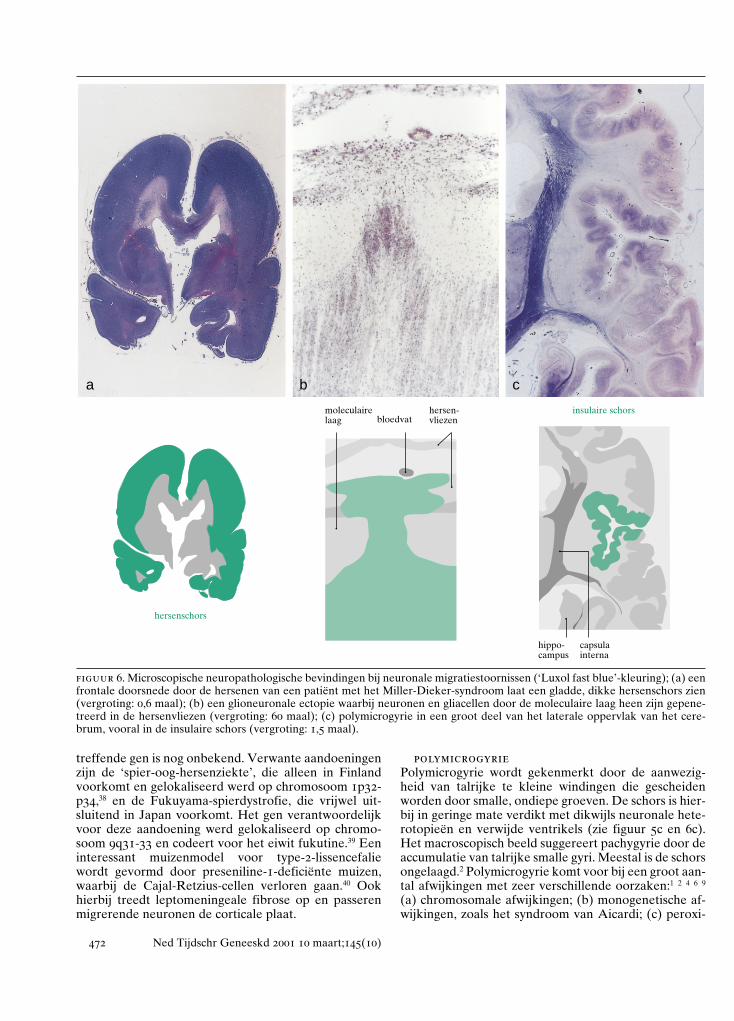
treffende gen is nog onbekend. Verwante aandoeningenzijn de ‘spier-oog-hersenziekte’, die alleen in Finlandvoorkomt en gelokaliseerd werd op chromosoom 1p32-p34,38 en de Fukuyama-spierdystrofie, die vrijwel uit-sluitend in Japan voorkomt. Het gen verantwoordelijkvoor deze aandoening werd gelokaliseerd op chromo-soom 9q31-33 en codeert voor het eiwit fukutine.39 Eeninteressant muizenmodel voor type-2-lissencefaliewordt gevormd door preseniline-1-deficiënte muizen,waarbij de Cajal-Retzius-cellen verloren gaan.40 Ookhierbij treedt leptomeningeale fibrose op en passerenmigrerende neuronen de corticale plaat.
polymicrogyriePolymicrogyrie wordt gekenmerkt door de aanwezig-heid van talrijke te kleine windingen die gescheidenworden door smalle, ondiepe groeven. De schors is hier-bij in geringe mate verdikt met dikwijls neuronale hete-rotopieën en verwijde ventrikels (zie figuur 5c en 6c).Het macroscopisch beeld suggereert pachygyrie door deaccumulatie van talrijke smalle gyri. Meestal is de schorsongelaagd.2 Polymicrogyrie komt voor bij een groot aan-tal afwijkingen met zeer verschillende oorzaken:1 2 4 6 9
(a) chromosomale afwijkingen; (b) monogenetische af-wijkingen, zoals het syndroom van Aicardi; (c) peroxi-
472 Ned Tijdschr Geneeskd 2001 10 maart;145(10)
figuur 6. Microscopische neuropathologische bevindingen bij neuronale migratiestoornissen (‘Luxol fast blue’-kleuring); (a) eenfrontale doorsnede door de hersenen van een patiënt met het Miller-Dieker-syndroom laat een gladde, dikke hersenschors zien(vergroting: 0,6 maal); (b) een glioneuronale ectopie waarbij neuronen en gliacellen door de moleculaire laag heen zijn gepene-treerd in de hersenvliezen (vergroting: 60 maal); (c) polymicrogyrie in een groot deel van het laterale oppervlak van het cere-brum, vooral in de insulaire schors (vergroting: 1,5 maal).
a b c
hersenschors
moleculaire laag
hersen-vliezenbloedvat
insulaire schors
hippo-campus
capsulainterna

somale aandoeningen, zoals het syndroom van Zell-weger; (d) destructieve laesies, zoals porencefalie ten ge-volge van vasculaire stoornissen, vaak in het vasculari-satiegebied van de A. cerebri media en na cytomegalo-virusinfecties; (e) tweelingschap en (f) overmatig alco-holgebruik tijdens de zwangerschap.
andere corticale afwijkingenOnder corticale dysplasie wordt verstaan dat op een ofmeer plaatsen in de hersenschors neuronen en gliacellenop abnormale plaatsen zijn gelegen. Deze afwijkingwerd beschreven bij dyslexie41 en vooral in de tempo-raalkwab van patiënten met epilepsie.42 Corticale dys-plasie is vaak focaal, maar kan ook op meerdere plaat-sen voorkomen,1 43 en kan worden beschouwd als eenlichte vorm van een neuronale migratiestoornis. Mo-gelijk zijn de betreffende neuronen al bij hun ontstaanin de ventriculaire zone afwijkend en hierdoor niet instaat normale lagen te vormen. Men onderscheidt welfocale corticale dysplasie, gegeneraliseerde corticale dys-plasie, focale Rolandische macrogyrie, dysplastischeafwijkingen van de temporaalkwab en hemimegalence-falie. Een intrigerende migratiestoornis wordt gevondenbij het overigens zeldzame syndroom van Kallmann, datwordt gekenmerkt door anosmie en infertiliteit. Hierbijbestaat er een stoornis in de migratie van gonadotro-pine-‘releasing’-hormoon(GnRH)-producerende neuro-nen vanuit de neusholte naar de hypothalamus. Het ei-wit KAL1 ontbreekt bij de X-gebonden vorm van dezeaandoening.44 Hoe het KAL1-eiwit de migratie vanGnRH-neuronen reguleert, is nog onduidelijk. De ge-nen die betrokken zijn bij de autosomaal dominante ende autosomaal recessieve vormen van deze aandoeningzijn nog onbekend. Hormonale substitutietherapie bijpatiënten met het Kallmann-syndroom is noodzakelijkom de secundaire geslachtskenmerken te induceren.
conclusiesParallel aan de snelle vooruitgang in ons begrip van demechanismen die een rol spelen bij de vorming van dehersenschors, vooral op basis van dierexperimenteelwerk, hebben de mogelijkheden van de moderne af-beeldingstechnieken onze kennis van de neuronale mi-gratiestoornissen bij de mens sterk vergroot. Nieuweneurochirurgische behandelingsmethoden bieden nusoms ruimte voor behandeling van de gevolgen vanneuronale migratiestoornissen, vooral de aandoeningendie gepaard gaan met ernstige, niet goed op medicatiereagerende vormen van epilepsie. Correlatie van feno-type en genotype heeft geleid tot nieuwe inzichten in hetontstaan van de hersenschors en de daarbij optredendeontwikkelingsstoornissen. Voor een correcte determi-natie van neuronale migratiestoornissen bij een indivi-duele patiënt blijft een zorgvuldige morfologische be-schrijving van belang, eens temeer omdat mede op basisvan deze determinatie advies gegeven moet worden overprognose, behandeling en herhalingsrisico.
Mw.M.de Leeuw, medisch illustrator, hielp bij het vervaardi-gen van de illustraties.
abstractDevelopment and development disorders of the human brain.III. Neuronal migration disorders of the cerebrum– Neuronal migration disorders of the cerebral cortex form aheterogeneous group of abnormalities, characterised by men-tal retardation, epilepsy and hypotonia.– They are prevalent in 1% of the population and in 20-40% ofthe untreatable forms of epilepsy.– Disorders at the start of the migration result in nodular hete-rotopias. Bilateral periventricular nodular heterotopias are X-linked disorders, in which cortical neurons are unable to leavetheir position at the ventricular surface due to the absence offilamin 1.– The large group of lissencephalies can be divided into a num-ber of syndromes, each of which is characterised by a gene mu-tation (LIS1, DCX, RELN). These mutations result in agyriaand pachygyria, which are characteristic for this group.– A number of these abnormalities, especially the smaller nod-ular heterotopias and focal cortical dysplasia, may be treatedby neurosurgical excision.
literatuur1 Norman MG, McGillivray BC, Kalousek DK, Hill A, Poskitt KJ.
Congenital malformations of the brain. Pathologic, embryologic,clinical, radiologic and genetic aspects. New York: Oxford Univer-sity Press; 1995.
2 Harding BN, Copp AJ. Malformations. In: Graham DI, Lantos PL,editors. Greenfield’s neuropathology. Londen: Arnold; 1996. p. 397-533.
3 Mischel PS, Nguyen LP, Vinters HV. Cerebral cortical dysplasia as-sociated with pediatric epilepsy. Review of neuropathologic featuresand proposal for a grading system. J Neuropathol Exp Neurol 1995;54:137-53.
4 Barth PG. Disorders of neuronal migration. Can J Neurol Sci 1987;14:1-16.
5 Aicardi J. Malformations of the CNS. In: Aicardi J, editor. Diseasesof the nervous system in childhood. Clinics in developmental medi-cine. Nr 115/118. Londen: MacKeith Press; 1992. p. 108-202.
6 Lammens M. Developmental neuropathology in etiology and patho-genesis of human malformations: a personal contribution. Acta Bio-med Lovaniensia 1997;154:1-175.
7 Copp AJ, Harding BN. Neuronal migration disorders in humans andin mouse models – an overview. Epilepsy Res 1999;36:133-41.
8 Gleeson JG, Walsh CA. Neuronal migration disorders: from genet-ic diseases to developmental mechanisms. Trends Neurosci 2000;23:352-9.
9 Dobyns WB, Truwit CL. Lissencephaly and other malformations ofcortical development: 1995 update. Neuropediatrics 1995;26:132-47.
10 Barkovich AJ, Kuzniecky RI, Dobyns WB, Jackson GD, Becker LE,Evrard P. A classification scheme for malformations of cortical de-velopment. Neuropediatrics 1996;27:59-63.
11 Kollias SS, Ball WS. In: Ball WS, editor. Pediatric neuroradiology.Philadelphia: Lippincott-Raven; 1997. p. 91-174.
12 Curran T, D’Arcangelo G. Role of reelin in the control of brain de-velopment. Brain Res Rev 1998;26:285-94.
13 Lambert de Rouvroit C, Goffinet AM. The reeler mouse as a modelof brain development. Adv Anat Embryol Cell Biol 1998;150:1-106.
14 D’Arcangelo G, Nakajima K, Miyata T, Ogawa M, Mikoshiba K,Curran T. Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. J Neurosci 1997;17:23-31.
15 Donkelaar HJ ten, Wesseling P, Lammens M, Renier WO, MullaartRA, Thijssen HOM. Ontwikkeling en ontwikkelingsstoornissen vanhet humane brein. I. Vroege ontwikkeling van de grote hersenen.Ned Tijdschr Geneeskd 2001;145:345-53.
16 Donkelaar HJ ten, Wesseling P, Lammens M, Thijssen HOM,Renier WO, Gabreëls FJM. Ontwikkeling en ontwikkelingsstoor-nissen van het humane brein. II. Ontwikkeling van de hersenschorsen de grote baansystemen. Ned Tijdschr Geneeskd 2001;145:401-10.
17 Falconer DS. Two new mutations, trembler and reeler with neuro-logical action in the mouse. J Genet 1951;50:192-201.
Ned Tijdschr Geneeskd 2001 10 maart;145(10) 473

18 Hatten ME. Central nervous system neuronal migration. Annu RevNeurosci 1999;22:511-39.
19 Pearlman AL, Faust PL, Hatten ME, Brunstrom JE. New directionsfor neuronal migration. Curr Opin Neurobiol 1998;8:46-54.
20 Molnár Z, Adams R, Goffinet AM, Blakemore C. The role of thefirst postmitotic cortical cells in the development of thalamocorticalinnervation in the reeler mouse. J Neurosci 1998;18:5746-65.
21 Terashima T. Course and collaterals of corticospinal fibers arisingfrom the sensorimotor cortex of the reeler mouse. Dev Neurosci1995;17:8-19.
22 Homayouni R, Curran T. Cortical development: Cdk5 gets intosticky situations. Curr Biol 2000;10:R331-4.
23 DeSilva U, D’Arcangelo G, Braden VV, Chen J, Miao GG, CurranT, et al. The human reelin gene: isolation, sequencing, and mappingon chromosome 7 [letter]. Genome Res 1997;7:157-64.
24 Hong SE, Shugari YY, Huang DT, Al Shahwan S, Grant PE,Hourihane JO, et al. Autosomal recessive lissencephaly with cer-ebellar hypoplasia is associated with human RELN mutations. NatGenet 2000;26:93-6.
25 Sisodiya SM. Surgery for malformations of cortical developmentcausing epilepsy. Brain 2000;123(Pt 6):1075-91.
26 Huttenlocher PR, Taravath S, Mojtahedi S. Periventricular hetero-topia and epilepsy. Neurology 1994;44:51-5.
27 Eksioglu YZ, Scheffer IE, Cardenas P, Knoll J, DiMario F, RamsbyG, et al. Periventricular heterotopia: an X-linked dominant epilepsylocus causing aberrant cerebral cortical development. Neuron 1996;16:77-87.
28 Fox JW, Lamperti ED, Eksioglu YZ, Hong SE, Feng Y, GrahamDA, et al. Mutations in filamin 1 prevent migration of cerebral cor-tical neurons in human periventricular heterotopia. Neuron 1998;21:1315-25.
29 Sisodiya SM, Free SL, Thom M, Everitt AE, Fish DR, Shorvon SD.Evidence for nodular epileptogenicity and gender differences inperiventricular nodular heterotopia. Neurology 1999;52:336-41.
30 Dobyns WB, Stratton RF, Greenberg F. Syndromes with lissen-cephaly. I: Miller-Dieker and Norman-Roberts syndromes and iso-lated lissencephaly. Am J Med Genet 1984;18:509-26.
31 Barkovich AJ, Ferriero DM, Barr RM, Gressens P, Dobyns WB,Truwit CL, et al. Microlissencephaly: a heterogeneous malformationof cortical development. Neuropediatrics 1998;29:113-9.
32 Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, DobynsWB, et al. Isolation of a Miller-Dieker lissencephaly gene con-taining G protein beta-subunit-like repeats. Nature 1993;364:717-21.
33 Dobyns WB, Andermann E, Andermann F, Czapansky-Beilman D,Dubeau F, Dulac O, et al. X-linked malformations of neuronal mi-gration. Neurology 1996;47:331-9.
34 Dobyns WB, Truwit CL, Ross ME, Matsumoto N, Pilz DT,Ledbetter DH, et al. Differences in the gyral pattern distinguishchromosome 17-linked and X-linked lissencephaly. Neurology 1999;53:270-7.
35 Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, SchefferI, et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a puta-tive signaling protein. Cell 1998;92:63-72.
36 Pinard JM, Feydy A, Carlier R, Perez N, Pierot L, Burnod Y.Functional MRI in double cortex: functionality of heterotopia.Neurology 2000;54:1531-3.
37 Lee KS, Schottler F, Collins JL, Lanzino G, Couture D, Rao A, etal. A genetic animal model of human neocortical heterotopia asso-ciated with seizures. J Neurosci 1997;17:6236-42.
38 Cormand B, Avela K, Pihko H, Santavuori P, Talim B, TopalogluH, et al. Assignment of the muscle-eye-brain disease gene to 1p32-p34 by linkage analysis and homozygosity mapping. Am J HumGenet 1999;64:126-35.
39 Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E,Nomura Y, et al. An ancient retrotransposal insertion causesFukuyama-type congenital muscular dystrophy. Nature 1998;394:388-92.
40 Hartmann D, De Strooper B, Saftig P. Presenilin-1 deficiency leadsto loss of Cajal-Retzius neurons and cortical dysplasia similar to hu-man type 2 lissencephaly. Curr Biol 1999;9:719-27.
41 Galaburda AM, Sherman GF, Rosen GD, Aboitiz F, Geschwind N.Developmental dyslexia: four consecutive patients with corticalanomalies. Ann Neurol 1985;18:222-33.
42 Hardiman O, Burke T, Phillips J, Murphy S, O’Moore B, StauntonH, et al. Microdysgenesis in resected temporal neocortex: incidenceand clinical significance in focal epilepsy. Neurology 1988;38:1041-7.
43 Meencke HJ. Diagnosis of cortical and subcortical dysplasias in epi-lepsy. In: Schmidt D, Schachter SC, editors. Epilepsy-problem solv-ing in clinical practice. Londen: Dunitz; 2000. p. 95-109.
44 Franco B, Guioli S, Pragliola A, Incerti B, Bardoni B, Tonlorenzi R,et al. A gene deleted in Kallmann’s syndrome shares homology withneural cell adhesion and axonal path-finding molecules. Nature1991;353:529-36.
Aanvaard op 31 januari 2001
474 Ned Tijdschr Geneeskd 2001 10 maart;145(10)
de aandoeningHet fragiele-X-syndroom is een X-chromosomaal ge-bonden verstandelijke handicap met bijkomende ken-merken, zoals een lang gelaat met vergrote, afstaandeoren, vergrote testikels en specifieke gedragskenmer-ken (figuur). Andere verschijnselen zijn onder meer een
hoog, smal verhemelte (52%), strabismus (36%), over-strekbare handgewrichten (67%), viervingerlijn (eentransversale lijn over de breedte van de hand; 25%),platvoeten (71%) en hartruis of klik (18%).1 2 Een kindmet het fragiele-X-syndroom is vaak hyperactief, ‘flad-dert’ met de armen/handen, herhaalt woorden (echola-lie) en heeft verminderd oogcontact. Vroegtijdige onder-kenning van het syndroom bij het kind leidt tot beterebegeleiding en gerichte medische follow-up van ondermeer epilepsie (20%), met bindweefsel verband hou-dende problemen (platvoeten en mitralisklepprolaps)en frequente middenoorontstekingen (85%).
Medische vignetten
Van gen naar ziekte; het fragiele-X-syndroom: erfelijke mentale retardatie dooreen groeiend gen
l.b.a.de vries en b.a.oostra
Erasmus Universiteit, afd. Klinische Genetica, Postbus 1738, 3000 DRRotterdam.Dr.L.B.A.de Vries, klinisch geneticus; prof.dr.B.A.Oostra, moleculairgeneticus.Correspondentieadres: prof.dr.B.A.Oostra ([email protected]).





























