Embed Size (px)
Citation preview

Nefrotik Sendromun Genetiği
Dr. Fatih ÖzaltınHacettepe Üniversitesi Tıp Fakültesi
Pediatrik Nefroloji Bilim Dalı
8.Ulusal Çocuk Nefroloji Kongresi30 Ekim 2014, Antalya
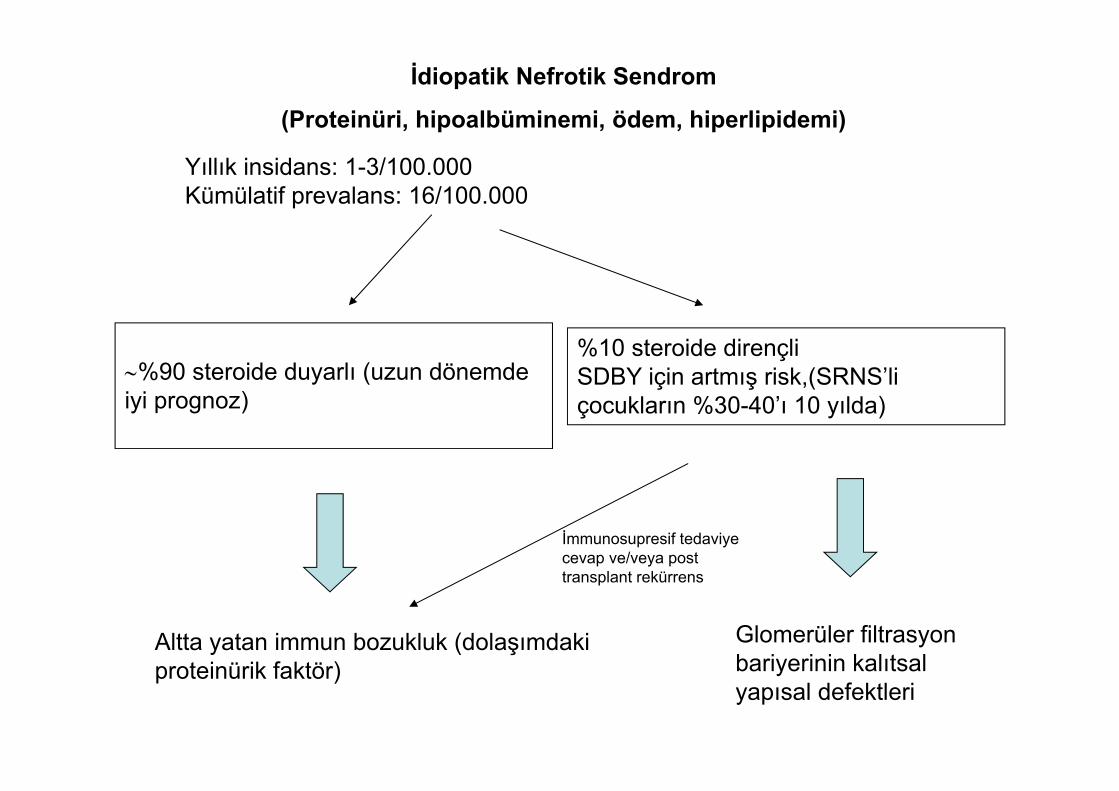
%90 steroide duyarlı (uzun dönemde iyi prognoz)
İdiopatik Nefrotik Sendrom
(Proteinüri, hipoalbüminemi, ödem, hiperlipidemi)
Yıllık insidans: 1-3/100.000 Kümülatif prevalans: 16/100.000
%10 steroide dirençli SDBY için artmış risk,(SRNS’li çocukların %30-40’ı 10 yılda)
Altta yatan immun bozukluk (dolaşımdaki proteinürik faktör)
İmmunosupresif tedaviye cevap ve/veya post transplant rekürrens
Glomerüler filtrasyon bariyerinin kalıtsal yapısal defektleri
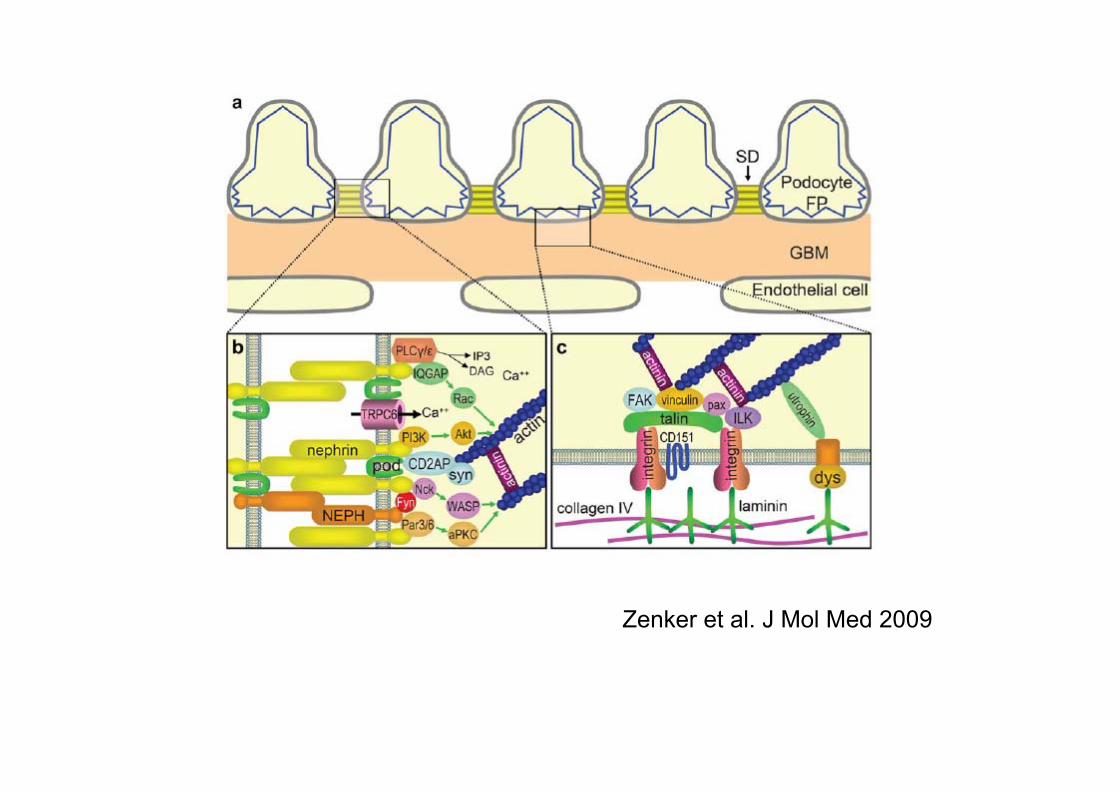
Zenker et al. J Mol Med 2009
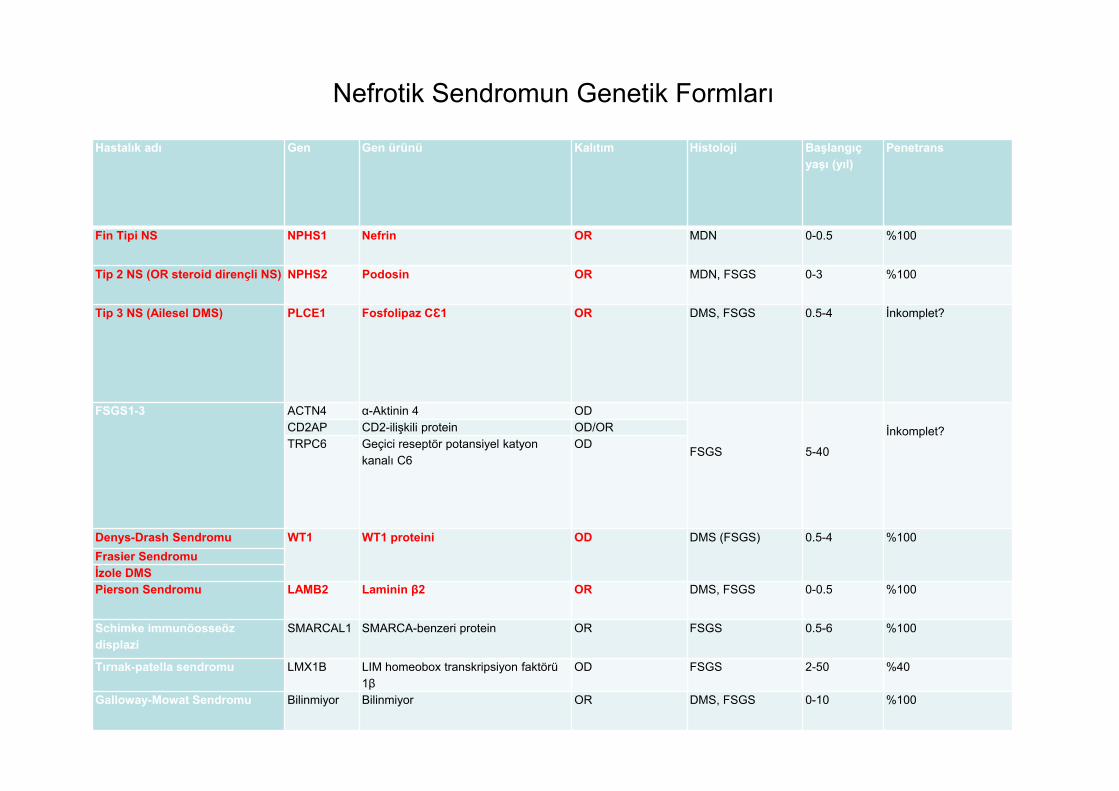
Nefrotik Sendromun Genetik Formları
Hastalık adı Gen Gen ürünü Kalıtım Histoloji Başlangıç yaşı (yıl)
Penetrans
Fin Tipi NS NPHS1 Nefrin OR MDN 0-0.5 %100
Tip 2 NS (OR steroid dirençli NS) NPHS2 Podosin OR MDN, FSGS 0-3 %100
Tip 3 NS (Ailesel DMS) PLCE1 Fosfolipaz CƐ1 OR DMS, FSGS 0.5-4 İnkomplet?
FSGS1-3 ACTN4 α-Aktinin 4 OD
FSGS 5-40
İnkomplet?CD2AP CD2-ilişkili protein OD/ORTRPC6 Geçici reseptör potansiyel katyon
kanalı C6OD
Denys-Drash Sendromu WT1 WT1 proteini OD DMS (FSGS) 0.5-4 %100Frasier Sendromuİzole DMSPierson Sendromu LAMB2 Laminin β2 OR DMS, FSGS 0-0.5 %100
Schimke immunöosseöz displazi
SMARCAL1 SMARCA-benzeri protein OR FSGS 0.5-6 %100
Tırnak-patella sendromu LMX1B LIM homeobox transkripsiyon faktörü 1β
OD FSGS 2-50 %40
Galloway-Mowat Sendromu Bilinmiyor Bilinmiyor OR DMS, FSGS 0-10 %100
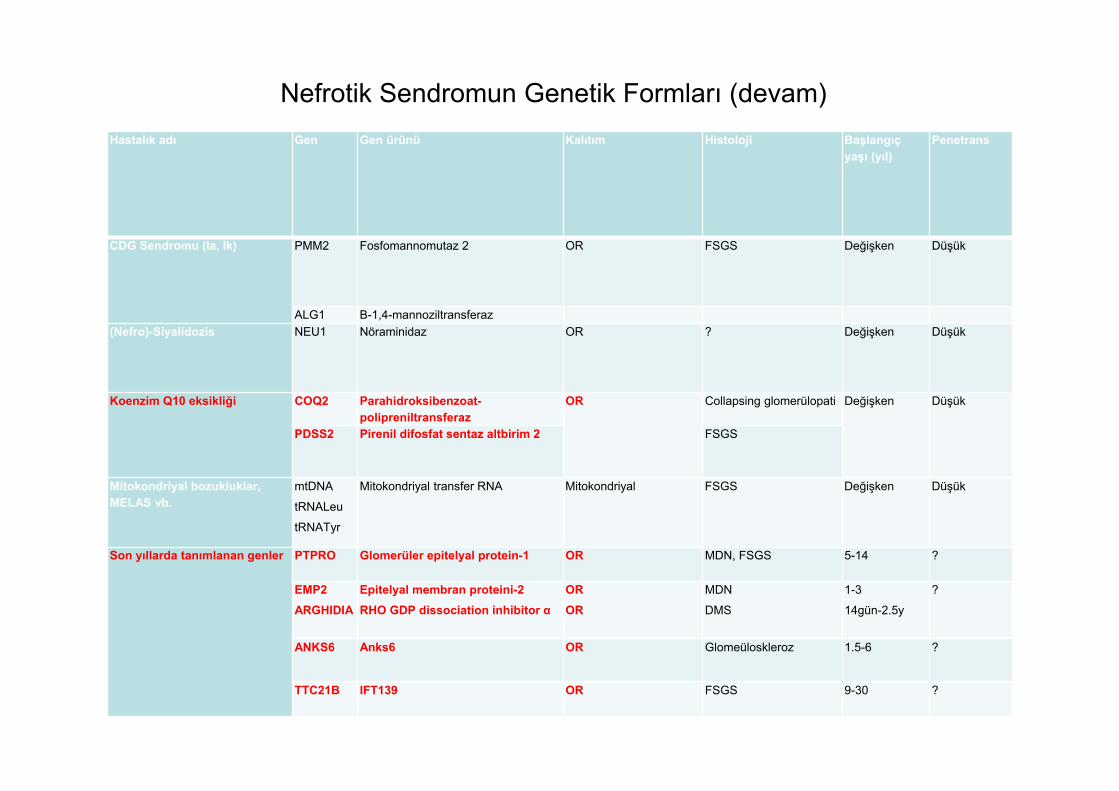
Hastalık adı Gen Gen ürünü Kalıtım Histoloji Başlangıç yaşı (yıl)
Penetrans
CDG Sendromu (Ia, Ik) PMM2 Fosfomannomutaz 2 OR FSGS Değişken Düşük
ALG1 Β-1,4-mannoziltransferaz(Nefro)-Siyalidozis NEU1 Nöraminidaz OR ? Değişken Düşük
Koenzim Q10 eksikliği COQ2 Parahidroksibenzoat-polipreniltransferaz
OR Collapsing glomerülopati Değişken Düşük
PDSS2 Pirenil difosfat sentaz altbirim 2 FSGS
Mitokondriyal bozukluklar, MELAS vb.
mtDNA
tRNALeu
tRNATyr
Mitokondriyal transfer RNA Mitokondriyal FSGS Değişken Düşük
Son yıllarda tanımlanan genler PTPRO Glomerüler epitelyal protein-1 OR MDN, FSGS 5-14 ?
EMP2ARGHIDIA
Epitelyal membran proteini-2RHO GDP dissociation inhibitor α
OROR
MDN
DMS
1-3
14gün-2.5y
?
ANKS6 Anks6 OR Glomeüloskleroz 1.5-6 ?
TTC21B IFT139 OR FSGS 9-30 ?
Nefrotik Sendromun Genetik Formları (devam)

Podosit/Slit Diafram Hastalıkları
Proteinüri-Nefrotik Sendrom
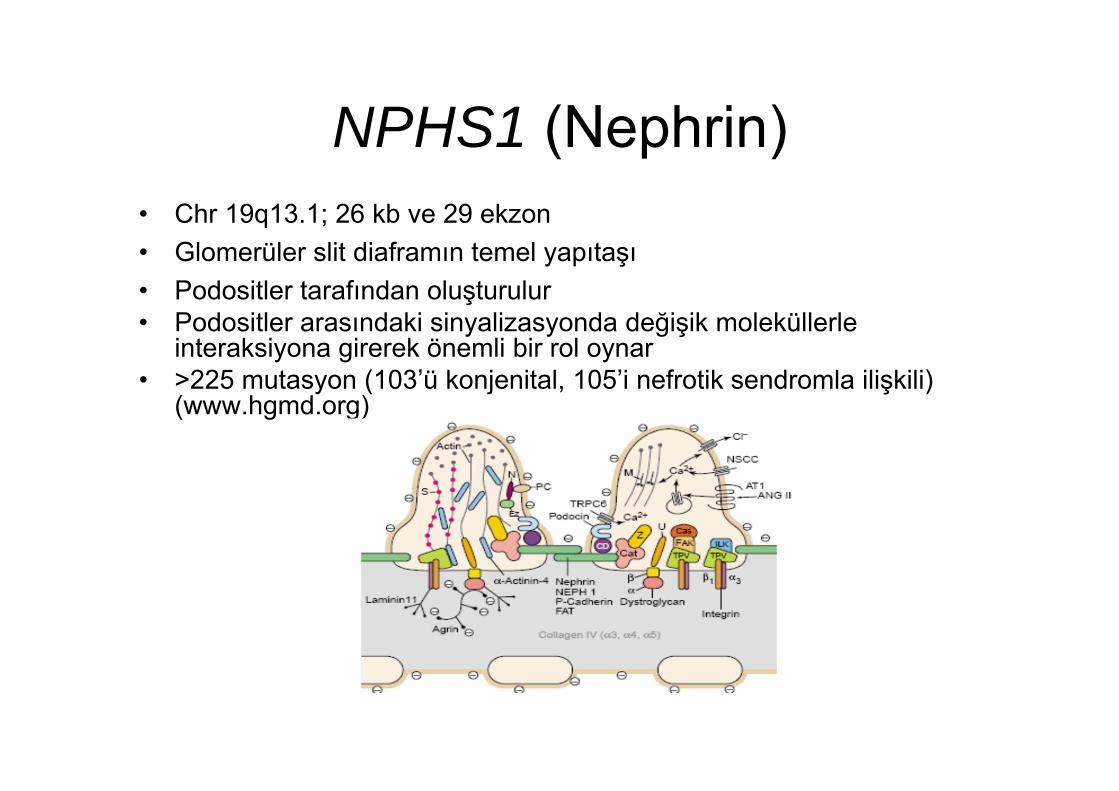
NPHS1 (Nephrin)• Chr 19q13.1; 26 kb ve 29 ekzon• Glomerüler slit diaframın temel yapıtaşı• Podositler tarafından oluşturulur• Podositler arasındaki sinyalizasyonda değişik moleküllerle
interaksiyona girerek önemli bir rol oynar• >225 mutasyon (103’ü konjenital, 105’i nefrotik sendromla ilişkili)
(www.hgmd.org)
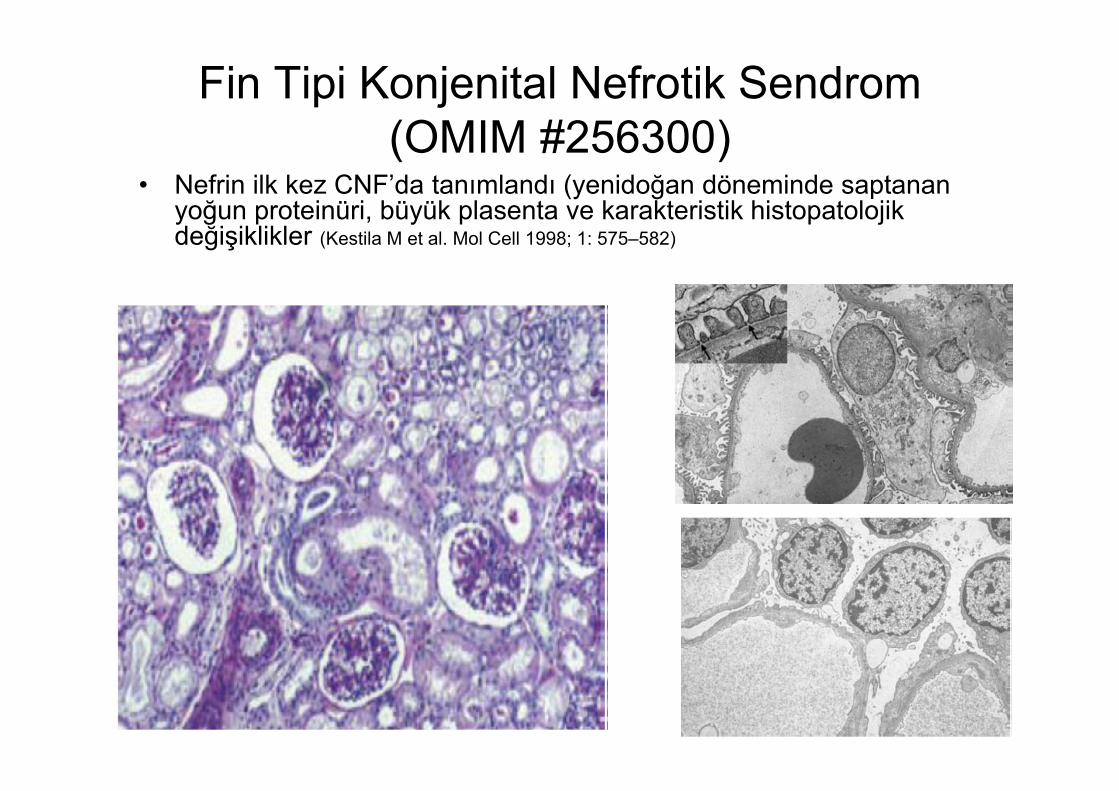
Fin Tipi Konjenital Nefrotik Sendrom (OMIM #256300)
• Nefrin ilk kez CNF’da tanımlandı (yenidoğan döneminde saptanan yoğun proteinüri, büyük plasenta ve karakteristik histopatolojik değişiklikler (Kestila M et al. Mol Cell 1998; 1: 575–582)

NPHS1• NPHS1 Fin major (p.L41fsX91), Fin minor (p.R1109X), (Fin
hastaların mutant allelerinni %78 ve %16’sından sorumlu) • Fin orijinli olmayan CNF’lu hastalarda NPHS1 mutasyon saptanma
oranı %66 (Lenkkeri U et al Am J Hum Genet 1999; 64:51–61)
• Konj. NS’lu tüm vakaların yaklaşık %40’ından sorumlu (Hinkes B, et al Pediatrics 2007; 119:e907–e919)
• İnfantil/çocukluk çağı NS’a da neden olabilir (Philippe A et al. J Am Soc Nephrol 2008; 19: 1871–1878)
farklı mutasyonlar farklı klinik şiddet spektrumuna neden olabilir• Diğer fenotipler: DMS, FSGS, MCNS

Tüm NPHS1 mutasyonları ağır bir fenotipe neden
olmayabilir
NPHS1 geninde homozigot p.R1160X mutasyonlu hastaların %50’sinde hafif fenotip. İlk 3 ayda ağır bir NS’u takiben spontan parsiyel ya da tam bir remisyonun eşlik ettiği daha iyi bir seyir.
Hastaların çoğu kız
(Koziell A et al. Hum Mol Genet 2002; 11:379–388)

NPHS1 ve İnfantil/Çocukluk Çağı NS
• Hayatın ilk 3 ayından sonra podosin mutasyonu saptanmamış NS’lu hastaların %7-14’ünde NPHS1 (Philippe A et al. J Am Soc Nephrol 2008; 19:1871–1878; Santin S et al. Kidney Int 2009;
76:1268–1276)
• MCD, FSGS, mesangioproliferative GN• 1 hafif mutasyonun varlığı geç başlangıçı açıklar• Nefrin mutasyonu olan (C265R/V822M) iki kardeşte spontan
parsiyel remisyon ve ÜSYE ile birlikte relapslar tanımlanmıştır (Kitamura A
et al. Kidney Int 2007; 71:946–951)
• Aynı NPHS1 mutasyonu olan (p.R827X and p.R976S) 27 yaşında FSGS ve normal renal fonksiyonlu yetişkin ile NS’lu infant düzenleyici genler veya çevresel faktörlerin rolü olduğunu düşündürmekte (Philippe A et al. J Am Soc Nephrol 2008)
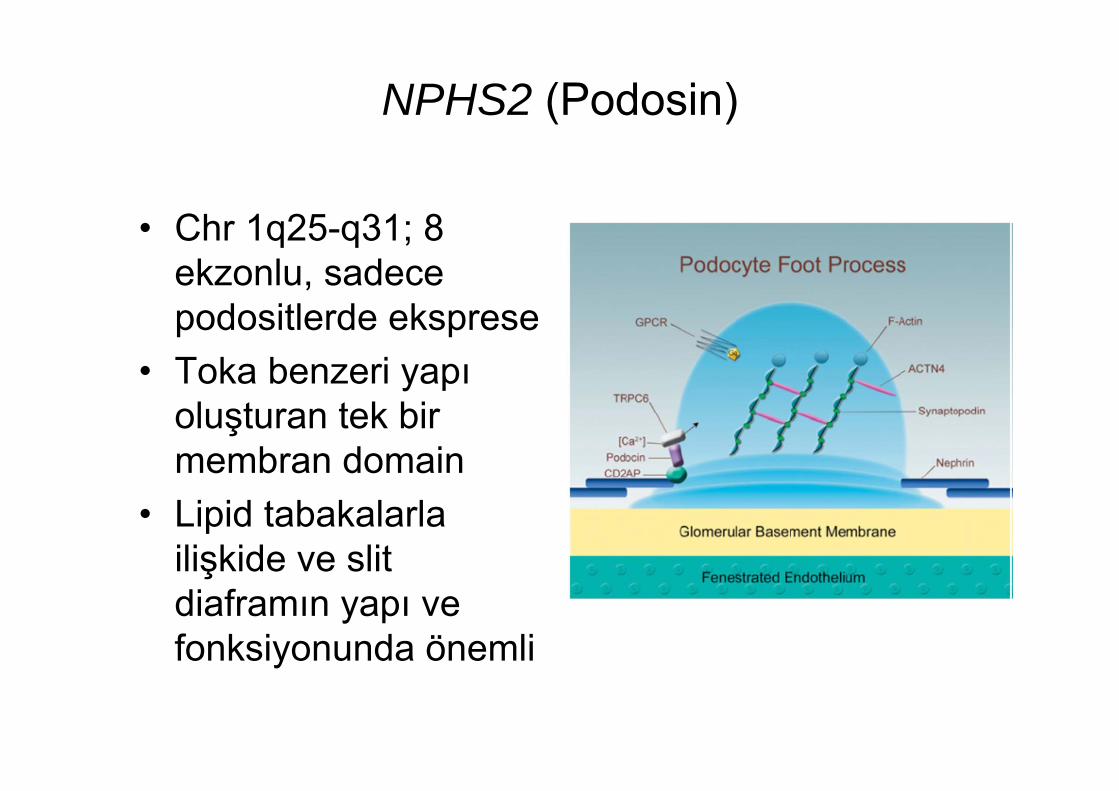
NPHS2 (Podosin)
• Chr 1q25-q31; 8 ekzonlu, sadece podositlerde eksprese
• Toka benzeri yapı oluşturan tek bir membran domain
• Lipid tabakalarla ilişkide ve slit diaframın yapı ve fonksiyonunda önemli

İlk tanım OR SRNS ve FSGS’li çocuklar (OMIM #600995) (Boute et al.,2000)
Sporadik çocukluk çağı SRNS (Caridi et al., 2001; Karle et al., 2002)
Adult başlangıçlı SRNS (Tsukaguchi et al., 2000, 2002)
Konjenital ve infantil nefrotik sendrom’da %15-39 (Hinkes et al., 2007, 2008;Machuca et
al., 2010; Santin et al., 2011). Bizim kohortta %15 (yayınlanmamış bilgi)
Familyal SRNS vakalarının %30-40’ından (Karle et al., 2002;Weber et al.,2004; Berdeli et al., 2007; Hinkes et al., 2007, 2008)
Sporadik SRNS vakalarının %10-30’undan sorumlu (Caridi et al., 2001, 2003; Karle et al., 2002; Ruf et al., 2004; Weber et al., 2004; Berdeli et al., 2007; Hinkes et al.,2008; Megremis et al., 2009; Jungraithmayr et al., 2011)
Homozigot veya birleşik heterozigot mutasyonlar6 yaşından önce SRNS (Weber et al., 2004; Hinkes et al., 2008)
Hızla SDBY’ne gidiş ancak düşük postransplant rekürrens riski (%9 vs %30-50) (Boute et al., 2000; Weber et al., 2004; Bouchireb et al, 2013)
NPHS2 mutasyonları

Ekim 1999-Eylül 2013 126 patojenik mutasyon (25’i yeni)
53 missense, 17 nonsense, 11 küçük insersiyon, 26 küçük delesyon, 16 splice, 2 indel
43 varyantın önemi bilinmiyor (çoğunlukla heterozigot ve in slico analizlerde benign)
http://databases.lovd.nl/shared/genes/NPHS2

R229Q varyasyonuArg229Gln podosin in vitro olarak nefrine anlamlı olarak düşük bağlanma göstermekte (Tsukaguchi et al., 2002)
Genel popülasyonda ve ince membran nefropati hastalarında proteinüriye artmış eğilim yaratmakta (Pereira et al., 2004, Voskarides et al., 2012)
Diğer taraftan birçok çalışmada R229Q varyasyonunun SRNS ve normal kontrollerde benzer sıklıkta olduğu gösterilmiştir (Ruf et al., 2004; Weber et al., 2004;
McKenzie et al., 2007) bu varyant heterozigot olduğunda SRNS için bir risk faktörü değil? (Karle et al., 2002; McKenzie et al., 2007)
Prevalans Avrupada 0.03–0.13 (Karle et al., 2002; Tsukaguchi et al., 2002; Caridi et al., 2003; Lowik et al., 2003; Pereira et al., 2004; Ruf et al., 2004; Weber et al., 2004; Aucella et al., 2005; Franceschini et al., 2006; Kottgen et al., 2008); Afrika orijinli olanlarda 0.005–0.025 (Tsukaguchi et al., 2002; Pereira et al., 2004; Dusel et al., 2005)
Sonuç: R229Q şimdilk non-nötral polimorfizm olarak kabul edilmekte
R229Q mutasyonu eğer 1 patojenik NPHS2 mutasyonu varsa geç başlangıçlı SRNS ile ilişkili (median 13 yaş; SDBY median 26yaş (10-50yıl) ) (Machuca E et al.
Kidney Int 2009; 75:727–735, Tory et al. Nat Genet 2014). Homozigot R229Q hastalıktan sorumlu değil


Genotip-Fenotip İlişkisiBaşlangıç yaşı: İki mutasyon varlığında ort 41.2 ± 5.9 ay iken mutasyon olmayanlarda ort 76.8 ay (Weber et al., 2004; Berdeli et al., 2007; Hinkes et al., 2008)
SDBY’ne gidiş: Homozigot-birleşik heterozigot NPHS2 mutasyonu taşıyanlarda SDBY 8.6 ± 5.2 yıl iken 1 patojenik mutasyon+R229Q taşıyanlarda ort 26.1 ± 18.9 yıl (Machuca et al., 2009)
Böbrek dışı bulgular: Podosin hemen tamamen podositlerde ifade edilir. Nadir kalp (ventrikül hipertrofisi ve PS) ve göz bulguları (Exotropia , anisometropic amblyopia ve Mittendorf ’s dot) tanımlanmıştır (Frishberg et al., 2006; Ozaltin et al., 2008; Sonmez et al., 2008; Machuca et al., 2010)
Tedaviye yanıt: NPHS’de 2 resesif mutasyonu olan hastalar standart steroid tedavisine yanıt vermez ancak posttransplant rekürrens oranı anlamlı derecede düşük (%35’e karşı %8) (Ruf R et al J Am Soc Nephrol 2004; 15: 722–32; Weber S et al Kidney Int 2004; 66: 571–79
.

NPHS2 mutasyonu ve Konjenital NS
• CNS’lu Avrupalı hastaların %51’inden sorumlu
• Ortanca başlangıç yaşı 4 hafta
(Hinkes B, et al Pediatrics 2007; 119:e907–e919)

PLC1
• Fosfolipaz C epsilon 1’i kodlar
• Membran fosfolipidlerinin hidrolizi ve ikincil haberciler
oluşmasını katalizler (IP3, DAG), böylelikle hücre
büyüme ve farklılaşmasına katılır

PLC1 ve İnfantil/Çocukluk Çağı NS (OMIM #610725)
• DMS’ye neden olan en önemli gen; FSGS’nin nadir nedeni
• İzole DMS’li ailelerde mutasyon saptanma oranı %28.6(Gbadegesin R et al
2008; Nephrol Dial Transplant 23:1291–1297)
• Çocukluk çağı FSGS’den oluşan 19 hastanın hiçbirisinde PLCE1
mutasyonu bulunmamış (Lowik M et al Nephrol Dial Transplant 2008;23:3146–3151)
• İdiopatik/herediter FSGS’li 69 hastanın hiçbirisinde mutasyon
saptanmamış (median başlangıç yaşı 26 yıl, range 1–66 yıl) (Gbadegesin
R, et al. Pediatr Nephrol 2009; 24:281–285)

WT1 ve İnfantil/Çocukluk Çağı NS
• Dominant veya de novo• Familyal olmayan izole SRNS’li hastaların
%9’undan sorumlu• Wilms tümör, WAGR send, Denys-Drash
send ve Frasier send ile ilişkili• İzole DMS’ye neden olur (başlangıç yaşı
hayatın ilk birkaç günü- 2 yaş; izole FSGS (1-14 yaş))

61 WT1 pozitif hasta vs 700 WT1 negatif hastaFSGS heriki grupta eşitDMS WT1 için spesifik (%34)Ambigus ve/veya ürogenital anomaliler (%52)Wilms tümör (%38) ve gonadoblastoma (%5)
DNA bağlanma bölgesini etkileyen missense mutDMS (%74), erken SRNS ve hızla SDBY’ne gidişTrunkasyon mutWilms tm (%78), tipik olarak geç başlangıçlı SRNSIntronik mut: İzole SRNS (%37) (median başlangıç 4.5y), FSGS ve SDBY’ne yavaş ilerleyiş (median 13.6y)
WT1 mut!

LAMB2• Laminin β2 kodlar
• Hücre adezyon, proliferasyon, farklılaşma ve migrasyonda önemli rolleri olan bazal
membranların vazgeçilmez yapıtaşı
• Laminin-521 (α5, β2, ve γ1 alt üniteler) β2 zinciri içeren en yaygın laminin izoformu
• Spesifik olarak belli yerlerde exprese edilir (GBM, değişik oküler yapılar ve
nöromüsküler sistem; Pierson sendromundaki organ tutulum paterni ile uyumlu)
• Pierson sendromu (CNS (DMS)+mikrokori; konj. Kas güçsüzlüğü/myasteni ve gelişme
geriliği dahil ağır nörolojik-gelişimsel defisit)
• Nadiren daha hafif ya da oligosemptomatik hastalık varyantları ile ilişkili olabilir

Matejas V et al. Hum Mutat 2010;31:992-1002
LAMB2
• Mutasyonların çoğu (nonsense, frameshift) trunke proteine neden olur
• Pierson sendromu heterojenöz değil (tipik vakalarda LAMB2 mutasyon saptama sıklığı %98-100)
• Homozigot ya da birleşik heterozigot mutasyonların çoğu hayatın ilk yılında NS ile kendini gösterir ve hızla SDBY’ne ilerler
• SDBY doğumda olabilir ve nefrotik semptomları baskılayabilir
• Genellikle LAMB2 mutasyonları izole NS’a nadiren neden olur

İlk 1 yıl içindeki NS’ların 2/3’ü ile CNS’ların %85’i 4 gen mutasyonları ile açıklanabilir
NPHS1 %39.8 NPHS2 %39.8 WT1 %2.2 LAMB2 %4.4
Hinkes B, et al. Pediatrics 2007; 119: e907–e919
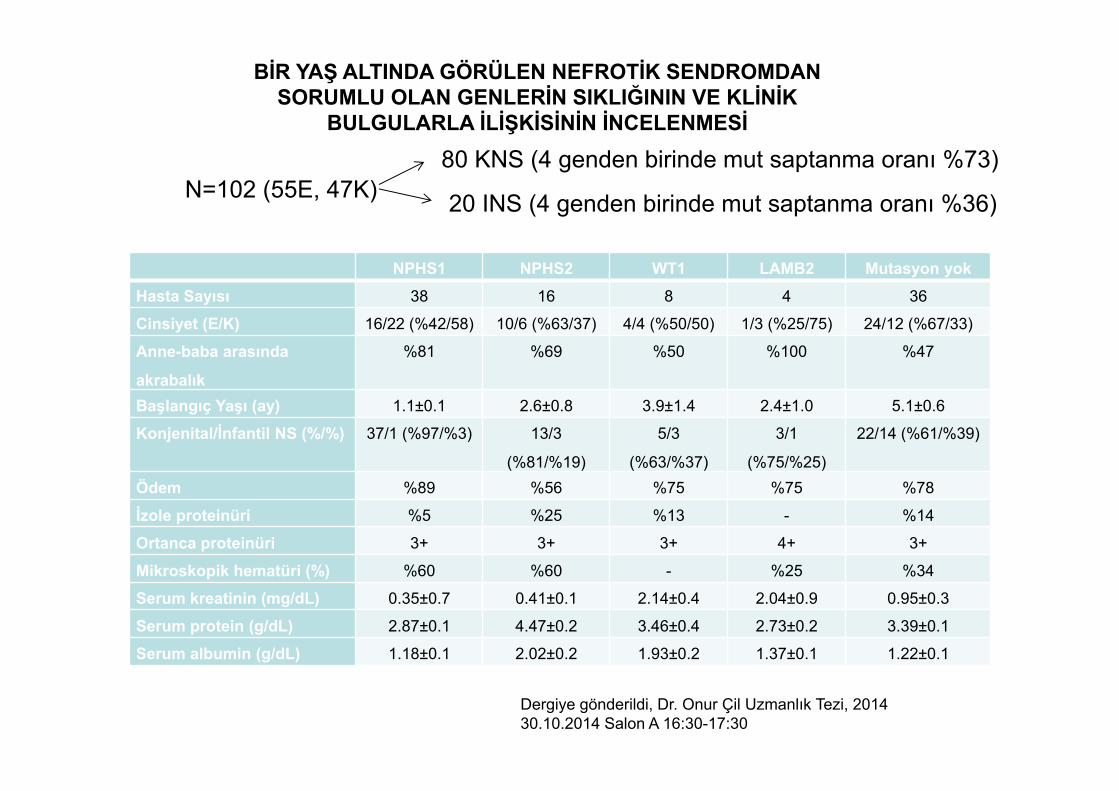
BİR YAŞ ALTINDA GÖRÜLEN NEFROTİK SENDROMDAN SORUMLU OLAN GENLERİN SIKLIĞININ VE KLİNİK
BULGULARLA İLİŞKİSİNİN İNCELENMESİ
N=102 (55E, 47K)80 KNS (4 genden birinde mut saptanma oranı %73)
20 INS (4 genden birinde mut saptanma oranı %36)
Dergiye gönderildi, Dr. Onur Çil Uzmanlık Tezi, 201430.10.2014 Salon A 16:30-17:30
NPHS1 NPHS2 WT1 LAMB2 Mutasyon yokHasta Sayısı 38 16 8 4 36
Cinsiyet (E/K) 16/22 (%42/58) 10/6 (%63/37) 4/4 (%50/50) 1/3 (%25/75) 24/12 (%67/33)
Anne-baba arasında
akrabalık
%81 %69 %50 %100 %47
Başlangıç Yaşı (ay) 1.1±0.1 2.6±0.8 3.9±1.4 2.4±1.0 5.1±0.6
Konjenital/İnfantil NS (%/%) 37/1 (%97/%3) 13/3
(%81/%19)
5/3
(%63/%37)
3/1
(%75/%25)
22/14 (%61/%39)
Ödem %89 %56 %75 %75 %78
İzole proteinüri %5 %25 %13 - %14
Ortanca proteinüri 3+ 3+ 3+ 4+ 3+
Mikroskopik hematüri (%) %60 %60 - %25 %34
Serum kreatinin (mg/dL) 0.35±0.7 0.41±0.1 2.14±0.4 2.04±0.9 0.95±0.3
Serum protein (g/dL) 2.87±0.1 4.47±0.2 3.46±0.4 2.73±0.2 3.39±0.1
Serum albumin (g/dL) 1.18±0.1 2.02±0.2 1.93±0.2 1.37±0.1 1.22±0.1

1. Konjenital ve infantil nefrotik sendrom hastalarının 2/3’ünde NPHS1,
NPHS2, WT1 ve LAMB2 genlerinin birisinde hastalığa neden olan
mutasyonlar bulunmaktadır.
2. Bu 4 genden birisinde mutasyon saptama oranı konjenital nefrotik
sendrom hastalarında infantil nefrotik sendrom hastalarına göre iki kat
daha yüksektir.
3. Türk toplumunda en sık mutasyon saptanan gen NPHS1’dir.
Sonuç;

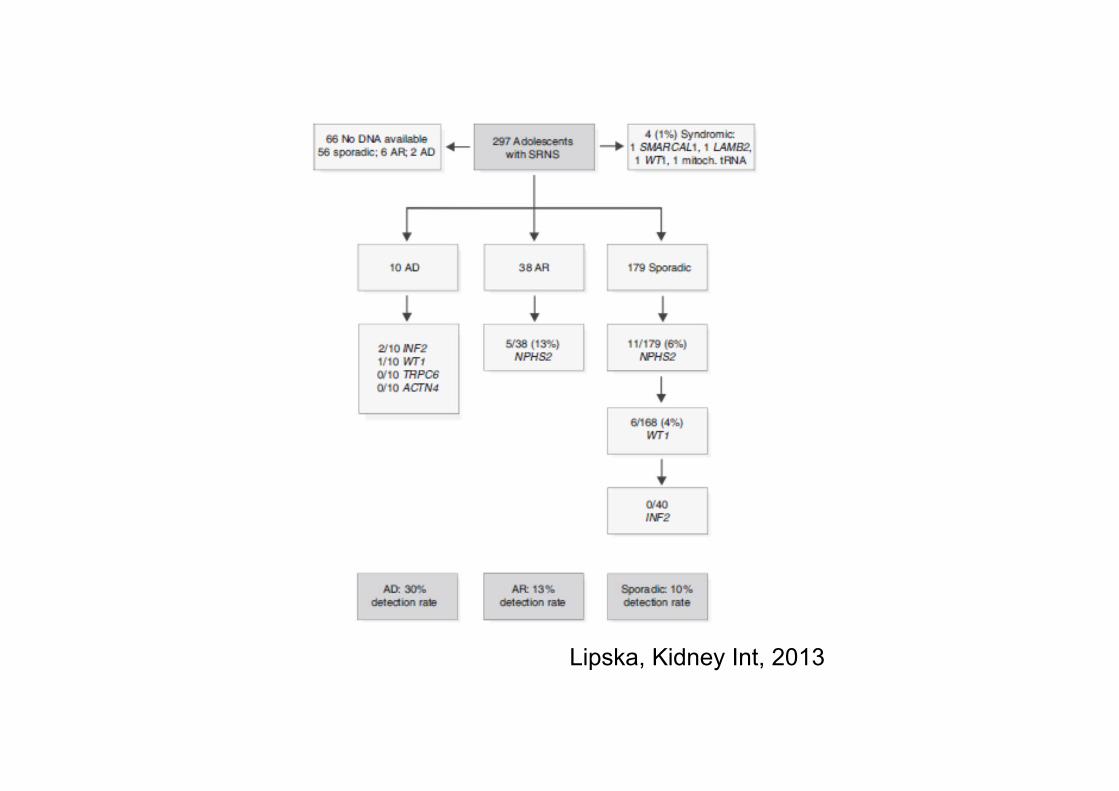
Lipska, Kidney Int, 2013

Overall mut saptama oranı %11
OR bireylerin %13’ü; sporadik bireylerin %6’sı podosin ilişkili
NPHS2 mutasyonu taşıyan bireylerin %56’sı R229Q ile birleşik heterozigot
Sporadik SRNS’lerin %4’ünde; OD’ların %10’unda WT1
OD’ların %20’sinde INF2 mutasyonu varken sporadiklerin hiçbirinde yok
Sporadik SRNS’li adölesanlarda NPHS2’nin tüm ekzonları ile WT1 ekzon 8 ve 9 taranmalı
Steroid direnci tanımlanır tanımlanmaz ikinci basamak tedavilere geçmeden önce genetik tarama yapılmalı ve mutasyon saptananlara bu tedaviler başlanmamalı

Am J Hum Genet. 2011;89(1):139-47
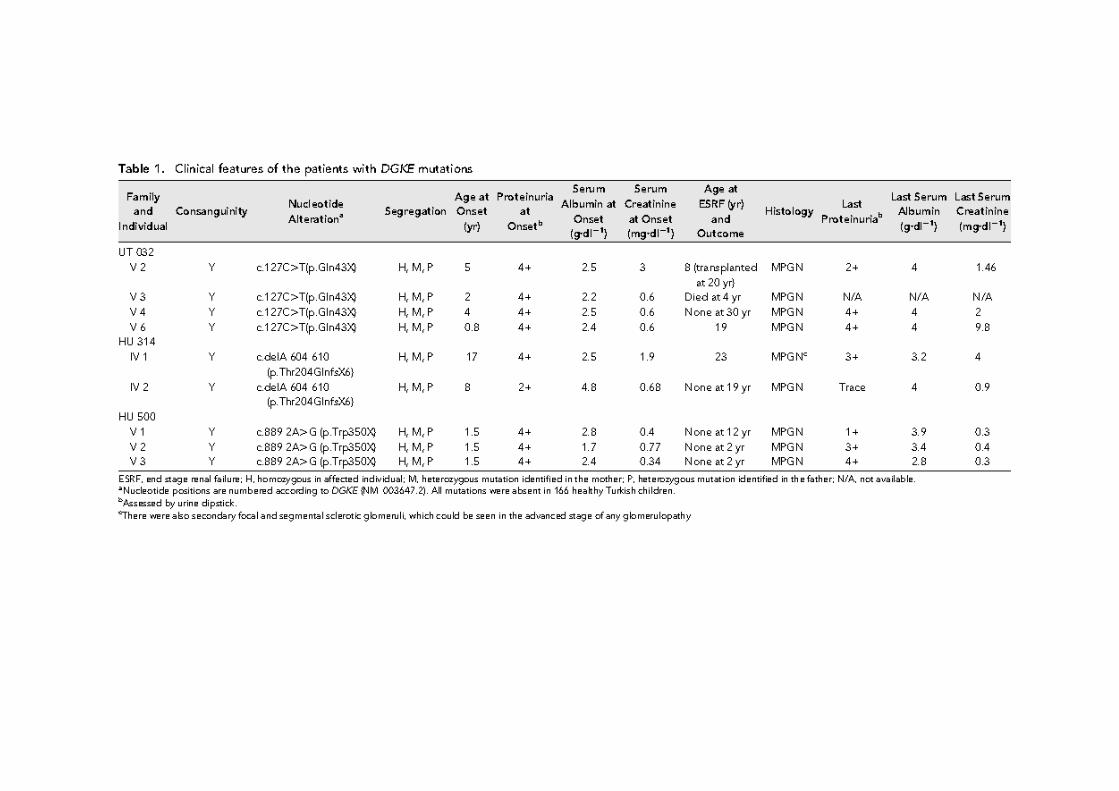
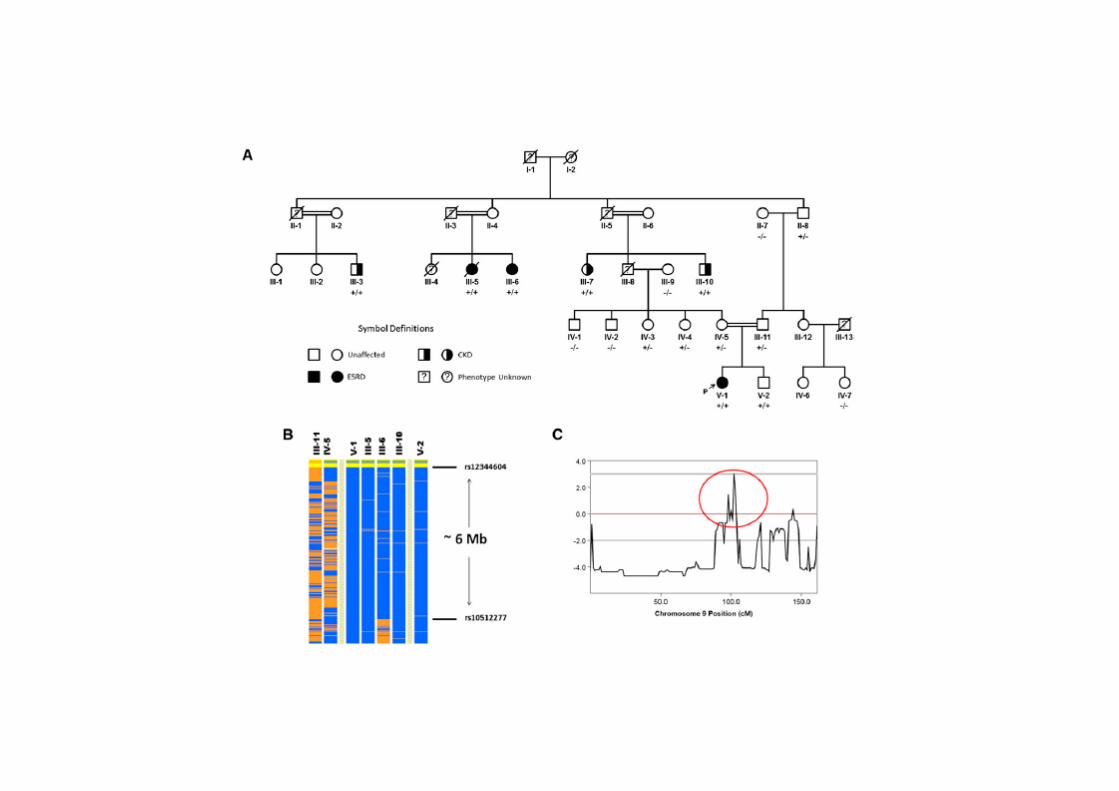
OR SRNS’li 17 aileden 29 etkilenmiş, 22 normal birey
MIM 614196, Nephrotic syndrome type 6
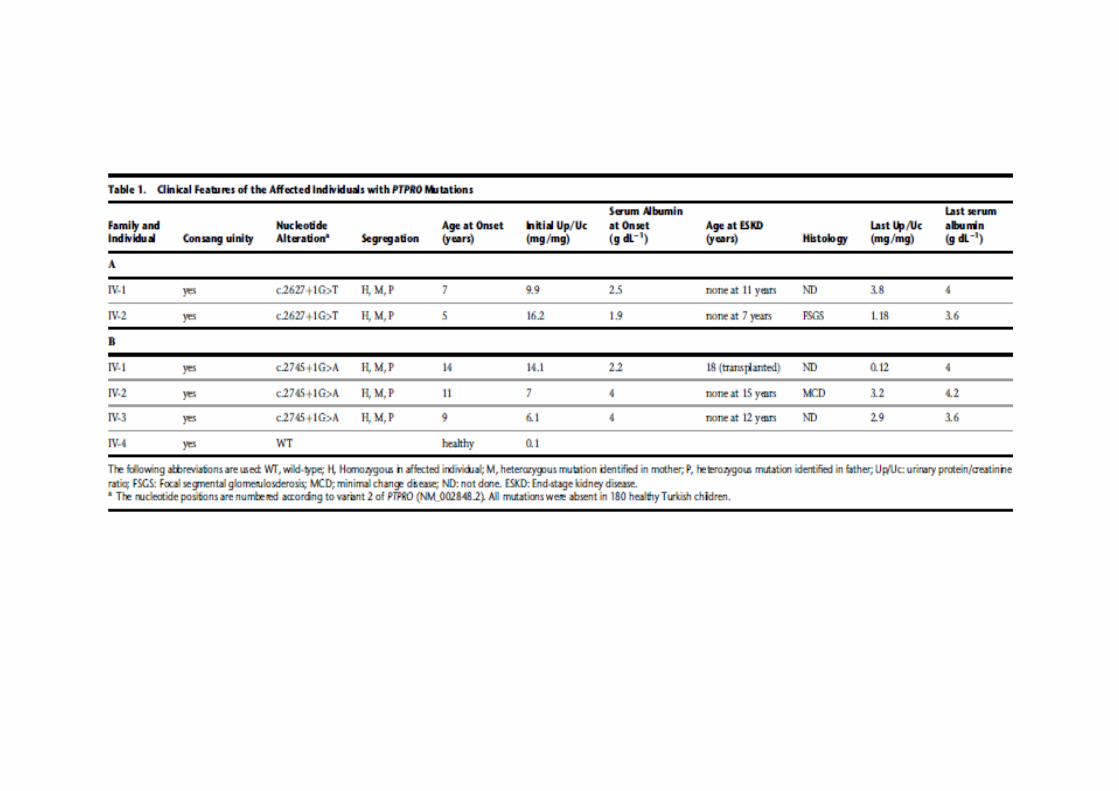

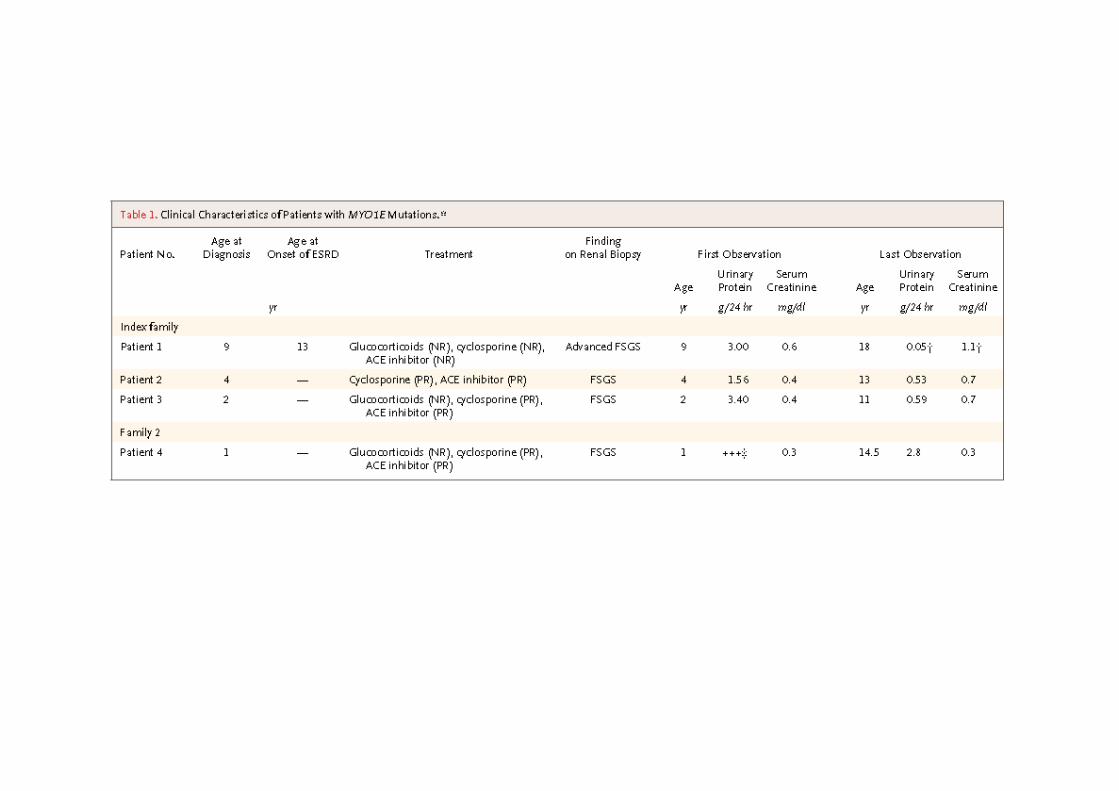

Mutasyonları RHO GTPazlarla ilişkiyi bozuyor, aktif GDP bağlı RAC1 ve CDC42 artışıpodosit migrasyonunda artış

MitokondrialUbikinon (CoQ10) eksikliği

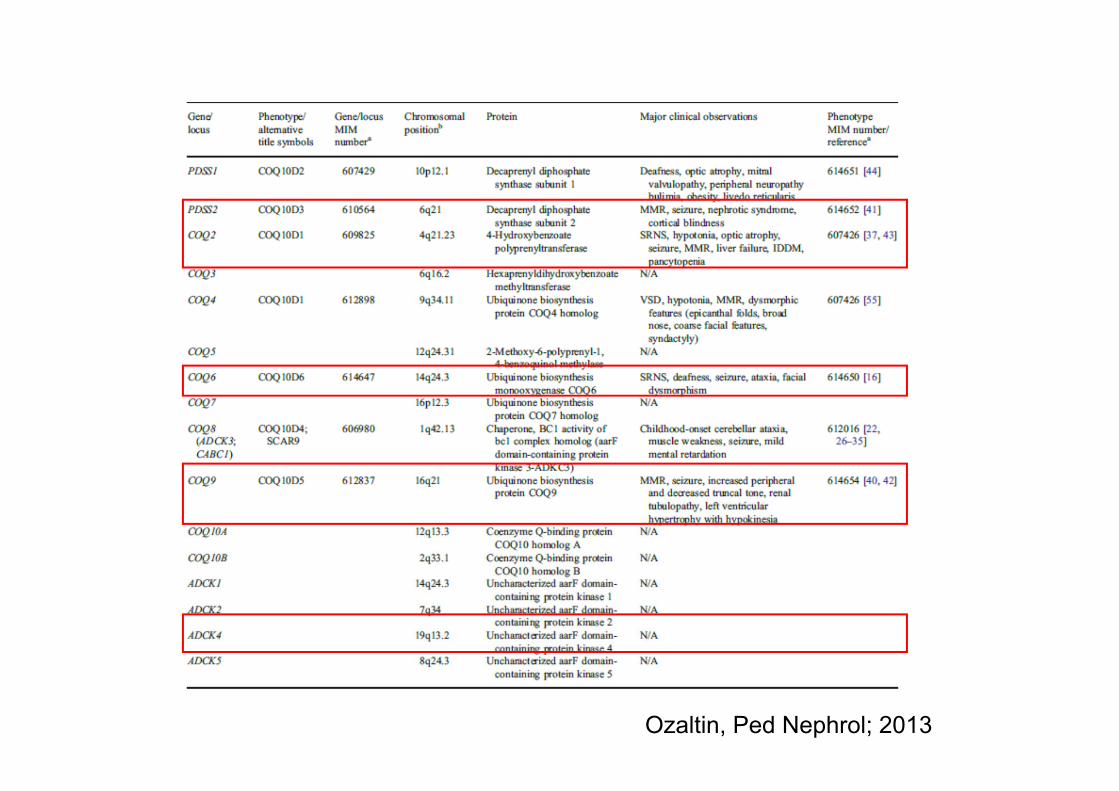
Ozaltin, Ped Nephrol; 2013
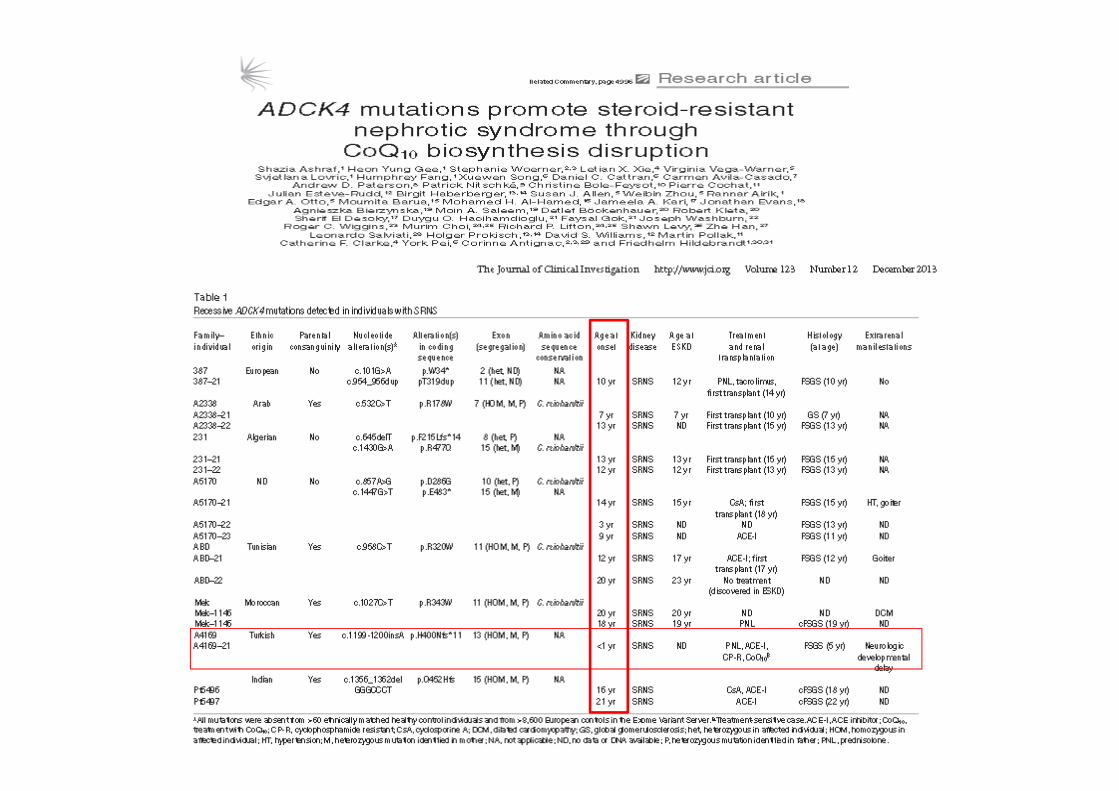







Dominant SRNS/FSGSÇoğunlukla geç başlangıçlı• CD2AP (OMIM #607832) (haploinsufficiency FSGS’e yatkınlık)• INF2 (OMIM #613237) (FSGS±Charcot- Marie-Tooth hast)
Yaş: 11-72y (mikroskopik hematüri, HT, Nefrotik proteinüri..)Mutasyonların çoğu ekzon 2-4’deAynı ailede farklı yaşlarda prezentasyon (inkomplet penet.)
• TRPC6 (OMIM #603965): (gain-of-functionIS Ca girişi)Inkomplet penetrans, 9y SRNS/FSGS9.5y SDBY; Annede FSGS prezentasyonu 30 yaşında; 7y FSGS olan çocuğun aynı mut. Taşıyan sağlıklı aile bireyleriCNI ile tedaviye kısmi cevap
• ACTN4 (OMIM #603278): Adölesan ve yetişkinlerd Nefrotik-nonnefrotik proteinüri . Çocukluk çağı FSGS’de de tanımlanmış

Nadir Sendromla• Schimke immuno-ossöz displazi (SMARCAL1-OR): Büyüme
geriliği, immun defekt, serebral infarkt, cilt pigmentasyonu, erken başlangıçlı SRNS
• Multisentrik karpo-tarsal osteolizis+proteinüri+ilerleyici böbrek yetm (MAFB-OR)
• Aksiyon miyoklonus-renal failure (AMRF): FSGS+progresif miyoklonus epilepsi (SCARB2/Limp2; lizozomal depo hast-OR)
• Tırnak-patella sendromu (LMX1B-OD)• Galloay Mowat• CD151 eksikliği (NS, SDBY, cilt lezyonları, S/N işitme kaybı,
talasemi)• Epidermolizis bülloza+FSGS (ITGB4, ITGA3)• MHY9 (FSGS+SDBY)

SRNS’in Konjenital ve İnfantil formlarında sıklıkla monogenik nedenler varken çocuk ve erişkin SRNS’de genetik, genetik dışı ya da multifaktöriyel patogenez olabilir
Genetik tarama endikasyonları
1- Başlangıç yaşı2- Tedaviye cevap3- Pozitif aile hikayesi4- Böbrek biyopsi bulguları
SRNS Hastalarında Mutasyon Saptanması
1- Spesifik mutasyonların varlığında gereksiz immunosupresif tedavilerden kaçınılması2- Transplantasyon sonrası rekürrens ihtimalinin tahmin edilmesi3- Prenatal tanıya imkan sağlar
Tedavinin bireyselleştirilmesiAkılcıl tedavi yaklaşımları





























