Embed Size (px)
Citation preview

Ž .Chemical Physics 244 1999 101–109www.elsevier.nlrlocaterchemphys
Near-infrared-induced proton transfer studied by electron spinresonance
Małgorzata Komorowska a, Jacek Lamperski b, Ludwik Komorowski b,)
a Institute of Physics, Wrocław UniÕersity of Technology, Wyb. Wyspianskiego 27, 50-370 Wrocław, Poland´b Institute of Physical and Theoretical Chemistry, Wrocław UniÕersity of Technology, Wyb. Wyspianskiego 27, 50-370 Wrocław, Poland´
Received 4 January 1999
Abstract
Ž . Ž .The effect of near-infrared NIR radiation 700–2000 nm on water solution containing the nitroxide spin probe andselected amines has been studied. The concentration of free radical reversibly decreased upon irradiation. Explanation of thisphenomenon has been based on the known properties of nitroxide radical in water. A reaction model for the process ofreversible reduction of the nitroxide in water in the presence of amine and oxygen has been elaborated. The key reactionproposed for this process is NIR-induced decomposition of a complex between amine and nitroxide: RXNHq PPP ON ØR.3
q 1999 Elsevier Science B.V. All rights reserved.
1. Introduction
The nitroxide free radical has been known toundergo a number of transformations into non-paramagnetic products: oxidation, disproportiona-
w xtion, radical recombination and reduction 1,2 . Onereaction is of particular interest in application of thenitroxide radicals as a spin probe in biological mate-
Ž .rials membranes :
aN ØyOqeyqH Oq™aNyOHqH O .3 2
This reaction has been studied in solution andw xdescribed as ‘acid catalysis of radical reactions’ 3,4 .
This first step of this process involves formation ofthe hydrogen bond between the hydronium ion and
Ž Ø .the nitroxide O–H PPP O–N b . Subsequently, theelectron transfer occurs. Similar mechanism has been
w xproposed for the disproportionation reaction 5 .
) Corresponding author. Fax: q48-71-203364
Products of the latter reaction have been identified ashydroxylamine derivatives. In the presence of oxy-
Ž . w xgen air , a reverse reaction has been observed 6and the nitroxyl radical is recovered.
Hydrogen-bond formation has been known as typ-w xical feature of the nitroxide group 7,8 . Zundel et al.
studied an easily polarizable hydrogen bonds be-tween various proton donors and acceptors. Theoreti-cal studies suggested that the shape of the potentialwell within the hydrogen bond can be considerably
w xmodeled by the external potential 9,10 .The purpose of this present work was to study the
effect of irradiation on a system containing the hy-drogen-bonded nitroxide radicals, thus amenable forthe proton transfer. Earlier studies in this laboratoryhave focused on the irradiation of m-nitroanilineŽ . Ž . w xmNA and m-nitrophenol mNP crystals 11–13 .
Ž .Crystals have been irradiated by near-infrared NIRin the range of the overtones of the stretching vibra-tions for OH, NH and CH. Generation of free radical
0301-0104r99r$ - see front matter q 1999 Elsevier Science B.V. All rights reserved.Ž .PII: S0301-0104 99 00075-0
( )M. Komorowska et al.rChemical Physics 244 1999 101–109102
species in these crystals upon irradiation presumablyinvolved the proton transfer within the hydrogenbond as a first step of the reaction. A preliminaryexperimental study performed in this laboratory haspreviously been reported for water solution contain-
Ž .ing 2,2,6,6-tetramethyl-piperidine-1-oxyl TEMPOand sodium glutaminate or polyglutaminate. Re-versible decay of TEMPO radical has been found in
w xthese solution upon irradiation by NIR 14 . In thispresent work, a systematic study has been attempted,to explain this effect observed on a number of mixedsolutions containing TEMPO and amines and aminoacids.
2. Experimental details
2.1. Materials
Ž2,2,6,6-Tetramethyl-1-piperydyl-N-oxyl TEM-. Ž .PO , DL-arginine hydrochloride ARG and amino-
Ž .acetic acid glycine, GLI were purchased fromSigma Chemical and used without purification. Thecommercial mNA was purified by zone refinement.Redistilled and deionized water was used as a sol-vent. An appropriate amount of TEMPO and aminewas dissolved in water and mixture was titrated by0.05 M NaOH solution to neutral pHs7.2"0.1.Typical concentration of both components was 0.3
mM. The ESR experiments were done using air-con-taining solutions. In a separate set of measurements,oxygen was removed from water by 15 min bubblingwith argon.
2.2. ESR measurements
The spectra were recorded on the standard SErX-28 electron spin resonance spectrometer operating inthe X-band. Typical spectrometer settings were 100kHz modulation with amplitude 0.04 mT, scan range5 mT, and scan speed 50 srspectrum. Minimumtime interval between two spectra recorded for thesame sample was 1 min.
2.3. Irradiation procedure
A halogen lamp equipped with filter of trans-parency range of 700–2000 nm was used as the lightsource. The light was focused on a flat glass tubewith solution inside of ESR spectrometer cavity withthe power density of incident light on the sample1.28 mWrmm2. The temperature of samples wasstabilized at Ts303"3 K. Illumination was ac-complished through a slotted grid in the front of thecavity.
The ESR spectra were taken up to 10 times for 30min in darkness for each sample, in order to checkstability of ESR signal, then the illumination has
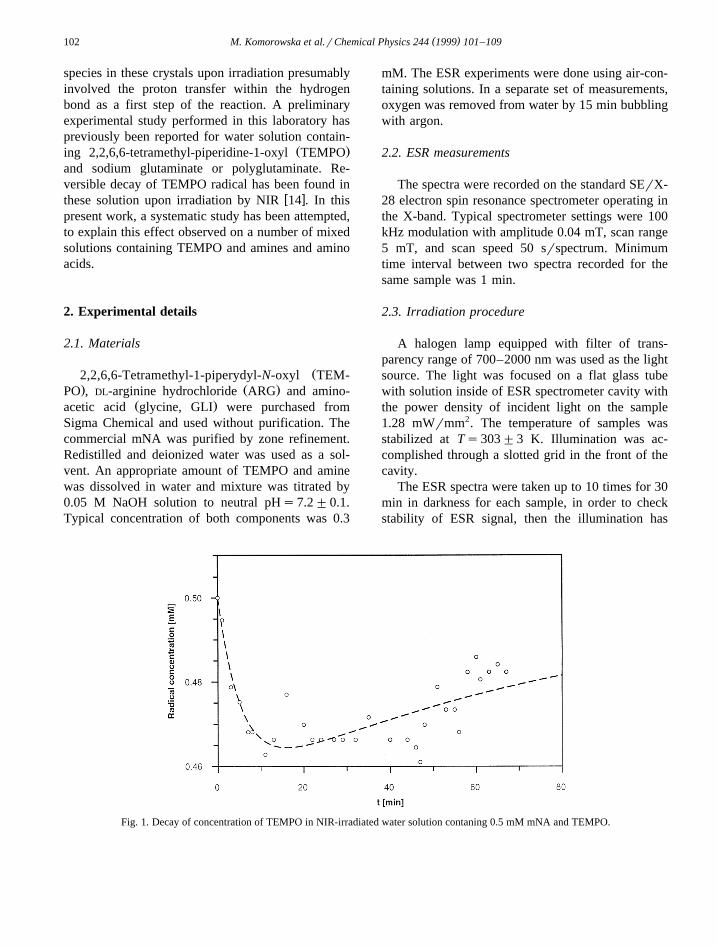
Fig. 1. Decay of concentration of TEMPO in NIR-irradiated water solution contaning 0.5 mM mNA and TEMPO.

( )M. Komorowska et al.rChemical Physics 244 1999 101–109 103
Fig. 2. Decay of concentration of TEMPO in NIR-irradiated water solution contaning 3.0 mM mNA and TEMPO.
been set. Spectra were recorded approximately everyminute during illumination, until a change in theESR signal was observed. Time was controlled bythe stopwatch and the moment of appearance of themaximum of the central line has been recorded.Total number of paramagnetic molecules is propor-tional to the area under absorption peak. No changesin line width have been detected upon irradiation,and therefore the signal amplitude alone was the
convenient measure of concentration of free radicalsin the sample; the height of the central line wasmonitored versus time.
2.4. Results
The decay of the free radical concentration hasbeen observed upon the onset of irradiation to the
Ž .solution. In the presence of oxygen air a minimum
Fig. 3. Decay of concentration of TEMPO in NIR-irradiated water solution contaning 0.3 mM mNA and TEMPO. Sample desoxidized withargon.

( )M. Komorowska et al.rChemical Physics 244 1999 101–109104
Ž .Fig. 4. Decay of concentration of TEMPO in NIR-irradiated water solution contaning 0.3 mM glycine GLI and TEMPO.
has been observed on the time dependence of theŽ .ESR intensity radical concentration . The minimum
appeared at ca. 5–20 min. after radiation has beenŽset. Systematic recovery of the signal increase of the
.radical concentration occurred at a time span longerthan the minimum. For a sample saturated with
Ž .argon oxygen removal no minimum has been ob-served, but the radical concentration was decreasing
upon illumination for hours. Examples of the experi-mental results are given in Figs. 1–5.
3. Quantum–chemical analysis of the hydrogen-bonded species involving the nitroxyl radical
Mixed water solution of the nitroxide radical andŽ .amine BH contains a number of hydrogen-bonded2
Ž .Fig. 5. Decay of concentration of TEMPO in NIR-irradiated water solution contaning 0.3 mM. Arginine ARG and TEMPO in 1:2 molarratio.

( )M. Komorowska et al.rChemical Physics 244 1999 101–109 105
species involving the nitroxide radical that have vari-ous concentration and variable structure of the hy-drogen bond. The list may include hydrogen-bondedcomplexes of the nitroxide as well as its protonatedform with H O, H Oq, BH , BHq. Stability and the2 3 2 3
structure of the energy barrier within the hydrogenbond has been studied for complexes with thesespecies for mNA and GLI in order to identify a
component potentially active in low-energy radiationapplied.
Calculation of the electronic structure of com-plexes of TEMPO, 2,2,6,6-tetramethyl-piperydylo-
Ž Ø . Ž Ø q.N-oxyl RN O and its protonated form RN OHwith partner protonated bases and bases, respec-tively, has been performed in two stages. First, theoptimized electronic structure of component mole-
Ž .Fig. 6. Calculated energy profile for the hydrogen bond between the oxygen atom in TEMPO radical and the nitrogen in A mNA cationŽ .and B GLI cation.

( )M. Komorowska et al.rChemical Physics 244 1999 101–109106
cules has been calculated by means of the RHF me-w xthod using GAUSSIAN 94 15 , with the basis set,
6-31G2UU. Then, the energy of the hydrogen-bondX–H PPP Y was analysed with the MOPACK pro-gram maintaining the rigid structure of componentmolecules. Two optimization parameters have beenused in this latter procedure: the total length of the
Ž .hydrogen bond R and the torsional angle be-X Y
tween the ring plane of the nitroxide and the selectedŽ .principal plane of the partner molecule a . R X Y
and a have been optimized first, keeping the posi-tion of hydrogen in its mother molecule rigid. Thenthe energy profile of the hydrogen bond was foundby varying the position of the hydrogen with R X Y
and a fixed. The energy profile for the mNA andGLI complexes are given in Fig. 6.
Although the results obtained for the gas phaseŽ .free molecule can hardly reproduce the situation ofthe molecule in solution, a set of important hints is
Ž . Ø qborn from the results. i The RN O PPP H B com-3
plex is stabilized in a shallow energy minimum. Thequantitative accuracy of the ab initio calculations isinsufficient to discuss the depth of this minimum;however, the energy barrier for the proton transferreaction within the hydrogen bond is in the range of
Ž . Ž .the radiation energy of NIR radiation Fig. 6 . iiEnergy minimum in the RN ØOHq PPP BH complex2
is by an order of magnitude deeper than the formerŽ .one. iii Proton transfer reaction between the two
minima is associated with the overall bond expan-Ž . Ž .sion by ca 0.06 nm ca. 50% . iv Hence, the proton
transfer reaction
RN ØO PPP HqBH ™RN ØOHq PPP BH 1Ž .2 2
produces energetically stabilized product and is irre-versible due to the associated bond expansion.
4. Reaction model
The analysis given in Section 3 motivates a con-Ž .clusion that reaction 1 be taken as an educated
guess for the initial stage of the effect of NIRradiation on the systems under study. The entire setof subsequent reactions may be built including thedissociation equilibria for the base and a series ofprocesses already described in the literature for thenitroxide radical in water. If the equilibrium con-
stants are denoted by ‘K ’ and the rate constants forelementary reactions by ‘k’, the key processes maybe summarized:
Equilibria:
BH qH OmHqBH qOHy K , 2Ž .2 2 2 b
RN ØO qHqBH mRN ØO PPP HqBH K , 3Ž .2 2 e
Kinetic processes:
RN ØO PPP HqBH 2
™RN ØOHqqBH hÕ , k , 4Ž . Ž .2 1
RN ØO qRNO ØHq™RNOqqRNOH k , 5Ž .2
RNOHqO H OŽ .2 2
™RN ØO qH OqqO Øy k , 6Ž .3 2 3
RNOqqO Øy™RN ØO qO k . 7Ž .2 2 4
The RN ØOHq PPP BH complex does not appear in2
equilibria as it may be considered as the secondŽ .intermediate in the reaction 4 . Such complex must
be unstable in water in the presence of RN ØO;Ø q Ž . Ž .RN OH is reoxidized in reactions 5 – 7 . Under
the laboratory conditions applied, i.e. rather weakbases used, pH(7 and 1:1 ratio of the concentrationof amine and nitroxide, it may safely be assumed:
q w x w xBH < BH ( BH and3 2 2 o
Ø q Øw xRN O PPP H BH < RN O . 8Ž .2
The equilibrium concentration of the complex,w Ø q xRN O PPP H BH may be approximated as:2
Ø qRN O PPP H BH 2
14 q Øw x w x(K K 10 H O BH RN O . 9Ž .e b 3 2 o
Ž .Postulated photochemical reaction 4 is under thesecircumstances pseudo first-order reaction whose rateconstant k is directly related to the radiation inten-1
Ž . Ž .sity I , quantum yield f , extinction coefficientoŽ . Ž .´ and the solution layer thickness l ; k sf I ´ l1 ow x16 . Under constant radiation intensity, the rate for
Ž .the reaction 4 becomes:14 q Øw x w xr sf I ´ l K K 10 H O BH RN O .1 o e b 3 2 o
Ž . Ž .Reactions 5 – 7 have been described in theliterature as the typical route of disproportionation ofthe unstable RN ØOHq radical in water in the pres-
w xence of oxygen 1 . Under constant rate of produc-

( )M. Komorowska et al.rChemical Physics 244 1999 101–109 107
Ø q Ž .tion RN OH in reaction 4 induced by NIRillumination, its concentration may be assumed as
w Ø qxstationary, d RN OH rd t s 0. The followingw Ø qx Ž .result is readily obtained: RNO H s k rk =1 2
14 w qxw xK K 10 H O BH . Hence, the overall dis-e b 3 2 o
proportionation of RN ØO radical to RNOq andŽ . Ž . Ž .RNOH in reactions 3 , 4 and 5 under constant
irradiation may then be summarized in one equation:
RN ØOqRN ØO PPP HqBH 2
hÕ q™ RNO qRNOHqBH , 10Ž .2
w Øxwith the rate rsk RNO , where k sf I ´ l K =1 1 o e14 w qxw xK 10 H O BH . The subsequent reproduc-b 3 2 o
tion of nitroxide radical occurs in two stages, in theŽ .presence of oxygen. Reaction 6 is known to be
Ž . 9slow while reaction 7 is 10 orders of magnitudew xfaster 1,17 . Thus, they may be summarized in one
equation with some effective rate constant related tothe concentration of oxygen.
O2q Ø qRNOHqRNO qH O ™ 2RN OqH O . 11Ž .2 3
Øy Ž Ž . Ž ..The concentration of unstable O Eqs. 6 and 72
is exceedingly low as a consequence of its effectiveŽ .annihilation in reaction 7 ; it may be assumed sta-
Ž .tionary. The rate of overall reaction 11 is expressedw x w qx w xas rsk RNOH q RNO , where k sk O .2 2 3 2
Solution of the kinetic problem for the entire processŽŽ . Ž ..of the NIR-induced reactions in water 2 – 7 can
now be reduced to simple autocatalytic kinetic modelŽ . Ž .for reactions 10 and 11 only.
SqA™PqC rsk S , 12Ž .1
P™2SqD rsk P , 13Ž .2
Žwhere S stands for the nitroxide radical initial con-.centration S , A for the hydrogen-bonded complex,o
�w x w qx4and P for the sum RNOH q RNO . The lattertwo products are the only non-magnetic NO contain-ing species within entire reaction chain. Hence, the
Ž .difference S yP is a measure of the concentrationo
of all magnetic molecules, indiscernible under exper-Ž .imental conditions. Reaction 12 is formally of the
Ž .first order, as the effect of approximately constantconcentration of the complex A is buried in k .1
The kinetic model described above produces thedifferential equation:
d2S dS 1q k qk q k k s0 . 14Ž . Ž .1 2 1 22 d t 2d t
w xIt is solvable analytically 18 with the general solu-tion function assumed in the double exponential
Ž . Ž . Ž .form: S t sC exp l t qC exp l t . The char-1 1 2 2Ž Ž .. Žacteristic equation Eq. 15 yields two roots Eq.
Ž ..16 and the explicit functions for concentrationŽ . Ž . Ž Ž ..S t and P t in time Eq. 17 with the boundary
Ž . Ž .conditions S ts0 sS , P ts0 s0:o
12 w xl q k qk lq k k s0 , 15Ž .2 1 1 22
1 2 2(l sy k qk " k qk , 16Ž . Ž .1,2 2 1 2 12
w x w x2S l qk l qko 1 1 2 1 l t l t2 1� 4P t s e ye . 17Ž . Ž .w xk l yl2 1 2
The above results may be used as a source ofexperimental parameters derived from the observed
Ž .radical concentration in time Figs. 1–5 . The ESRintensity is a measure of the concentration of mag-
Ž . Ž .netic species in the solution, here F t sS yP t .o
Two quantitative parameters are provided directlyŽ . XŽ .from the experiment: i F 0 , the initial slope, and
Ž .ii t , the position of the minimum.min
5. Results and discussion
The global rate constants k and k have been1 2
calculated from the experimental data through tworelations obtained by the analysis of the double
Ž .exponential relation given by Eq. 17 :
FX ts0 syk S , 18Ž . Ž .1 o
1 l1t s ln . 19Ž .min
l yl l1 2 2
Experimental data have been fitted with the doubleŽ .exponential curve F t by means of the least-squares
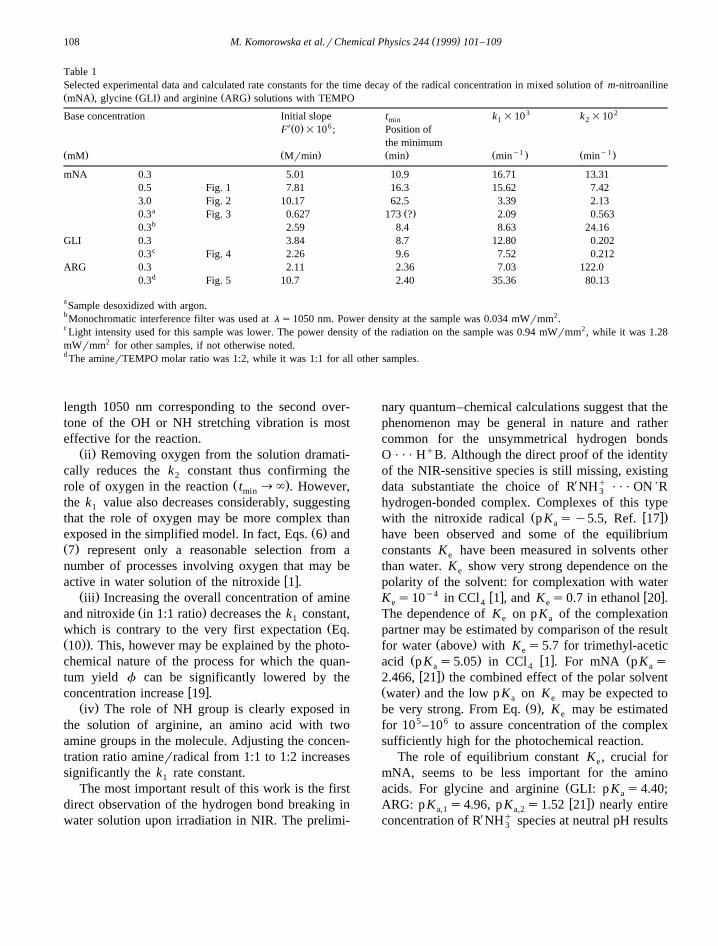
procedure. Calculated curves have been indicated bydashed lines in Figs. 1–5. Calculated fitting parame-ters k and k are collected in Table 1. The data1 2
confirm the reaction model proposed for this photo-chemical process. This may be summarized in thefollowing observation that emphasize the photo-chemical character of the process under study:
Ž .i Decreasing the light intensity reduces k but1
leaves k unaffected, exactly corresponding to the2Ž Ž . Ž ..proposed interpretation of k Eqs. 10 and 12 .1
Out of the entire NIR spectrum explored, the wave-
( )M. Komorowska et al.rChemical Physics 244 1999 101–109108
Table 1Selected experimental data and calculated rate constants for the time decay of the radical concentration in mixed solution of m-nitroanilineŽ . Ž . Ž .mNA , glycine GLI and arginine ARG solutions with TEMPO
3 2Base concentration Initial slope t k =10 k =10min 1 2X 6Ž .F 0 =10 ; Position of
the minimumy1 y1Ž . Ž . Ž . Ž . Ž .mM Mrmin min min min
mNA 0.3 5.01 10.9 16.71 13.310.5 Fig. 1 7.81 16.3 15.62 7.423.0 Fig. 2 10.17 62.5 3.39 2.13
a Ž .0.3 Fig. 3 0.627 173 ? 2.09 0.563b0.3 2.59 8.4 8.63 24.16
GLI 0.3 3.84 8.7 12.80 0.202c0.3 Fig. 4 2.26 9.6 7.52 0.212
ARG 0.3 2.11 2.36 7.03 122.0d0.3 Fig. 5 10.7 2.40 35.36 80.13
aSample desoxidized with argon.b Monochromatic interference filter was used at ls1050 nm. Power density at the sample was 0.034 mWrmm2.c Light intensity used for this sample was lower. The power density of the radiation on the sample was 0.94 mWrmm2, while it was 1.28mWrmm2 for other samples, if not otherwise noted.d The aminerTEMPO molar ratio was 1:2, while it was 1:1 for all other samples.
length 1050 nm corresponding to the second over-tone of the OH or NH stretching vibration is mosteffective for the reaction.
Ž .ii Removing oxygen from the solution dramati-cally reduces the k constant thus confirming the2
Ž .role of oxygen in the reaction t ™` . However,min
the k value also decreases considerably, suggesting1
that the role of oxygen may be more complex thanŽ .exposed in the simplified model. In fact, Eqs. 6 and
Ž .7 represent only a reasonable selection from anumber of processes involving oxygen that may be
w xactive in water solution of the nitroxide 1 .Ž .iii Increasing the overall concentration of amine
Ž .and nitroxide in 1:1 ratio decreases the k constant,1Žwhich is contrary to the very first expectation Eq.
Ž ..10 . This, however may be explained by the photo-chemical nature of the process for which the quan-tum yield f can be significantly lowered by the
w xconcentration increase 19 .Ž .iv The role of NH group is clearly exposed in
the solution of arginine, an amino acid with twoamine groups in the molecule. Adjusting the concen-tration ratio aminerradical from 1:1 to 1:2 increasessignificantly the k rate constant.1
The most important result of this work is the firstdirect observation of the hydrogen bond breaking inwater solution upon irradiation in NIR. The prelimi-
nary quantum–chemical calculations suggest that thephenomenon may be general in nature and rathercommon for the unsymmetrical hydrogen bondsO PPP HqB. Although the direct proof of the identityof the NIR-sensitive species is still missing, existingdata substantiate the choice of RX NHq PPP ON ØR3
hydrogen-bonded complex. Complexes of this typeŽ w x.with the nitroxide radical pK sy5.5, Ref. 17a
have been observed and some of the equilibriumconstants K have been measured in solvents othere
than water. K show very strong dependence on thee
polarity of the solvent: for complexation with watery4 w x w xK s10 in CCl 1 , and K s0.7 in ethanol 20 .e 4 e
The dependence of K on pK of the complexatione a
partner may be estimated by comparison of the resultŽ .for water above with K s5.7 for trimethyl-acetice
Ž . w x Žacid pK s5.05 in CCl 1 . For mNA pK sa 4 aw x.2.466, 21 the combined effect of the polar solvent
Ž .water and the low pK on K may be expected toa eŽ .be very strong. From Eq. 9 , K may be estimatede
for 105–106 to assure concentration of the complexsufficiently high for the photochemical reaction.
The role of equilibrium constant K , crucial fore
mNA, seems to be less important for the aminoŽacids. For glycine and arginine GLI: pK s4.40;aw x.ARG: pK s4.96, pK s1.52 21 nearly entirea,1 a,2
concentration of RX NHq species at neutral pH results3

( )M. Komorowska et al.rChemical Physics 244 1999 101–109 109
from the autodissociation and the formation of azwitterion. The effect of sensitivity to NIR studied inthis work vanishes at pH)8; this additionally cor-roborates the role of RX NHq rather than RX NH as3 2
the complex partner.The ESR method applied in this work provides
merely a tool for the direct observation of the effectof bond breaking on the subsequent process of reduc-tion of the nitroxide radical. The analysis of theproposed reaction model provides explanation whyobservation of this phenomenon in ESR is limited toweak bases and amino acids in nearly neutral solu-tions, pH(7, in the presence of oxygen. pH higherŽ .stronger base by 1 unit will result in exponential
Ž .decreasing of the rate constant k ca. =0.1 . With1
the k approximately unaffected, the time position of2
the minimum intensity, t , will increase quicklymin
beyond the time scale of typical laboratory measure-ments, and also the initial slope will turn unde-
Žtectably small. On the other hand, at pH low acidic. Øsolution , the reduction of RN O in the presence of
oxygen goes spontaneously even in darkness.The discovered effect of NIR on the hydrogen
bond system in water solution does not seem to belimited to a presence of radical. However, its obser-vation in other systems, especially those of physio-logical importance, may prove to be difficult; theeffect of infrared on the hydrogen bond systems innearly neutral water solutions is a challenging prob-lem for future studies.
Acknowledgements
This work has been sponsored by the PolishŽ .National Committee for Scientific Research KBN
under the Wrocław University of Technology statu-tory funds.
References
w x1 N. Kocherginsky, M.M. Schwartz, Nitroxide Spin Labels,Reactions in Biology and Chemistry, CRC Press, Boca Ra-ton, FL, 1995.
w x2 V. Pokhodenko, A.A. Beloded, V.G. Koshechko, RedoxReactions of Free Radicals, Naukova Dumka, Kiev, 1977, p.275.
w x3 A.L. Buchachenko, A.H. Vasserman, Stable Radicals, Elec-tronic Structure, Reactivity and Application, Khimya,Moscow, 1973, p. 375.
w x4 A.A. Medzhidov, A.L. Buchachenko, M.B. Neiman, Dok.Ž .Akad. Nauk. SSSR 161 1965 878.
w x5 V.A. Golubev, Yu.N. Kozlov, A.N. Petrov, A.P. Pourmal, in:Ž .E.G. Rosantsev, R.I. Zhdanov Eds. , Nitroxyl Radicals,
Synthesis, Chemistry and Applications, Nauka, Moscow,1987, p. 271.
w x6 E.G. Rosantsev, Free Nitroxyl Radicals, Plenum Press, NewYork, 1970.
w x7 N.A. Malik, E.A. Smith, M.C.R. Symons, J. Chem. Soc.Ž .Faraday. Trans. II 85 1989 3246.
w x8 U. Starr, W. Muller-Warmuth, Ber. Bunsenges. Phys. Chem.Ž .94 1990 168.
w x9 R. Janoschek, E.G. Weidemann, G. Zundel, J. Chem. Soc.Ž .Faraday Trans. II 69 1973 505.
w x Ž .10 G. Zundel, in: L. Pacher Ed. , Methods in Enzymology, Vol.127, Academic Press, 1989, p. 439.
w x Ž .11 J. Fritsch, G. Zundel, J. Phys. Chem. 85 1981 556.w x12 M.M. Szostak, B. Jakubowski, M. Komorowska, Mol. Cryst.
Ž .Liq. Cryst. 229 1993 7.w x13 M. Komorowska, G. Wojcik, M.M. Szostak, J. Mol. Struct.´
Ž .348 1995 445.w x14 M. Komorowska, V. Norek, in: J.C. Merlin, S. Turrelle, J.P.
Ž .Huvenne Eds. , Spectroscopy of Biological Molecules,Kluwer Academic, London, 1995, p. 49.
w x15 M.J. Frisch, G.W. Trucks, H.B. Schlegel, P.M.W. Gill, B.G.Johnson, M.A. Robb, J.R. Cheeseman, T.A. Keith, G.A.Petersson, J.A. Montgomery, K. Raghavachari, M.A. Al-Laham, V.G. Zakrzewski, J.V. Ortiz, J.B. Foresman, J.Cioslowski, B.B. Stefanov, A. Nanayakkara, M. Challa-combe, C.Y. Peng, P.Y. Ayala, W. Chen, M.W. Wong, J.L.Andres, E.S. Replogle, R. Gomperts, R.L. Martin, D.J. Fox,J.S. Binkley, D.J. Defrees, J. Baker, J.P. Stewart, M. Head-Gordon, C. Gonzalez, J.A. Pople, GAUSSIAN94, RevisionA.1, Gaussian, Pittsburgh, PA, 1995.
w x16 M. Klessinger, J. Michl, Excited States and Photochemistryof Organic Molecules, VCH, 1995, p. 365.
w x17 E.G. Rozantsev, V.D. Sholle, Organic Chemistry of FreeRadicals, Khimiya, Moscow, 1979, p. 343.
w x18 S.K. Scott, Oscillations, Waves and Chemical Kinetics, Ox-ford University Press, 1994.
w x19 P. Suppan, Chemistry and Light, Royal Society of Chem-istry, Cambridge, 1994, p. 70.
w x20 N.A. Sysoeva, A.Yu. Karnilov, A.L. Bucharenko, Chem.Ž .Phys. 15 1976 313.
w x Ž .21 D.R. Lide Ed. , CRC Handbook of Chemistry and Physics,CRC Press, Boca Raton, FL, 1994.





























