Embed Size (px)
Citation preview

Molekularbiologie in der klinischen Diagnostik
1. Zytogenetik
2. Marker für Krankheiten
1

Ultraschall
• Proben werden unter Ultraschallsicht entnommen
• Indikation zur Methode der Wahl durch US bestätigt
• Nachsorge/Kontrolle mit Ultraschall
• ....
2

Studenten und Ausbildung
3
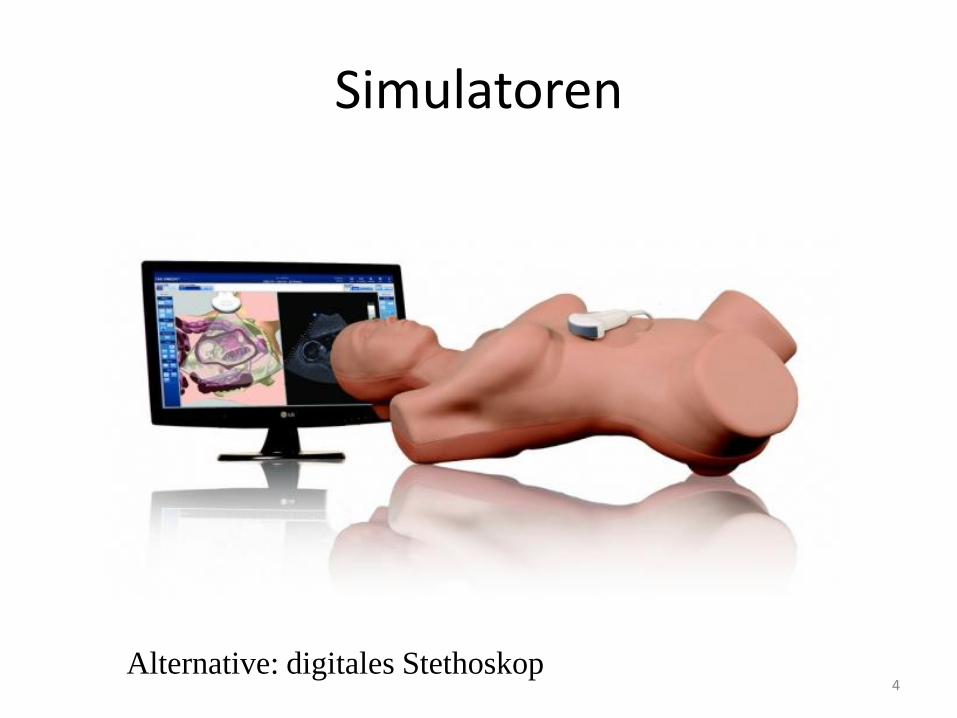
Simulatoren
4 Alternative: digitales Stethoskop
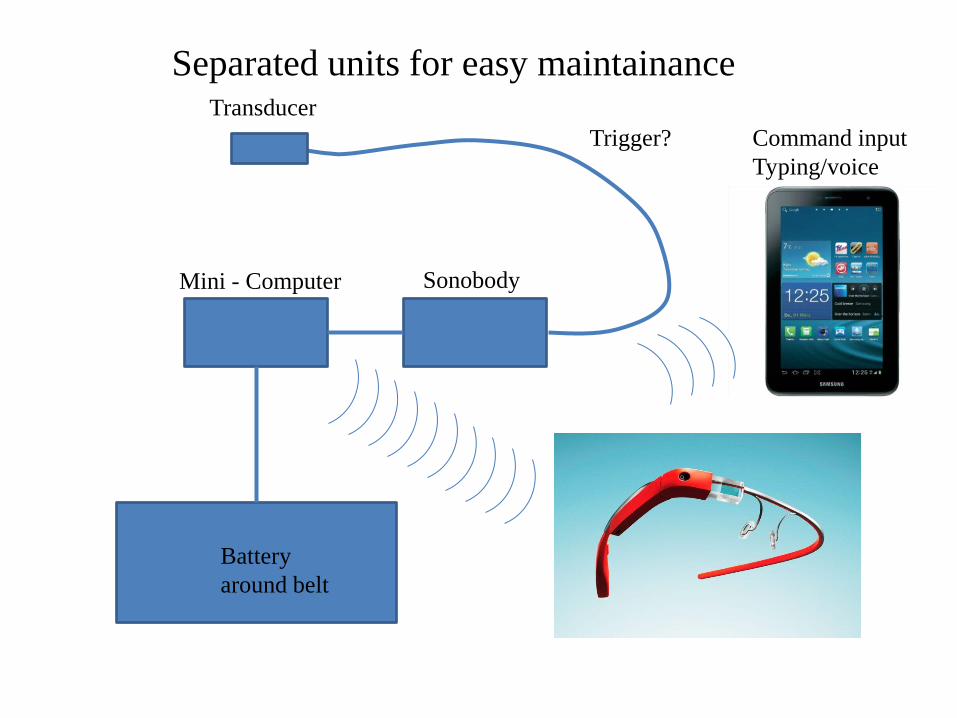
Transducer
Sonobody Mini - Computer
Battery
around belt
Separated units for easy maintainance
Command input
Typing/voice
Trigger?

Live-transmission to expert consultant, worldwide
Telemedicine:
Executive and expert center
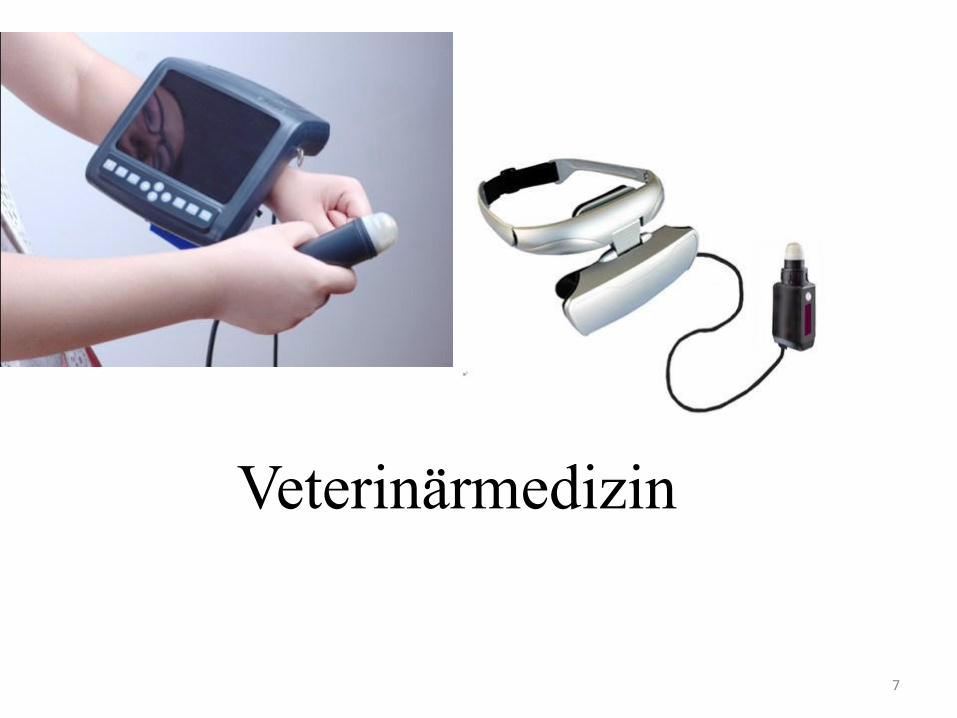
7
Veterinärmedizin
8
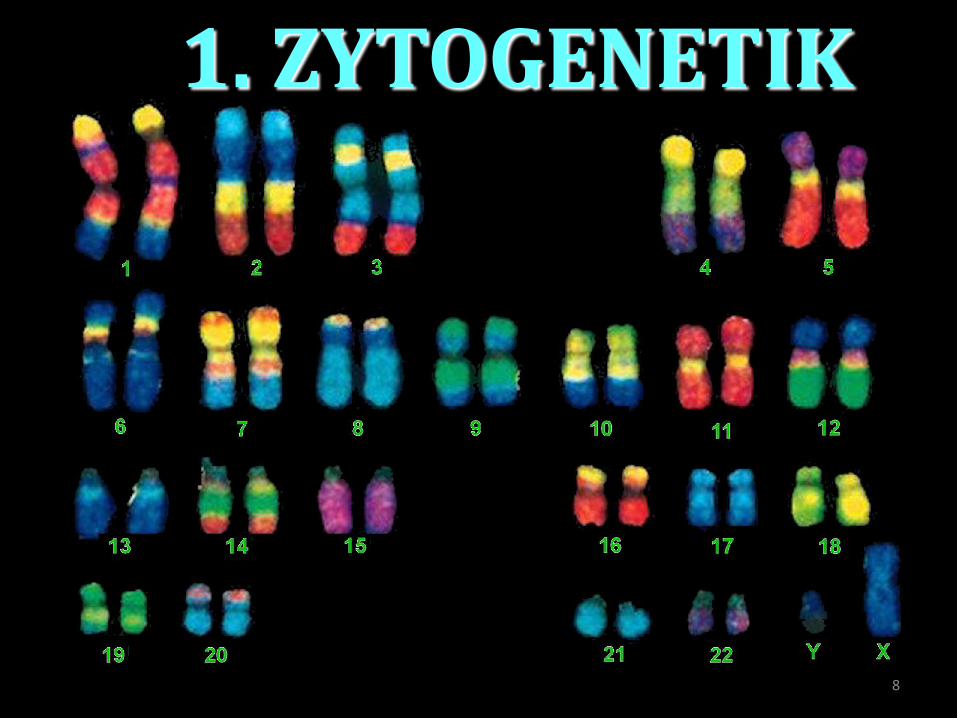
1. ZYTOGENETIK

9
Cytogenetik/Zytogenetik
untersucht die Chromosomen und ihre Rolle
während der Vererbung.
Zytogenetik beschäftigt sich mit der:
1. Diagnostik der Chromosomenstörungen.
2. Lokalisierung der chromosomalen Regionen /
DNA-Sequenzen .
Cytogenetik

10
Chromatin: ist die nicht kondensierte DNA, die werden mit Proteinen beigefügt
(während der Interphase des Zellzyklus)
Chromosom: ist die kondensierte DNA mit Proteinen beigefügt (während der
M-Phase des Zellzyklus)
Karyotyp: bezeichnet alle Eigenschaften der Chromosomenansatz eines Individuums/
Zellkernes und die Anzahl der Chromosomen (zB 46 XY, 47 XYY)
Karyogramm: ist die paarweise und angeordnete zu photografierende
Chromosomenansatz
Idiogramm: ist die schematische Darstellung der Chromosomen nach abnehmende
Grösse und Form
Die menschliche Zellen enthalten normalerweise 46 Chromosomen:
44 Autosomen und
2 Geschlechtschromosomen.
Cytogenetische Begriffsdefinitionen
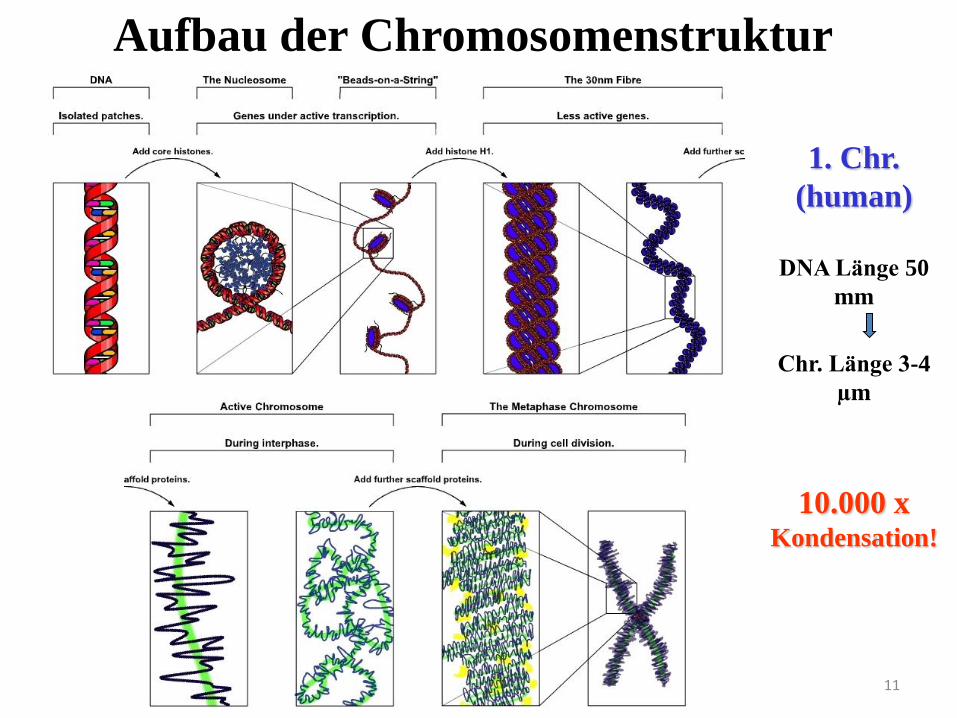
11
1. Chr.
(human)
DNA Länge 50
mm
Chr. Länge 3-4
µm
10.000 x Kondensation!
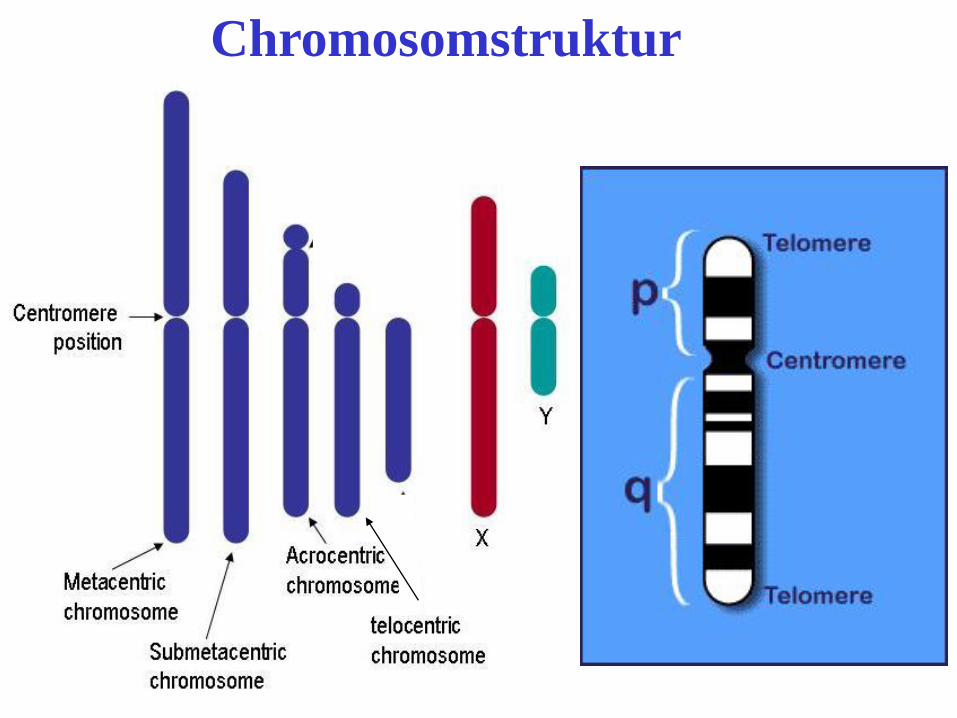
Aufbau der Chromosomenstruktur

12
Die Geschichte der Identifizierung des menschlichen Chromosoms
1879. Arnold: Erste Visualisierung der menschlichen Chromosomen.
1888. Waldeyer: Das Geburt des Wortes Chromosom (Chroma: Farbe, soma: Körper)
1882. Walther Flemming: 20-28 Chromosomen in den Zellen der Hornhaut
1921. T.S. Painter: 48 menschlichen Chromosomen, X & Y-Chromosomen (Wissenschaft)
1956. Jo Hin Tijo und Albert Levan: 46 menschlichen Chromosomen (Hereditas)
1959. Lejeune: Trisomie 21 = Down-Syndrom
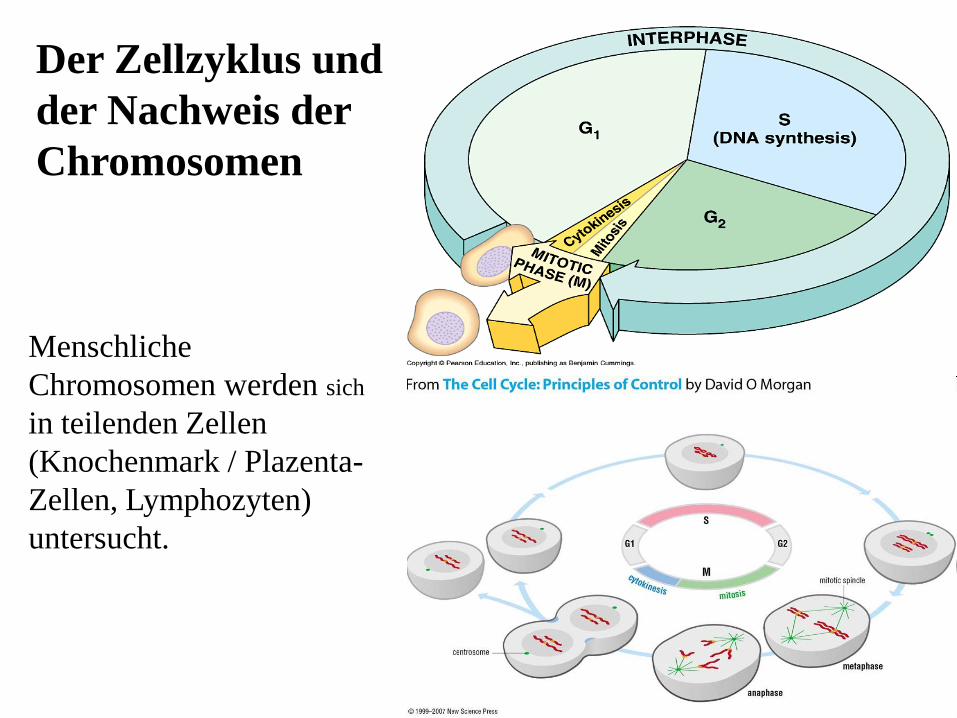
13
Menschliche
Chromosomen werden sich
in teilenden Zellen
(Knochenmark / Plazenta-
Zellen, Lymphozyten)
untersucht.
Der Zellzyklus und
der Nachweis der
Chromosomen

14
1960. Denver: Die menschlichen Chromosomen wurden von 1 bis 22 nummeriert, in der
Reihenfolge der Größe, mit Ausnahme der Geschlechtschromosomen. Die 22 Chromosomen
wurden in 7 Gruppen eingeteilt.
1963. London: der Schriftzug der Gruppe (AG) wurde angenommen
1966. Chicago: chromosomale Syndrome wurden markiert
1971. Paris, die 1976. Mexiko, 1978. Stockholm: Chromosom Banding
1995. ISCN: International System (für Menschen) Cytogenetische Nomenklatur
Karyotyping conferences
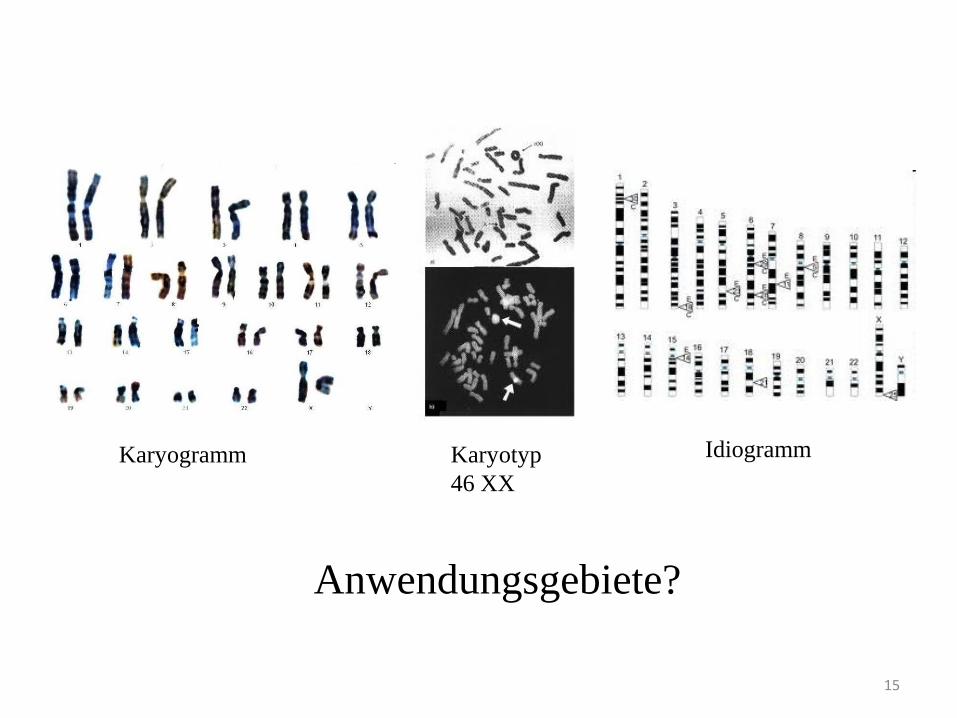
15
Karyotyp
46 XX
Karyogramm Idiogramm
Anwendungsgebiete?
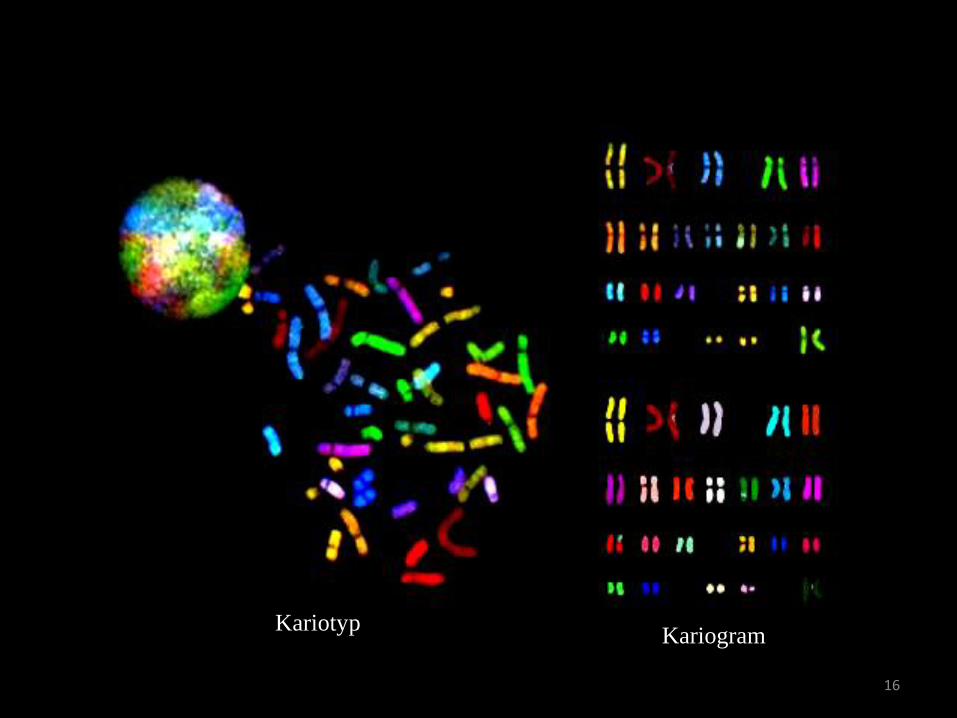
16
Kariotyp Kariogram

Anwendungsgebiete
• Pränataldiagnostik
• Gynäkologie
• Pädiatrie
17
18
Chromosomstruktur
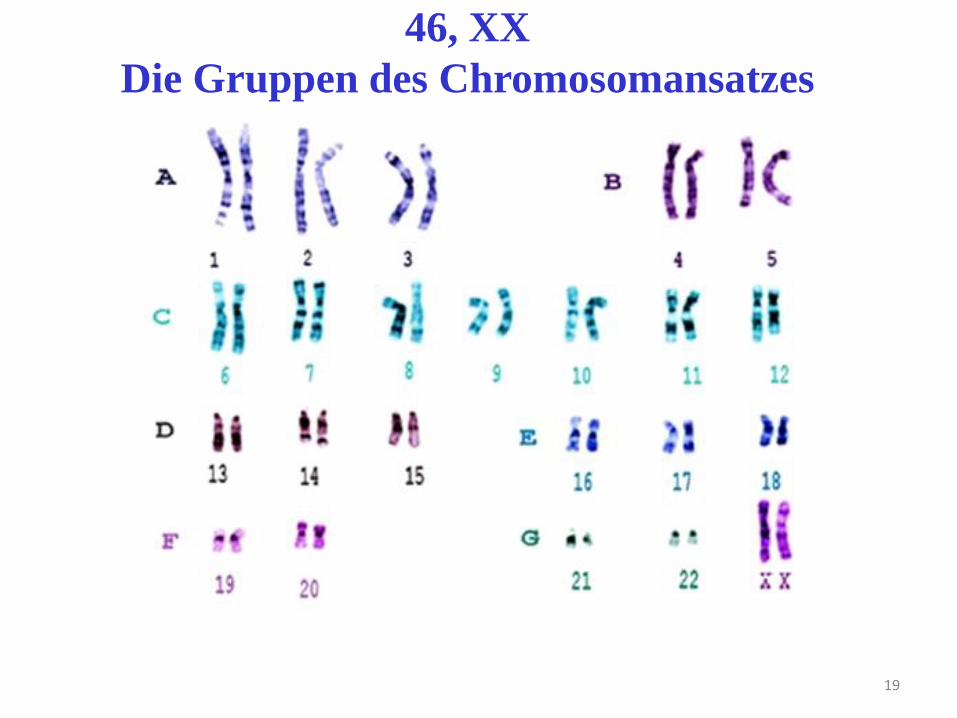
19
46, XX
Die Gruppen des Chromosomansatzes
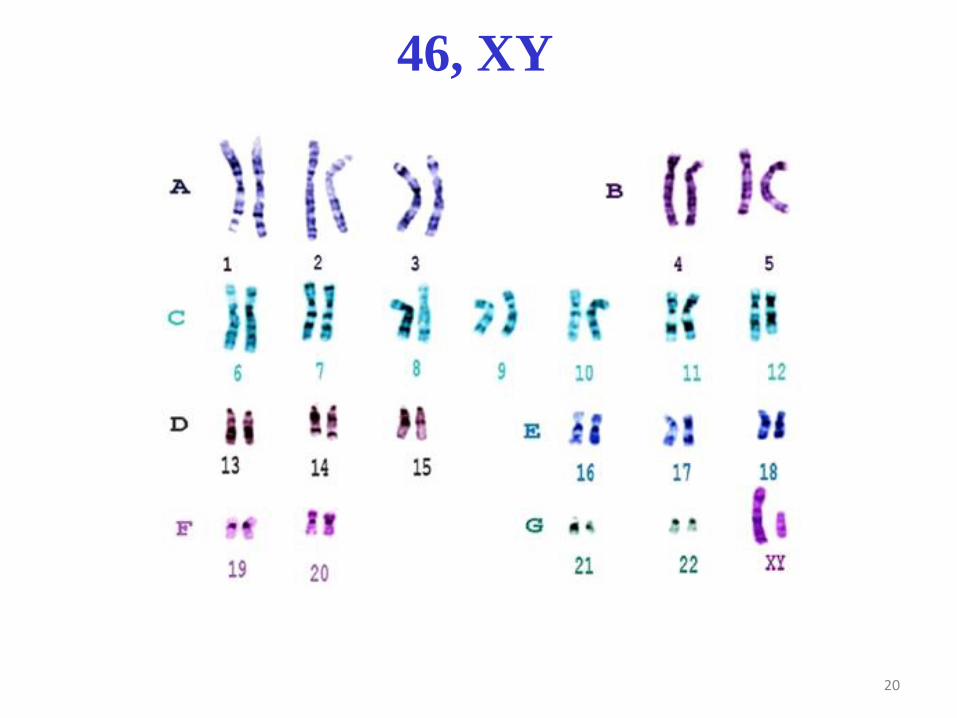
20
46, XY

21
Chromosomenbänderung Die Chromosombänderungstechniken verwenden verschiedene
Färbungen um die Chromosomen und
Chromosomenstrukturänderungen zu zeigen.
Die folgende Bänderungstechniken sind oft verwendet:
G-bänderung: Giemsa Färbung
R-Bänderung: (rückwärts) geändert Giemsa Färbung
C-Bänderung: Centromer spezifische Färbung
T-Streifen: Telomer spezifische Färbung
Q-Streifen: Quinacrin Färbung (fluoreszierend)
Der kurze und lange
Chromosomarmen
werden aufgrund der
Färbungsmuster
nummeriert.

22
Q-banding G-banding
R-banding C-banding T-banding
Chromosomenbänderung
23
Die verschiedenen Krankheitsbilder werden durch Aneuploidien, Translokationen
oder Deletionen verursacht.
Chromosomale Aberrationen
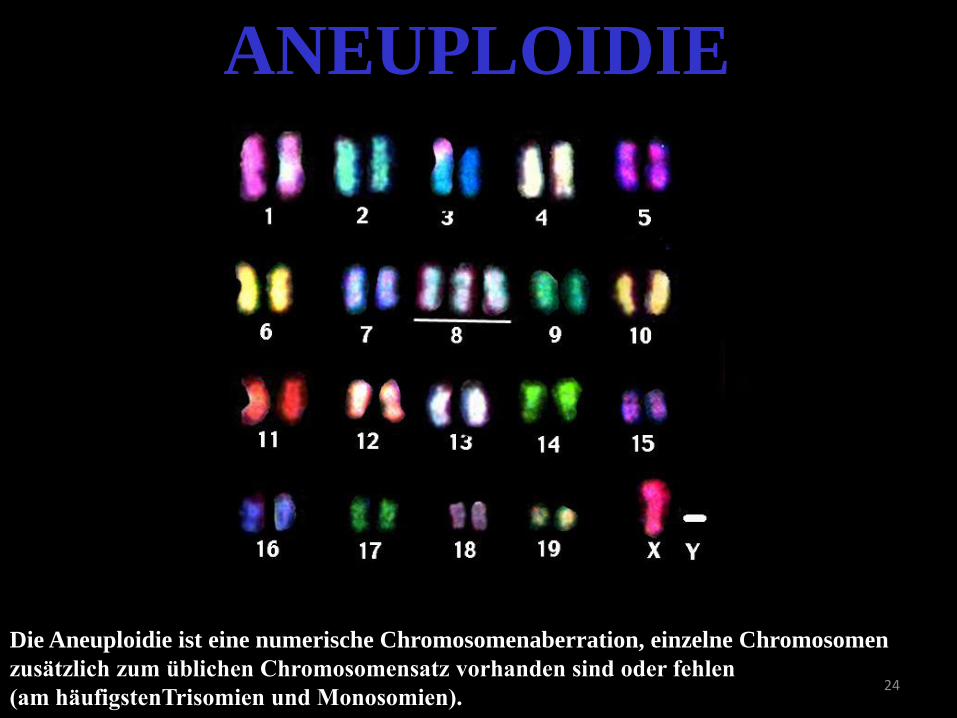
24
ANEUPLOIDIE
Die Aneuploidie ist eine numerische Chromosomenaberration, einzelne Chromosomen
zusätzlich zum üblichen Chromosomensatz vorhanden sind oder fehlen
(am häufigstenTrisomien und Monosomien).
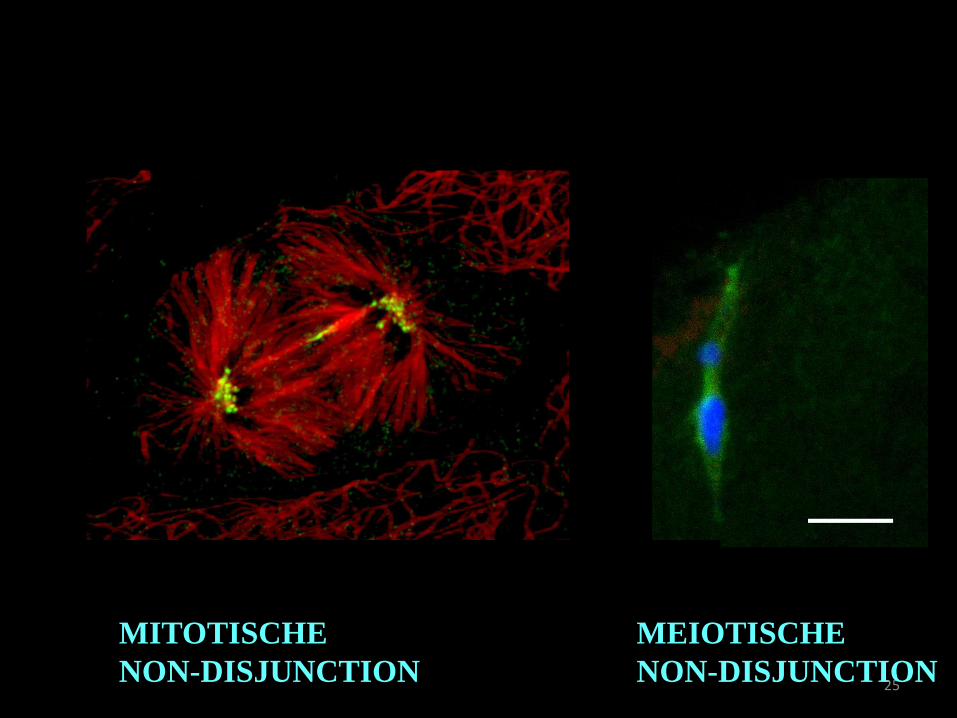
25
MITOTISCHE
NON-DISJUNCTION
MEIOTISCHE
NON-DISJUNCTION
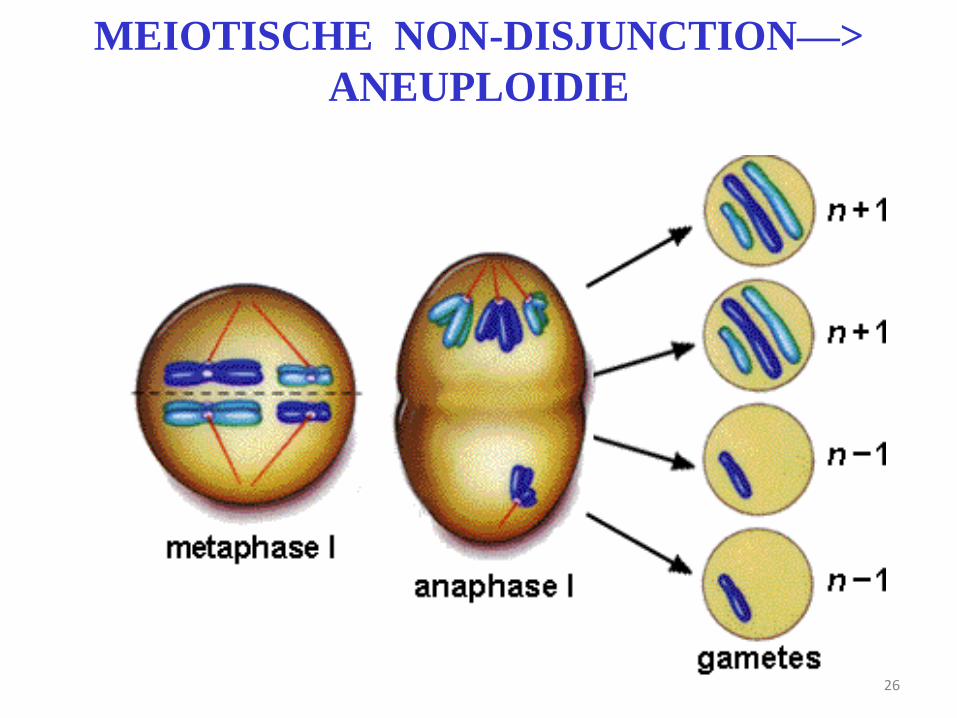
26
MEIOTISCHE NON-DISJUNCTION—>
ANEUPLOIDIE
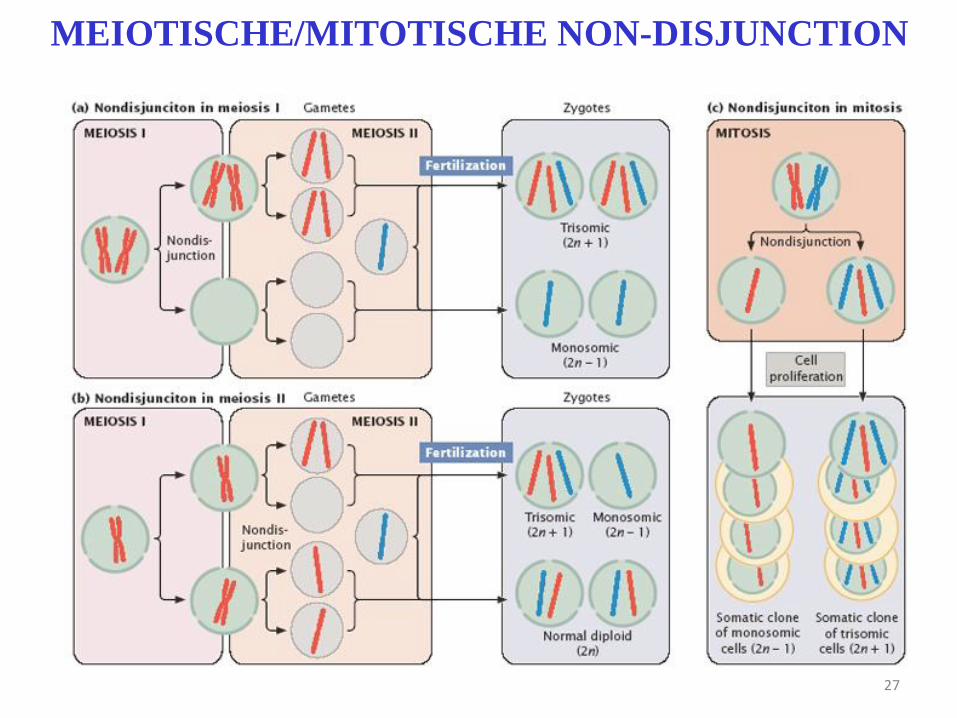
27
MEIOTISCHE/MITOTISCHE NON-DISJUNCTION

28
Numerische Chromosomenaberrationen
Euploid Chromosomenmutationen: Triploidie (3n), Polyploidie
Aneuploid Chromosomenmutation
Autosomale Häufigkeit Geschlechtschro
mosomale
Häufigkeit
Trisomie
Trisomie des 21. Chr.
Down-Syndrom 1/700 47, XXX Triplo-
X-Syndrom 1/1000 in
Frauen
Trisomie des 18. Chr.
Edwards- Syndrom 1/13000 47, XXY
Klinefelter-
Syndrom
1/1000 in
Männer
Trisomie des 13. Chr.
Patau-Syndrom 1/15000 47, XYY
Doppel-Y-
Syndrom
1/1000 in
Männer
Selte Trisomien 3-,7-,8-,9-,12-
,14-,15-,19-,22-es
Monosomie 45, X0 Turner-
Syndrom 1/2500 in
Frauen

29
AUTOSOMALE ANEUPLOIDIE:
DOWN-SYNDROM

Down Syndrom
• 3x 21 od. Teile (freie Trisomie, Mosaik)
• 1:650 (häufigste Chr.anomalie)
• Risk ab 35. Jahr der Mutter, dennoch 70% der Mütter jünger!
• Sonographie!
– Verdickte Nackenfalte 11.-14.SSW
– Quotient biparietaler Schädeldurchmesser
– Femurlänge >50% erhöht
30

Down Syndrom
• Labor: Triple Test (a-Fetoprotein, Estriol, HCG)
• PAPP-A (Pregnancy-associated plasma protein)
• Körperliche Befunde:
– Körperbau/Bewegungsapparat
– Extremitäten/Akren
– Kopf/Augen
• Geistig/psychisch: IQ unterschiedlich, ca.-50 vom Normalwert
31

Down Syndrom
• Fehlbildungen innerer Organe
– Herz 50% , Septum, AV-Kanal, Fallot-tetralogie,etc.
– GI Atresien/Stenosen, Pancreas anulare, Morbus Hirschsprung, Eingeweidebrüche
– Hypogonadismus, Infertilität
– Immundefizienz URTI
– Leukämie
– OSA
• Ca.45% werden älter als 60 Jahre (Demenz!) 32
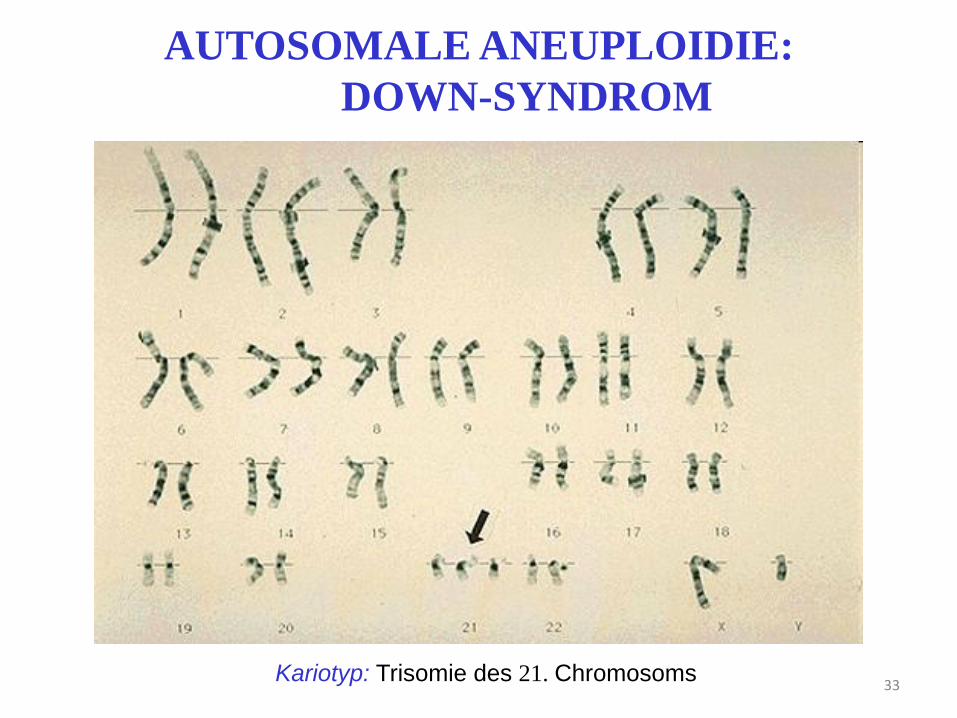
33 Kariotyp: Trisomie des 21. Chromosoms
AUTOSOMALE ANEUPLOIDIE:
DOWN-SYNDROM
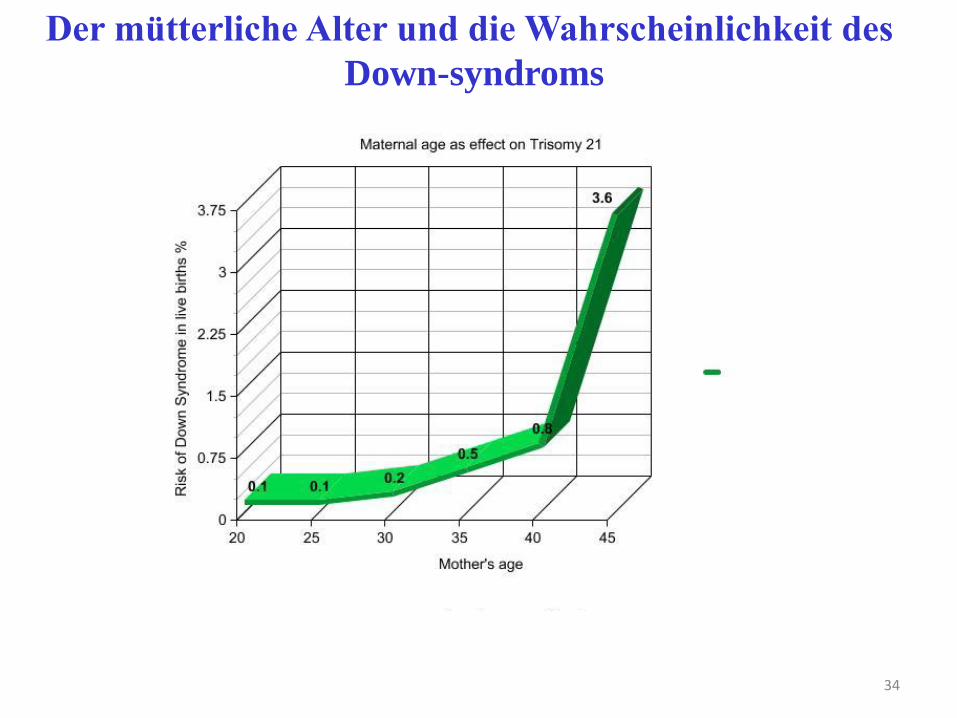
34
Der mütterliche Alter und die Wahrscheinlichkeit des
Down-syndroms

35
Karyotyp: Trisomie des 18. Chromosoms
AUTOSOMALE ANEUPLOIDIE:
EDWARDS-SYNDROM

Edwards Syndrom • Verdreifachtes Chr. 18 od. Teile davon
• 1:3000-5000, häufiger Mädchen
• Kurzer Stamm, kleine Mamillen
• Langer, schmaler Schädel
• Ohrmuscheldysplasie, Mikrognathie
• Extremitätenanomalien
• Herzfehler 90%
• Nierenfehl. und GI Atresien
• Keine Therapie (nur 10% überleben 1.Jahr)
36
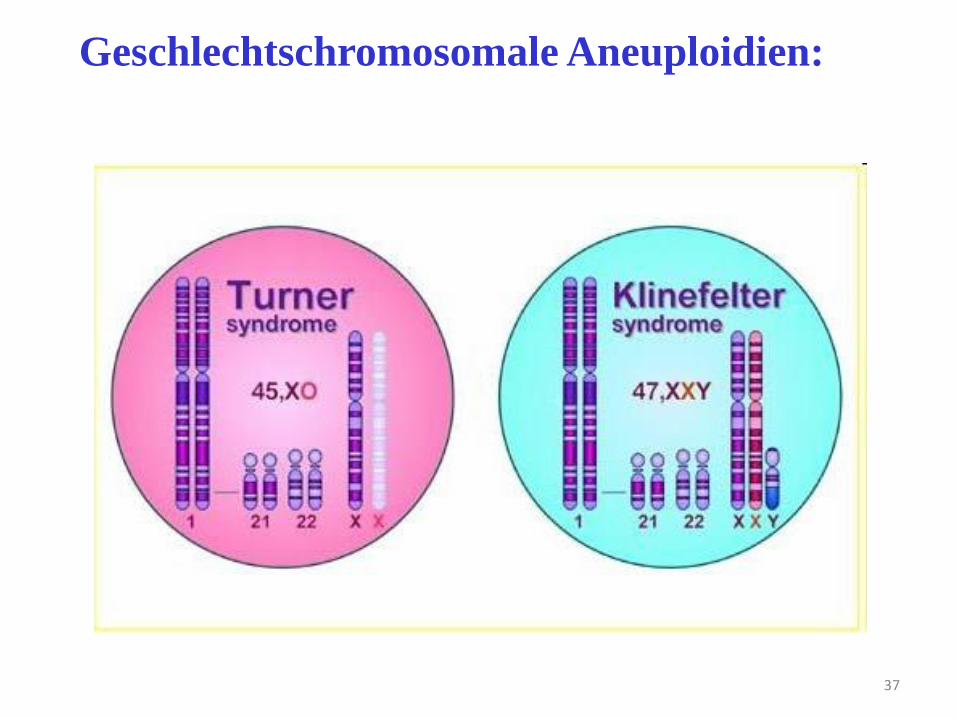
37
Geschlechtschromosomale Aneuploidien:
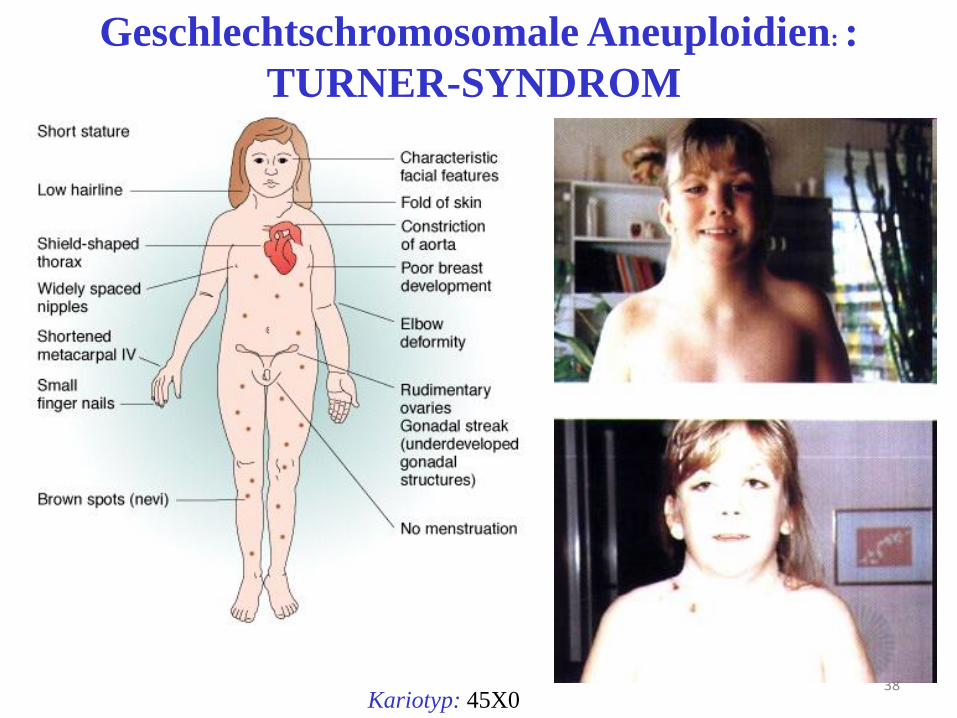
38 Kariotyp: 45X0
Geschlechtschromosomale Aneuploidien: :
TURNER-SYNDROM

Turner Syndrom
• 45, X
• 1:2500 aller weiblichen Neugeboren
• Bei Geburt: Lymphödeme an Hand/Fußrücken
• Kleinwuchs, kurzer Hals m. Pterygium colli
• Epikanthus, Hypertelorismus, tiefer Haaransatz
• Schildthorax
• Breiter Mamillenstand
39

Turner Syndrom
• Metacarpalia Iv u. V. verkürzt
• Gonadendysgenesie, 1° Amenorrhö, hypoplastisches inneres u. Äußeres Genitale
• Infertilität
• Nierenfehlbildungen (Hufeisen)
40
41
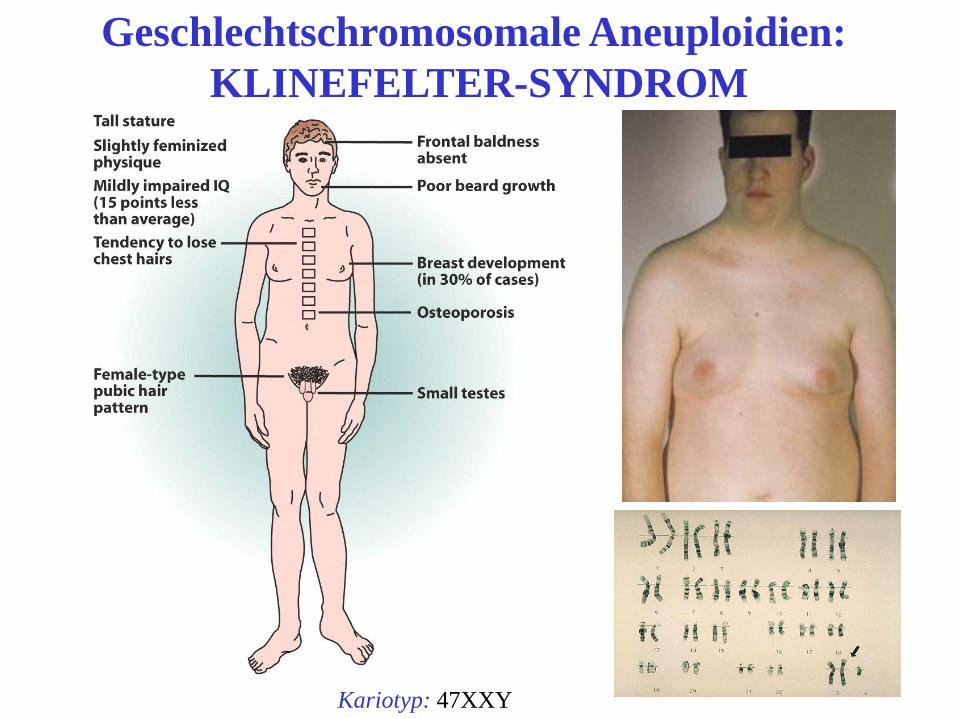
Geschlechtschromosomale Aneuploidien:
KLINEFELTER-SYNDROM
Kariotyp: 47XXY

Klinefelter Syndrom
• 47, XXY, selten 48, XXXY
• 1:1000 aller männlichen Neugeborenen
• Erstdiagnose häufig in der Pubertät
• Euchnuchoider Hochwuchs
• Adipositas, Gynäkomastie
• Hypergonadotroper Hypogonadismus
• Hodenatrophie Aspermie
• Fehlen der sek. Männlichen Behaarung 42
43
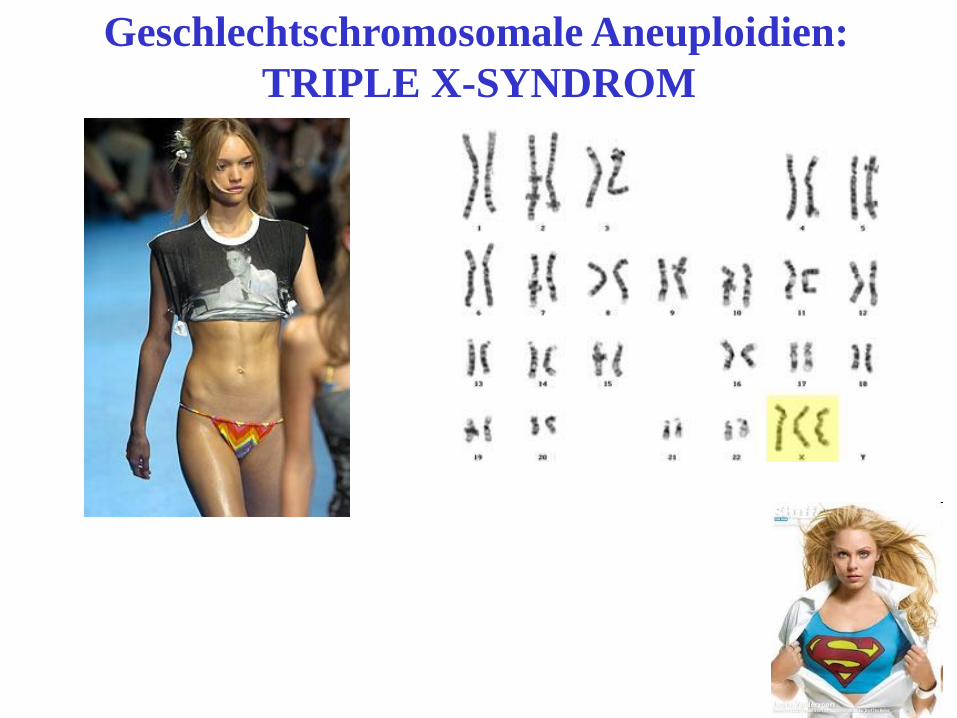
Geschlechtschromosomale Aneuploidien:
TRIPLE X-SYNDROM
44
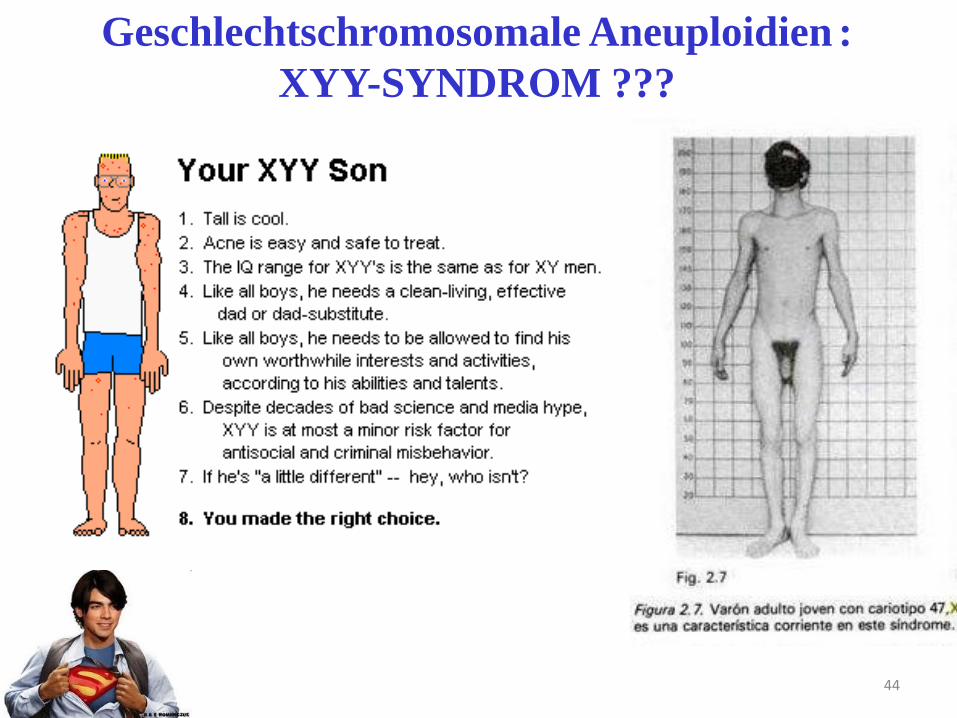
Geschlechtschromosomale Aneuploidien :
XYY-SYNDROM ???
45
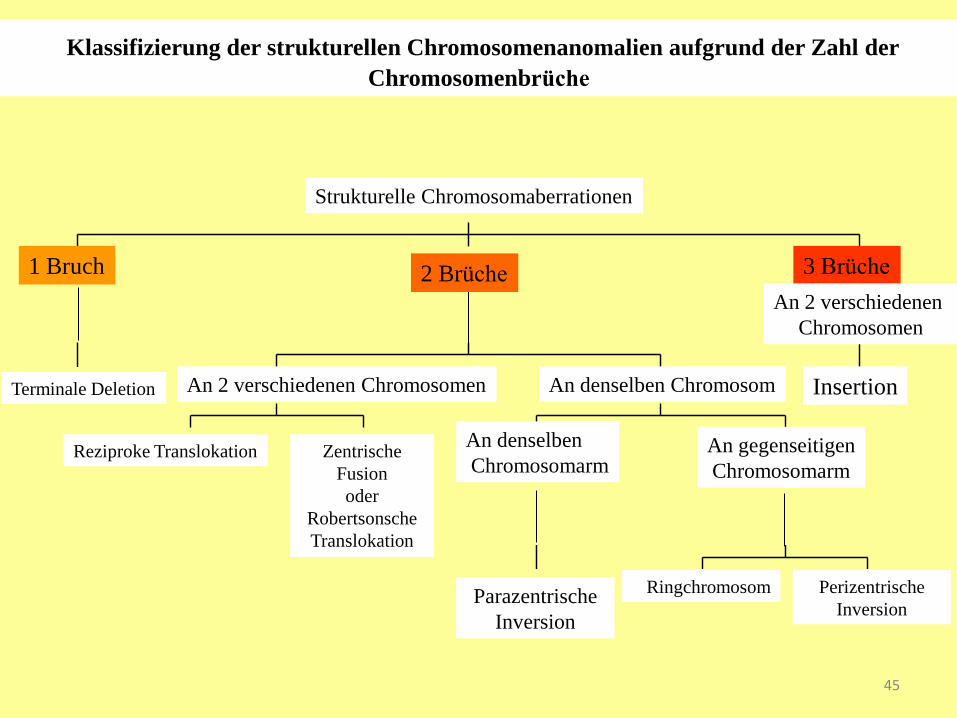
Klassifizierung der strukturellen Chromosomenanomalien aufgrund der Zahl der
Chromosomenbrüche
terminális deléció
1 törés
reciprok transzlokáció centrikus fúzió
vagy
Robertson-féle
transzlokáció
2 különböző kromoszómán
paracentrikus inverzió
uazon a kromoszóma
karon
gyűrű kromoszóma pericentrikus inverzió
ellentétes kromoszóma
karon
uazon a kromoszómán
2 törés
inszerció
3 törés2 különböző
kromoszómán
szerkezeti kromoszóma aberráció
1 Bruch 2 Brüche 3 Brüche
An denselben
Chromosomarm An gegenseitigen
Chromosomarm
An denselben Chromosom An 2 verschiedenen Chromosomen
Strukturelle Chromosomaberrationen
An 2 verschiedenen
Chromosomen
Terminale Deletion Insertion
Reziproke Translokation Zentrische
Fusion
oder
Robertsonsche
Translokation
Parazentrische
Inversion
Ringchromosom Perizentrische
Inversion
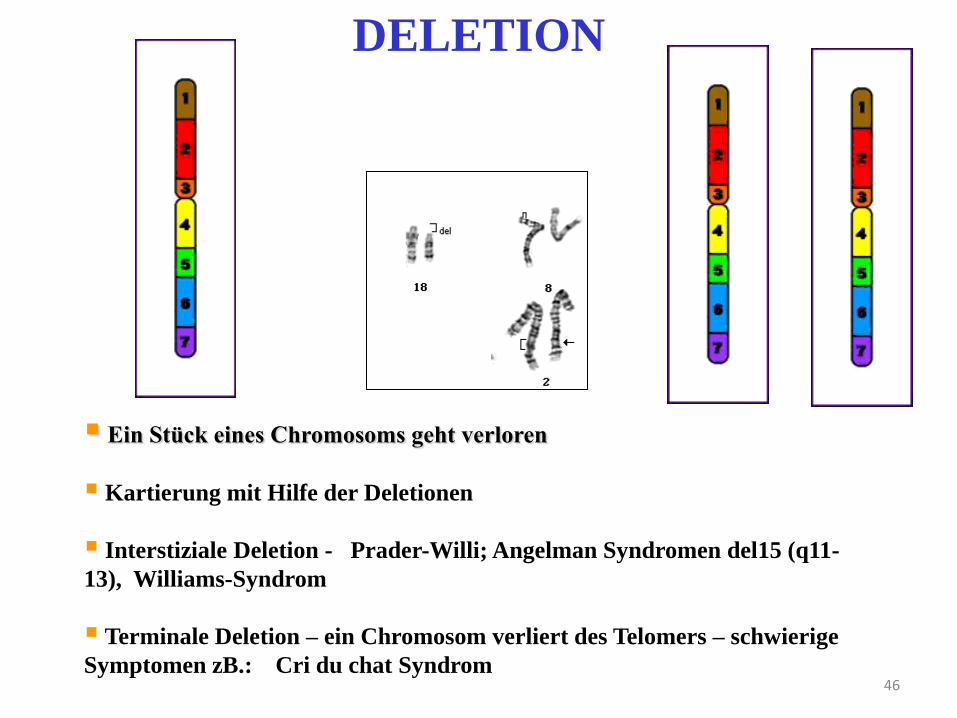
46
DELETION
Ein Stück eines Chromosoms geht verloren
Kartierung mit Hilfe der Deletionen
Interstiziale Deletion - Prader-Willi; Angelman Syndromen del15 (q11-
13), Williams-Syndrom
Terminale Deletion – ein Chromosom verliert des Telomers – schwierige
Symptomen zB.: Cri du chat Syndrom
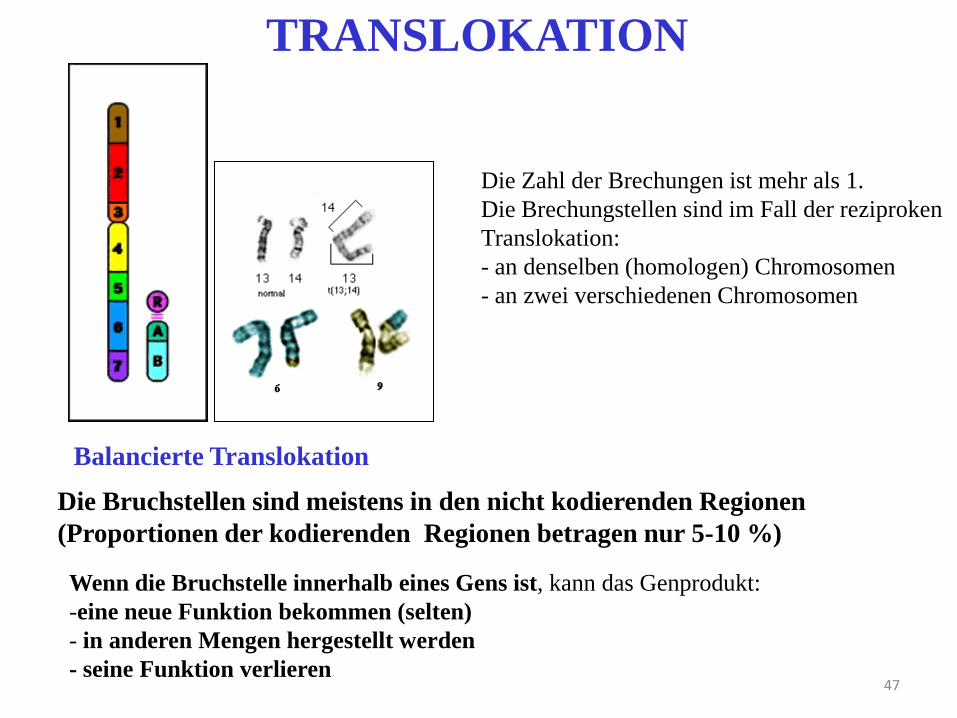
47
TRANSLOKATION
Die Zahl der Brechungen ist mehr als 1.
Die Brechungstellen sind im Fall der reziproken
Translokation:
- an denselben (homologen) Chromosomen
- an zwei verschiedenen Chromosomen
Die Bruchstellen sind meistens in den nicht kodierenden Regionen
(Proportionen der kodierenden Regionen betragen nur 5-10 %)
Balancierte Translokation
Wenn die Bruchstelle innerhalb eines Gens ist, kann das Genprodukt:
-eine neue Funktion bekommen (selten)
- in anderen Mengen hergestellt werden
- seine Funktion verlieren
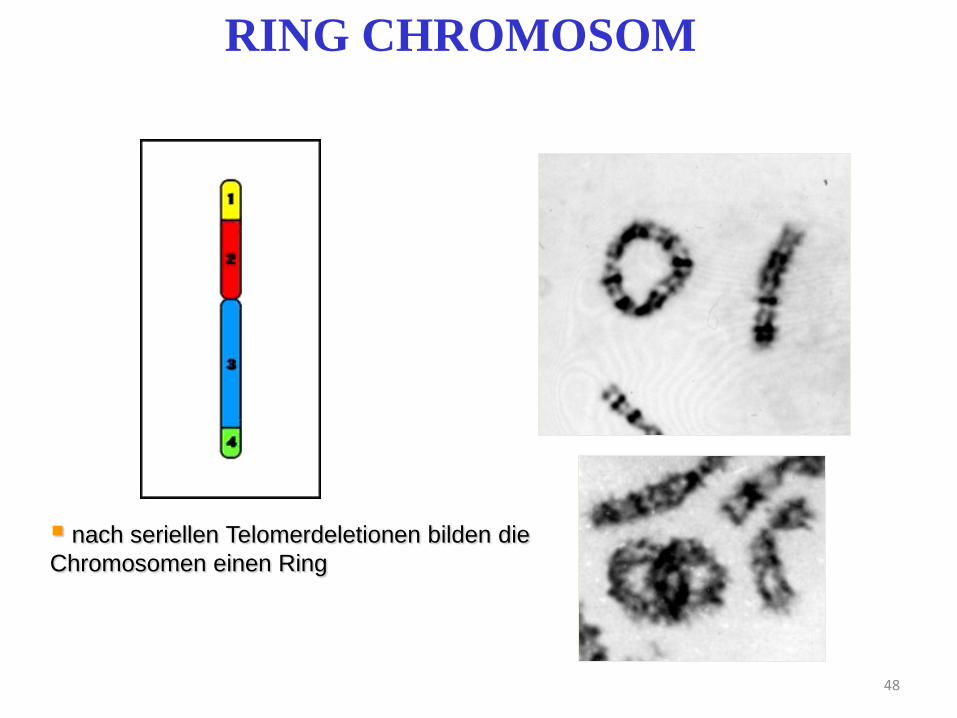
48
RING CHROMOSOM
nach seriellen Telomerdeletionen bilden die
Chromosomen einen Ring
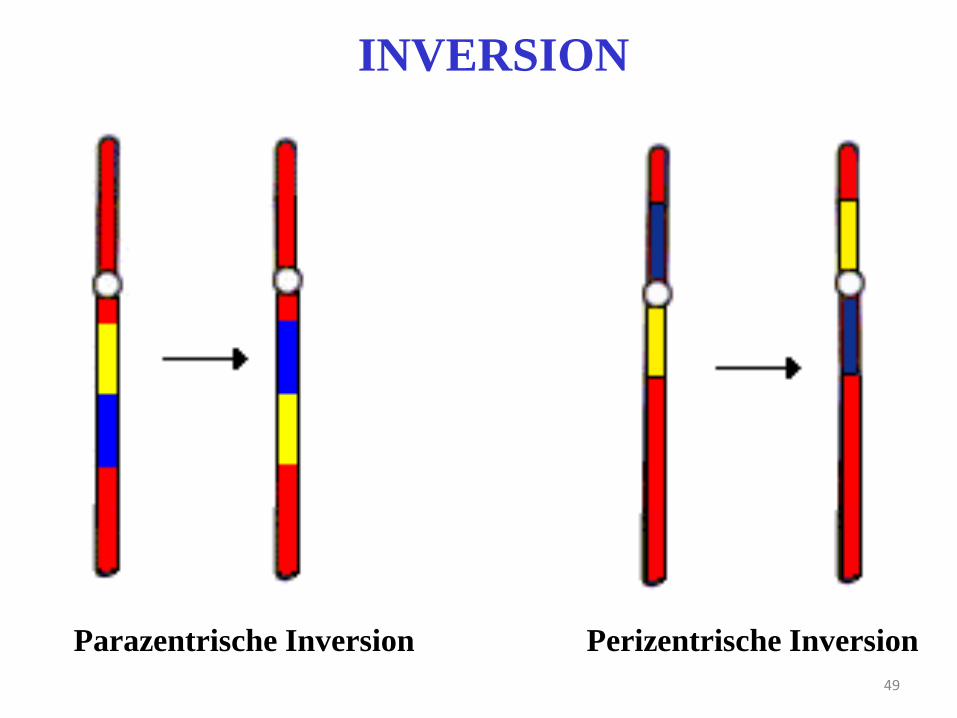
49
INVERSION
Parazentrische Inversion Perizentrische Inversion
50
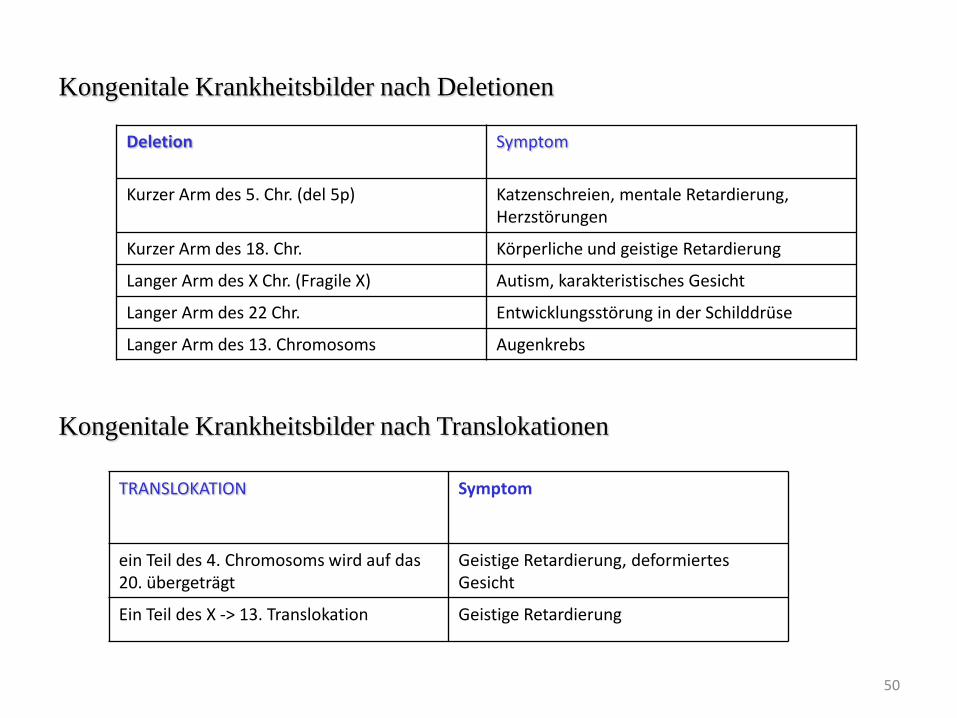
Kongenitale Krankheitsbilder nach Deletionen
Deletion Symptom
Kurzer Arm des 5. Chr. (del 5p) Katzenschreien, mentale Retardierung, Herzstörungen
Kurzer Arm des 18. Chr. Körperliche und geistige Retardierung
Langer Arm des X Chr. (Fragile X) Autism, karakteristisches Gesicht
Langer Arm des 22 Chr. Entwicklungsstörung in der Schilddrüse
Langer Arm des 13. Chromosoms Augenkrebs
Kongenitale Krankheitsbilder nach Translokationen
TRANSLOKATION Symptom
ein Teil des 4. Chromosoms wird auf das 20. übergeträgt
Geistige Retardierung, deformiertes Gesicht
Ein Teil des X -> 13. Translokation Geistige Retardierung
51
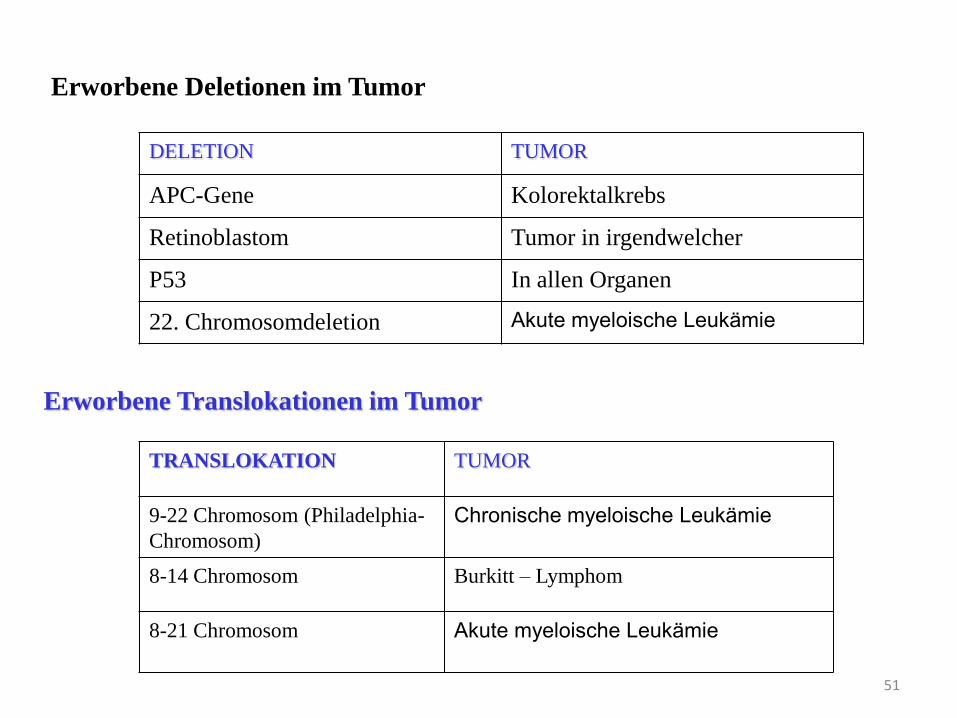
Erworbene Deletionen im Tumor
DELETION TUMOR
APC-Gene Kolorektalkrebs
Retinoblastom Tumor in irgendwelcher
P53 In allen Organen
22. Chromosomdeletion Akute myeloische Leukämie
TRANSLOKATION TUMOR
9-22 Chromosom (Philadelphia-
Chromosom)
Chronische myeloische Leukämie
8-14 Chromosom Burkitt – Lymphom
8-21 Chromosom Akute myeloische Leukämie
Erworbene Translokationen im Tumor
52
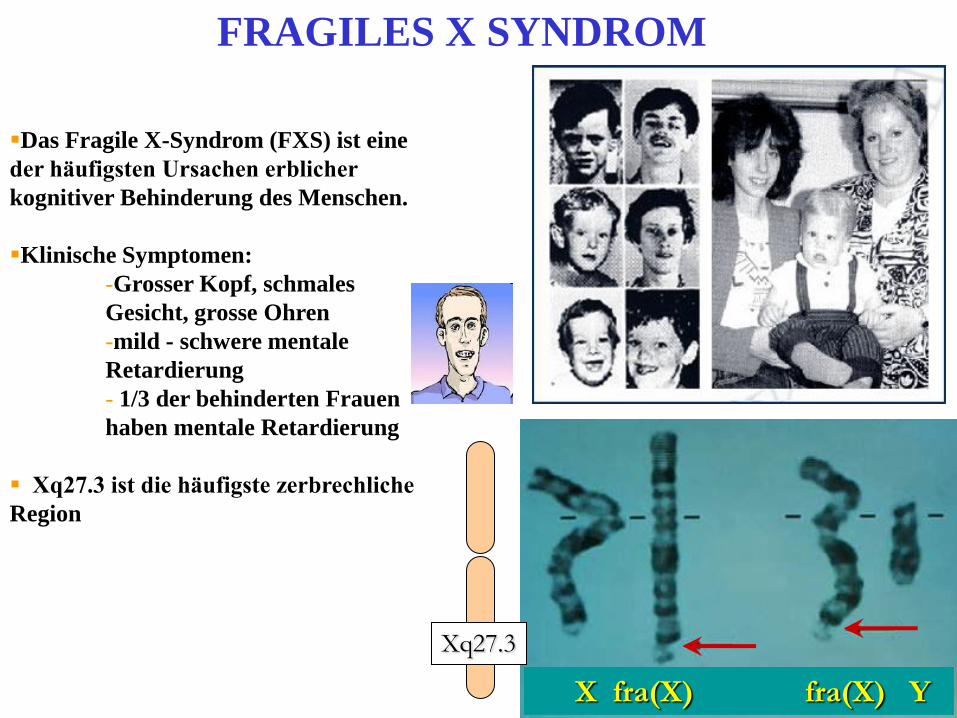
Das Fragile X-Syndrom (FXS) ist eine
der häufigsten Ursachen erblicher
kognitiver Behinderung des Menschen.
Klinische Symptomen:
-Grosser Kopf, schmales
Gesicht, grosse Ohren
-mild - schwere mentale
Retardierung
- 1/3 der behinderten Frauen
haben mentale Retardierung
Xq27.3 ist die häufigste zerbrechliche
Region
X fra(X) fra(X) Y
FRAGILES X SYNDROM
Xq27.3

Fragiles X Syndrom
• Monogen vererbt, Xq27.3
• 1:1200-2500
• Mädchen: geringer ausgeprägte Symptome
• Kleinkindesalter Hochwuchs, Makrozephalie
• Gesichtsdysmorphien
• Große dysplastische Ohren
• Jungen: große Hoden
53

Fragiles X Syndrom
• Mentale Retardierung IQ < 60
• Hyperkinesie
• Hypersensibilität
• Autismus?
• Überstreckbarkeit der Gelenke
• Dilatierter Aortenbogen
• Mitralklappenprolaps
54

55
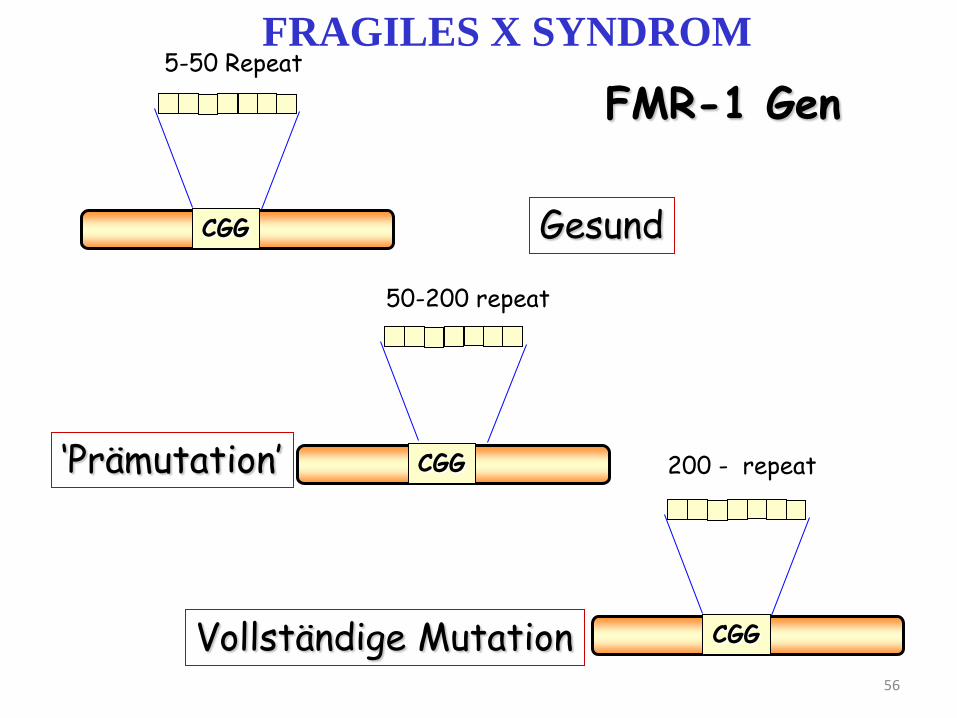
Die Mutation wirkt die CGG Trinukleotid Repeats des Gens FRAXA in der Region Xq28
Die Expansion der CGG Repeaten entsteht beim „mütterlichen Übertragung”
Es ist den 5’ Teil der nicht transkribierenden Region begleitet
die Verlängerung dieser Region folgt die Hemmung der Expression durch Methylieung
Das FMR1 Protein ist ein RNA-bindendes Protein
Für die mentale Retardation ist wahrscheinlich das mGluR5 (metabotrope glutamate receptor)
verantwortlich
FRAGILES X SYNDROM
56
CGG
CGG
5-50 Repeat
CGG
50-200 repeat
200 - repeat
Gesund
‘Prämutation’
Vollständige Mutation
FMR-1 Gen
FRAGILES X SYNDROM
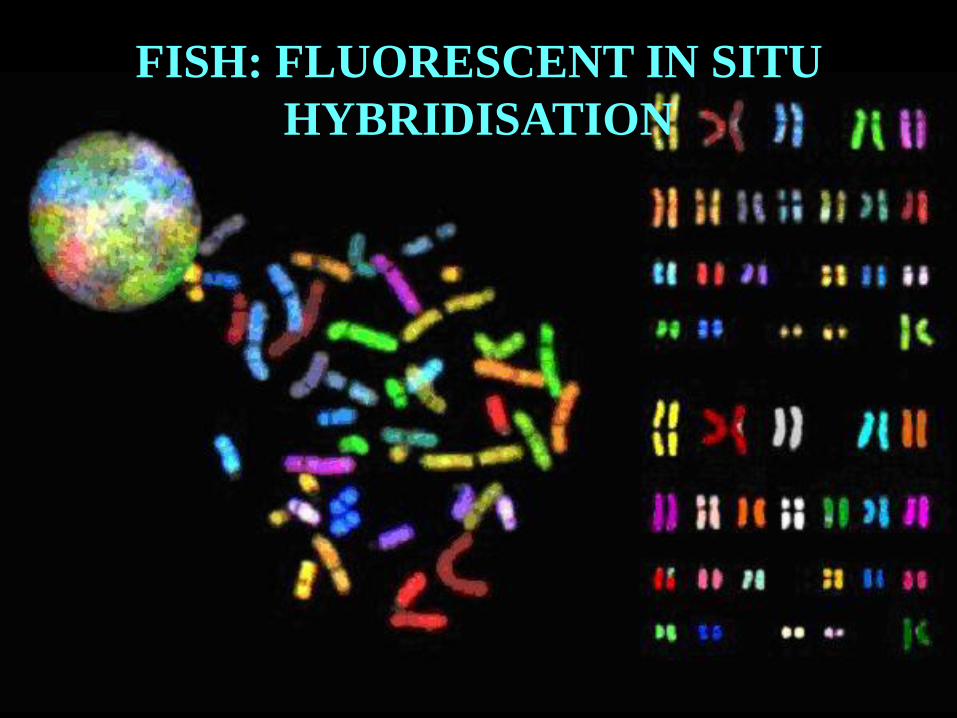
57
FISH: FLUORESCENT IN SITU
HYBRIDISATION
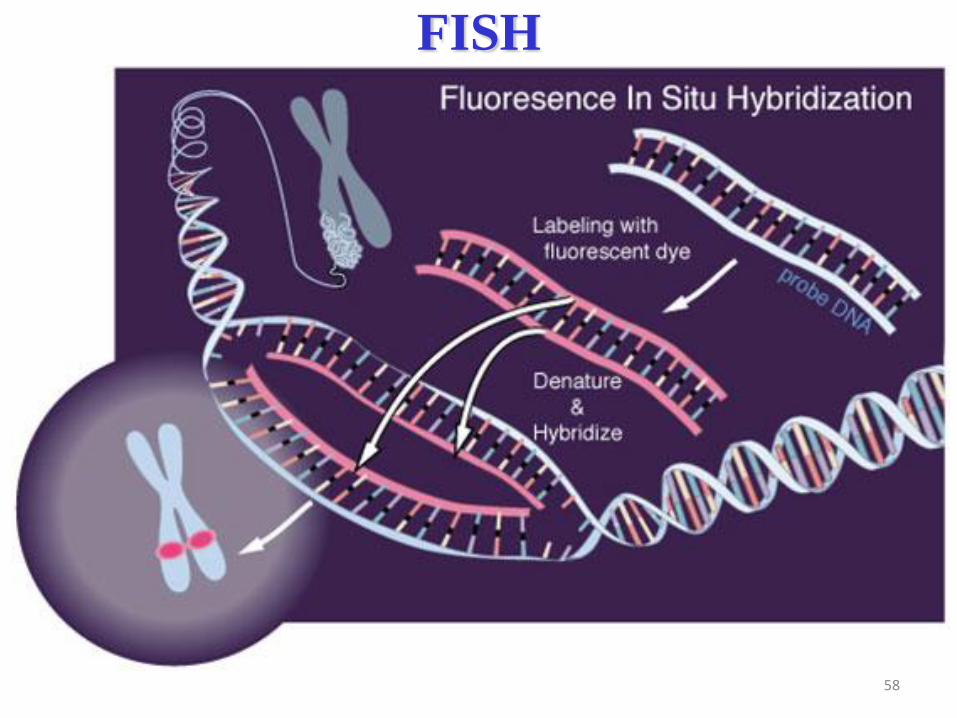
58
FISH
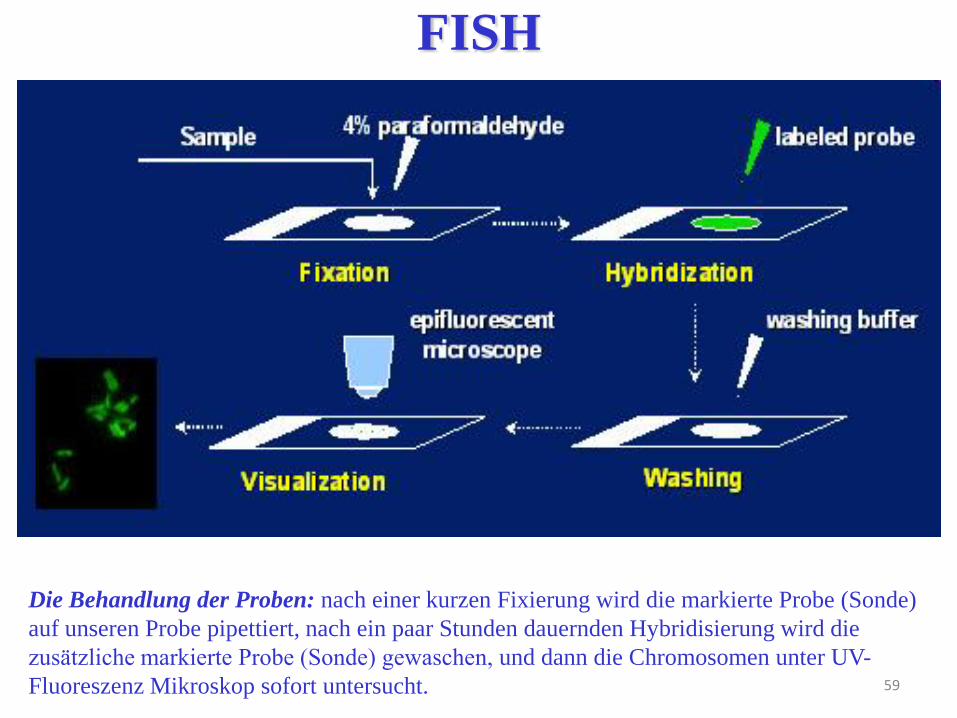
59
Die Behandlung der Proben: nach einer kurzen Fixierung wird die markierte Probe (Sonde)
auf unseren Probe pipettiert, nach ein paar Stunden dauernden Hybridisierung wird die
zusätzliche markierte Probe (Sonde) gewaschen, und dann die Chromosomen unter UV-
Fluoreszenz Mikroskop sofort untersucht.
.
FISH
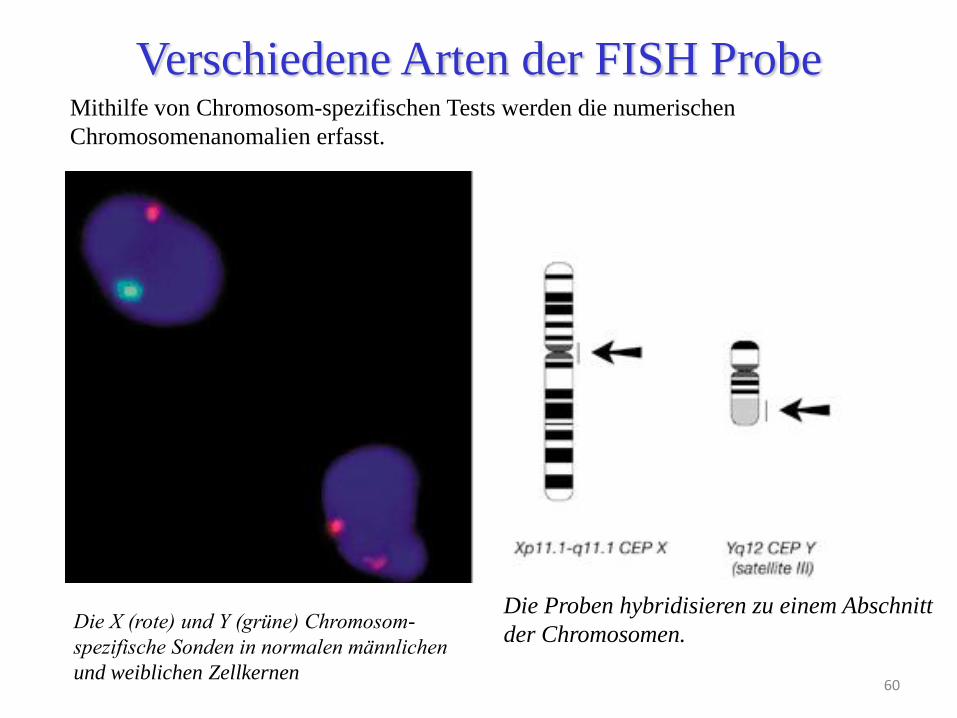
60
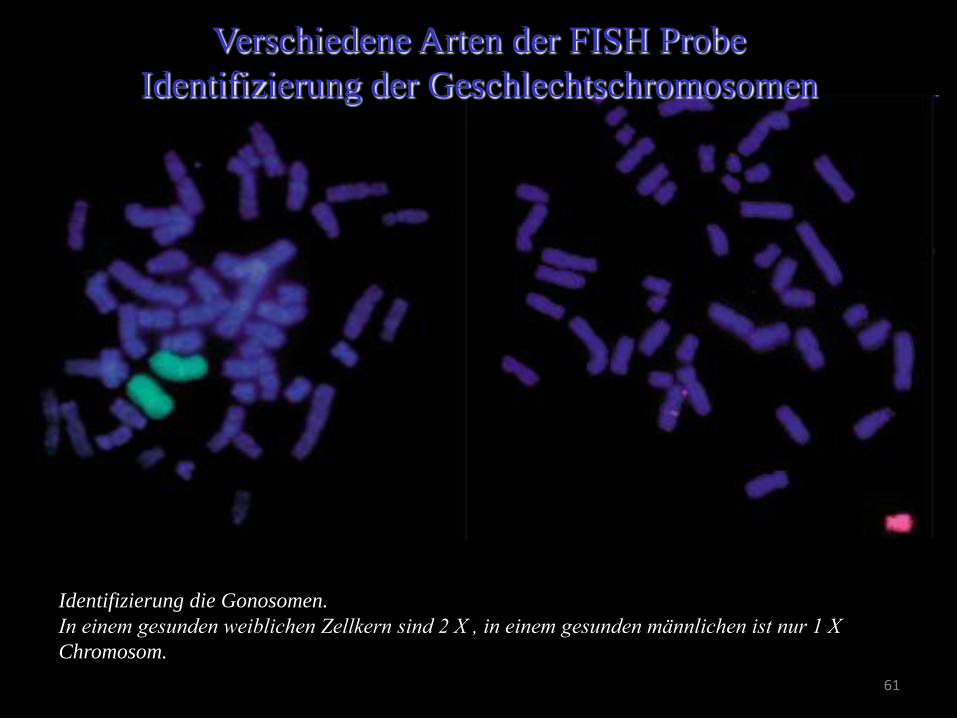
Verschiedene Arten der FISH Probe Mithilfe von Chromosom-spezifischen Tests werden die numerischen
Chromosomenanomalien erfasst.
Die X (rote) und Y (grüne) Chromosom-
spezifische Sonden in normalen männlichen
und weiblichen Zellkernen
Die Proben hybridisieren zu einem Abschnitt
der Chromosomen.
61
Identifizierung die Gonosomen.
In einem gesunden weiblichen Zellkern sind 2 X , in einem gesunden männlichen ist nur 1 X
Chromosom.
Verschiedene Arten der FISH Probe
Identifizierung der Geschlechtschromosomen
62
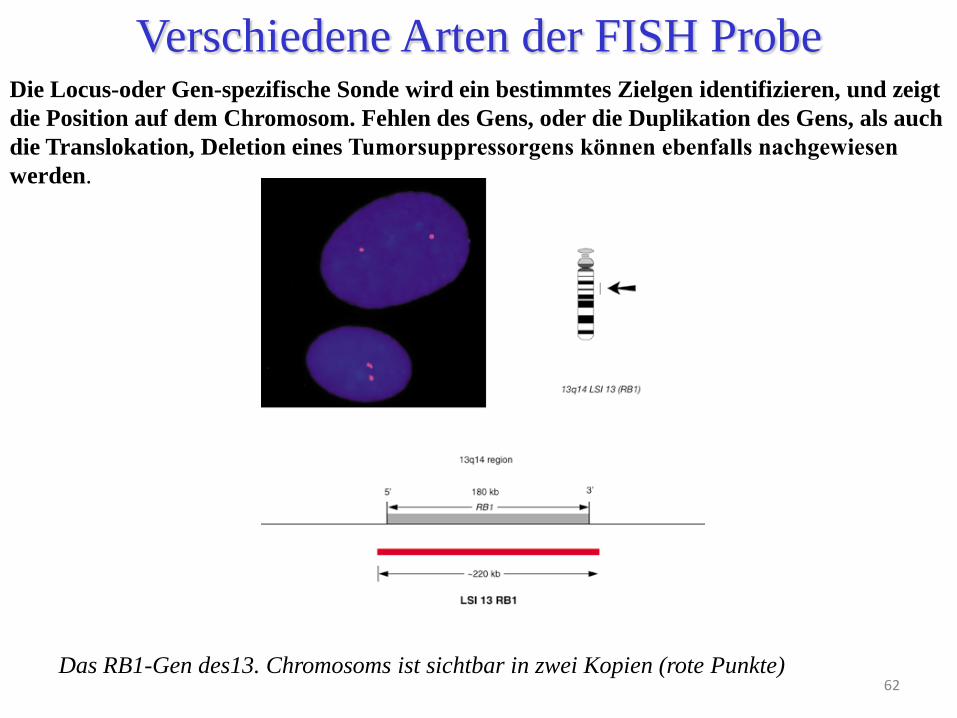
Die Locus-oder Gen-spezifische Sonde wird ein bestimmtes Zielgen identifizieren, und zeigt
die Position auf dem Chromosom. Fehlen des Gens, oder die Duplikation des Gens, als auch
die Translokation, Deletion eines Tumorsuppressorgens können ebenfalls nachgewiesen
werden.
Das RB1-Gen des13. Chromosoms ist sichtbar in zwei Kopien (rote Punkte)
Verschiedene Arten der FISH Probe

63
Locus spezifische FISH Probe für
verschiedene Syndromen • Prader-Willi Syndrom 15q11-13
• Angelman Syndrom 15q11-13
• Di-George Syndrom /VCFS 22q11.2
• Williams Syndrom 7q11.23
• Wolf-Hirschhorn Syndrom 4p16.3
• Cri du Chat Syndrom 5p15.2
• Kallmann Syndrom Xp22.3
• SRY Gen Yp11.3
• X-verbundene Ichthyosis Xp22.3
• Retinoblastoma (RB1-Gen) 13q14
• Smith-Magenis Syndrom 17p11.2
• Miller-Dieker Syndrom 17p13.3
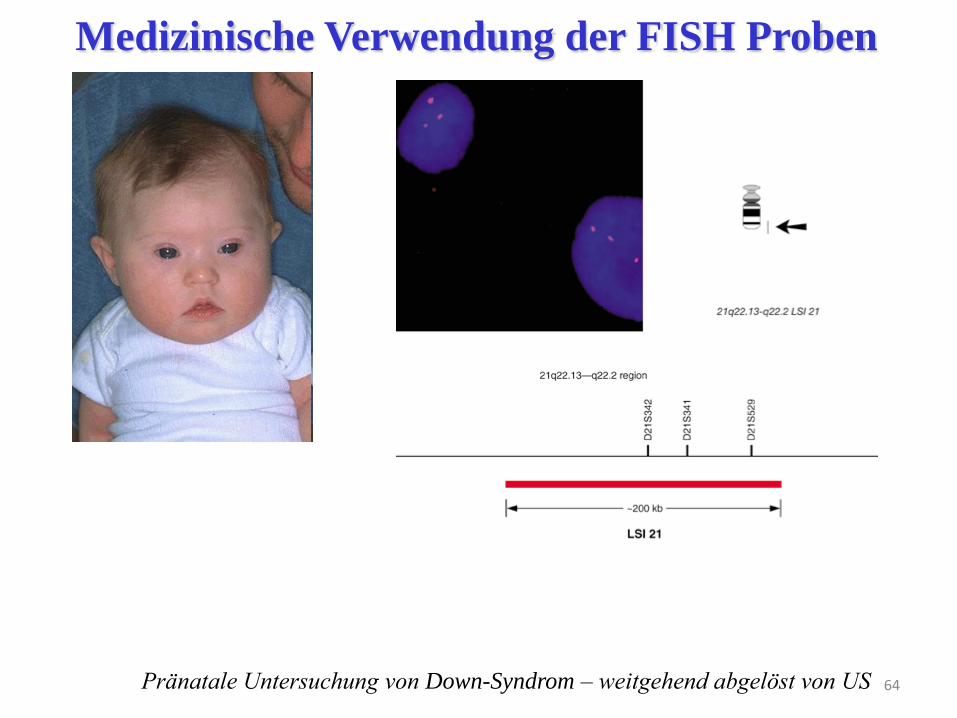
64 Pränatale Untersuchung von Down-Syndrom – weitgehend abgelöst von US
Medizinische Verwendung der FISH Proben
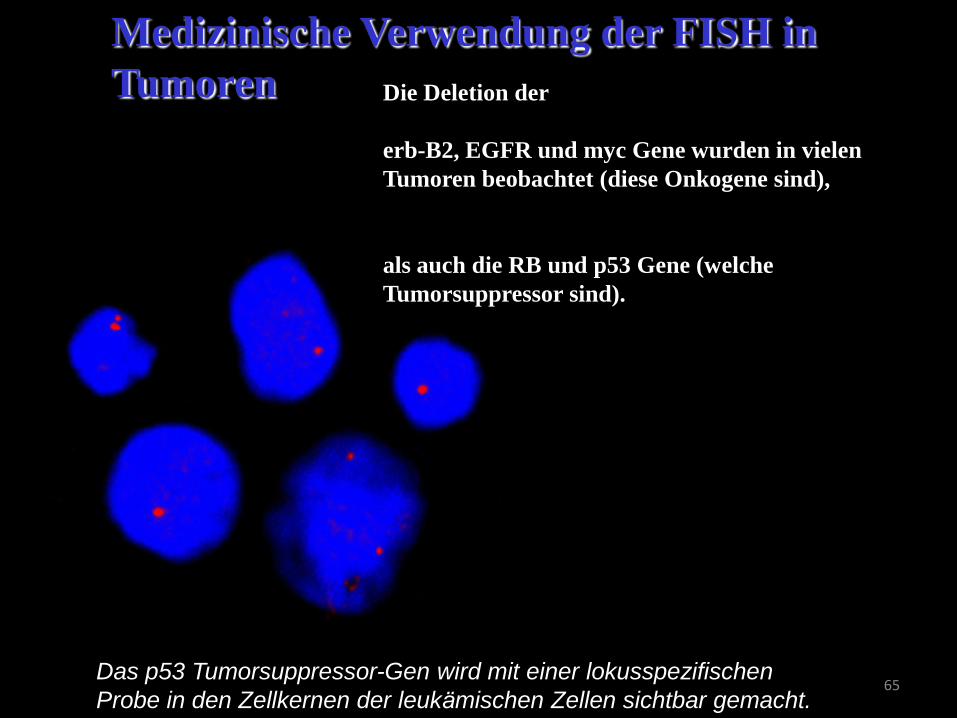
65 Das p53 Tumorsuppressor-Gen wird mit einer lokusspezifischen
Probe in den Zellkernen der leukämischen Zellen sichtbar gemacht.
Medizinische Verwendung der FISH in
Tumoren Die Deletion der
erb-B2, EGFR und myc Gene wurden in vielen
Tumoren beobachtet (diese Onkogene sind),
als auch die RB und p53 Gene (welche
Tumorsuppressor sind).
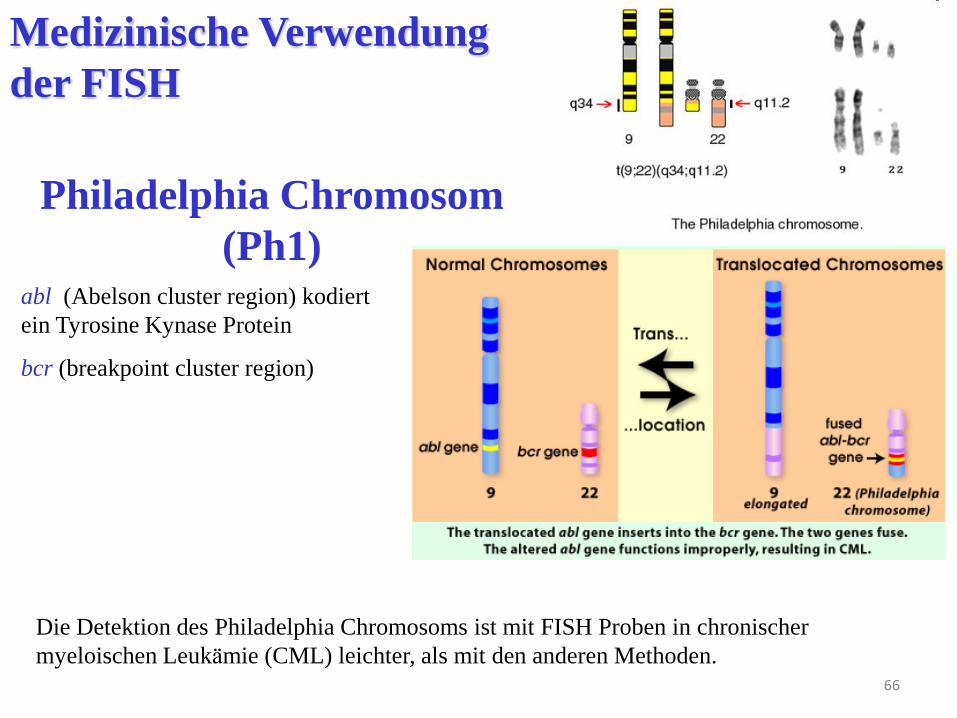
66
Die Detektion des Philadelphia Chromosoms ist mit FISH Proben in chronischer
myeloischen Leukämie (CML) leichter, als mit den anderen Methoden.
Philadelphia Chromosom
(Ph1) abl (Abelson cluster region) kodiert
ein Tyrosine Kynase Protein
bcr (breakpoint cluster region)
Medizinische Verwendung
der FISH

67
das Gen BCR liegt normalerweise auf dem 22. Chromosom,
das ABL Gen auf dem 9. Chromosom.
Nach der Translokation werden die zwei Gene fusioniert.
Medizinische Verwendung des FISH

68
Verschiedene Arten der FISH
Telomerspezifischen Probe
Telomer
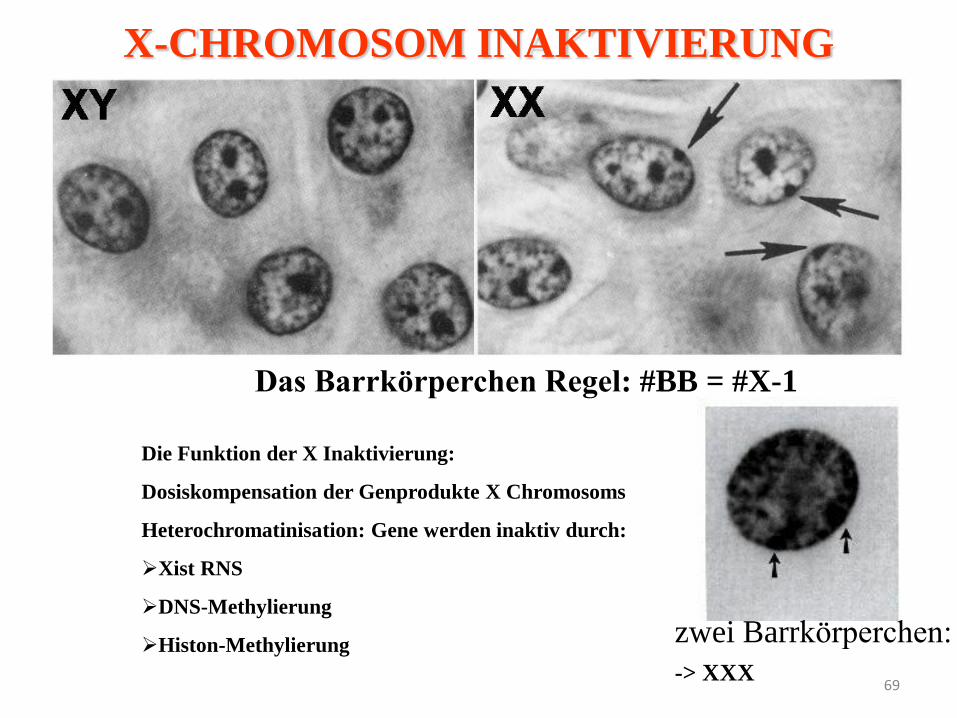
69
Das Barrkörperchen Regel: #BB = #X-1
zwei Barrkörperchen:
-> XXX
Die Funktion der X Inaktivierung:
Dosiskompensation der Genprodukte X Chromosoms
Heterochromatinisation: Gene werden inaktiv durch:
Xist RNS
DNS-Methylierung
Histon-Methylierung
X-CHROMOSOM INAKTIVIERUNG

70
- Es ist ein random Prozess
- das Prozess ist während der frühen Embryogenese schon aktiv
- Das Ergebnis ist unumkehrbar
X-CHROMOSOM INAKTIVIERUNG
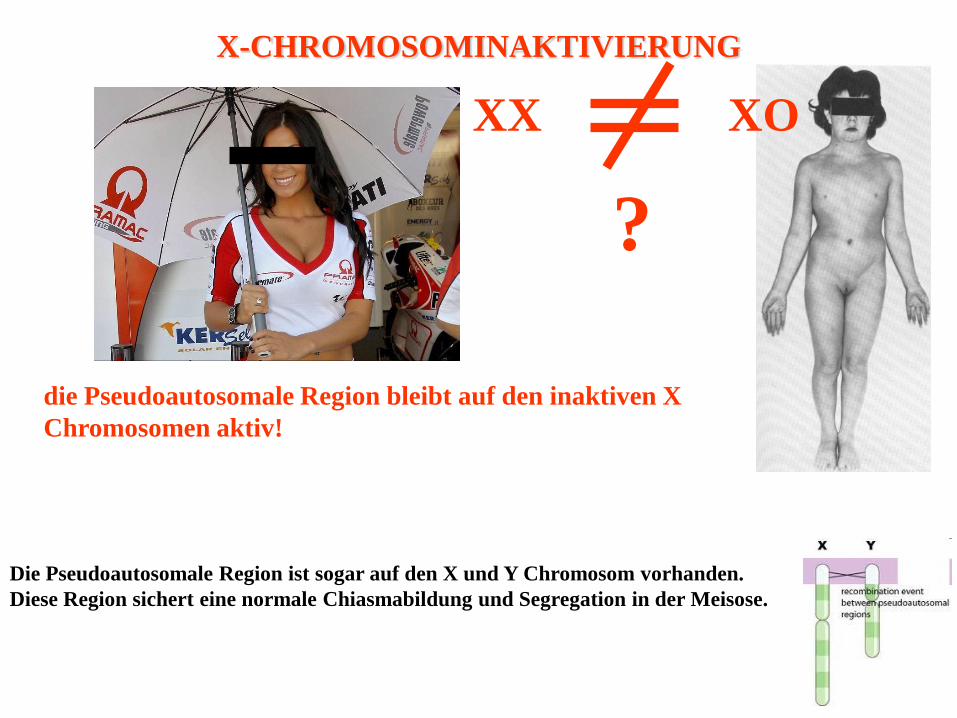
71
die Pseudoautosomale Region bleibt auf den inaktiven X
Chromosomen aktiv!
XX XO
?
Die Pseudoautosomale Region ist sogar auf den X und Y Chromosom vorhanden.
Diese Region sichert eine normale Chiasmabildung und Segregation in der Meisose.
X-CHROMOSOMINAKTIVIERUNG
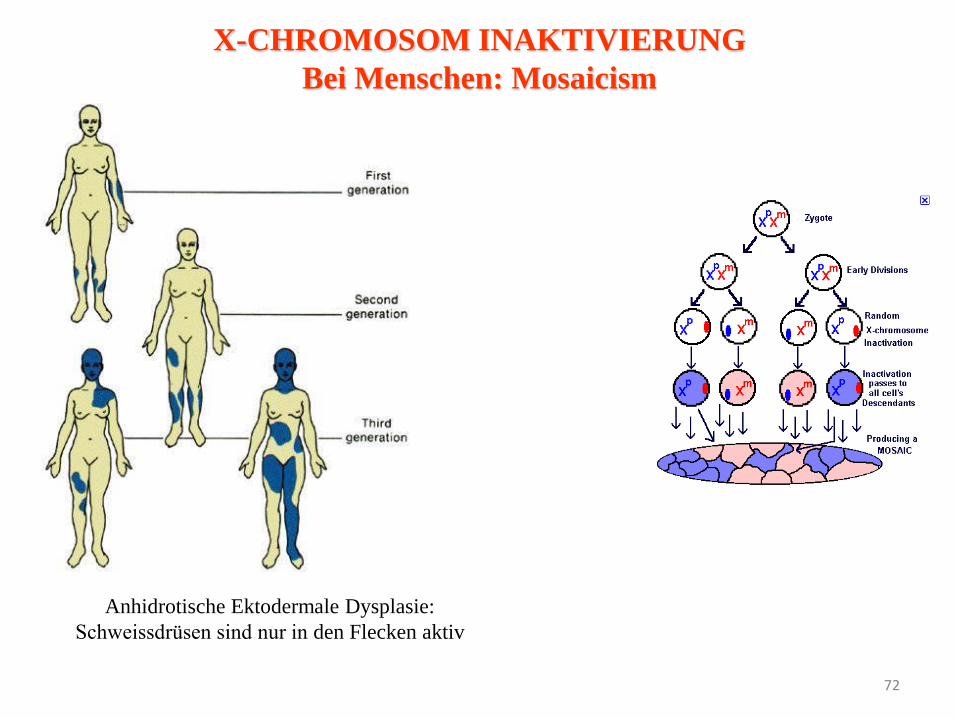
72
Anhidrotische Ektodermale Dysplasie:
Schweissdrüsen sind nur in den Flecken aktiv
X-CHROMOSOM INAKTIVIERUNG
Bei Menschen: Mosaicism

2. Marker für Krankheiten
• Geburtshilfe
• Neurologie
• Geriatrie
73

74
Danke für die Aufmerksamkeit!
























![cytogenetics eng final [Kompatibilitási mód]web.med.u-szeged.hu/mdbio/eng/materials/2013-2014/2nd_semester/... · encodes a tirosin kinase bcr(breakpoint cluster region) Medical](https://img.dokumen.tips/doc/110x75/5b03069e7f8b9a3c378b91f3/cytogenetics-eng-final-kompatibilitsi-mdwebmedu-a-tirosin-kinase-bcrbreakpoint.jpg)



