Embed Size (px)
Citation preview

Dispatch R63
Molecular evolution: Please release me, genetic code Niles Lehman
The genetic code is no longer universal, even in non-mitochondrial genomes. Recent studies have implicatedthe eukaryotic release factor eRF1 in mediating codingchanges that are not as inconceivable as once thought.Specific residues in eRF1 proteins can be correlatedwith specific code changes in a wide variety of taxa.
Address: Department of Biological Sciences, The University at Albany,State University of New York, 1400 Washington Avenue, Albany, NewYork 12222, USA.
Current Biology 2001, 11:R63–R66
0960-9822/01/$ – see front matter © 2000 Elsevier Science Ltd. All rights reserved.
Once thought universal, the specific relationships betweenamino acids and codons that are collectively known as thegenetic code are now proving to be variable in many taxa[1,2]. While this realization has been disappointing tosome — the genetic code was often hailed as the ultimateevolutionary anchor in that its universality was perhapsthe indisputable piece of evidence that all life shared acommon ancestor at some point — it has also opened up arich field of evolutionary analysis, by forcing us to considerwhat sequence of molecular events in a cell could possiblyallow for codon reassignment. A good example of such astudy, published in this issue of Current Biology [3], hasprovided compelling evidence that the translationalrelease factor eRF1 has played an important role in theevolution of the genetic code in many taxa.
The standard view of the evolution of the genetic codehad been that, once the code became fixed in someprimitive lineage of organisms, then any coding changewould be precluded because the transitory coding stagethat a population must experience to change its codewould be lethal [4,5]. Consider, for example, mutationsthat change the charging specificity of a tRNA aminoacylsynthetase, such that it charged a glycyl-tRNA witharginine instead. Suddenly glycines are replaced byarginines thoughout the genome, which would undoubt-edly cause irreparable cellular chaos. This could bethought of as the quintessential case of stabilizing selec-tion: a ‘Death Valley’ in the adaptive landscape.
A pattern is emerging from the study of beasts that do notuse the universal code — let us now call it the canonicalcode [6] — that makes code evolution not only compre-hendible, but exciting. The first examples of non-canonical codes were found in mitochondria, and couldeasily be rationalized by the fact that mitochondria have atranslation system partially independent of the nucleus,
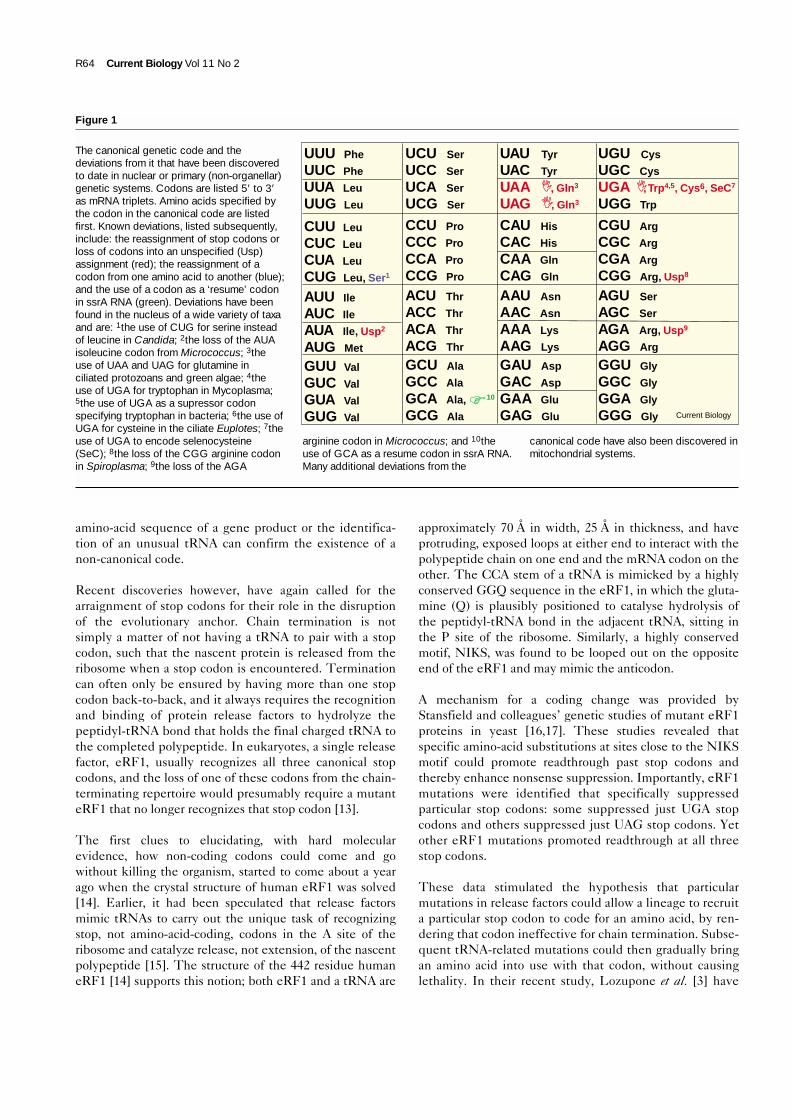
such that all sorts of nefarious genetics are possible [7,8].Since 1985 however, coding changes have been spotted inthe nuclear systems of dozens of organisms, many of whichare ciliated protozoans, but also Mycoplasma and otherFirmicutes, some Diplomonads other than Giarardia, theyeast Candida and some other fungi, and Acetabularia andsome other green algae [1]. The striking feature of a greatpreponderance of these coding changes is that theyinvolve codons that are either untranslated or cause chaintermination (see Figure 1). Only the reassignment of theCUG codon from leucine to serine in Candida is knownto depart from this trend. Most mitochondrial codingchanges also involve untranslated or stop codons. On topof all this, the ‘twenty-first amino acid’, selenocysteine [9]is even incorporated into some proteins in organisms asdiverse as Escherichia coli and humans via the ‘umber’codon UGA.
What is it about these noncoding codons that puts them atrisk for coding changes? In most cases a stop codon hasbeen recruited to specify an amino acid. In these situa-tions, the evolution is mediated by the appearance of amutation in the anticodon of a tRNA or its cognate syn-thetase, such that the tRNA, charged with an amino acid,recognizes and base pairs with a canonical mRNA stopcodon — UAA, UAG, or UGA — during translation on theribosome. An important characteristic of stop codons maybe that they are A+U rich, weakening the stability of thecodon–anticodon base-pairing, at least when unchaper-oned. It has been proposed that the relative instability ofthis interaction relegated these three codons to a termina-tion role during the primordial development of the code[10], in conjunction with thermodynamic arguments thatthe earliest versions of the code may have been dominatedby G and C containing codons [11,12]. It is interesting tonote that many of the other evolutionary labile codons areA+U rich, such as the AUA and AAA codons that seem toflip-flop between assigned amino acids in severalmitochondrial lineages [2].
A curious problem with the discovery of non-canonicalcodes — and many such discoveries are certain to be forth-coming with the proliferation of genome studies — is thatthey may be biased to reveal changes involving stopcodons, because these are the easiest to detect fromnucleotide sequence data. Without corresponding proteinsequences, the coding relationships of a gene are usuallyonly found aberrant when canonical stop codons appear inthe midst of the gene, and when these codons can bematched with amino acids appearing at the same positionsin orthologous sequences from other organisms. Only the
amino-acid sequence of a gene product or the identifica-tion of an unusual tRNA can confirm the existence of anon-canonical code.
Recent discoveries however, have again called for thearraignment of stop codons for their role in the disruptionof the evolutionary anchor. Chain termination is notsimply a matter of not having a tRNA to pair with a stopcodon, such that the nascent protein is released from theribosome when a stop codon is encountered. Terminationcan often only be ensured by having more than one stopcodon back-to-back, and it always requires the recognitionand binding of protein release factors to hydrolyze thepeptidyl-tRNA bond that holds the final charged tRNA tothe completed polypeptide. In eukaryotes, a single releasefactor, eRF1, usually recognizes all three canonical stopcodons, and the loss of one of these codons from the chain-terminating repertoire would presumably require a mutanteRF1 that no longer recognizes that stop codon [13].
The first clues to elucidating, with hard molecularevidence, how non-coding codons could come and gowithout killing the organism, started to come about a yearago when the crystal structure of human eRF1 was solved[14]. Earlier, it had been speculated that release factorsmimic tRNAs to carry out the unique task of recognizingstop, not amino-acid-coding, codons in the A site of theribosome and catalyze release, not extension, of the nascentpolypeptide [15]. The structure of the 442 residue humaneRF1 [14] supports this notion; both eRF1 and a tRNA are
approximately 70 Å in width, 25 Å in thickness, and haveprotruding, exposed loops at either end to interact with thepolypeptide chain on one end and the mRNA codon on theother. The CCA stem of a tRNA is mimicked by a highlyconserved GGQ sequence in the eRF1, in which the gluta-mine (Q) is plausibly positioned to catalyse hydrolysis ofthe peptidyl-tRNA bond in the adjacent tRNA, sitting inthe P site of the ribosome. Similarly, a highly conservedmotif, NIKS, was found to be looped out on the oppositeend of the eRF1 and may mimic the anticodon.
A mechanism for a coding change was provided byStansfield and colleagues’ genetic studies of mutant eRF1proteins in yeast [16,17]. These studies revealed thatspecific amino-acid substitutions at sites close to the NIKSmotif could promote readthrough past stop codons andthereby enhance nonsense suppression. Importantly, eRF1mutations were identified that specifically suppressedparticular stop codons: some suppressed just UGA stopcodons and others suppressed just UAG stop codons. Yetother eRF1 mutations promoted readthrough at all threestop codons.
These data stimulated the hypothesis that particularmutations in release factors could allow a lineage to recruita particular stop codon to code for an amino acid, by ren-dering that codon ineffective for chain termination. Subse-quent tRNA-related mutations could then gradually bringan amino acid into use with that codon, without causinglethality. In their recent study, Lozupone et al. [3] have
R64 Current Biology Vol 11 No 2
Figure 1
The canonical genetic code and thedeviations from it that have been discoveredto date in nuclear or primary (non-organellar)genetic systems. Codons are listed 5′ to 3′as mRNA triplets. Amino acids specified bythe codon in the canonical code are listedfirst. Known deviations, listed subsequently,include: the reassignment of stop codons orloss of codons into an unspecified (Usp)assignment (red); the reassignment of acodon from one amino acid to another (blue);and the use of a codon as a ‘resume’ codonin ssrA RNA (green). Deviations have beenfound in the nucleus of a wide variety of taxaand are: 1the use of CUG for serine insteadof leucine in Candida; 2the loss of the AUAisoleucine codon from Micrococcus; 3theuse of UAA and UAG for glutamine inciliated protozoans and green algae; 4theuse of UGA for tryptophan in Mycoplasma;5the use of UGA as a supressor codonspecifying tryptophan in bacteria; 6the use ofUGA for cysteine in the ciliate Euplotes; 7theuse of UGA to encode selenocysteine(SeC); 8the loss of the CGG arginine codonin Spiroplasma; 9the loss of the AGA
arginine codon in Micrococcus; and 10theuse of GCA as a resume codon in ssrA RNA.Many additional deviations from the
canonical code have also been discovered inmitochondrial systems.
UUU Phe
UUC Phe
UUA Leu
UUG Leu
CUU Leu
CUC Leu
CUA Leu
CUG Leu, Ser1
AUU Ile
AUC Ile
AUA Ile, Usp2
AUG Met
GUU Val
GUC Val
GUA Val
GUG Val
UCU Ser
UCC Ser
UCA Ser
UCG Ser
CCU Pro
CCC Pro
CCA Pro
CCG Pro
ACU Thr
ACC Thr
ACA Thr
ACG Thr
GCU Ala
GCC Ala
GCA Ala, 10
GCG Ala
UAU Tyr
UAC Tyr
UAA , Gln3
UAG , Gln3
CAU His
CAC His
CAA Gln
CAG Gln
AAU Asn
AAC Asn
AAA Lys
AAG Lys
GAU Asp
GAC Asp
GAA Glu
GAG Glu
UGU Cys
UGC Cys
UGA , Trp4,5, Cys6, SeC7
UGG Trp
CGU Arg
CGC Arg
CGA Arg
CGG Arg, Usp8
AGU Ser
AGC Ser
AGA Arg, Usp9
AGG Arg
GGU Gly
GGC Gly
GGA Gly
GGG
Gly Current Biology
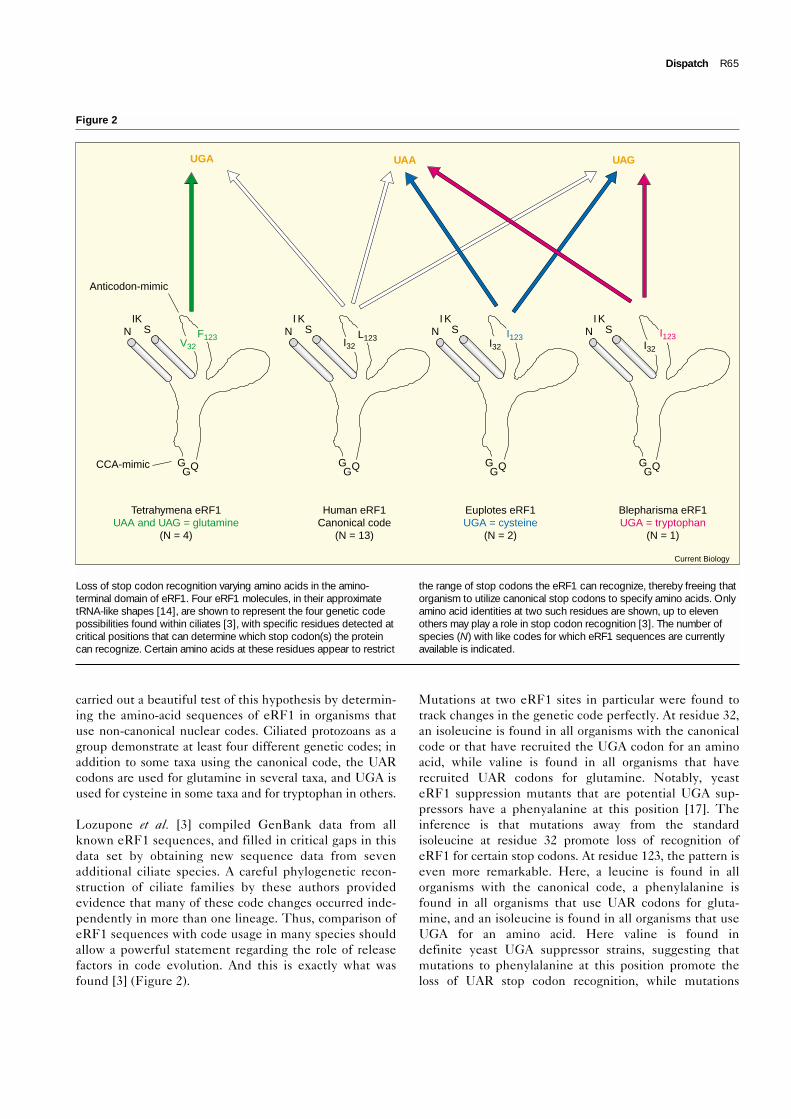
carried out a beautiful test of this hypothesis by determin-ing the amino-acid sequences of eRF1 in organisms thatuse non-canonical nuclear codes. Ciliated protozoans as agroup demonstrate at least four different genetic codes; inaddition to some taxa using the canonical code, the UARcodons are used for glutamine in several taxa, and UGA isused for cysteine in some taxa and for tryptophan in others.
Lozupone et al. [3] compiled GenBank data from allknown eRF1 sequences, and filled in critical gaps in thisdata set by obtaining new sequence data from sevenadditional ciliate species. A careful phylogenetic recon-struction of ciliate families by these authors providedevidence that many of these code changes occurred inde-pendently in more than one lineage. Thus, comparison ofeRF1 sequences with code usage in many species shouldallow a powerful statement regarding the role of releasefactors in code evolution. And this is exactly what wasfound [3] (Figure 2).
Mutations at two eRF1 sites in particular were found totrack changes in the genetic code perfectly. At residue 32,an isoleucine is found in all organisms with the canonicalcode or that have recruited the UGA codon for an aminoacid, while valine is found in all organisms that haverecruited UAR codons for glutamine. Notably, yeasteRF1 suppression mutants that are potential UGA sup-pressors have a phenyalanine at this position [17]. Theinference is that mutations away from the standardisoleucine at residue 32 promote loss of recognition ofeRF1 for certain stop codons. At residue 123, the pattern iseven more remarkable. Here, a leucine is found in allorganisms with the canonical code, a phenylalanine isfound in all organisms that use UAR codons for gluta-mine, and an isoleucine is found in all organisms that useUGA for an amino acid. Here valine is found indefinite yeast UGA suppressor strains, suggesting thatmutations to phenylalanine at this position promote theloss of UAR stop codon recognition, while mutations
Dispatch R65
Figure 2
UAA UAGUGA
NI K
S
GGQ
I32 I32L123 I123
I32
I123NIK
S
GGQ
V32
F123N
I KS
GGQ
NI K
S
GGQ
Human eRF1Canonical code
(N = 13)
Euplotes eRF1UGA = cysteine
(N = 2)
Tetrahymena eRF1UAA and UAG = glutamine
(N = 4)
Blepharisma eRF1UGA = tryptophan
(N = 1)
CCA-mimic
Anticodon-mimic
Current Biology
Loss of stop codon recognition varying amino acids in the amino-terminal domain of eRF1. Four eRF1 molecules, in their approximatetRNA-like shapes [14], are shown to represent the four genetic codepossibilities found within ciliates [3], with specific residues detected atcritical positions that can determine which stop codon(s) the proteincan recognize. Certain amino acids at these residues appear to restrict
the range of stop codons the eRF1 can recognize, thereby freeing thatorganism to utilize canonical stop codons to specify amino acids. Onlyamino acid identities at two such residues are shown, up to elevenothers may play a role in stop codon recognition [3]. The number ofspecies (N) with like codes for which eRF1 sequences are currentlyavailable is indicated.

to isoleucine or valine promote the loss of UGA stopcodon recognition.
Surprisingly the NIKS motif remained essentiallyinvariant across codes, although several other mutationsnear the NIKS motif showed correlations similar to thosefound at positions 32 and 123, indicating that manyresidues in the first amino-terminal domain of eRF1 areresponsible for stop-codon recognition specificity. Itwould seem that, once mutations become established atthese sites, then the release factor is restricted to recogniz-ing a smaller set of stop codons, ‘releasing’ them from onlycoding for chain termination.
Other molecular clues to the significance of stop codonscome from considerations of cellular elements thatpromote the exact opposite response to release factors.One such element is small stable RNA (ssrA RNA) of bac-teria. This multifunctional 360-nucleotide molecule, bindsto the P-site of ribosomes in which translation has stalledbecause the mRNA is prematurely broken at its 3′ endand no codon exists for the next amino acid (see [18] forreview). As a consequence of its unique secondary struc-ture, ssrA RNA is able to provide both the ‘resume’ codonand eventually the stop codon, after a short tag sequenceof about 10 amino acids has been added to the carboxy-terminal end of the polypeptide. This tag, which targetsthe abortive gene product for cellular degradation, con-tains several alanine residues, including an alanine at thefirst, or ‘resume’ position. Alanine is perhaps a goodchoice; its codons, like those of proline, arginine andglycine, are G+C rich and thus serve as the antithesis ofstop codons... go codons.
Our ever-expanding list of nonstandard genetics is notserving to unravel the unity of biology. Instead theyare strengthening our understanding of the mechanismsof evolution.
References1. Osawa S, Jukes TH, Watanabe K, Muto A: Recent evidence for the
evolution of the genetic code. Microbiol Rev 1992, 56:229-264.2. Knight RD, Freeland SJ, Landweber LF: Rewiring the keyboard:
evolvability of the genetic code. Nat Rev Gen 2001, in press.3. Luzopone CA, Knight RD, Landweber LF: The molecular basis of
nuclear genetic code change in ciliates. Curr Biol 2001, this issue.4. Crick FH: The origin of the genetic code. J Mol Biol 1968,
38:367-379.5. Wong JT-F: The evolution of a universal genetic code. Proc Natl
Acad Sci USA 1975, 73:2336-2340.6. Keeling PJ, Doolittle WF: A non-canonical genetic code in an early
diverging eukaryotic lineage. EMBO J 1996, 15:2285-2290.7. Kroon AM, Saccone C: The non-universality of the genetic code. In
Genes: Structure and Expression. Edited by Kroon AM. Springfield:John Wiley and Sons; 1983:347-355.
8. Jukes TH: Evolution of the amino acid code: inferences frommitochondrial codes. J Mol Evol 1983, 19:219-225.
9. Hatfield D, Diamond A: UGA: a split personality in the universalgenetic code. Trends Genet 1993, 9:69-70.
10. Lehman N, Jukes TH: Genetic code development by stop codontakeover. J Theor Biol 1988, 135:203-214.
11. Eigen M: Self organization of matter and the evolution of biologicalmacromolecules. Naturwissenschaften 1971, 58:465-526.
12. Turner DH, Bevilacqua PC: RNA thermodynamics. In The RNAWorld. Edited by Gesteland RF, Atkins JF. Plainview, NY: Cold SpringHarbor Press; 1993:447-464.
13. Osawa S, Jukes TH: Codon reassignment (codon capture) inevolution. J Mol Evol 1989, 28:271-278.
14. Song H, Mugnier P, Das AK, Webb HM, Evans DR, Tuite MF,Hemmings, BA, Barford D: The crystal structure of humaneukaryotic release factor eRF1 - mechanisms of stop codonrecognition and peptidyl-tRNA hydrolysis. Cell 2000, 100:311-321.
15. Ito K, Ebihara K, Uno M, Nakamura Y: Conserved motifs inprokaryotic and eukaryotic polypeptide release factors: tRNA-protein mimicry hypothesis. Proc Natl Acad Sci USA 1996,93:5443-5448.
16. Stansfield I, Akhmaloka, Tuite MF: A mutant allele of the SUP45(SAL4) gene of Saccharomyces cerevisiae shows temperature-dependent allosupressor and omnipotent supressor phenotypes.Curr Genet 1995, 27:417-426.
17. Bertram G, Bell HA, Richie DW, Fullerton G, Stansfield I: Terminatingeukaryotic translation: Domain I of release factor eRF1 functionsin stop codon recognition. RNA 2000, 6:1236-1247.
18. Karzai AW, Roche ED, Sauer RT: The SsrA-SmpB system forprotein tagging, directed degradation and ribosome rescue. NatStruct Biol 2000, 7:449-455.
R66 Current Biology Vol 11 No 2



![[MP] 02 - Phylogenetics - biologia.campusnet.unito.it · Molecular Phylogenetics Basis of Molecular Phylogenies Overview ¾Phylogenetics Definitions ¾Genetic Variation and Evolution](https://img.dokumen.tips/doc/110x75/5c6216d809d3f238158b4601/mp-02-phylogenetics-molecular-phylogenetics-basis-of-molecular-phylogenies.jpg)

























