Embed Size (px)
Citation preview

Molecular biology

Course structure
Polymerase Chain Reaction
RNA Isolation
cDNA Synthesis
Amplification
Electrophoresis
Transfection

Genomic DNA mRNA
Which molecules can be analyzed?

Nucleic acid chemistry
DNA and RNA store and transfer genetic information in living organisms.
DNA:– major constituent of the nucleus– stable representation of an organism’s complete genetic makeup
RNA:– found in the nucleus and the cytoplasm– key to information flow within a cell

Why purify genomic DNA?
Many applications require purified DNA. Purity and amount of DNA required depends on intended
application.
Tissue typing for organ transplant Detection of pathogens Human identity testing Genetic research

DNA purification challenges1. Separating DNA from other cellular components such as
proteins, lipids, RNA, etc.
2. Avoiding fragmentation of the long DNA molecules by mechanical shearing or the action of endogenous nucleases.
Effectively inactivating endogenous nucleases and preventing them from digesting the genomic DNA is a key early step in the purification process. DNases can usually be inactivated by use of heat or chelating agents.

DNA purification
There are many DNA purification methods. In all cases, one must:
1. Effectively disrupt cells or tissues(usually using detergent)
2. Denature proteins and nucleoprotein complexes(a protease/denaturant)
3. Inactivate endogenous nucleases(chelating agents)
4. Purify nucleic acid target selectively(could involve RNases, proteases, selective matrix and alcohol precipitations)

Total Cellular RNA
•Messenger RNA (mRNA): 1-5%Serves as a template for protein synthesis
• Ribosomal RNA (rRNA): >80%Structural component of ribosomes
• Transfer RNA (tRNA): 10-15%Translates mRNA information into the appropriate amino acid

What RNA is needed for?
Messenger RNA synthesis is a dynamic expression of the genome of an organism. As such, mRNA is central to information flow within a cell.
• Size – examine differential splicing• Sequence – predict protein product• Abundance – measure expression levels• Dynamics of expression – temporal, developmental, tissue specificity
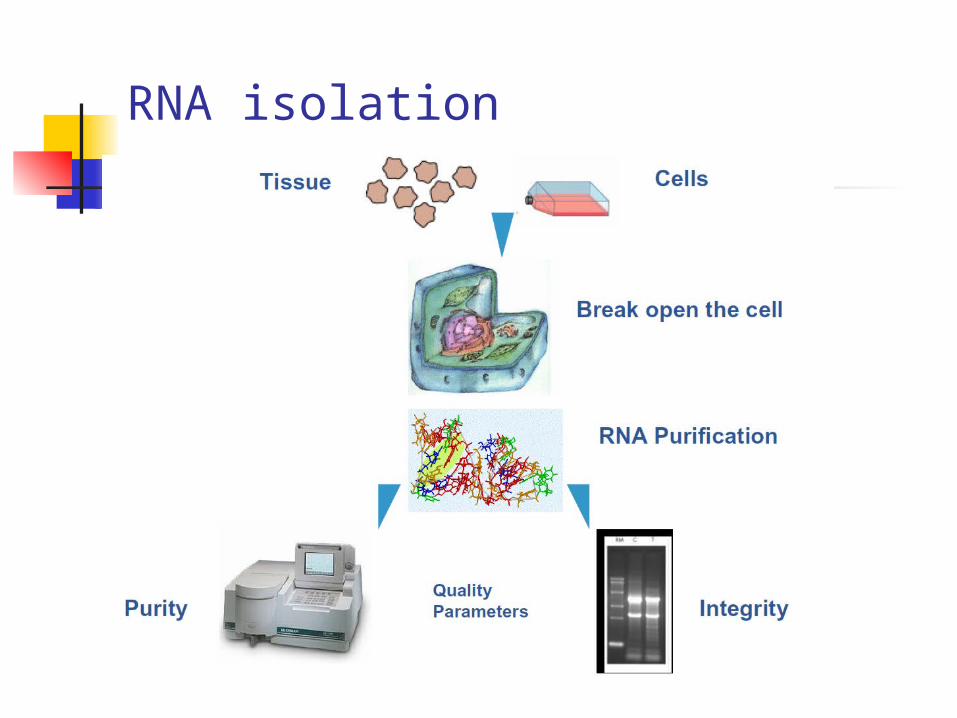
RNA isolation

RNA purification
•Total RNA from biological samples– Organic extraction– Affinity purification
• mRNA from total RNA– Oligo(dT) resins
• mRNA from biological samples– Oligo(dT) resins

Total RNA purification
• Cells or tissue must be rapidly and efficiently disrupted
• Inactivate RNases
• Denature nucleic acid-protein complexes
• RNA selectively partitioned from DNA and protein

RNases
• RNases are naturally occurring enzymes that degrade RNA
• Common laboratory contaminant (from bacterial and human sources)
• Also released from cellular compartments during isolation of RNA from biological samples
• Can be difficult to inactivate

Protecting against RNases
• Wear gloves at all times• Use RNase-free tubes and pipette tips• Use RNase-free chemicals• Pre-treat materials with extended heat (180°C for several hours), wash with DEPC-treated water• Supplement reactions with RNase inhibitors• Include a chaotropic agent (guanidine) in the procedure that inactivate and precipitate RNases and other proteins

Organic extraction of total RNA

Advantages• Versatile - compatible with a variety of sample types• Scalable - can process small and large samples• Established and proven technology• Inexpensive
Disadvantages• Organic solvents• Not high-throughput• RNA may contain contaminating genomic DNA
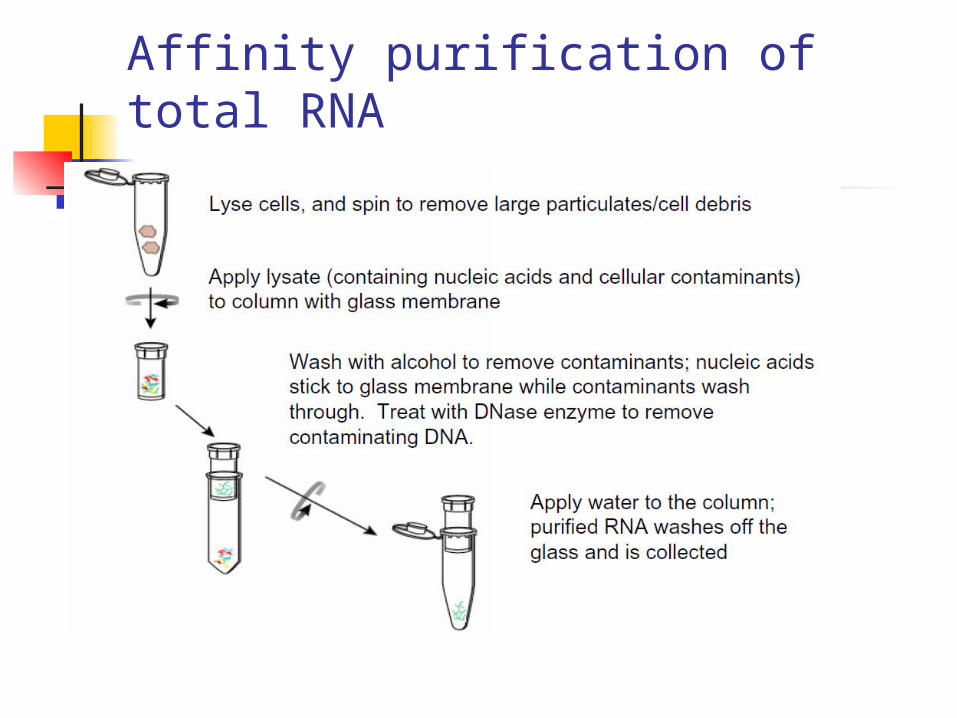
Affinity purification of total RNA

Advantages• Eliminates need for organic solvents• Compatible with a variety of sample types (tissue, tissue culture cells, white blood cells, plant cells, bacteria, yeast, etc.)• DNase treatment eliminates contaminating genomic DNA• Excellent RNA purity and integrity
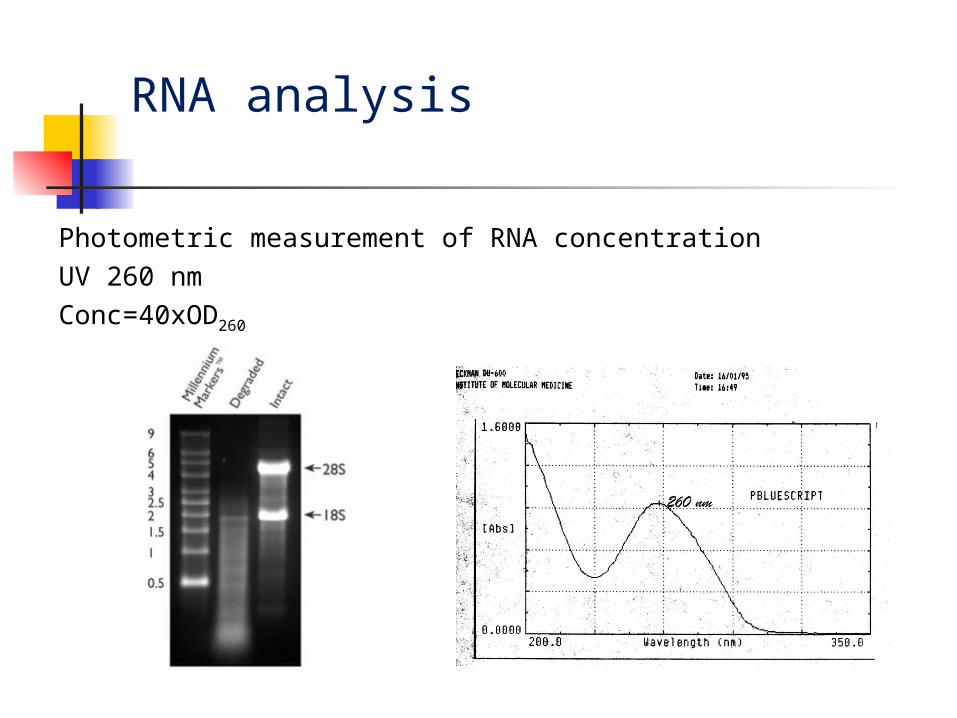
RNA analysis
Photometric measurement of RNA concentration
UV 260 nm
Conc=40xOD260

Determination of the RNA concentration
[RNA] μg/ml = A260 x dilution x 40.0
where
A260 = absorbance (in optical densities) at 260 nm
dilution = dilution factor (200)
40.0 = average extinction coefficient of
RNA.

cDNA Synthesis

Template:- Total RNA - Poly (A)+ RNA- Specific RNA
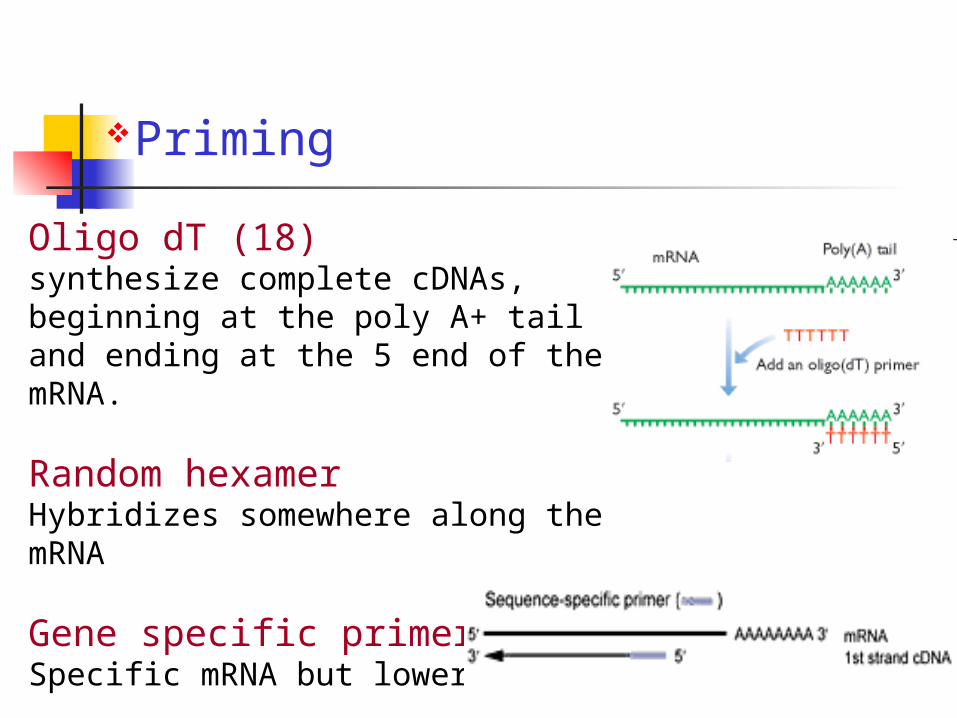
Priming
Oligo dT (18)synthesize complete cDNAs, beginning at the poly A+ tail and ending at the 5 end of the mRNA.
Random hexamerHybridizes somewhere along the mRNA
Gene specific primersSpecific mRNA but lower yields

Reverse Transcriptases (RTs)
The reverse transcriptase from the avian myoblastosis virus (AMV-RT): Temperature optimum of activity at 45-50°C. Highly thermostable, can be used at temperatures up to 60°C. Demonstrates DNA exonuclease and RNase activities.
The reverse transcriptase from the Moloney murine leukemia virus (MMLV-RT):The optimal working temperature is about 37°C, with a maximum of 42°C.Weaker RNase activity.

Normalization
Most gene expression assays are based on the comparison of two or more samples and require uniform sampling conditions for comparison to be valid.
Many factors can contribute to variability in the analysis of samples, making the results difficult to reproduce between experiments: Sample degradation, extraction efficiency, contamination, Sample concentration, RNA integrity, reagents, reverse transcription
Housekeeping genes: ß-actin, ß-tubulin, GAPDH, have often been used as reference genes for normalization. The expression of these genes is constitutively high and that a given treatment will have no effect on the expression level.

Amplification (PCR)
The Polymerase Chain Reaction

PCR – first described in mid 1980’s, Mullis (Nobel prize in 1993)
An in vitro method for the enzymatic synthesis of specific DNA sequences
Selective amplification of target DNA from a heterogeneous, complex DNA/cDNA population
RequiresTwo specific oligonucleotide primersThermostable DNA polymerase (Taq DNA polymerase from Thermus aquaticus)dNTP’sTemplate DNAMgCl2Sequential cycles of (generally) three steps (temperatures)

MgCl2 (mM) 1.5 2 3 4 5
Magnesium Chloride (usually 0.5-5.0mM)
Magnesium ions have a variety of effects
Mg2+ acts as cofactor for Taq polymerase
Mg2+ binds DNA - affects primer/template interactions
Mg2+ influences the ability of Taq pol to interact with primer/template sequences
More magnesium leads to less stringency in binding

Thermal cyclersThermal cyclers
thin walled tube, volume, cost evaporation & heat transfer concerns0
Introduced in 1986.
PCR cyclers available from many suppliers.
Reactions in tubes or 96-well micro-titre plates
29
heated lidsadjustable ramping timessingle/multiple blocksgradient thermocycler blocks

72 0C - primer extension
94 0C - denaturation
Temperature control in a PCR thermocycler
94 0C - denaturation
50 – 70 0C - primer annealing
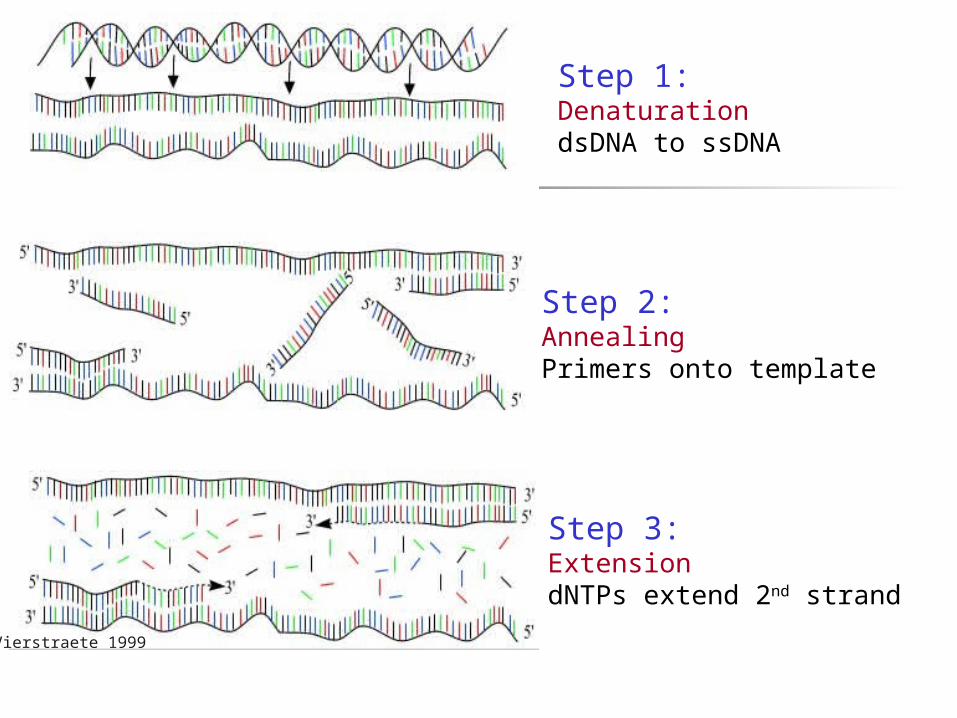
Vierstraete 1999
Step 1: DenaturationdsDNA to ssDNA
Step 2: AnnealingPrimers onto template
Step 3: ExtensiondNTPs extend 2nd strand

PCR Problems
Mis-priming
Set up reactions on ice
Hot-start PCR –holding one or more of the PCR components until the first heat denaturation
Manually - delay adding polymerasePolymerase antibodies
Touch-down PCR – set stringency of initial annealing temperature high, incrementally lower with continued cycling
PCR additives0.5% Tween 20 5% polyethylene glycol 400 DMSO

Primer Design
1. Typically 20 to 30 bases in length
2. Annealing temperature dependent upon primer sequence (~ 50% GC content)
3. Avoid secondary structure, particularly 3’
4. Avoid primer complementarity (primer dimer)

Primer Design Software
Many free programs available online
OLIGO
PRIMER
PrimerQuest

Many free programs available online
•OLIGOwww.oligo.net
•PrimerQuestwww.eu.idtdna.com/scitools/applications/primerquest
•Primer3www.frodo.wi.mit.edu/primer3
•GeneWorkswww.geneworks.com.au
•Yeast genome primer design www.yeastgenome.org/cgi-bin/web-primer
35Primer Design Software
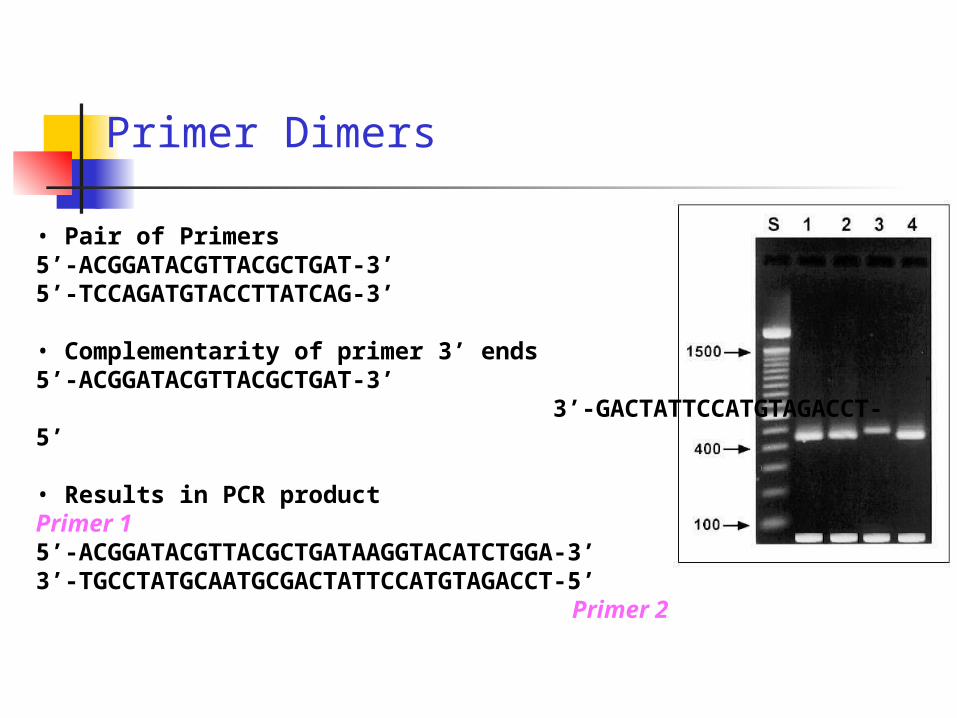
Primer Dimers
• Pair of Primers5’-ACGGATACGTTACGCTGAT-3’5’-TCCAGATGTACCTTATCAG-3’
• Complementarity of primer 3’ ends5’-ACGGATACGTTACGCTGAT-3’ 3’-GACTATTCCATGTAGACCT-5’
• Results in PCR productPrimer 15’-ACGGATACGTTACGCTGATAAGGTACATCTGGA-3’3’-TGCCTATGCAATGCGACTATTCCATGTAGACCT-5’
Primer 2

Rules of thumb for PCR conditions
Add an extra 3-5 minute (longer for Hot-start Taq) to your cycle profile to ensure everything is denatured prior to starting the PCR reaction
Approximate melting temperature (Tm) = [(2 x (A+T)) +(4 x (G+C))]ºC
If GC content is < 50% start 5ºC beneath Tm for annealing temperature
If GC content ≥ 50% start at Tm for annealing temperature
Extension @ 72ºC: rule of thumb is ~500 nucleotide per minute. Use 3 minutes as an upper limit without special enzymes

Typical PCR Temps/Times
Initial denaturation 90o – 95o C 1 – 3 min
Denature 90o – 95o C 0.5 – 1 min
Primer annealing 45o – 65o C 0.5 – 1 min
Primer extension 70o – 75o C 0.5 – 2 min
Final extension 70o – 75o C 5 – 10 min
Stop reaction 4o C or 10 mM EDTA Hold
25 – 40 cycles
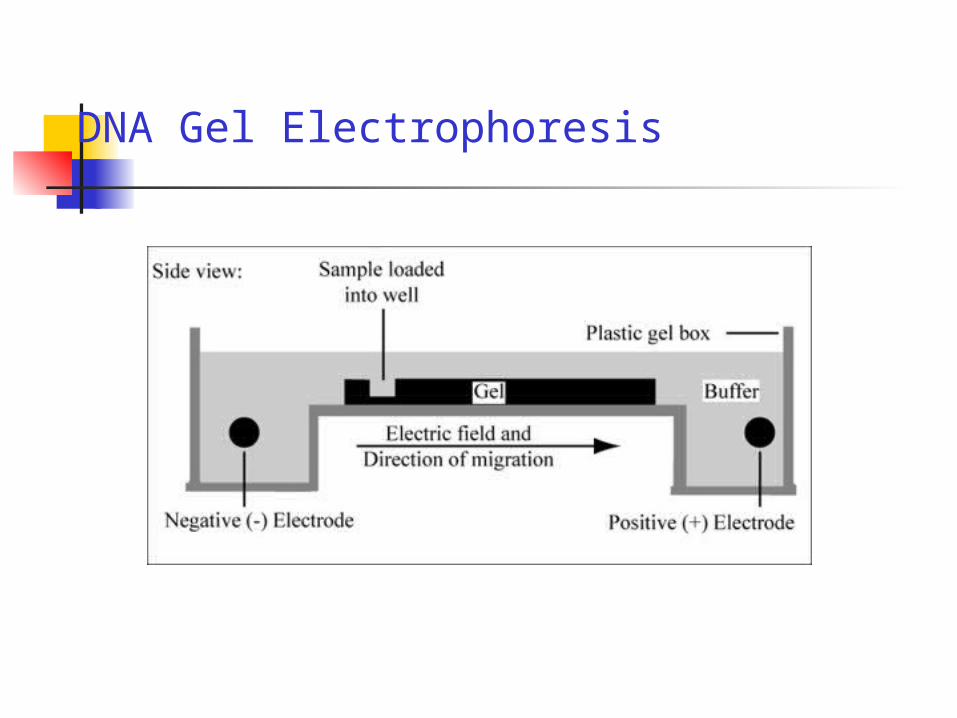
DNA Gel Electrophoresis

Agarose
Agarose is a polysaccharide consisting of a linear polymer (repeating units) of D-galactose and 3,6-anhydro L-galactose
The movement of molecules through an agarose gel is dependent on the size and charge of molecules and the pore sizes present in the agarose gel.
At neutral pH, molecules migrate toward the anode when an electric field is applied across the gel.
Small, highly negatively charged molecules migrate faster through agarose gels than large, less negatively charged molecules.
Rates can also be effected by the pore size of the agarose gel. Decreasing pore sizes increases the separation of small and large molecules during electrophoresis. Pore size can be decreased by increasing the percentage of agarose in the gel.

TBE or TAE BuffersTris helps to maintain a consistent pH of the solution. EDTA chelates divalent cations like magnesium. This is important because most nucleases require divalent cations for activity.

Ethidium Bromide A fluorescent dye is added to agarose gels.
It intercalates between the bases of DNA, allowing DNA fragments to be located in the gel under UV light.
The intensity of the band reflects the concentration
Because of its interaction with DNA, ethidium bromide is a powerful mutagen. Always handle all gels and gel equipment with gloves. Agarose gels with Ethidium Bromide must be disposed as hazardous waste in the labelled container.

Add loading dye to all samples. Loading dye contains xylene cyanol as a tracking dye to follow the progress of the electrophoresis as well as glycerol to help the samples sink into the well.






























