Embed Size (px)
Citation preview

: REVIEW
Mechanisms of Nonsteroidal Anti-Inflammatory Drug-Induced Gastric Damage ROBERTT. SCHOEN, M.D., F.A.c.P., RONALD J. VENDER, M.D. NewHaven, Connecticut
The effects of nonsteroidal anti-inflammatory drugs (NSAIDs) on the gastric mucosa are well documented. The complex mechanisms of gastric damage, however, are not fully understood. This review examines current knowledge about the nor- mal function of the gastric mucosal barrier; the role of prostaglandins in cytoprotection and repair; the mechanisms by which aspirin and other weak organic acids are absorbed by the stomach; and the subsequent cascade of events-including ion trap- ping and back diffusion of hydrogen ions-that leads to gastric erosion and bleeding. A hypothesis describing NSAIDs’ dual insult on the stomach is advanced.
From the Department of Internal Medicine, Yale University School of Medi- cine, New Haven, Connecticut. Requests for reprints should be addressed to Robert T. Schoen. M.D., F.A.C.P.. Internal Medicine and Rheumatology, P.C., 60 Temple Street, Suite 6A. New Haven, Connecticut 06510. Manu- ;c$ submitted May 12, 1988, and accepted in revised form January 13,
T he use of sodium salicylate in 1875 to treat pa- tients with rheumatic disease [l] was followed by
reports of “dyspeptic indigestion” in patients receiv- ing salicylates and other nonsteroidal anti-inflamma- tory drugs (NSAIDs) [2]. Gastric mucosal damage is now recognized as the most important adverse effect of NSAIDs, causing gastrointestinal symptoms, ero- sions, ulcers, and upper gastrointestinal bleeding [3]. The synthesis of aspirin (acetylsalicylic acid) by Hoff- man in 1899 from the more toxic salicylic acid reduced toxicity and introduced the modern era of NSAID therapy [4]. Investigation continues in regard to the development of potent anti-inflammatory drugs that avoid gastric mucosal damage.
The problem of NSAID gastrointestinal toxicity has stimulated investigation of normal gastric defense mechanisms. Exactly how the stomach protects itself from the hydrochloric acid and pepsin it produces is not known. The protective mechanisms that must ex- ist, however, are grouped together by the term “gastric mucosal barrier” [5]. The study of how aspirin and other NSAIDs induce gastric damage has been the study of how these agents “break” this barrier [6].
Gastric Defense: The Gastric Mucosal Barrier More than two centuries ago, Reaumur recognized
that gastric juice is capable of digesting meat [?I. This observation led to the concept of a gastric mucosal barrier. Claude Bernard believed that the layer of mu- cus that covers the surface epithelium of the stomach acts as a “porcelain-like shell” separating the epitheli- urn from its gastric contents [B]. Although mucus may indeed be important [9], the concept of a gastric muco- sal barrier must include modern observations of nor- mal gastric physiology, including hydrogen ion, bicar- bonate, and electrolyte secretion. For example, the electrolyte concentration in gastric juice varies with the rate of gastric secretion. At low secretory rates, gastric juice has a high sodium ion concentration and a low hydrogen ion concentration. At high secretory rates, these concentrations are reversed [lo]. This dis- covery suggested that the gastric mucosa is a diffusion barrier in which a sodium ion is exchanged for a hydro- gen ion at the mucosal cell surface [II].
Gastric Defense: Mucus and Bicarbonate Secretion In 1954, Hollander [12] proposed a two-component
gastric mu.cosal barrier consisting of the mucus layer together with the epithelial cells beneath it. In this model, hydrogen ion concentration is reduced at the epithelial cell surface by sodium secreted from inter- stitial fluid. Heatley [13] suggested that an “un- stirred” mucus layer allows the development of a pH gradient between the epithelial cell and the lumen. Allen and Garner [9] proposed that bicarbonate is se- creted by the mucosa and that mucus has gel proper- ties that limit the diffusion of hydrogen ion, creating a
April 1989 The American Journal of Medicine Volume 86 449

MECHANISMS OF NSAID-INDUCED GASTRIC DAMAGE / SCHOEN AND VENDER
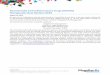
Mltcosal cells (=retion)
HCO; -
Na+-
mucus giycoprctein -
I pH7 -pHgraclient- pH l-2
Cl-“Na+
rpeps’n i k desr=k+d
glycoprotein subunits
Lumen (mixing)
-H+
- ci-
pH gradient across the mucus layer (Figure 1). Dav- enport [7] viewed the mucus layer as having merely a lubricating function. He emphasized anatomic proper- ties of the lipid and protein gastric cell membrane and tight junctions between cells in limiting penetration of water-soluble compounds, including hydrogen ion. Gastric mucus is in fact a high-viscosity gel composed of glycoprotein polymers that reduce the rate at which hydrogen ion from the lumen and bicarbonate from the epithelial cells can mix [9]. Recently, a pH gradient across the mucus layer has been demonstrated using pH-sensitive microelectrodes [14-161.
Bicarbonate is secreted by receptor-mediated active transport by the gastric mucosa [9,17,18]. In addition, vagus nerve stimulation increases bicarbon- ate output during the cephalic phase of gastric secre- tion [19]. Although bicarbonate is actively secreted, the rate of secretion is only about 5 to 10 percent of maximal acid output [9]. If a mucus-bicarbonate mod- el is correct, the gel properties of mucus must signifi- cantly limit the rate of reaction between secreted bi- carbonate and luminal hydrogen ion. In addition, other factors must maintain a gradient in which hy- drogen ion is secreted in much higher concentration than bicarbonate. For example, it has been shown that gastric mucus significantly retards penetration of pep- sin from the lumen [20]. It has also been suggested that the mucus layer may thicken in response to injury, forming a framework for re-epithelialization of the mucosa [21].
Gastric Defense: Surface Hydrophobicity Hills and associates [22] found canine gastric muco-
sa to be exceptionally hydrophobic in contrast to the duodenal mucosa, which is hydrophilic. They postu- lated that phospholipids, high levels of which have been identified in gastric mucosa and gastric secre- tions [23], are concentrated on the luminal surface of the gastric epithelium. This phospholipid layer cre-
450 April 1989 The American Journal of Medicine VOhne 86
Figure 1. Surface neutralization. A mod- el in which the mucosa secretes bicar- bonate, and an “unstirred” viscous mu- cus layer on the epithelial cell surface permits the development of a pH gradi- ent that limits diffusion of hydrogen ion from the lumen. (Adapted with permis- sion from [9].)
ates a hydrophobic surface, which may significantly limit the diffusion of hydrogen ion from the lumen into the mucosa [24].
Gastric Defense: Mucosal Blood Flow and Reconstitution Several studies show that gastric mucosal blood flow
protects the mucosa from acid injury [3]. If the gastric mucosal barrier is disrupted by acid, intracellular hy- drogen ion is removed by a compensatory increase in mucosal blood flow. If this compensatory increase is prevented, cell death occurs [25]. There is also evi- dence that within the gastric microcirculation, trans- port of bicarbonate from the interstitium to surface epithelial cells helps to prevent acid injury [26].
Mucosal re-epithelialization within 30 minutes fol- lowing injury (“reconstitution”) with agents such as hypertonic saline, ethanol, and aspirin occurs in the amphibian gallbladder, duodenum, and colon [27], as well as in the amphibian and mammalian stomach [28]. This process may be an important repair mecha- nism throughout the entire gastrointestinal tract. In an in vitro system, reconstitution is associated with a net alkalinization of the luminal solution during the first four hours, changing to a net acid secretion com- parable to that in control tissues by six hours. If an acidic luminal pH is maintained, reconstitution does not occur [29]. Rapid epithelial reconstitution is prob- ably important in maintaining the anatomic integrity of the gastric mucosal barrier [29].
Gastric Defense: Prostaglandins Prostaglandins are a vital component of gastric mu-
cosal defense. These short-acting, widely distributed, 20-carbon chain, unsaturated fatty acids are found throughout the gut in locally high concentration. A major stimulus for their synthesis is perturbation of cell membranes, including cell trauma by acid or alkali [30]. Prostaglandins have an anti-secretory effect on gastric acid production [31], but the ability of prosta-

MECHANISMS OF NSAID-INDUCED GASTRIC DAMAGE / SCHOEN AND VENDER
glandins to defend the stomach against injury by lumi- nal acid and other noxious agents at concentrations that do not inhibit gastric acid secretion is called “gas- tric cytoprotection” [32]. In addition, a variety of irri- tants, including ethanol, hydrochloric acid, alkali, hy- pertonic saline, and thermal injury, stimulates prostaglandin production. In association with such stimulation, gastric necrosis is prevented when ani- mals are subsequently challenged with agents that would ordinarily damage the stomach. This phenome- non, termed “adaptive cytoprotection,” may protect the mucosa from the damaging effect of gastric lumi- nal contents [30].
Adaptive cytoprotection may result from prosta- glandin-mediated mechanisms, but this is not proven. Mild irritants that are protective do not always stimu- late endogenous prostaglandin synthesis, and prosta- glandin suppression by indomethacin has not univer- sally abrogated adaptive cytoprotection [33]. Hawkey and associates [34] showed that the protective effect of 20 percent ethanol is present even after prostaglandin Es release is 86 percent inhibited by indomethacin. They found extensive superficial disruption of the sur- face epithelium following 20 percent ethanol adminis- tration and postulated that this desquamated debris may form a protective covering that could account for adaptive cytoprotection.
Although it is uncertain that adaptive cytoprotec- tion critically depends on prostaglandin synthesis, it is clear that prostaglandins enhance many of the postu- lated components of gastric mucosal defense. Prosta- glandins stimulate bicarbonate secretion [33] and the synthesis of mucus [35]. Prostaglandins increase mu- cus gel thickness [36]. As a result, prostaglandin Es enhances the pH gradient in mucus between the lu- men and epithelial cell surface [37].
Prostaglandin Es increases the surface hydropho- bicity of gastric mucosa [38] by increasing surface- active phospholipids [39]. Both prostaglandin E and I enhance mucosal blood flow following gastric acid pro- duction [40], preventing erosions that are seen when such vasodilatation does not occur [41]. Prostaglan- dins may also protect the mucosa by stimulating sodi- um-active transport [42].
Prostaglandins probably also have a repair function, stimulating rapid resolution of disrupted surface epi- thelium [43], although this has not always been found [44]. Repair mechanisms may include migration of basal cells toward the lumen to repair mucosal injury [45]. Also possible is enhanced DNA, RNA, protein, and collagen synthesis, which has been demonstrated in cutaneous tissue [46], but not for the gastric mucosa [471.
Gastric Defense: Sulfhydryl Compounds Ethanol induces a decrease in gastric non-protein
sulfhydryl compounds in association with mucosal erosions [48]. Aspirin- [49] and ethanol- [48] induced erosions can be prevented by preadministration of sulfhydryl compounds. These observations suggest that sulfhydryl compounds may mediate gastric cyto- protection. It is possible that prostaglandin-induced cytoprotection requires sulfhydryl compounds, since sulfhydryl blocking agents such as N-ethylmaleiamide prevent the mucosal cytoprotective effect of prosta- glandin Fsn [48]. In addition, ethanol-induced gastric damage is greater following administration of both a prostaglandin inhibitor, indomethacin, and a sulfhy-
dry1 blocker, N-ethylmaleiamide, than following ei- ther compound alone [50].
Sulfhydryl compounds, such as reduced glutathi- one, which is found in high concentration in the stom- ach [51], bind free radicals that form following tissue injury by noxious agents [52]. Sulfhydryl compounds may also protect mucus, since mucus subunits are joined by disulfide bridges that, if reduced, render mucus water-soluble [53]. A vasoprotective effect for sulfhydryl compounds following ethanol-induced in- jury is also postulated [50].
Much of the current understanding of the gastric mucosal barrier has been gained from investigating changes in mucosal integrity caused by acute manipu- lation with NSAIDs such as aspirin. Several studies, however, show that the stomach may become less sus- ceptible to these compounds over time [54,55] by com- pensatory mechanisms, which may include increased cell turnover and the emergence of younger cell popu- lations [56]. This phenomenon is called “gastric adap- tation” [55].
NSAID Damage: Breaking the Gastric Mucosal Barrier Aspirin and most other NSAIDs are weak organic
acids. A solution of two aspirin tablets in 100 ml of drinking water has a pH of about 2.5 [57]. Aspirin damages the gastrointestinal tract in the absence of hydrochloric acid [58] and induces lesions in the buc- cal mucosa by direct acid damage [59]. Aspirin and indomethacin also increase basal [60] and maximally stimulated gastric acid secretion [61,62], which may contribute to NSAID-induced damage.
NSAID Damage: Ion Trapping Aspirin and other NSAIDs also damage the gastric
mucosal barrier by altering cell membrane permeabili- ty, allowing “back diffusion” of hydrogen ion. Weak organic acids such as aspirin are concentrated in the mucosal cell as a result of “ion trapping” [63-651. Be- cause of its lipid and protein membrane, the mucosal cell absorbs lipid-soluble compounds such as aspirin more readily than it does water-soluble compounds.
For example, acetic acid, which is lipid-soluble, dif- fuses across the mucosa three times faster than hydro- chloric acid, which is not [7]. In the strongly acid envi- ronment of normal gastric juice (pH 2.5), aspirin (pKa 3.5) is mostly non-ionized. As an undissociated acid, aspirin freely diffuses into the mucosal cell. Once in- side the cell, the much higher pH of the intracellular environment (pH 7) favors acid dissociation. In this ionized state, aspirin is water-soluble and “trapped” inside the cell. These events favor a strong concentra- tion gradient, moving dissociated ions of weak organic acids such as aspirin (pKa 3.5) [66] (Figure 2), indo- methacin (pKa 5.2), phenylbutazone (pKa 4.8), and other acidic NSAIDs into the gastric mucosa. This rapid absorption has been demonstrated for a variety of weak organic acids, including acetic acid (pKa 4.78), propionic acid (pKa 4.86), and butyric acid (pKa 3.90): as well as acetylsalicylic acid [7,67].
NSAID Damage: Back Diffusion The rapid intracellular penetration of aspirin and
its entrapment as an ionized salt are followed by alter- ation in cell membrane permeability and damage by luminal hydrogen ion.
Davenport [5] examined this damaging effect on the canine gastric pouch after exposure to 20 mM of aspi-
April 1989 The American Journal of Medicine Volume 86 451

MECHANISMS OF NSAID-INDUCED GASTRIC DAMAGE / SCHOEN AND VENDER
Ionized, Water Soluble UN-ionized, Fat Soluble
0 0
Hf + -O-C-CHa7b
T -F-O-
0 0
From HCI
? -0-C-CH,
-$-OH
0
Removal of Blood
Figure 2. Absorption of weak organic acids. Un-ionized aspirin (pKa 3.5) dif- fuses into the gastric mucosa, dissoci- ates, and accumulates in the cells. (Adapted with permission from [66].)
rin. The rate of absorption of aspirin into the mucosa is dependent on the luminal pH, as would be expected, with a low pH favoring more rapid absorption. The absorption of aspirin is associated within minutes with abnormal ion fluxes across the mucosa. Sodium and potassium enter the luminal fluid, and hydrogen ion disappears from the lumen into the mucosa (an obser- vation termed “back diffusion of hydrogen ion”). In these experiments, frank mucosal bleeding is not ob- served when the gastric pouch is irrigated with 20 mM aspirin in relatively weak acid (1 mM hydrochloric acid or 10 mM hydrochloric acid), but gross bleeding occurs when aspirin in 100 mM hydrochloric acid is instilled. Davenport concluded that aspirin damages the mucosal barrier as it is absorbed by an unknown mechanism that renders the mucosa abnormally per- meable to water-soluble hydrogen ion. Back diffusion of strongly acid gastric juice then leads to mucosal damage, including erosion and bleeding [5] (Figure 3). These experiments may explain why achlorhydric pa- tients are less susceptible to aspirin-induced gastric injury than normal persons [68] and why damage from aspirin is markedly reduced when gastric secretions are buffered to pH 6 to 7 [69].
Figure 3. Aspirin-induced back diffusion. Davenport [5] found that absorption (l), followed by ionization and entrap- ment (2), of aspirin is associated with abnormal ion flux across the gastric mu- cosa (3). Back diffusion of hydrogen ion from the lumen leads to gastric erosion and bleeding.
The aspirin-induced increase in hydrogen ion per- meability observed experimentally [5] has been con- firmed in normal volunteers [70-721. In addition, it has been demonstrated for other NSAIDs, including indomethacin [6,73,74] and fenoprofen [75]. Fenopro- fen causes less hydrogen ion flux than indomethacin or aspirin, but more than a control preparation. Indo- methacin causes sustained back diffusion associated with bleeding at both neutral and acidic pH [6].
NSAID Damage: Transmucosal Potential Difference The increased permeability to hydrogen ion caused
by aspirin occurs within minutes after exposure and is associated with additional evidence of damage to the gastric mucosal cell. Transmucosal electrical potential difference is a sensitive index of gastric mucosal func- tion [75]. In numerous studies, aspirin (Figure 4) and indomethacin induce an immediate decrease in gastric potential difference [71,74,76,77]. Both the change in potential difference [77] and the increase in ion per- meability [67] typically recover within 90 minutes, consistent with the hypothesis that weak organic acid NSAIDs are rapidly trapped, but diffuse out of the cell more slowly [67].
452 April 1989 The American Journal of Medicine Volume 86

MECHANISMS OF NSAID-INDUCED GASTRIC DAMAGE / SCHOEN AND VENDER
Figure4. Potential difference. Aspirin in- duces an immediate decrease in gastric transmucosal electrical potential differ- ence, a sensitive index of gastric muco- sal function. (Adapted with permission from [73].)
Figure 5. Aspirin-induced injury. A pho- tomicrograph (toluidine blue stain; origi- nal magnification X 620, reduced by 25 percent) shows damage to surface mu- cous cells by 20 mM aspirin. Changes were observed within one minute of as- pirin administration, with maximum damage occurring by eight minutes, (Adapted with permission from [78].)
Time-3 Min. Intervals
NSAID Damage: Ultrastructure The aspirin-induced injury that Davenport demon-
strated physiologically has been confirmed by electron microscopy. Hingson and Ito [78] administered 20 mM of aspirin to mice under experimental conditions simi- lar to those of Davenport [5]. They found “sudden and drastic morphological alterations in many of the ex- posed surface mucus cells” [78] at all concentrations of aspirin and hydrochloric acid (Figure 5). These changes are seen within one minute after aspirin ad- ministration, with maximum damage occurring by eight minutes. Ten to 85 percent of the total surface areas show injury, including karyolysis, erosions, and bleeding. By four hours, there is marked resolution of these lesions [78]. Rainsford [79] observed similar changes and showed that damage was greatest for the
parietal cell and could be prevented if aspirin is buff- ered with bicarbonate. Using a freeze-fracture tech- nique, Meyer et al [80] showed that aspirin disrupts the tight junctions between cells. Hingson and Ito ex- perimented with other weak organic acids to demon- strate that the mechanism of damage is not unique to aspirin. Severe ultrastructural damage is seen with 20 mM solutions of acetic acid, benzoic acid (pKa 4.2), and salicylic acid (pKa 3.0). Salicylamide, a non-acid, does not induce damage (Table I) [78].
NSAID Damage: Autoradiographic Techniques Autoradiographic techniques also provide evidence
that gastric injury caused by aspirin and other acidic NSAIDs occurs because these compounds are selec- tively trapped within mucosal cells, whereas non-acid-
April 1989 The American Journal of Medicine Volume 86 453

MECHANISMS OF NSAIDdNDUCED GASTRIC DAMAGE / SCHOEN AND VENDER
TABLE I
Frequency of Damage to the Gastric Surface Epithelium of Mice following Oral Administration of Aspiriri Analogues
Agent
Agent Electrolytes (mEq/liter)
Percent ml/liter H+ Na+ CI- Time (minutes) Frequency (D/n)
Salicylic acid
Benzoic acid Acetic acid
0.275 0.14
0.245 0.12 0.12
20
:oo :i
145 135
145 135 145
155 145
155 145 155
1:
1: 8
616 2/3
y: 3/a
Salicylamide 0.275 20 10 140 150 8 * D/n = number of stomachs with damaged surface mucosal cells/total number of stomachs. (Adapted with permission from [63].)
O/7
ic compounds are not. In a series of experiments, Brune and colleagues [Bl] demonstrated accumulation of radiolabeled aspirin, salicylic acid, phenylbutazone, and indomethacin within the stomach wall, but no such accumulation of the non-acidic compounds anti- pyrine, aminopyrine, or paracetamol. In addition, di- flunisal, a derivative of salicylic acid reported to cause less gastric irritation than other salicylates [82], and proquazone, a non-acid anti-inflammatory agent with a low incidence of gastric irritation [83], are not detec- tably absorbed by the stomach [84,85]. Within one minute after drug administration, thin slices of the stomach wall show high concentrations of tritiated sal- icylic acid, which persists in the surface mucus and the mucus-producing superficial cells [84]. There is also selective trapping of salicylate within parietal cells [84]. After 20 minutes, when tritiated drug is concen- trated in the stomach wall, gastric cell decay is demon- strated by electron microscopy [85]. No such changes are seen with proquazone [85]. These studies provide additional evidence that acidic NSAIDs reach high concentrations within the mucosa, and that this bio- distribution of drug is important in the development of gastric toxicity [85].
These different lines of evidence suggest that NSAIDs damage the mucosal cell by altering mem- brane permeability, allowing back diffusion of hydro- gen ion. The mechanisms by which the accumulation of acidic NSAIDs within mucosal cells leads to these changes are unknown. The dissociated acid may in- crease the osmolarity of the intracellular fluid and cause cell swelling [86]. Such changes in cell shape may increase membrane permeability [87]. Membrane per- meability changes could also result from inhibition of active transport. Several experiments demonstrate that aspirin at low concentration inhibits active trans- port of sodium and hydrogen ion across the mucosal cell without an increase in membrane permeability [87,88]. At higher concentrations, both ion transport and permeability are affected, suggesting that inhibi- tion of ion transport either by enzyme inhibition [89,90] or oxidative uncoupling [91,92] is the initial insult caused by aspirin, and that changes in mem- brane permeability are a secondary consequence [88]. Indomethacin also inhibits sodium-active transport [42]. Spenney [93] has demonstrated an acetylsalicylic e&erase in gastric mucosa that hydrolyzes aspirin (acetylsalicylic acid) to salicylic acid. Intracellular conversion of aspirin to salicylate could be important in aspirin-induced injury, since salicylate potently un- couples and/or inhibits mitochondrial oxidative phos- phorylation whereas nonhydrolyzed aspirin may not WI * 454 April 1989 The American Journal of Medicine Volume 86
NSAID Damage: MUCUS, Bicarbonate, and Surface Hy- drophobic@
Aspirin [95], indomethacin [96], and phenylbuta- zone 1971 inhibit gastric mucus secretion. Aspirin de- creases the thickness of mucus [98], inhibits the incor- poration of radiolabeled precursors into the glycoprotein component of mucus, and inhibits the activity of enzymes necessary for mucus biosynthesis [99,100]. Aspirin increases pepsin-mediated proteoly- sis of mucus, decreases mucus viscosity, and increases the permeability of mucus to hydrogen ion [ 1011. Aspi- rin also diminishes the pH gradient across the mucus layer [16]. Aspirin induces ulcers in rats with a low percentage of high-molecular-weight glycoprotein in mucus, while sparing rats with a high percentage [102].
Aspirin [103] indomethacin [104,105], and fenclo- fenac [106] inhibit active bicarbonate secretion by the gastric mucosa. The restoration of ambient bicarbon- ate protects against aspirin-mediated erosions [ 1071. Aspirin also reduces the surface hydrophobic proper- ties of the gastric mucosa, probably by altering surface phospholipids [39].
NSAID Damage: Mucosal Blood Flow and Cellular Regen- eration
The reported effects of NSAIDs on mucosal blood flow have been inconsistent. Aspirin has been found both to increase [108] and to decrease [61] mucosal blood flow. Indomethacin has been reported to de- crease mucosal blood flow [41,61]. Such reduction in mucosal blood flow could increase NSAID-induced gastric damage.
Aspirin may retard the ordinarily rapid regenera- tion [log] and migration [llO] of normal gastric epi- thelium through diverse effects on normal cell metab- olism [log]. Croft and Wood [ill] found that aspirin increases epithelial cell loss measured by the DNA content of gastric lavage, and postulated that rapid cell turnover and inability to replete cell populations at normal rates may lead to mucosal erosions.
NSAID Damage: Prostaglandin Inhibition In 1971, Vane [112] proposed that the biologic ac-
tions of aspirin-like drugs result from inhibition of prostaglandin synthesis, a theory that has gained widespread acceptance. Not only are prostaglandins “cytoprotective” [32] as described previously, but prostaglandins administered exogenously protect against NSAID-induced gastric injury [113,114] and accelerate the healing of gastric and duodenal ulcers [1X]. The doses of NSAIDs required to inhibit cyclo- oxygenase, the prostaglandin synthesizing enzyme, are well within the therapeutic range of these drugs

MECHANISMS OF NSAID-INDUCED GASTRIC DAMAGE / SCHOEN AND VENDER
[116]. Since prostaglandins play an important, if as yet imperfectly understood, role as cytoprotective agents [32], it is likely that inhibition of prostaglandin syn- thesis by NSAIDs disrupts the functional integrity of the gastric mucosa. In experiments evaluating exoge- nously administered prostaglandins, however, it is un- clear whether such administration compensates for a drug-induced decrease in the mucosal prostaglandins or induces additional pharmacologic effects, unrelated to the correction of an NSAID-induced prostaglandin deficiency [74]. For example, exogenous prostaglandin therapy for peptic ulcer disease employs compounds that potently inhibit gastric acid production. This anti-secretory effect may be more pharmacologically important than any cytoprotective action [117].
A primary role for prostaglandin inhibition in NSAID-induced gastric injury is supported by studies that demonstrate gastric damage with parenteral as well as oral administration of NSAIDs [118,119]. These studies have been criticized, however, as using toxic doses of aspirin [120], or adding hydrochloric acid or acid-stimulating agents such as histamine [121]. More recent parenteral animal [122] and human [123,124] studies fail to demonstrate NSAID-induced damage when these drugs are given alone in therapeu- tic intravenous doses.
In addition, the finding that highly buffered aspirin [57], enteric-coated aspirin [125,126], and enteric- coated naproxen [127] are significantly less damaging to the stomach than the plain drug indicates that a direct topical effect of NSAIDs contributes to their gastric toxicity.
The potency of prostaglandin inhibition by differ- ent NSkIDs can be correlated with the degree of gas- tric damage in several studies. Whittle and associates [128] demonstrated that prostacyclin (PGIz) inhibi- tion for six NSAIDs parallels the extent to which these drugs induce gastric erosions. Potent inhibitors of prostacyclin, including indomethacin, flurbiprofen, and naproxen, induce comparable gastric damage, which correlates with their inhibition of prostaglandin production. Aspirin produces more gastric erosions, which the authors attribute to a “synergistic interac- tion” between topical irritation and cycle-oxygenase inhibition. Sodium salicylate and the experimental drug BW755C, which do not inhibit mucosal cyclo- oxygenase, cause little gastric damage [128]. In a simi- lar study, carboprofen is a less potent prostaglandin inhibitor than indomethacin, but also less toxic to the stomach [ 1291.
Rainsford and Willis [74] found that significant prostaglandin inhibition by aspirin, indomethacin, su- lindac, and diclofenac parallels significant mucosal damage. Flufenamic acid, azapropazone, and fenclo- fenac are intermediate. The non-acidic NSAID mese- clazone potently inhibits both mucosal and plasma levels of prostaglandins, but causes little gastric irri- tancy. A similar result is found for proquazone, a non- acidic NSAID that is less ulcerogenic than indometha- tin [83], but that is an equally effective inhibitor of prostaglandin synthesis [130]. Rainsford and Willis [74] conclude that NSAID-induced gastric damage is mediated by two components: “(1) The direct effects of the acidic drug on membrane permeability (which in turn is influenced by the rate of absorption of the drug), and (2) drug-related effects on prostaglandin production, which may be of particular importance in relation to the ischemia which appears to be of partic-
Figure 6. Dual-insult hypothesis. Current information suggests that most NSAlDs induce gastric damage by a dual-insult mechanism.
ular significance in the early stages of NSAID drug- induced injury” [74].
Several studies fail to correlate NSAID-induced gastric damage with prostaglandin suppression. Red- fern and associates [131] found significant individual variability in prostaglandin concentration following indomethacin suppression and no correlation between the degree of prostaglandin suppression and endo- scopic evidence of mucosal damage. Ligumsky and as- sociates [132] observed that 95 percent inhibition of cycle-oxygenase by parenteral aspirin is not by itself sufficient to induce gastric mucosal damage. They conclude that in addition to a high degree of cyclo- oxygenase inhibition, NSAID-induced gastric damage also involves direct mucosal toxicity.
Rainsford and Willis [74] evaluated the temporal relationship between changes in mucosal cell perme- ability and changes in prostaglandin levels associated with indomethacin-induced gastric lesions. They dem- onstrated a decrease in potential difference, an in- crease in hydrogen ion flux, and marked reductions in mucosal prostaglandins, as would be expected after indomethacin administration. In addition, although these changes occur rapidly in all variables, the in- crease in gastric cell permeability precedes the de- crease in prostaglandin levels. They concluded that direct contact of the acidic drug with the mucosa initi- ates permeability changes and that the additive effect of prostaglandin inhibition follows secondarily.
NSAID-Induced Gastric Mucosal Damage: A Dual-Injury Hypothesis
NSAIDs are a chemically diverse group of com- pounds, most, but not all, of which are organic acids and most, but not all, of which mediate their anti- inflammatory effects through prostaglandin inhibi- tion. How these drugs cause gastric mucosal injury has been extensively studied. Aspirin and its effects after short-term administration have received the most at- tention. Although information gained from the study of the effects of short-term aspirin administration cannot be extrapolated to the long-term clinical use of all NSAIDs (for example, phenylbutazone does not alter gastric potential difference [133]), a review of mechanisms of NSAID-induced gastric toxicity sug-
April 1989 The American Journal of Medicine Volume 86 455

MECHANISMS OF NSAID-INDUCED GASTRIC DAMAGE / SCHOEN AND VENDER
gests that these drugs share common pathogenic fea- tures.
There is strong evidence for an acid-mediated topi- cal effect of these compounds on the gastric mucosa. Highly buffered NSAIDs [57], enteric-coated NSAIDs [125-1271, and non-acidic NSAIDs [74,83,130] do not induce significant gastric damage, despite potent prostaglandin inhibition and therapeutic effect. Most NSAIDs, however, are organic acids administered as the plain drug. Ion trapping of these weak organic acids facilitates their absorption by mucosal cells, set- ting off a cascade in which gastric mucosal cell perme- ability is increased, exposing these cells to the toxic effect of luminal hydrogen ion and pepsin. Back diffu- sion of hydrogen ion is caught up in this vicious cycle, whether as the cause or the effect.
Once this topical acid damage begins, it is com- pounded by prostaglandin inhibition. Were prosta- glandin inhibition not present, the cell might recover through such prostaglandin-mediated mechanisms as increased mucosal blood flow [40], decreased acid se- cretion [31], or increased ATP-mediated ion transport [42]. Sodium salicylate and the experimental com- pound BW755C, which lower membrane potential dif- ference but do not inhibit prostaglandin synthesis, are significantly less likely to induce gastric damage than aspirin [128], The success of exogenous prostaglandin therapy [113,114,134], although at least in part anti- secretory [117], and of treatment with sucralfate, which may [135] or may not [136] protect the stomach via a prostaglandin-mediated mechanism, is addition- al evidence of the importance of prostaglandin inhibi- tion in NSAID-induced gastric damage.
It is therefore possible that NSAID-induced gastric damage occurs as a result of a dual insult-first, by NSAID-mediated direct acidic damage, which is fol- lowed almost simultaneously by the deleterious effect of prostaglandin inhibition (Figure 6). This hypothe- sis incorporates currently available information about mechanisms by which NSAIDs induce gastric injury. In this hypothesis, both insults are necessary. Strate- gies that avoid this dual insult may significantly di- minish gastric damage, the most serious toxicity of this important group of drugs.
REFERENCES 1. Flower RJ, Moncada S, Vane JR: Analgesic-antipyretics and anti-inflammatory agents; drugs employed in the treatment of gout. In: Gilman AG, Goodman LS, Rail TW, et al, eds. The pharmacological basis of therapeutics, 7th ed. New York: Macmillan Publishing, 1985; 674-679. 2. Gross M, Greenberg LA: The salicylates. A critical bibliographic review. New Haven: Hillhouse Press, 1948; 93-97. 3. Semble EL, Wu WC: Antiinflammatory drugs andgastric mucosal damage. Semin Arthritis Rheum 1987; 16: 271-286. 4. Roth SH, Bennett RE: Nonsteroidal anti-inflammatory druggastropathy: recog- nition and response. Arch Intern Med 1987; 147: 2093-2100. 5. Davenport HW: Damage to the gastric mucosa: effects of salicylates and stimula- tion. Gastroenterology 1965; 49: 189-196. 6. Chvasta TA, Cooke AR: The effect of several ulcerogenic drugs on the canine gastric mucosal barrier. J Lab Clin Med 1972; 79: 302-315. 7. Davenport HW: Salicylate damage to the gastric mucosal barrier. N Engl J Med 1967; 276 1307-1312. 8. Menguy R: Gastric mucus and the gastric mucous barrier: a review. Am J Surg 1969; 117: 806-812. 9. Allen A, Garner A: Progress report: mucus and bicarbonate secretion in the stomach and their possible role in mucosal protection. Gut 1980; 21: 249-262. 10. i)brink KJ: Studies on the kinetics of the parietal secretion of the stomach. Acta Physiol Stand 1948; 15(suppl 51): l-106. 11. Teorell T: Electrolyte diffusion in relation to the acidity regulation of the gastric juice. Gastroenterology 1947; 9: 425-443.
456 April 1989 The American Journal of Medicine Volume 86
12. Hollander F: The two-component mucous barrier. Arch Intern Med 1954; 93: 107-120. 13. Heatley NG: Mucosubstance as a barrier to diffusion. Gastroenterology 1959; 37: 313-317. 14. Williams SE, Turnberg LA: Studies of the “protective” properties of gastric mucus: evidence for a ‘mucus-bicarbonate’ barrier. Gut 1980; 20: A922-A923. 15. Flemstrdm G, Kivilaakso E: Demonstration of a pH gradient at the luminal surface of rat duodenum in viva and its dependence on mucosal alkaline secretion. Gastroenterology 1983: 84: 784-794. 16. Bahari HMM, Ross IN, Turnberg LA: Demonstration of a pH gradient across the mucus layer on the surface of human gastric mucosa in vitro. Gut 1982: 23: 513- 516. 17. Flemstrdm G, Sachs G: Ion transport by amphibian antrum in vitro. I. General characteristics. Am J Physiol 1975; 228: 1188-1198. 18. Garner A, Flemstrdm G: Gastric HCOar secretion in the guinea pig. Am J Physiol 1978; 234: 1535-1541. 19. Forssell H, Stenquist B. Olbe L: Vagal stimulation of human gastric bicarbonate secretion. Gastroenterology 1985; 89: 581-586. 20. Allen A: The structure and function of gastrointestinal mucus. In: Harmon JW, ed. Basic mechanisms of gastrointestinal mucosal cell injury and protection. Balti- more: Williams and Wilkins, 1981; 351-367. 21. Morris GP, Harding PK, Wallace JL: A functional model for extracellular gastric mucus in the rat. Virchows Arch [B] 1984; 46: 239-251. 22. Hills BA, Butler BD, Lichtenberger LM: Gastric mucosal barrier: hydrophobic lining to the lumen of the stomach. Am J Physiol 1983; 244(Gastrointest Liver Physiol 7): G561-G568. 23. Butler RD. Lichtenberger LM, Hills BA: Distribution of surfactants in the canine gastrointestinal tract and their ability to lubricate. Am J Physiol 1983; 244(Gas- trointest Liver Physiol 7): G645-6651. 24. Goddard PJ, Hills BA, Lichtenberger LM: Does aspirin damage canine gastric mucosa by reducingitssurface hydrophobicity! Am J Physiol 1987; 252(Gastroin- test Liver Physiol 15): G421-G430. 25. Guth PH: Local metabolism and circulation in mucosal defense. In: Allen A, Flemstrom G, Garner A, eds. Mechanisms of mucosal protection in the upper gastrointestinal tract. New York: Raven Press, 1984; 235-258. 26. Gannon B, Browning J. O’Brien P. Rogers P: Mucosal microvascular architec- ture of the fundus and body of human stomach. Gastroenterology 1984; 86: 866- 875. 27. Hudspeth AJ: The recovery of local transepithelial resistance following single- cell lesions. Exp Cell Res 1982; 138: 331-342. 28. Rutten MJ, Ito S: Morphology and electrophysiology of guinea pig gastric mucosal reoair in vitro. Am J Phvsiol 1983: 244(Gastrointest Liver Phvsiol 7): G171-G18i
. _
29. Silen W: Gastric mucosal defense and repair. In: Johnson LR, ed. Physiology of the gastrointestinal tract, 2nd ed. New York: Raven Press, 1987; 1055-1069. 30. Robert A, Nezamis JE, Lancaster C, Davis JP, Field SO, Hanchar AJ: Mild irritants prevent gastric necrosis through ‘adaptive cytoprotection’ mediated by prostaglandins. Am J Physiol 1983; 245(Gastrointest Liver Physiol 8): G113- G121. 31. Robert A, Nezamis JE, Phillips JP: Effect of prostaglandin El on gastric secre- tion and ulcer formation in the rat. Gastroenterology 1968; 55: 481-487. 32. Robert A, Nezamis JE. Lancaster C, Hanchar Al: Cytoprotection by prostaglan- dins in rats: prevention of gastric necrosis produced by alcohol, HCI, NaOH, hyper- tonic NaCl and thermal injury. Gastroenterology 1979; 77: 433-443. 33. Rees WDW. Turnberg LA: Mechanisms of gastric mucosal protection: a role for the ‘mucus-bicarbonate’ barrier. Clin Sci 1982; 62: 343-348. 34. Hawkey CJ, Kemp RT, Walt RP. Bhaskar NK, Davie J, Filipowitz B: Evidence that adaptive cytoprotection in rats is not mediated by prostaglandins. Gastroen- terology 1988; 94: 948-954. 35. Domschke W, Domschke S, Hornig D, Demling L: Prostaglandinstimulated gastric mucus secretion in man. Acta Hepato-Gastroenterol 1978; 25: 292-294. 36. McQueen S, Hutton D. Allen A, Garner A: Gastric and duodenal surface mucus gel thickness in rat: effects of prostaglandins and damaging agents, Am J Physiol 1983; 245(Gastrointest Liver Physiol 8): G388G393. 37. Kivilaakso E, FlemstrBm G: Surface pH gradient and prostaglandin cytoprotec- tion in gastroduodenal mucosa. In: Allen A, Flemstriim G. Garner A, et a/, eds. Mechanisms of gastric mucosal protection in the upper gastrointestinal tract. New York: Raven Press, 1984; 227-232. 38. Lichtenberger LM, Richards JE, Hills BA: Effect of 16,16-dimethyl prostaglan- din Es on the surface hydrophobicity of aspirin-treated canine gastric mucosa. Gastroenterology 1985; 88: 308-314. 39. Lichtenberger LM, Graziani LA, Dial EJ, Butler BD, Hills BA: Role of surface- active phospholipids in gastric cytoprotection. Science 1983; 219: 1327-1329. 40. Kauffman GL. Whittle BJR, Aures D, Vane JR, Grossman Ml: Effects of prosta- cyclin and a stable analogue, 6~-PGlr on gastric secretion, mucosal blood flow, and blood pressure in conscious dogs. Gastroenterology 1979; 77: 1301-1306. 41. Kauffman GL, Aures D. Grossman Ml: Intravenous indomethacin and aspirin reduce basalgastric mucosal blood flow in dogs. Am J Physioll980; 238(Gastroin- test Liver Physiol 1): G131-6134. 42. Chaudhury TK, Jacobson ED: Prostaglandin cytoprotection of gastric mucosa. Gastroenterology 1978; 74: 59-63. 43. Hawkey CJ, Rampton DS: Prostaglandins and the gastrointestinal mucosa: are they important in its function, disease, or treatment? Gastroenterology 1985; 89:

MECHANISMS OF NSAID-INDUCED GASTRIC DAMAGE / SCHOEN AND VENDER
1162-l 188. 44. Svanes K, Critchlow J, Takeuchi K, Magee D, Ito S, Silen W: Factors influencing reconstitution offroggastric mucosa: role of prostaglandins. In: Allen A, Flemstrom G, Garner A, et a/, eds. Mechanisms of mucosal protection in the upper gastroin- testinal tract. New York: Raven Press, 1984; 33-39. 45Tarnawski A, Hollander D, Stachura J, Krause WJ, Gergely H: Prostaglandin protection of the gastric mucosa against alcohol injury-a dynamic time-related process: role of the mucosal proliferative zone. Gastroenterology 1985; 88: 334- 352. 46. Lupulescu A: Effect of prostaglandins on protein, RNA, DNA, and collagen synthesis in experimental wounds. Prostaglandins 1975; 10: 573-579. 47. Miller TA, Gum ET, Guinn EJ, Henagan JM: Prostaglandin prevents alterations in DNA, RNA, and protein in damaged gastric mucosa. Dig Dis Sci 1982; 27: 776- 781. 48. Szabo S. Trier JS, Frankel PW: Sulfhydryl compounds may mediate gastric cytoprotection. Science 1981; 214: 200-202. 49. Szabo S, Trier JS. Brown A, LaRocque M: Protection against aspirin-induced hemorrhagic gastric erosions and vascular injury by coadministration of sulfhydryl drugs (abstr). Gastroenterology 1985; 88: 1604. 50. Szelenyi I, Bruno K: Possible role of sulfhydryls in mucosal protection induced by aluminum hydroxide. Dig Dis Sci 1986; 31: 1207-1210. 51. Boyd SC, Sasame HA, Boyd MR: High concentrations of glutathione in glandu- lar stomach-possible implications for carcinogenesis. Science 1979; 205: lOlO- 1012. 52. Kosower NS, Kosower EM: The glutathione status of cells. Int Rev Cytoll978; 54: 109-160. 53. Allen A: Structure of gastrointestinal mucus glycoproteins and the viscous and gel-forming properties of mucus. Br Med Bull 1978; 34: 28-33. 54. Lev R. Seigel HI, Glass GBJ: Effects of salicylates on the canine stomach: a morphologic and histochemical study. Gastroenterology 1972; 62: 970-980. 55. Graham DY, Smith JL, Dobbs SM: Gastric adaptation occurs with aspirin ad- ministration in man. Dig Dis Sci 1983; 28: l-6. 56. Eastwood GL, Quimby GF: Effect of chronic aspirin ingestion on epithelial proliferation in rat fundus, antrum and duodenum. Gastroenterology 1982; 82: 852-856. 57. lvey KJ: Gastrointestinal intolerance and bleeding with non-narcotic analge- sics. Drugs 1986; 32(suppl 4): 71-79. 58. lvey KJ, Baskin WN, Krause WJ, Terry B: Effect of aspirin and acid on human jejunal mucosa: an ultrastructural study. Gastroenterology 1979; 76: 50-56. 59. Roth JLA, Valdes-Dapena A, Pieses P, Buchman E: Topical action of salicylates in gastrointestinal erosion and hemorrhage. Gastroenterology 1963; 44: 146-158. 60. Feldman M, Colturi TJ: Effect of indomethacin on gastric acid and bicarbonate secretion in humans. Gastroenterology 1984: 87: 1339-1343. 61. Gerkens JF, Shand DG, Flexner C, Nies A, Oates J, Data J: Effect of indometha- tin and aspirin on gastric blood flow and acid secretion. J Pharmacol Exp Ther 1977; 203: 646-652. 62. Levine RA, Schwartzel EH: Effect of indomethacin on basal and histamine stimulated human gastric acid secretion. Gut 1984; 25: 718-722. 63. Teorell T: On the permeability of the stomach mucosafor acids and some other substances. J Gen Physiol 1939; 23: 263-274. 64. Schanker LS: On the mechanism of absorption of drugs from the gastrointesti- nal tract. J Med Pharm Chem 1960; 2: 343-359. 65. Martin BK: Accumulation of drug anions in gastric mucosal cells. Nature 1963; 198: 896-897. 66. Davenport HW: Physiology of the digestive tract. Chicago: Yearbook Medical Publishers, 1971; 169. 67. Flemstrijm G: Intracellular accumulation and permeability effects of some weakacids in the isolatedfroggastricmucosa. Acta Physiol Stand 1971; 82: l-16. 68. Jabbari M. Valberg LS: Role of acid secretion in aspirin-induced gastric mucosal injury. Can Med Assoc J 1970; 102: 178-181. 69. lvey KJ: Drugs, gastritis, and peptic ulcer. Clin Gastroenterol 1981; 3(suppl2): 29-34. 70. lvey KJ, Morrison S, Grey C: Effect of oral and intravenous salicylates on the gastric mucosal barrier. Aust N Z J Med 1971; 1: 300-301. 71. Baskin W, lvey KJ. Krause WJ, Jeffrey GE, Gemmell RT: Aspirin-induced ultra- structural changes in human gastric mucosa: correlation with potential difference. Ann Intern Med 1976; 85: 299-303. 72. Stern Al: The gastric mucosal barrier. Med J Aust 1985; 142(suppl): S9-SlO. 73. Lin TM, Warrick MW. Evans DC, Nash JF: Action of the anti-inflammatory agents, acetylsalicylic acid, indomethacin and fenoprofen on the gastric mucosa of dogs. Res Commun Chem Pathol Pharmacol 1975; 11: 1-14. 74. Rainsford KD, Willis C: Relationship of gastric mucosal damage induced in pigs by antiinflammatory drugs to their effects on prostaglandin production. Dig Dis Sci 1982; 27: 624-635. 75. Cooke AR: The role of the mucosal barrier in drug-induced gastric ulceration and erosions. Dig Dis Sci 1976; 21: 155-164. 76. Murray HS. Strottman MP. Cooke AR: Effect of several drugs on gastric poten- tial difference in man. Br Med J 1974; 1: 19-21. 77. Bowen BK. Krause WJ, lvey KJ: Effect of sodium bicarbonate on aspirin-in: duced damage and potential difference changes in human gastric mucosa. Br Med J 1977; 2: 1052-1055. 78. Hingson DJ. Ito S: Effect of aspirin and related compounds on the fine structure of mouse gastric mucosa. Gastroenterology 1971; 61: 156-177.
79. Rainsford KD: Electron microscopic observations on the effects of orally ad- ministered aspirin and aspirin-bicarbonate mixtures on the development of gastric mucosal damage in the rat. Gut 1975: 16: 514-517. 80. Meyer RA, McGinley D, Posalaky Z: Effects of aspirin on tight junction structure of the canine gastric mucosa. Gastroenterology 1986; 91: 351-359. 81. Brune K. Rainsford KD. Schweitzer A: Biodistribution of mild analgesics. Br J Clin Pharmacol 1980; 10: 279S-2845. 82. Brogden RN, Heel RC, Pakes GE, Speight TM, Avery GS: Diflunisal: a review of its pharmacological properties and therapeutic use in pain and musculoskeletal strains and sprains and pain in osteoarthritis. Drugs 1980; 19: 84-106. 83. Takesue El, Perrine JW, Trapold JH: The anti-inflammatory profile of proqua- zone. Arch Int Pharmacodyn Ther 1976; 221: 122-131. 84. Brune K, Schweitzer A, Lanz R: Importance of drug biodistribution and metabo- lism in the development of side-effects by antiinflammatory/analgesic drugs. In: Rainsford KD. Velo GP, eds. Advances in inflammation research, ~016. New York: Raven Press, 1984; 9-15. 85. Schweitzer A, Brune K: Salicylic acid and proquazone: the differences in ab- sorption and biodistribution explain their different profile of side-effects. in: Wil- lougby DA, Giroud JP, Velo GP, eds. Perspectives in inflammation: future trends and developments. Proceedings of the 3rd International Meeting, European Bio- logical Research Association. Lancaster, United Kingdom: MTP Press, 1977; 353- 360. 86. Frenning B, dbrink KJ: The effects of acetic and acetylsalicylic acids on the appearance of the gastric mucosal surface epithelium in the scanning electron microscope. Stand J Gastroenterol 1971; 6: 605-612. 87. Flemstrom G, Marsden NVB: Dextran permeability, electrical properties, and H+ secretion in isolated frog gastric mucosa after acetylsalicylic acid. Gastroenter- ology 1973; 64: 278-284. 88. Kuo YJ, Shanbour LL: Mechanism of action of aspirin on canine gastric muco- sa. Am J Physiol 1976; 230: 762-767. 89. Kasbekar DK: Effects of salicylate and related compoundson gastric HCI secre- tion. Am J Physiol 1973; 225: 521-527. 90. Tague LL. Shanbour LL: Effects of ethanol on mucosal adenosine 3’5’ mono- phosphate (CAMP). Life Sci 1974; 14: 1065-1073. 91. Brody TM: Action of sodium salicylate and related compounds on tissue metab- olism in vitro. J Pharmacol Exp Ther 1956; 117: 39-51. 92. Sachs G, Collier RH, Shoemaker RL. Hirschowitz El: The energy source for gastric H+ secretion. Biochim Bioohvs Acta 1968: 162: 210-219. 93. Spenney JG: Acetylsalicylic acid hydrolase of gastric mucosa. Am J Physiol 197% 234: Efi06-E610. 94. Spenney JG. Brown M: Effect of acetylsalicylic acid on gastric mucosa. II: Mucosal ATP and phosphocreatine content and salicylate effects on mitochondrial metabolism. Gastroenterology 1977; 73: 995-999. 95. Menguy R, Masters YF: Effects of aspirin on gastric mucous secretion. Surg Gynecol Obstet 1965; 120: 92-98. 96. Menguy R, Desbaillets L: Role of inhibition of gastric mucous secretion in the phenomenon of gastric mucosal injury by indomethacin. Am J Dig Dis 1967; 12: 862-866. 97. Menguy R, Desbaillets L: Influence of phenylbutazone on gastric secretion of mucus. Proc Sot Exp Biol Med 1967; 125: 1108-1111. 98. Berrisford RG, Wells M, Dixon MF: Gastric epithelial mucus-a densitometric histochemical study of aspirin-induced damage in the rat. Br J Exp Pathol 1985; 66: 27-33. 99. Dekanski JB, MacDonald A, Sacra P: Effects of fasting, stress and drugs on gastric glycoprotein synthesis in the rat. Br J Pharmacol 1975; 55: 387-392. 100. Kent PW, Allen A: The biosynthesis of intestinal mucins: the effect of salicylate on glycoprotein biosynthesis by sheep colonic and human gastric mucosal tissues in vitro. Biochem J 1968; 106: 645-658. 101. Sarosiek J, Mizuta K, Slomiany A, Slomiany B: Effect of acetylsalicylic acid on gastric mucin viscosity, permeability to hydrogen ion, and susceptibility to pepsin. Biochem Pharmacol 1986; 35: 4291-4295. 102. Bagshaw PF, Munster DJ, Wilson JG: Molecular weight of gastric mucus glycoprotein is a determinant of the degree of subsequent aspirin induced chronic gastric ulceration in the rat. Gut 1987; 28: 287-293. 103. Garner A: Mechanisms of action of aspirin on thegastric mucosa of the guinea pig. Acta Physiol Stand 1978; (special suppl): 101-l 10. 104. Garner A, Flemstrom G, Heylings JR: Effects of antiinflammatory agents and prostaglandins on acid and bicarbonate secretions in the amphibian-isolated gas- tric mucosa. Gastroenterology 1979; 77: 451-457. 105. Rees WDW. Gibbons LC. Turnbern LA: Effects of non-steroidal anti-inffamma- tory drugs and prostaglandins on alkalisecretion by rabbit gastric fundus in vitro. Gut 1983; 24: 784-789. 106. Garner A: Assessment of gastric mucosal damage: comparative effects of aspirin and fenclofenac on the gastric mucosa of the guinea pig. Toxicol Appl Pharmacol 1977; 42: 477-486. 107. Rowe PH, Lange R, Marrone G. Matthews JB, Kasdon E, Silen W: In vitro protection of amphibian gastric mucosa by nutrient HCO$ against aspirin injury. Gastroenterology 1985; 89: 767-778. 108. Johnson LR, Overholt BF: Release of histamine into gastric venous blood following injury by acetic or salicylic acid. Gastroenterology 1967; 52: 505509. 109. Rainsford KD, Smith MJH: Inhibition of protein biosynthesis by salicylate in gastric-mucosal scrapings of pig and man in vifro (abstr). Biochem J 1969; 111: 37p.
April 1989 The American Journal of Medicine Volume 86 457

MECHANISMS OF NSAID-INDUCED GASTRIC DAMAGE / SCHOEN AND VENDER
110. Morris GP, Harding PL: Mechanisms of mucosal recovery from acute gastric damage: roles of extracellular mucus and cell migration. In: Allen A, Flemstrom G, Garner A, eds. Mechanisms of mucosal protection in the upper gastrointestinal tract. New York: Raven Press, 1984; 209-214. 111. Croft DN, Wood PHN: Gastric mucosa and susceptibility to occult gastrointes- tinal bleeding caused by aspirin. Br Med J 1967; 1: 137-141. 112. Vane JR: Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature New Biol 1971; 231: 232-235. 113. Johansson C, Kollberg 6, Nordemar R, Samuelson K, Bergstrom S: Protec- tive effect of prostaglandin Ea in the gastrointestinal tract during indomethacin treatment of rheumatic diseases. Gastroenterology 1980; 78: 479-483. 114. Rainsford KD: Prostaglandins and the development of gastric mucosal dam- age by anti-inflammatory drugs. Agents Actions 1979; 9(suppl 6): 193-210. 115. Konturek SJ: Prostaglandins in pathophysiology of peptic ulcer disease. Dig Dis Sci 1985; 3O(suppl): 105S-108s. 116. Whittle BJR, Vane JR: A biochemical basis for the gastrointestinal toxicity of non-steroid antirheumatoid drugs. Arch Toxicol 1984; (suppl 7): 315-322. 117. Wilson DE: Antisecretory and mucosal protective actions of misoprostal: po- tential role in the treatment of peptic ulcer disease. Am J Med 1987; 83(suppllA): 2-7. 118. Grossman MI, Matsumoto KK, Lichter RJ: Fecal blood loss produced by oral and intravenous administration of various salicylates. Gastroenterology 1961; 40: 383-388. 119. Main IHM, Whittle BJR: Investigation of the vasodilator and antisecretory role of prostaglandins in the rat gastric mucosa by use of non-steroidal anti-inflamma- tory drugs. Br J Pharmacol 1975; 53: 217-224. 120. Rainsford KD: The biochemical pathology of aspirin-induced gastric damage. Agents Actions 1975; 5: 326-344. 121. Hansen DG, Aures D, Grossman Ml: Histamine augments gastric ulceration produced by intravenous aspirin in cats, Gastroenterology 1978; 74: 540-543. 122. Pfeiffer CJ, Lewandowski LG: Comparison of gastric toxicity of acetylsalicylic acid with route of administration in the rat. Arch Int Pharmacodyn 1971; 190: 5- 13. 123. Cooke AR, Goulston K: Failure of intravenous aspirin to increasegastrointesti- nal blood loss. Br Med J 1969; 3: 330-332.
124. lvey KJ. Paone DB, Krause WJ: Acute effect of systemic aspirin on gastric mucosa in man. Dig Dis Sci 1980; 25: 97-99. 125. Hoftiezer JW. Burks M, Silvoso GR. lvey KJ: Comparison of the effects of regular and enteric-coated aspirin on gastroduodenal mucosa of man. Lancet 1980; II: 609612. 126. Lanza FL, Rack MF, Wagner GS, Balm TK: Reduction in gastric mucosal hemorrhage and ulceration with chronic high-level dosing of enteric-coated aspirin granules two and four times a day. Dig Dis Sci 1985; 30: 509-512. 127. Trondstad RI. Aadland E. Holler T, Olaussen B: Gastroscopic findings after treatment with enteric-coated and plain naproxen tablets in healthy subjects. Stand J Gastroenterol 1985; 20: 239-242. 128. Whittle BJR. H&s GA, Eakins KE, Moncada S. Vane JR: Selective inhibition of prostaglandin production in inflammatory exudates and gastric mucosa. Nature 1980; 284: 271-273. 129. Konturek SJ, Kwiecien N, Obtulowicz W. et al: Effect of carprofen and indo- methacin ongastricfunction, mucosal integrityandgeneration of prostaglandins in men. Hepatogastroenterol 1982; 29: 267-270. lfO.Aehringhaus U. Weiler H, Peskar BA, Peskar BM: Molecular mechanisms of the gastric toxicity of antirheumatic drugs. Arch Toxicol1984; (suppl7): 323-327. 131. Redfern JS, Lee E, Feldman M: Effect of indomethacin on gastric mucosal prostaglandins in humans: correlation with mucosal damage. Gastroenterology 1987; 92: 969-977. 132. Ligumsky M, Golanska EM, Hansen DG, Kauffman GL Jr: Aspirin can inhibit gastric mucosal cycle-oxygenase without causing lesions in rat. Gastroenterology 1983; 84: 756-761. 133. Cooke AR, Kienzle MG: Studies of anti-inflammatory drugs and aliphatic alco- hols on antral mucosa. Gastroenterology 1974; 66: 56-62. 134. Gilbert DA, Surawicz CM, Silverstein FE, et al: Prevention of acute aspirin- inducedgastric mucosal injury by 15-R-15 methyl prostaglandin ES: an endoscopic study. Gastroenterology 1984; 86: 339-345. 135.Guth PH, Paulsen G: Aspirin-induced gastric injury in the rat: histologic changes and sucralfate cytoprotection. Proc Sot Exp Biol Med 1987; 184: 423- 428. 136. Shea-DonohueT, Steel L, Montcalm E. DuboisA: Gastric protection bysucral- fate: role of mucus and prostaglandins. Gastroenterology 1986; 91: 660-666.
458 April 1989 The American Journal of Medicine Volume 86