Embed Size (px)
Citation preview

lable at ScienceDirect
Journal of Human Evolution 79 (2015) 105e118
Contents lists avai
Journal of Human Evolution
journal homepage: www.elsevier .com/locate/ jhevol
DNA analysis of ancient dogs of the Americas: Identifying possiblefounding haplotypes and reconstructing population histories
Kelsey E. Witt a, Kathleen Judd b, Andrew Kitchen c, Colin Grier d, Timothy A. Kohler d, e, f,Scott G. Ortman g, Brian M. Kemp d, h, Ripan S. Malhi a, i, *
a School of Integrative Biology, University of Illinois Urbana-Champaign, Urbana, IL 61801, USAb Kemp Lab of Molecular Anthropology and Ancient DNA, Washington State University, Pullman, WA 99164, USAc Department of Anthropology, University of Iowa, Iowa City, IA 52242, USAd Department of Anthropology, Washington State University, Pullman, WA 99164, USAe Santa Fe Institute, Santa Fe, NM 87501, USAf Crow Canyon Archaeological Center, 23390 Road K, Cortez, CO 81321-9408, USAg Department of Anthropology, University of Colorado, Boulder, CO 80309, USAh School of Biological Sciences, Washington State University, Pullman, WA 99164, USAi Department of Anthropology and Institute for Genomic Biology, University of Illinois Urbana-Champaign, Urbana, IL 61802, USA
a r t i c l e i n f o
Article history:Received 22 January 2014Accepted 22 October 2014Available online 18 December 2014
Keywords:Ancient DNADomesticationPopulation geneticsCanis lupus familiarisMitochondrial DNANew World
* Corresponding author.E-mail address: [email protected] (R.S. Malhi).
http://dx.doi.org/10.1016/j.jhevol.2014.10.0120047-2484/© 2014 Elsevier Ltd. All rights reserved.
a b s t r a c t
As dogs have traveled with humans to every continent, they can potentially serve as an excellent proxywhen studying human migration history. Past genetic studies into the origins of Native American dogshave used portions of the hypervariable region (HVR) of mitochondrial DNA (mtDNA) to indicate thatprior to European contact the dogs of Native Americans originated in Eurasia. In this study, we sum-marize past DNA studies of both humans and dogs to discuss their population histories in the Americas.We then sequenced a portion of the mtDNA HVR of 42 pre-Columbian dogs from three sites located inIllinois, coastal British Columbia, and Colorado, and identify four novel dog mtDNA haplotypes. Next, weanalyzed a dataset comprised of all available ancient dog sequences from the Americas to infer the pre-Columbian population history of dogs in the Americas. Interestingly, we found low levels of geneticdiversity for some populations consistent with the possibility of deliberate breeding practices. Further-more, we identified multiple putative founding haplotypes in addition to dog haplotypes that closelyresemble those of wolves, suggesting admixture with North American wolves or perhaps a seconddomestication of canids in the Americas. Notably, initial effective population size estimates suggest atleast 1000 female dogs likely existed in the Americas at the time of the first known canid burial, and thatpopulation size increased gradually over time before stabilizing roughly 1200 years before present.
© 2014 Elsevier Ltd. All rights reserved.
Introduction
The domestic dog (Canis lupus familiaris) holds a unique place inthe history of animal domestication, in that this species was notonly the first to be domesticated, but was also domesticated for avariety of purposes: as guards, hunting aids, and even as com-panions (Clutton-Brock, 1995). Dog remains dating to10,000e14,000 years before present (BP) have been discoveredacross Eurasia, and genetic studies suggest that dogs weredomesticated from gray wolves between 11,000 and 20,000 yearsago (Germonpr�e et al., 2009; Pang et al., 2009; Ding et al., 2012;
Freedman et al., 2014). Recent analysis of an ancient Siberiancanid with a morphology suggestive of a ‘transitional dog’ and amitochondrial DNA (mtDNA) haplotype found in contemporary dogpopulations suggests that domestication could have taken place inexcess of 33,000 years BP (Druzhkova et al., 2013). The exact originof domestic dogs is uncertain, though suggested geographic originsinclude the Middle East, Southeast Asia, and Europe (Pang et al.,2009; Vonholdt et al., 2010; Ardalan et al., 2011; Ding et al., 2012;Thalmann et al., 2013). Most recently, however, results suggestthat modernwolf populations diverged from one another at aroundthe same time as dog domestication, and therefore modern pop-ulations cannot be used to determine where dogs were firstdomesticated (Freedman et al., 2014).
Dogs are found in a variety of archaeological contexts in theAmericas that date as early as 10,500 years BP, with the first

K.E. Witt et al. / Journal of Human Evolution 79 (2015) 105e118106
unequivocal dog burial dating to roughly 9000 years BP (Morey andWiant, 1992). Interestingly, genetic analysis of ancient dog mtDNAindicates that many of these dogs were domesticated from Eurasianwolves, suggesting that these ancient dogs likely came to theAmericas with humans (Leonard et al., 2002). However, someancient dogs in the Americas have mitochondrial haplotypes eithershared with or nearly identical to those of North American wolves,suggesting either post-domestication admixture between dogs andwolves or even a separate domestication of canids in the Americas(Koop et al., 2000; Van Asch et al., 2013). Ethnohistorical recordsindicate that Native American peoples used dogs as hunters,herders, haulers, sources of food, and companions, and this practicelikely spans into prehistory (Schwartz, 1997).
Dogs have evolved to live with humans, and some of their adap-tations provide a historical record of human activity as well. Forexample, recent studies have examined how genes governing starchdigestion differ between dogs and wolves. As dogs were domesti-cated before the advent of agriculture, it would have been importantfor themto adapt to thechanginghumandiet andbemore efficient atdigesting starchy crops. One gene in particular, which codes for theenzyme alpha-2B-amylase (AMY2B), has two copies inwolves but asmany as 30 copies in dogs (Axelsson et al., 2013). These differences incopynumber correlatewith thehistoryof agriculture in a dogbreed'sregion of origin. For example, the saluki, which derives from theFertile Crescent, has 30 copies of the gene while the Siberian husky,which arose in a regionwith no agriculture, has only two (Freedmanet al., 2014). Likewise, humanpopulations have higher copy numbersof salivary amylase (AMY1) in regions with high-starch diets (Perryet al., 2007). Another example can be found in the Tibetan Mastiff,which has adapted to live with humans at high altitudes. A recentstudy has identified multiple candidate genes for this adaptation (Liet al., 2014), some of which (such as EPAS1, a transcription factor thatregulates cell response tohypoxia) are the samegenes that havebeenimplicated in human high-altitude adaptation (Xu et al., 2011) andothers (such as PLXNA4, a gene that promotes angiogenesis) thatshare similar functions (Scheinfeldt et al., 2012). Changes in genesexpressed in the brain are also commonly found when comparingdogs and wolves (Saetre et al., 2004; Li et al., 2013), suggesting thatbehavioral differencesbetweenwolves anddogshave a genetic basis.These changes in behavior are thought to have arisen early indomestication (Kukekova et al., 2012).
Given the close bond that dogs and humans have sharedthroughout history, dogs canprovide complementary data sources instudies of human populations. Notably, in cases where ancient hu-man remains are inaccessible for use in genetic analysis, dogs can beused as a proxy to examine the population history of humans (Barta,2006). Critically, organisms that are closely involved with humanshave likely moved across the earth following similar routes and atsimilar times, thus the genetic structure of these populations mayreflect upon that of the humans they followed. Like dogs, rats havebeen distributed worldwide by humans, and they have been used totrace worldwide migration patterns. For example, Polynesian ratpopulationshave been used to informmultiple hypotheses about theorigins of humans in Oceania (Matisoo-Smith and Robins, 2004).
Prior to AD 1492, dogs were widespread across the Americasand have likely been present since humans arrived on the continent(Leonard et al., 2002), so they potentially present an ideal studysystem for supplementing the reconstruction of human populationhistory in the Americas. Dog remains have been recovered from theJaguar Cave in Idaho and Agate Basin in Wyoming that date to over10,000 years BP (Yohe and Pavesic, 2000). By studying dogs inparallel with humans, we may also learn more about the history ofthe peopling of the Western Hemisphere than we can by studyinghumans alone, as dogs can help further test hypotheses about hu-man migration. For example, as dogs have a shorter generation
time (four years) than humans (20e30 years) (Fuller, 1995), theyhave a higher substitution rate than humans, and theoretically theyshould have accumulated more variation, and possibly geneticstructure, over a shorter timescale than possible with human ge-netic data (Walberg and Clayton,1981). However, artificial selection(such as breeding) may result in reduced genetic variation ormagnified population structure between regional populations.
Using genetic data from contemporary dogs alone to answerquestions about the history of pre-contact Native American dogscan be problematic. Genetic studies indicate that much of the di-versity of dogs in the early Americas was lost after European con-tact (Castroviejo-Fisher et al., 2011). A broad sampling of modernvillage dogs in the Americas demonstrated that nearly all haplo-types identified were shared with dogs introduced to the Americasthrough European colonization. However, some isolated pop-ulations like the Carolina dog and Alaskan Eskimo dog show con-tinuity with ancient samples and retain much of their indigenousdiversity, with a maximum of 30% European admixture in thematernal line of extant populations (Van Asch et al., 2013). Due tothe collapse of Native American dog populations after AD 1492 andthe subsequent replacement of indigenous dog lineages duringEuropean colonization, sampling ancient dog remains may provideamuch clearer picture of the ancient population history of dogs andtheir Native American domesticators in the early Americas.
Humans first entered the Americas roughly 15,000e20,000 yearsago (KempandSchurr, 2010;Meltzer, 2010). Subsequentmovementsor expansions into North America from Northeast Asia followed theinitial peopling before the Bering Land Bridge became submergedand separated Siberia from theAmericas by10,000e11,000 years ago(Forster et al., 1996; Tamm et al., 2007; Fagundes et al., 2008; Kempand Schurr, 2010). The initial foundersmay have followed the PacificCoast and rapidly spread southward, establishing the Monte Verdesite in Chile, for example, approximately 15,000 years ago (Dillehayand Collins, 1988; Schurr and Sherry, 2004; Erlandson, 2007;Tamm et al., 2007; Fagundes et al., 2008). Expansion further inlandlikelyoccurredonceglaciationwithdrewandan ice-freecorridorwasopened (Hoffecker et al., 1993).
A topic of particular importance in studies of Native Americanpopulation history is identifying founding mitochondrial haplo-types that were carried by the initial population that peopled theAmericas. Accurate estimation of the initial diversity found inAmerican populations is key to finding their geographic originoutside of the continents and estimating the founding populationsize. Torroni et al. (1993a) suggested the use of three criteria indetermining which haplotypes of a haplogroup represent foundinglineages. First, a founding haplotype is expected to be geographi-cally widespread, cross-cutting linguistic and cultural divisionsbetween Native American populations. Second, the foundinghaplotype should be central to the phylogeny of the haplogroup, asall other haplotypes in the haplogroup evolved from the foundinghaplotype, and are thus derived. Lastly, the haplotype should alsobe found in Siberia or elsewhere in Asia. Initially using these pa-rameters, one founding haplotype was identified in each of the fourhaplogroups recognized at the time: A, B, C and D (Torroni et al.,1993b). Later, haplogroup X and D4h3a were identified as addi-tional founder lineages using these criteria (Smith et al., 1999;Kemp et al., 2007). Both of these haplogroups have been identi-fied in ancient skeletal remains (Malhi and Smith, 2002; Kempet al., 2007; Cui et al., 2013; Rasmussen et al., 2014), with D4h3adating to at least 12,600 years BP. As argued by Kemp et al. (2007),mtDNA types observed in individuals of great antiquity in theAmericas are likely to be founding lineages. A unique form ofhaplogroup M, observed in two ~5000 year old skeletons from theinterior of British Columbia (Malhi et al., 2007), while not known tobe widespread, may also represent a founder lineage.

Figure 1. Map depicting the locations of all archaeological sites in North and SouthAmerica containing dog remains from which mtDNA sequences were obtained. All reddots indicate sites from the literature, and blue dots indicate sites added in this pub-lication. Numbers next to each dot indicate population size, and multiple numbers nextto a single dot indicate that multiple archaeological sites in a single location weresampled. (For interpretation of the references to colour in this figure legend, the readeris referred to the web version of this article.).
K.E. Witt et al. / Journal of Human Evolution 79 (2015) 105e118 107
More recently, mitogenome data has been used to infer thatthere were at least 14 founding mitochondrial lineages carried tothe Americas: A2, B2, C1b, C1c, C1d, C4c, D1b, D1c, D1d, D2a, D3,D4h3, X2a, and X2g (Tamm et al., 2007; Fagundes et al., 2008;Achilli et al., 2008, 2013; Perego et al., 2009; Malhi et al., 2010;Hooshiar Kashani et al., 2012). Most of these haplogroups aregeographically widespread. Notably, each of these haplogroups canbe traced to a single ancestral haplotype that is derived by multiplesubstitutions relative to haplotypes present in Asia, suggesting aperiod of isolation for the Asia-to-Americas migrants from theirsource population (Tamm et al., 2007; Fagundes et al., 2008). Thisidea is commonly referred to as the Beringian Incubation Model(BIM) or Beringian Standstill Hypothesis, for which support can alsobe found in the nuclear genome (Tamm et al., 2007; Schroederet al., 2009; Villanea et al., 2013).
Estimating the population size of the first humans to enter theAmericas is also of particular interest. Changes in the effectivepopulation size of Native Americans have been estimated from theinitial peopling of the Americas to the present using a Bayesianskyline plot (BSP) analysis incorporating a large number of com-plete mitogenomes from geographically and linguistically diversepopulations (Kitchen et al., 2008; Mulligan et al., 2008). Thisanalysis suggested a three-stage model for colonization, in whichthere was an initial period of divergence of the migrant populationfrom their Central Asian source population, a period of7000e15,000 years of population stability in Beringia and a finalperiod of rapid population expansion upon entry into the Americas,with a founder population with an effective size (Ne) of 500e1000females. Critically, this model complements the BIM (Tamm et al.,2007) and suggests that while the initial founding populationcontained many founder haplogroups that diverged from Asianprogenitor haplotypes during the Beringian occupation, the Ne ofthe founding population of the peopling of the Americas is sur-prisingly small. Given that the population history of dogs should besimilar to that of humans, dogs may show a similarly small effectivefounding population size.
In this study, we have expanded the sampling of dogs in theearly Americas by sequencing individuals recovered from threearchaeological sites in North America. Using methods similar tothose used for humans, we aim to characterize the populationhistory of dogs in the early Americas by defining founding lineagesand examining changes in the dog effective population size overtime. In an attempt to move towards such a goal, in this study wesequence a portion of the hypervariable region (HVR) of mtDNAfrom ancient dog remains from these three archaeological sites. Wethen combine the sequence data with previously publishedsequence data from both ancient and modern dogs to identifyfounding dog mtDNA haplotypes in the Americas as well as inferpopulation history of dogs in the Americas.
Methods
Sampling information and context
Samples were taken from three distinct archaeological sitesacross multiple temporal horizons. A map of the approximatelocation of the archaeological sites from which dogs were previ-ously studied, as well as the location of archaeological sites incor-porated in this study can be found in Fig. 1.
Janey B. Goode (JBG)
The Janey B. Goode site (11S1232) is a large, prehistoric settle-ment covering over 6 ha in the American Bottom near Brooklyn,Illinois (Galloy, 2010). The site was occupied approximately
660e1350 years BP, and was most intensively occupied during theTerminal Late Woodland and Mississippian periods. Over 5400 dogremains were recovered, with 103 individual dogs identified(Kuehn, Personal communication). Approximately 80 dog burialswere identified, with animals interred individually or in groups oftwo or three near houses (Borgic and Galloy, 2004). Based onskeletal pathologies, most dogs were used as transport or packanimals. A subset of 39 individuals was identified for geneticanalysis, and 35 of those samples were successfully extracted,amplified and sequenced to obtain mtDNA haplotypes.
Dionisio Point (DP)
Dionisio Point includes two settlements: a large five-plankhouse village (DgRv-3) that was occupied between 1500and 1300 years BP and a single plankhouse (DgRv-6) that dates tobetween 1000 and 700 years BP. Substantial shell middens sur-round the houses at both sites. Extensive excavations have beencompleted at both sites over the last two decades (Grier, 2006;Grier et al., 2013) and abundant dog remains have been recoveredfrom both house contexts and midden areas.
The sample of eight dogs we analyzed is derived from the shellmidden behind the single, later plankhouse at DgRv-6. Dog remainsare particularly abundant in this location. Articulated dogs wererecovered from various midden layers, both in direct associationwith human burials and on their own. Isolated or fragmented dogremains were also frequently encountered in the deposits. Dogs ofall ages are represented. The association of dog and human burialssuggests more than haphazard deposition, and the midden mayhave been a highly symbolic place. An accurate minimum numberof individuals (MNI) for the dogs represented at Dionisio Point has

Table 1A list of primers used in sequencing a portion of the hypervariable region of the mitochondrial genome of the individuals used in the study.
Amplicon Forward Reverse Source
15421-15617 GCACCCAAAGCTGAGATTCT GAGTTAATATGTCCTATGTAAGG This study15451-15744 TTCCCTGACACCCCTACATTCATATATT GCCCGGAGCGAGAAGAGGGAC Druzhkova et al., 201315485-15694 CCCCTACTGTGCTATGTCAGTATCTCCAG GGCATGGTGATTAAGCCCTTATTGGAC Druzhkova et al., 201315595-15796 CCTTACATAGGACATATTAACTC AGAACCAGATGCCAGGTATAG This study15668-15863 GTCCAATAAGGGCTTAATCACCATGCC TCCATCGAGATGTCCCATTTGCGA Druzhkova et al., 201315776-15982 CTATACCTGGCATCTGGTTCT TTAGAGTTAGTGCCGTTGCG This study15963-16138 CGCAACGGCACTAACTCTAA TACGTGTACCCTAAAACTATAT This study
Table 2Samples extracted at WSU, indicating amount of each element processed and thenumber of repeat silica extractions performed.
Site Sample ID Element Amountextracted
(mg)
# Repeatsilica
DP DgRu-6 B1 SF10 PLM Tooth-P3 28 0DP DgRu-6 B1 SF11 PLM Tooth-Unerupted M3 35 0DP DgRu-6 B2 West 1 PLM Tooth-M1 51 0DP DgRu-6 B2 West 2 PLM Tooth-M2 32 0DP DgRu-6 B2 West 3 PLM Mandible 55 2DP DgRu-6 B3 East 45 Tooth-P4 40 0DP DgRu-6 B3 E 58 Tooth-M2 47 0DP DgRu-6 B4 SE 2 PLM Tooth-M2 31 0APP 5MT123 PD 1305 FS42 Tooth-Unknown 54 0APP 5MT123 PD 1936 FS2 Tooth-Incisor 42 1
Dionisio Point is abbreviated DP and Albert Porter Pueblo as APP.
K.E. Witt et al. / Journal of Human Evolution 79 (2015) 105e118108
not yet been generated; a full analysis of the dog remains is inprogress.
Albert Porter Pueblo (APP)
A large Prehispanic Pueblo village in the central Mesa Verderegion of southwestern Colorado, Site 5MT123 was occupiedintermittently as early as 1400 years BP, but most of its occupationdates from the Pueblo II (1100e850 years BP) and Pueblo III(720e850 years BP) periods (Ryan, 2004). The two canid specimenssampled here were excavated between 2001 and 2004 by CrowCanyon Archaeological Center and are part of a larger on-goingstudy of dog mtDNA variation in the American Southwest. Archi-tectural details and associated ceramic materials place thesespecimens within the 940e740 years BP interval (S. Ryan, Personalcommunication).
DNA extraction and sequencing e University of IllinoisUrbanaeChampaign (UIUC)
Samples from the Janey B. Goode (JBG) site were extracted in aclean room environment dedicated to the extraction of DNA fromancient organisms at the University of Illinois UrbanaeChampaign(UIUC), using a protocol developed previously by the Malhi lab (Cuiet al., 2013). No modern dog samples have been processed in theancient DNA lab, to ensure there is no cross-contamination be-tween modern DNA and the ancient samples. Briefly, all teeth werewiped down with 6% sodium hypochlorite using a Kimwipe for atleast 1 min to remove surface contaminants, rinsed with moleculargrade DNA-free water and dried under UV light, and were drilledusing a Dremel drill to produce 0.2 g powder. The powder was thendigested in a solution of 4 mL EDTA, 300 mL 10% w/v N-lauryl sar-cosine, and 100 mL 3.3% w/v proteinase K. The digestion wasconcentrated down using a centrifuge to a volume of 250 mL, andthen extracted using the QIAQuick PCR Purification kit by Qiagen.All individual samples were extracted at least twice at differenttimes to confirm all DNA sequences.
Multiple primers were used to amplify a portion of the hyper-variable region of mtDNA (15421e15691 bp), as listed in Table 1(Druzhkova et al., 2013). Samples were amplified using the poly-merase chain reaction (PCR), with a mix as follows: 2.00 mL DNA,13.25 mL molecular-grade water, 2.00 mL 10� PCR buffer, 1.20 mL50 mM MgCl2, 0.80 mL 100 mM dNTPs, 0.30 mL of each primer,concentration 20 mM, and 0.15 mL Platinum Taq DNA Polymerase(Life Technologies). The program used for PCR amplificationinvolved an initial step at 94 �C for 2 min, followed by 40 cycles of94 �C for 15 s, 55 �C for 15 s, and 72 �C for 12 s, with a final step at72 �C for 5 min, and successful amplification was verified with gelelectrophoresis. Sanger sequencing of the PCR products was per-formed at the Roy J. Carter Biotechnology Center at UIUC. All in-dividuals were sequenced at least twice for each extraction, and if aconsensus was not reached between the extractions, a thirdextraction was performed to confirm the DNA sequences of
individuals. A consensus between two extractions, in which se-quences amplified from multiple primers for each of two extrac-tions matched exactly, confirmed the individual's sequence, and noindividual required more than three extractions for sequenceconfirmation.
DNA extraction and sequencing e Washington State University(WSU)
Eight samples from DP and two samples from APP wereextracted in the ancient DNA lab at WSU (Table 2), following theWSU method described in Cui et al. (2013). Samples were firsttested for the presence of PCR inhibitors following Kemp et al.(2014) and subjected to repeat silica extraction until they weredeemed inhibitor-free.
Two mitochondrial DNA fragments were PCR-amplified usingthe following primer sets: 1) D15401F (30-AAGCTCTTGCTCCAC-CATCA-50) and D15595R (30-GATATAATATTATGTACATGCTTAT-50),2) D15534F (30-CTATGTACGTCGTGCATTAATG-50) and D15711R (30-GGTTGATGGTTTCTCGAGGC-50). Fifteen microliter reactions usingOmni Klentag LAwere conducted following Kemp et al. (2014) withan annealing temperature of 60 �C. Successful amplification wasconfirmed via gel electrophoresis and amplicons were preparedand sequenced according to Kemp et al. (2014).
All sequences from this study are available on Genbank (acces-sion numbers KJ189495-KJ189536).
Data analysis
The DNA sequences obtained from the JBG, DP, and APP dogswere combined with other ancient and modern North Americandog and wolf mtDNA haplotypes reported in previous studies(Table 3). Ancient DNA haplotypes include samples from 8000 yearold dog burials in Siberia, which may have come from the samesource population as ancient dogs in the Americas (Losey et al.,2013). Also used were ancient DNA haplotypes from Bolivia, Peru,

Table 3A list of mtDNA data of ancient dogs in the Americas in the literature.
Origin Age (years BP) # Individuals Source
Siberia 8000 4 Losey et al., 2013Bolivia >1000 5 Leonard et al., 2002Peru 1000 3 Leonard et al., 2002Argentina 1000 1 Thalmann et al., 2013Mexico 1400e800 5 Leonard et al., 2002Alaska ~400e600 11 Leonard et al., 2002Alaska ~200e800 7 Brown et al., 2013California ~900e400 3 Byrd et al., 2013Koster, Illinois 9000 1 Thalmann et al., 2013Florida 1000 1 Thalmann et al., 2013Western Canada Unknown 5 Koop et al., 2000
The general location of the archaeological site(s) is included, as well as the date(if the samples were dated) and the number of individuals.
K.E. Witt et al. / Journal of Human Evolution 79 (2015) 105e118 109
Mexico, Argentina, western Canada, and the United States (Koopet al., 2000; Leonard et al., 2002; Brown et al., 2013; Byrd et al.,2013; Thalmann et al., 2013). Modern haplotypes of North Amer-ican and Eurasian wolves and all published dog haplotypes werealso used for comparison (Tsuda et al., 1997; Vila et al., 1997;Okumura et al., 1999; Savolainen et al., 2002; Takahasi andMiyahara, 2002; Pang et al., 2009; Klütsch and de Caprona, 2010;Castroviejo-Fisher et al., 2011; Thalmann et al., 2013; Van Aschet al., 2013). The DNA sequences obtained from the literature variedin length, so sequences were trimmed to produce a dataset thatmaximizes the number of individuals incorporated while showingvariation along a shorter segment of 229 base pairs [nucleotidepositions (nps) 15458-15687, according to Genbank AccessionNC002008] of the mitochondrial genome.
Putative founding haplotypes were identified using thefollowing criteria, modeled after similar criteria used for inferringfounding haplotypes of Native Americans (Torroni et al., 1993a). Afounding haplotype should be present in multiple geographic re-gions of the Americas and should be central to a phylogeny of dogmtDNA sequences. Additionally, a founding haplotype may befound both in the Americas and in Asia. However, if a haplotype isfound to be infrequent or geographically localized in the Americasand also differs by multiple substitutions from other dog haplo-types, it may also be a putative founding haplotype. It would bemore likely for a sequence that differs by five or more substitutionsfrom other founding haplotypes to be another founding haplotypethan for it to be derived from a much more distant haplotype.
The dataset of ancient dog and modern dog and wolf sequenceswas aligned using Bioedit, and the program Network was used toconstruct a median-joining network for the DNA sequences(Bandelt et al., 1999). A network is a visual representation of howhaplotypes relate to one another and is useful to determine ifcertain haplotypes are shared among populations. The median-joining method constructs trees that minimize the genetic dis-tance between haplotypes by linking clusters of closely-relatedsequences, and then resamples those clusters to produce themost parsimonious network (Bandelt et al., 1999). For eachgeographic region studied as well as for the wolf samples, addi-tional calculations weremade using Arlequin (Excoffier and Lischer,2010) to compare diversity within and between groups. For eachgroup, the number of haplotypes and qs, the number of segregatingsites in the sample corrected by the number of individuals in asample (Watterson, 1975), were calculated for each group. Nucle-otide diversity was calculated for each group as a measure ofsequence diversity weighted both by haplotype frequency and thenumber of substitutions that differ from other haplotypes in thegroup. These measures of genetic diversity are often used tocompare populations and estimate how populations differ from
one another. A population with lower diversity might indicate abottleneck event or deliberate breeding, and a population withhigher diversity might indicate a larger and more stable populationsize over a longer period of time or a population that experiencedgene flow. Since only two samples were analyzed from the AlbertPorter Pueblo population, they were omitted from this analysis. Insome cases, such as in Alaska andMexico, samples are derived frommultiple archaeological sites, in which case measures of diversitywere also calculated for each archaeological site containing multi-ple dogs. Finally, an analysis of molecular variance (AMOVA) wasalso performed to estimate groupings that explained the mostvariation, to estimate what populations were most closely related.
Some mtDNA haplotypes were identified as ‘outliers’. Theseoutliers were ancient dog haplotypes that differed from otherancient dog haplotypes by at least four substitutions, and are eitherhaplotypes shared with wolves or putative founding haplotypesdue to their genetic distance from the other sequences. In additionto what was reported in the literature, these haplotypes wereidentified as wolf or dog haplotypes by constructing a distance tree.Using all modern dog haplotypes, all published wolf haplotypesand all ancient dog haplotypes, a phylogeny was constructed inMEGA, using a maximum likelihood criterion with an HKY þ I þ Gmodel of substitutions (Tamura et al., 2011). This method of treeconstruction starts with a neighbor-joining tree (incorporatingsequences into the tree one at a time in order of similarity until allare part of the tree), with branch lengths and substitution ratesoptimized to their maximum likelihood values to produce the finaltree.
A dataset consisting of ancient dog haplotype sequences notclosely related to wolf haplotypes (i.e., those not likely of wolf-dogadmixture) and all modern dog haplotype sequences found in dogbreeds originating in the Americas was analyzed under a Bayesiancoalescent framework. This analysis was performed using theBayesian Markov chain Monte Carlo (MCMC) methods imple-mented in BEAST (version 1.8.0; Drummond et al., 2012). For thisanalysis, an HKY þ G þ I substitution model, a strict molecularclock, and an extended Bayesian skyline plot (EBSP) demographicmodel (with linear changes in population size; Heled andDrummond, 2008) was used to infer the historical dynamics ofdog populations. Importantly, the family of Bayesian skyline plotdemographic models provides a means to infer past populationhistories using both ancient and contemporary genetic sampleswithout a priori definition of a parametric model of past populationdynamics (i.e., constant or exponentially growing population size).Mean dates for archaeological sites were used as sampling dates(Table 4 and Supplementary Online Material [SOM] Table 1) of theaDNA sequences and provided independent calibrations for themolecular clock; a CMTC prior was used as a prior for the substi-tution rate (Ferreira and Suchard, 2008). Unless otherwise noted,default priors and operator values were used. Markov chains wererun for 200 million generations with samples taken every 10,000generations; convergence of three independent MCMC runs wasassessed using Tracer (version 1.6; Rambaut and Drummond,2007), and all MCMC samples were combined after the first 10%of samples were discarded as burn-in. The combined data files wereused for final inferences about the historical population dynamicsof Native American dogs and to produce a summary genealogy ofthe HVR1 sequences in this dataset.
To assess the accuracy of our coalescent inference and deter-mine if our punctuated sampling of ancient dog HVR1 sequencesintroduced a bias to our analyses, two sets of simulations wereperformed. First, ten subsets of the dataset consisting of a randomsample of 25 aDNA sequences across all times were also analyzed inBEAST using the same models and priors from the analysis of thefull dataset. This was done to assess any possible bias in the

Table 4Measures of genetic diversity of ancient dog populations by region.
Population # Individuals # Haplotypes Theta S Nucleotide diversity,weighted by haplotype
frequency
Nucleotide diversity, not weightedby haplotype frequency
Janey B. Goode 34 7 0.00534 (0.00278) 0.00272 (0.00243) 0.0103 (0.00728)Dionisio Point 5 2 0.00629 (0.00444) 0.00783 (0.0063) 0.0130 (0.0151)Siberia 4 3 0.00476 (0.00383) 0.00435 (0.00431) 0.00580 (0.00596)Alaska 18 11 0.0267 (0.0107) 0.0198 (0.0114) 0.0208 (0.0124)Mexico 5 5 0.0272 (0.015) 0.0235 (0.0159) 0.0235 (0.0159)California 3 3 0.0204 (0.0137) 0.0217 (0.0180) 0.0217 (0.0180)Western Canada 5 4 0.0189 (0.0108) 0.0182 (0.0126) 0.0224 (0.0163)Bolivia 5 3 0.00419 (0.00331) 0.00349 (0.00348) 0.00582 (0.00598)Peru 3 3 0.0146 (0.0101) 0.0145 (0.0126) 0.0145 (0.0126)Wolf 7 6 0.0224 (0.00714) 0.0231 (0.0145) 0.0237 (0.0153)Dogs 83 36 0.0315 (0.00932) 0.0126 (0.0007) 0.0195 (0.0110)
For each population, the number of individuals and haplotypes is recorded, as well as Theta S, a measure of the number of segregating sites per nucleotide weighted by samplesize. Nucleotide diversity is also included, a measure of diversity that incorporates pairwise comparisons between samples, and was calculated both with equal haplotypefrequencies and with weighted haplotype frequencies.
K.E. Witt et al. / Journal of Human Evolution 79 (2015) 105e118110
estimation of past population dynamics introduced by both non-random space (i.e., the inter-relatedness of dogs at sites) and time(i.e., multiple samples come from the same horizon). Second, weperformed simulations to produce synthetic datasets with a knownconstant demographic history and substitution and clock modelparameters identical to those estimated in the analysis of the fulldataset. These simulations were performed in BEAST (Bielejec et al.,2014) and analyzed with the HKY þ G þ I substitution, strict clock,and EBSP demographic (linear variant) models. One hundred sim-ulations were performed to assess whether punctuated sampling oflow information genetic data might produce artifacts in recon-structed skyline plots.
To compare shifts in dog population size with shifts in humanpopulation size, an EBSP was constructed using the datasetemployed in O'Fallon and Fehren-Schmitz (2011), which consists ofliving individuals representing all five Native American hap-logroups and ancient individuals from Ontario, Illinois, and Peru,and is thus similar to our dataset in having both modern andancient sequences. To allow for direct comparison, only the HVR1 ofthese individuals was used in the analysis. This analysis was alsoperformed in BEAST 1.8.0 using an HKY þG þ I substitution model,an uncorrelated log-normally distributed relaxed molecular clock(Drummond et al., 2006) with a fixed mean substitution rate of1.64� 10�7 substitutions/site/year (Soares et al., 2009) and an EBSPdemographic model. Sampling dates for the ancient individualswere taken from the original analysis (O'Fallon and Fehren-Schmitz, 2011). To assess the accuracy of demographic recon-struction using short HVR1 sequences, 100 simulations were per-formed in which synthetic datasets were produced from known
Table 5Measures of genetic diversity of ancient dog populations by archaeological site.
Population # Individuals # Haplotypes Theta S
Janey B. Goode 34 7 0.00534 (0.00278)Dionisio Point 5 2 0.00629 (0.00444)Siberia 4 3 0.00476 (0.00383)Fairbanks, AK 11 9 0.0143 (0.0078)W. Alaska 7 3 0.0239 (0.0108)Tula, Mexico 3 3 0.032 (0.0206)California 3 3 0.0204 (0.0137)Western Canada 5 4 0.0189 (0.0108)Bolivia 3 3 0.00419 (0.00331)Peru 3 3 0.0146 (0.0101)
For each population, the number of individuals and haplotypes is recorded, as well as Thetsize. Nucleotide diversity is also included, a measure of diversity that incorporates pairwfrequencies and with weighted haplotype frequencies.
demographic history and analyzed in BEAST. Synthetic data weresimulated using the same substitutionmodel parameters estimatedin the empirical analysis and under a demographic history similarto that inferred from the empirical data: an initial population size of103 at 15,000 years BP, and a sudden decrease in population size to5 � 104 years BP. These synthetic datasets were analyzed with theHKY þ G þ I substation, strict clock (rate ¼ 1.64 � 10�7 sub-stitutions/site/year), and EBSP demographic (linear variant)models. All graphs were produced using R.
Results
The individuals sequenced in this study represent a total of ninedifferent HVR1 haplotypes, four of which are novel. Novel haplo-types were authenticated by confirming identical sequences fromtwo extractions, as well as by amplifying and sequencing eachextract twice, to ensure that the novel substitutions are not due tomiscoding lesions, a common problem in ancient DNA analysis(Gilbert et al., 2003). All haplotypes are shown in SOM Table 1. Dueto the short length of the sequence, nearly all of the individualswith haplotypes that are not novel have identical sequences tomultiple modern dog haplotypes. The measures of genetic diversityare shown in Table 4 for all geographical regions, and in Table 5 aresubdivided further by archaeological site. Taking a regionalperspective, Alaska andMexico have the highest diversity, followedby California and western Canada. Siberia, Bolivia, Illinois (repre-sented by JBG) and coastal British Columbia (represented by DP)have much lower levels of diversity. When subdivided intoarchaeological sites, Tula, Mexico, has the highest diversity,
Nucleotide diversity, weighted byhaplotype frequency
Nucleotide diversity, not weightedby haplotype frequency
0.00272 (0.00243) 0.0103 (0.00728)0.00783 (0.0063) 0.0130 (0.0151)0.00435 (0.00431) 0.00580 (0.00596)0.0199 (0.0127) 0.00177 (0.0111)0.0162 (0.00999) 0.0232 (0.0191)0.0318 (0.0256) 0.0318 (0.0256)0.0217 (0.0180) 0.0217 (0.0180)0.0182 (0.0126) 0.0224 (0.0163)
0.00349 (0.00348) 0.00582 (0.00598)0.0145 (0.0126) 0.0145 (0.0126)
a S, a measure of the number of segregating sites per nucleotide weighted by sampleise comparisons between samples, and was calculated both with equal haplotype
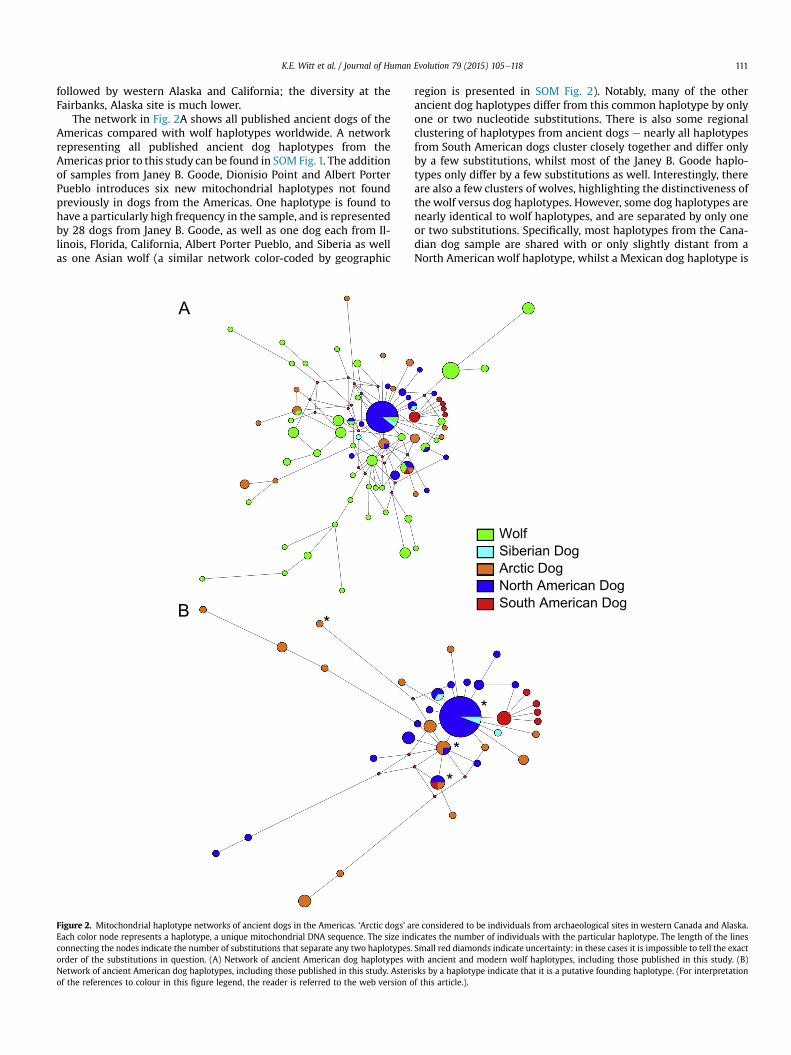
K.E. Witt et al. / Journal of Human Evolution 79 (2015) 105e118 111
followed by western Alaska and California; the diversity at theFairbanks, Alaska site is much lower.
The network in Fig. 2A shows all published ancient dogs of theAmericas compared with wolf haplotypes worldwide. A networkrepresenting all published ancient dog haplotypes from theAmericas prior to this study can be found in SOM Fig.1. The additionof samples from Janey B. Goode, Dionisio Point and Albert PorterPueblo introduces six new mitochondrial haplotypes not foundpreviously in dogs from the Americas. One haplotype is found tohave a particularly high frequency in the sample, and is representedby 28 dogs from Janey B. Goode, as well as one dog each from Il-linois, Florida, California, Albert Porter Pueblo, and Siberia as wellas one Asian wolf (a similar network color-coded by geographic
A
B*
Figure 2. Mitochondrial haplotype networks of ancient dogs in the Americas. ‘Arctic dogs’ aEach color node represents a haplotype, a unique mitochondrial DNA sequence. The size indconnecting the nodes indicate the number of substitutions that separate any two haplotypes.order of the substitutions in question. (A) Network of ancient American dog haplotypes wNetwork of ancient American dog haplotypes, including those published in this study. Asterof the references to colour in this figure legend, the reader is referred to the web version o
region is presented in SOM Fig. 2). Notably, many of the otherancient dog haplotypes differ from this common haplotype by onlyone or two nucleotide substitutions. There is also some regionalclustering of haplotypes from ancient dogs e nearly all haplotypesfrom South American dogs cluster closely together and differ onlyby a few substitutions, whilst most of the Janey B. Goode haplo-types only differ by a few substitutions as well. Interestingly, thereare also a few clusters of wolves, highlighting the distinctiveness ofthe wolf versus dog haplotypes. However, some dog haplotypes arenearly identical to wolf haplotypes, and are separated by only oneor two substitutions. Specifically, most haplotypes from the Cana-dian dog sample are shared with or only slightly distant from aNorth American wolf haplotype, whilst a Mexican dog haplotype is
WolfSiberian DogArctic DogNorth American DogSouth American Dog
*
*
*
re considered to be individuals from archaeological sites in western Canada and Alaska.icates the number of individuals with the particular haplotype. The length of the linesSmall red diamonds indicate uncertainty: in these cases it is impossible to tell the exactith ancient and modern wolf haplotypes, including those published in this study. (B)isks by a haplotype indicate that it is a putative founding haplotype. (For interpretationf this article.).

K.E. Witt et al. / Journal of Human Evolution 79 (2015) 105e118112
identical to a European wolf haplotype that differs by only a singlesubstitution from a North American wolf haplotype.
A second network was constructed that consists solely ofancient dog samples, to provide an illustration of how the doghaplotypes relate to one another without incorporating the wolfhaplotypes, and is represented by Fig. 2B (a similar network color-coded by geographic region is presented in SOM Fig. 3). It is moreapparent here that there are multiple outlier haplotypes. As dis-cussed above, four of the western Canadian dogs and the Mexicandog in the figure are likely the result of admixture between dogsand wolves. These wolf-like lineages may indicate that dogs wereseparately domesticated in the Americas. A single haplotype rep-resented by an Alaskan dog seems dissimilar to both dogs andwolves on the network in Fig. 2A, but all individuals withmore thanfour substitutions different from other dog haplotypes in Fig. 2B areconsidered to be outliers.
A phylogeny was constructed to infer the relationship of theoutlier sequences to all published dog and wolf haplotypes (Fig. 3).Overall, the wolf and dog haplotypes seem to be mixed throughoutthe tree. The haplotypes from the ancient dogs that are identified asoutliers in Fig. 2B are likely (i) founding haplotypes present in the
A175
A176
A166
A144
A114
AB007375
KC776175A
39A137
JBG13
A132AY163888
A159
A113
A183AY163893
A1A157EU816492
A121A3
A75
A145
A76A122
AY163891
AB007376
AB007378
KF661088A131A56A149KF66
1038A107
C11
C2AF098152C8
C18AF531683
A31C10
C16KF661090
AY163894
AF115697
AB007379E1B45
B7
B28CA5359
AF098116
B30
AF098115
AF098121
B34AF098117
AF005309
B25B11
B36B17
B19B27 AY163889AF008136KF661047
B1
0.005
Figure 3. Phylogeny of modern and ancient dogs and wolves of the Americas. If multiple anon the tree, they were omitted to simplify the tree for presentation. Triangles represent wohaplotypes and diamonds indicate outlier ancient dog haplotypes that differ by at least fivekey. All samples that are neither contemporary dog haplotypes nor part of this study are maran identical sequence.
dog population, (ii) haplotypes that show admixture with NorthAmerican wolves, or possibly (iii) the result of a separate domes-tication of dogs in the Americas. Notably, multiple outlier haplo-types are more closely related to wolves than dogs, suggestingadmixture or independent domestication. One haplotype from anAlaskan dog is not closely related to modern dog or wolf haplo-types, whilst two other outlier haplotypes clustermore closely withmodern dog haplotypes than wolf haplotypes. Interestingly, thelatter two outliers cluster with some haplotypes that are only foundin Siberia, the Americas or eastern Asia (Pang et al., 2009), possiblyindicative of a Northeast Asian origin, consistent with the origin ofNative Americans (Forster et al., 1996).
The EBSP of the dogs shows a relatively stable population fromthe present to the time of the most recent common ancestor, ~9000years BP, with the exception of a small dip in population size around1000 years BP (Fig. 4A). A summary genealogy relating the dogsamples in the dataset produced from the posterior distribution ofgenealogies sampled in the Bayesian coalescent analysis is pre-sented in SOM Fig. 5. The simulations using random subsets of theancient samples show no decrease in effective population size overtime, suggesting that the population decline is likely an artifact of
A83
A66
AY163890
AY163896
AY163887
A93
A112
A156
A160
A68
A69
A64
A164
AY163886
A81
A170
DQ421806
AF008140
KF661071
KF661044 AF11568
8KF661046
AF098151
AF098122AF098150AF098148AF098149A124
A181A29A101A147AY163878AY163884AY163885A111DP1
A86A102A103DP2
A104A95
A187
A77A177
AY163896
A22A82
A108A139A148A152A163A172D9
cient dogs, modern dog haplotypes or wolves from a particular region shared a branchlf haplotypes, squares represent modern dog haplotypes, circles represent ancient dogsubstitutions from other founding haplotypes identified in this study, as shown in theked with a Genbank accession number belonging to the individual sampled or one with
0 5000 10000 15000 20000
12
34
5
Years Before Present
log1
0(N
e *
gen
time)
0 5000 10000 15000 20000
12
34
5
Years Before Present
log1
0(N
e *
gen
time)
0 5000 10000 15000 20000
34
56
7
Years Before Present
log1
0(N
e *
gen
time)
0 5000 10000 15000 20000
34
56
7
Years Before Present
log1
0(N
e *
gen
time)
A B
C D
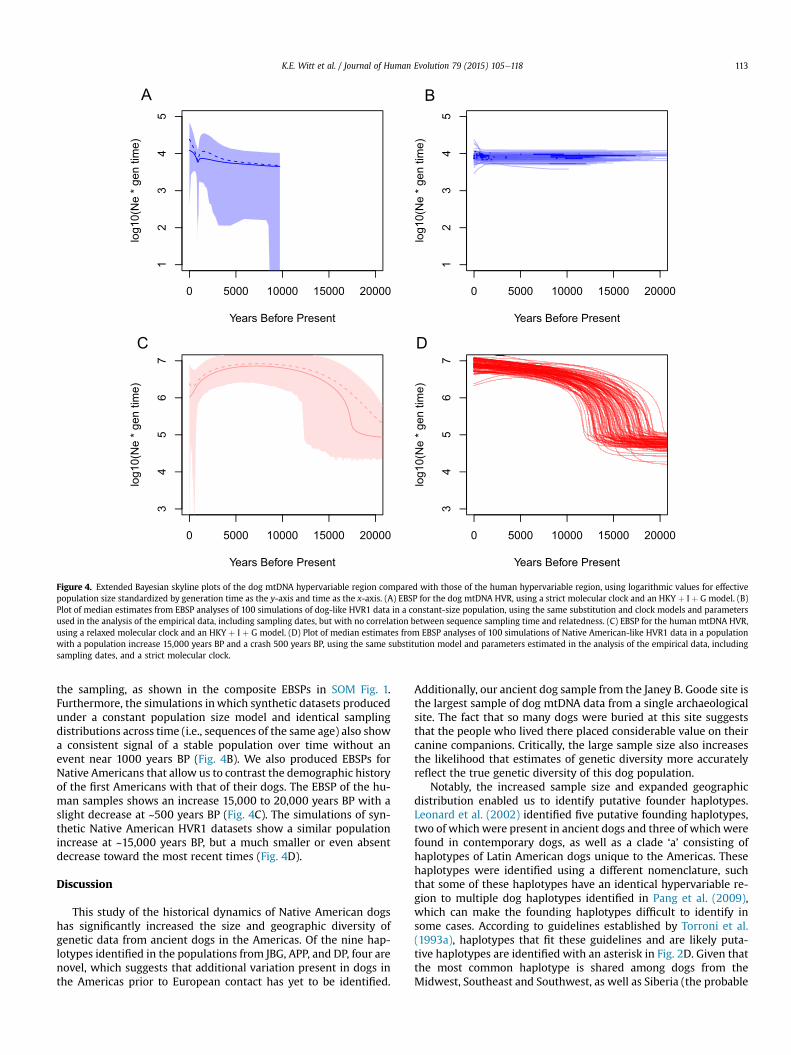
Figure 4. Extended Bayesian skyline plots of the dog mtDNA hypervariable region compared with those of the human hypervariable region, using logarithmic values for effectivepopulation size standardized by generation time as the y-axis and time as the x-axis. (A) EBSP for the dog mtDNA HVR, using a strict molecular clock and an HKY þ I þ G model. (B)Plot of median estimates from EBSP analyses of 100 simulations of dog-like HVR1 data in a constant-size population, using the same substitution and clock models and parametersused in the analysis of the empirical data, including sampling dates, but with no correlation between sequence sampling time and relatedness. (C) EBSP for the human mtDNA HVR,using a relaxed molecular clock and an HKY þ I þ G model. (D) Plot of median estimates from EBSP analyses of 100 simulations of Native American-like HVR1 data in a populationwith a population increase 15,000 years BP and a crash 500 years BP, using the same substitution model and parameters estimated in the analysis of the empirical data, includingsampling dates, and a strict molecular clock.
K.E. Witt et al. / Journal of Human Evolution 79 (2015) 105e118 113
the sampling, as shown in the composite EBSPs in SOM Fig. 1.Furthermore, the simulations inwhich synthetic datasets producedunder a constant population size model and identical samplingdistributions across time (i.e., sequences of the same age) also showa consistent signal of a stable population over time without anevent near 1000 years BP (Fig. 4B). We also produced EBSPs forNative Americans that allow us to contrast the demographic historyof the first Americans with that of their dogs. The EBSP of the hu-man samples shows an increase 15,000 to 20,000 years BP with aslight decrease at ~500 years BP (Fig. 4C). The simulations of syn-thetic Native American HVR1 datasets show a similar populationincrease at ~15,000 years BP, but a much smaller or even absentdecrease toward the most recent times (Fig. 4D).
Discussion
This study of the historical dynamics of Native American dogshas significantly increased the size and geographic diversity ofgenetic data from ancient dogs in the Americas. Of the nine hap-lotypes identified in the populations from JBG, APP, and DP, four arenovel, which suggests that additional variation present in dogs inthe Americas prior to European contact has yet to be identified.
Additionally, our ancient dog sample from the Janey B. Goode site isthe largest sample of dog mtDNA data from a single archaeologicalsite. The fact that so many dogs were buried at this site suggeststhat the people who lived there placed considerable value on theircanine companions. Critically, the large sample size also increasesthe likelihood that estimates of genetic diversity more accuratelyreflect the true genetic diversity of this dog population.
Notably, the increased sample size and expanded geographicdistribution enabled us to identify putative founder haplotypes.Leonard et al. (2002) identified five putative founding haplotypes,two of which were present in ancient dogs and three of which werefound in contemporary dogs, as well as a clade ‘a’ consisting ofhaplotypes of Latin American dogs unique to the Americas. Thesehaplotypes were identified using a different nomenclature, suchthat some of these haplotypes have an identical hypervariable re-gion to multiple dog haplotypes identified in Pang et al. (2009),which can make the founding haplotypes difficult to identify insome cases. According to guidelines established by Torroni et al.(1993a), haplotypes that fit these guidelines and are likely puta-tive haplotypes are identified with an asterisk in Fig. 2D. Given thatthe most common haplotype is shared among dogs from theMidwest, Southeast and Southwest, as well as Siberia (the probable

K.E. Witt et al. / Journal of Human Evolution 79 (2015) 105e118114
source of the American dog populations; Forster et al., 1996;Leonard et al., 2002), this haplotype is likely a founding haplo-type. Another haplotype, one shared with dogs from JBG, Alaska,and Peru, and identified by Leonard et al. (2002) as a putativefounding lineage, is likely also a founder haplotype because it iswidespread across the Americas. A third haplotype, also identifiedby Leonard et al. (2002), is shared between three Alaskan dogs andone dog from JBG, and could also be a putative founding lineage.Given that these founding haplotypes are characterized by only aportion of the mtDNA HVR, the haplotypes cannot be identifieddefinitively unless complete mitogenomes of these dogs aresequenced. The clade ‘a’ identified by Leonard et al. (2002), how-ever, seems to be a subhaplogroup localized to South America.
There are multiple outliers in the network that are distantlyrelated to most of the other dog haplotypes from the early Amer-icas. Some of them are closely related or identical to wolf haplo-types, as shown in Fig. 2A and B. These samples could indicateadmixture with North American wolves or a separate domestica-tion (or events) from North American wolves. This is most clearlydemonstrated by the western Canadian haplotypes, which arealmost identical to a North American wolf haplotype. Interestingly,the Mexican sample has an identical haplotype to a European wolf,but that haplotype is also only one base pair different from a NorthAmerican wolf haplotype. Given that mtDNA only captures a frac-tion of an individual's ancestry (i.e., maternal only) and only aportion of the hypervariable region of the mitochondrial genomewas analyzed in our study, our results do not provide the resolutionnecessary to distinguish between the two possibilities of admixtureand separate domestication in the Americas. Sequencing of otherregions of the genome would be required to determine if thesesequences derive from a single female wolf that interbred withdomesticated dogs in the Americas, or if this haplotype representsthe ancestor of domestic dogs in the Americas.
There are a total of eight outlier haplotypes shown in Fig. 2B thatdiffer significantly from other ancient dog haplotypes, and there aremultiple reasons why such outliers might be present in our dataset.First, these haplotypes represent dogs that came to the Americasvia Siberia that exhibited haplotypes different from the otherfounding haplotypes, and represent additional founder lineages.Second, these dog haplotypes are shared with wolf haplotypes thathave either gone extinct or have yet to be sampled and published inthe literature. If they are shared with wolves, admixture or separatedomestication are both viable possibilities. Interestingly, some ofthese haplotypes cluster more closely with dog haplotypes, sug-gesting that these dogs represent different founding haplotypes ethey are likely too distantly related to the other haplotypes in theAmericas to have diverged from another founding haplotype, butare more closely related to contemporary dog haplotypes thanwolfhaplotypes. Notably, some of the modern dog mtDNA haplotypesthat are most closely related to the outliers in Fig. 3, such as A31,A121, and B28, are exclusively found in Asian dogs, further sug-gesting that these were founding haplotypes (Pang et al., 2009).However, it is surprising that wolf and dog haplotypes are sothoroughly intermixed in Fig. 3, when in Fig. 2 the wolf haplotypesseem far more distant from the dog haplotypes. The cases ofadmixture identified in the network explain some of this inter-mixing, but the similarities may also be due to the use of only thehypervariable region in this study. The relatedness between the dogand wolf haplotypes may be much lower than the tree indicates ifthe complete mitogenome were incorporated into the tree.
When comparing the two measures of genetic diversity (theta Sand nucleotide diversity), the values are strongly positively corre-lated (R2¼ 0.66, p¼ 0.0024, see also values in Tables 4 and 5) acrosspopulations. Both values were calculated on a per nucleotide po-sition scale. Interestingly, the correlation is stronger when
excluding the sample from Tula, Mexico, which has a high theta Sestimate relative to its estimated of nucleotide diversity. The dogsfrom Tula, Mexico, havemitochondrial sequences that differ greatlyfrom one another in a very small sample size, which accounts forthis difference and is unusual amongst the regional datasets.
Surprisingly, the genetic diversity measurements of the dogpopulations studied do not seem to follow any geographic pattern.For example, the Alaska, western Canada, Mexico, and Peru pop-ulations display high levels of diversity, whereas the sites in Bolivia,and the North American Southwest and Midwest have much lowerlevels of diversity. This may be due to sampling e some of the re-gions used for analysis had multiple archaeological sites, whichmight inflate the genetic diversity as compared with regions withone or few. However, as shown in Table 5, evenwhen samples fromonly individual archaeological sites are used in the analysis, di-versity levels remain high in Mexico and Alaska, while Illinois andcoastal British Columbia have low levels of diversity. The measuresof nucleotide diversity for JBG and DP are also lowered, reflectingthe high frequencies of common haplotypes found at these sites,though diversity estimates remain low even when the haplotypefrequency bias is removed because all of the haplotypes sampledfrom each site are closely related. This could indicate that thepopulation arose from a small number of closely-related femalefounders, or that variable breeding regimens of domesticated dogswere practiced very early across the Americas.
Examining the skeletal remains for morphological similarities orsimilar skeletal modifications could further support the possibilityof selective breeding. Measurements of femora and humeri can beused as a reliable proxy for carnivore size (Von Valkenburgh, 1990).Femoral and humeral length in the JBG individuals have standarddeviations of only 8 and 6 mm, respectively, indicating phenotypichomogeneity (data not shown). This homogeneity could beexplained by inbreeding or selection, whether natural or artificial.Observations of village dogs that scavenge at human settlementsbut do not directly interact with humans suggest that they are allvery similar in size, likely because a balance must be struck be-tween being small enough to thrive on limited nutrients and beinglarge enough to defend against other dogs (Coppinger andCoppinger, 2001). Humans could have also been directly selectingfor a particular size of dog as well; given that JBG dogs were likelyused for hauling supplies (Borgic and Galloy, 2004), perhaps thehumans living there were selecting for dogs well-suited for haulinga given weight for long distances. An inbred population would behighly genetically similar and therefore phenotypically similar aswell. Additionally, the dogs sampled from the DP site in this studycome from a single shell midden, which represents only a smallportion of the dogs recovered from the DP site that are available forstudy (Barta, 2006). These dogs could have been closely related andwere therefore buried in the same shell midden, but the dog pop-ulation at DP could have been much more genetically diverseoverall. In future studies, comparing the phenotypic measurementsand genetic data from other dog populations to genetic data willhelp determine the correlation between phenotypic and geneticvariability in Native American dogs.
We performed Bayesian coalescent analysis to estimate thehistorical demography of dogs in the Americas using the methodsimplemented in BEAST. The EBSP shows a stable dog populationsize across time, with a small dip around 1000 years BP. However,our subsequent analyses suggest that the genetic signal of the EBSPis biased by the sampling. Specifically, large numbers of dogs weresampled from the same time period with identical haplotypes(mostly at JBG), and this likely causes the ‘dip’ in the plot roughly1000 years BP. We have two lines of evidence that support thisconclusion. First, we analyzed simulated datasets consisting ofrandom subsets of ancient samples (all modern samples were

K.E. Witt et al. / Journal of Human Evolution 79 (2015) 105e118 115
retained), which theoretically eliminated large numbers of dupli-cate sequences from the same ancient sampling period, whichproduced EBSPs with no discernible decrease in population size(SOM Fig. 1B). Second, our analysis of synthetic datasets simulatedunder a constant population size and with the same samplingregime (i.e., with the same sampling dates) but without any cor-relation between sampling time and relatedness (i.e., samples fromthe same period were not more closely related than those fromother sampling periods) also produced EBSPs without any consis-tent deviation from a constant population (Fig. 4B). Combined,these simulations suggest that the decrease in population size at~1000 years BP is an artifact introduced by sampling bias (i.e., thecorrelation between sampling time and relatedness).
As posited above, there are many reasons why we should expecthuman and dog population dynamics to be correlated, as the his-tories of dogs and humans are intertwined. To investigate humanhistory of the Americas at the same genetic resolution used in ouranalysis of dog history, we performed Bayesian coalescent analysisof the human HVR1 data used in O'Fallon and Fehren-Schmitz(2011) using the same demographic model we applied to theanalysis of the dog HVR1 dataset. As expected, the human EBSPshows an increase ~15,000 years BP, followed by a stable populationsize lasting nearly to the present day, with a late, non-significantdip in effective population size near the present. Interestingly,this EBSP differs from the EBSP produced from the complete humanmitogenome, in which there is a clear population decline aroundthe time of European contact (O'Fallon and Fehren-Schmitz, 2011).Our analysis of synthetic data simulated under a demographicmodel with a recent-post-Columbian population collapse of 50%also did not produce EBSPs that consistently reflect this event.Combined, these contrasting results suggest that the hypervariableregion of mtDNA may not be expected to reveal fine-grainedchanges in recent population history, though it may be possiblethat larger samples of HVR1 sequences might contain enoughsignal to reliably reflect recent population histories.
When considering both the human and dog EBSPs, the popu-lation stability found in the dog plot is unexpected, as one wouldexpect the dog population to increase in size over time, as thehuman population did. Intriguingly, this unexpected finding mightindicate that humans were controlling dog matings and effectivelybreeding them, or that the population of dogs in the Americasquickly reached long-term carrying capacity. The stable populationof dogs had a median effective population size of ~1000 femaleindividuals at the time of the first dog burial in the Americas. This isconsistent with other estimates of dog effective population sizethat suggest a global population of roughly 10,000 female dogs atthe same time (Thalmann et al., 2013). Additionally, the coalescencedate estimate for our dataset of American dogs is also surprising,given that by ~9000 years BP dogs should have been establishedacross the Americas as they are thought to have arrived in theAmericas ~15,000 years BP with humans. However, it is interestingthat from 9000 years BP to the present, the population of dogs inthe Americas roughly mirrors that of humans, in that both arestable for long periods of time.
Importantly, sequencing the hypervariable region alone cap-tures a lot of diversity in a short stretch of sequence, but does notprovide a full picture of mitogenome diversity. As demonstrated bySOM Table 1, multiple dog haplotypes have identical sequences inthe regionwe studied, so it is possible that we are underestimatingthe diversity of dog mitogenomes present in the Americas, as wasfound true of human mitochondrial diversity as mitogenomesequencing became more routine (Tamm et al., 2007; Achilli et al.,2008; Fagundes et al., 2008). This underestimation, combined withthe possibility of breeding practices mentioned above, may meanthat some of the putative founding haplotypes we have identified
are not as frequent or as widespread as our results seem to indicate.Furthermore, if such population structure does exist, widespreadsampling of dogs from multiple locations will be necessary toaccurately characterize the diversity of dog founding mtDNA line-ages. Calculating measures of population diversity using a shortsegment of DNA has revealed some interesting differences in di-versity between populations, but it is pertinent now to sequencecomplete mitogenomes of these dogs to ensure that short nucleo-tide sequences have not biased these estimates. Ultimately, theseanalyses have confirmed that using complete mitogenomes canalso provide a clearer picture of dog population history.
Given that dogs and humans have lived interdependently forthousands of years, dogs have potential for use as a proxy to testhuman models of migration. Comparison of human populationswith the dog populations in this study could demonstrate similar-ities that show the utility of dogs as a complementary dataset fortesting migration models. Notably, the JBG site also has humanremains, and DNA analysis of these individuals is currently inprogress. However, the human remains postdate the dog remainsby at least 200 years at this site, and so the populations may not bedirectly comparable. In the cases of APP and DP as well, there are nohuman remains that have been found to be contemporaneous withthe dog remains sampled in this study. However, this absence ofhuman remains highlights the utility of using dogs as a proxy tolearn more about how people lived in regions and time periodsfrom which no human burials have been found.
Additionally, the haplotype distribution and diversity of doghaplotypes in a given region can allow us to infer human in-teractions with dogs, which can tell us more about the humanpopulation. Deliberate burial of dogs indicates that humans tookcare of the animals, and low levels of diversity can suggestdeliberate breeding practices. If the dogs were being bred for aspecific purpose, such as to haul sledges as in the case of the JBGdogs, it is possible that their owners were selecting for specifictraits. If a single haplotype is frequent in multiple regions, it ispossible that dogs migrated with humans to multiple locations.Haplotype distributions can also reveal instances of geneticdrift, as seems to be the case in South America. Of the nine in-dividuals currently sequenced from South America, eight ofthem are all part of the same derived clade. This founder effecthas also been identified in human populations (Wang et al.,2007), supporting the idea that dogs and humans have trav-eled together and their populations have changed over time insimilar ways.
Conclusion
Combining our data generated in this study with all publishedancient dog haplotypes has revealed new diversity and multipleshared haplotypes across broad regions of the Americas. Somearchaeological sites exhibit low levels of genetic diversity, sug-gesting the possibility of deliberate breeding practices in the area.The most frequently identified haplotype in the sample is likely afounding haplotype, especially since it is identical to that of anancient Siberian dog, possibly from the same source population.Two additional haplotypes that are not as frequent or as wide-spread, but still had a broad sampling, are also putative foundinghaplotypes. Additional outlier haplotypes that are infrequent butshare similarities with modern dog haplotypes from Eurasia mayalso be possible putative founding haplotypes. This provides aminimum of three to five founding haplotypes. Given that each ofthese haplotypes is identical in sequence to multiple contemporarydog haplotypes, determining the nomenclature of the foundinghaplotypes should likely incorporate whole mitogenome se-quences. Of the ~273 dog haplotypes that have currently been

K.E. Witt et al. / Journal of Human Evolution 79 (2015) 105e118116
identified, 29 can be found in dogs of pre-European contactAmericas.
Multiple DNA sequences were identified that were identical tohaplotypes of North American wolves, and could representadmixture with wolves or a separate domestication event(s).However, the hypervariable region of the mitochondrial genomelacks power to draw certain conclusions (i.e., poor resolution of finehaplotype structure) and as mitochondrial DNA represents only thedirect maternal line of numerous ancestors, it is not possible todistinguish between admixture with wolves and a separatedomestication event. Examination of other regions of the genome,such as autosomal SNPs or even the exome (the complete codingregion of the genome) could better elucidate the evolutionaryhistory of these dogs, as they are a result of multiple ancestrallineages.
The analysis of mitochondrial DNA haplotypes in this studyprovides insight to the population history of dogs in the Americasand brings us closer to a comparison between population historiesof Native Americans and their dogs. However, the hypervariableregion analyzed in this study is only a short segment of the mito-chondrial genome and likely subject to recurrent mutations, whichcan obscure identifications of founding haplotypes in the Americas(Malhi et al., 2002). Additionally, the high frequency of specific doghaplotypes in certain archaeological sites, if the result of breedingpractices, can bias estimates of diversity as shown in the EBSP es-timates of dog population histories. Sequencing complete mito-chondrial genomes in pre-European contact dogs will likelyprovide a less biased view of mtDNA diversity of dogs in the earlyAmericas. For example, although the dog dated to 9000 years BPfrom Illinois and the dog uncovered from Florida dated to 1000years BP share a hypervariable region haplotype, their completemitochondrial genomes differ at 12 nucleotide sites (Thalmannet al., 2013). Given that their shared haplotype is the mostnumerous in ancient American dogs sampled thus far, it will beimportant to sequence additional complete mitochondrial ge-nomes for comparison purposes and to gain a more nuanced viewof the shared history of dogs and humans in the Americas.
Acknowledgments
We would like to thank S. Kuehn for the faunal analysis per-formed on the JBG dogs. We would also like to acknowledge theIllinois State Archaeological Survey (ISAS) and the Illinois Depart-ment of Transportation (IDOT) for their permission to use theircollections. Extraction of aDNA from the Albert Porter Pueblo ca-nids was supported by the National Science Foundation underGrant DEB-0816400 to the Village Ecodynamics Project, and wethank S. Ryan of Crow Canyon Archaeological Center for her assis-tance. Dog specimens from Dionisio Point were collected duringfieldwork funded by the National Science Foundation (Grant1062615) and with the permission of the Penelakut First Nation.This research was supported in part through computational re-sources provided by The University of Iowa, Iowa City, Iowa. Thanksto Cara Monroe for assistance in the laboratory at WSU.
Appendix A. Supplementary data
Supplementary data related to this article can be found online atdoi:10.1016/j.jhevol.2014.10.012.
References
Achilli, A., Perego, U.A., Bravi, C.M., Coble, M.D., Kong, Q.-P., Woodward, S.R.,Salas, A., Torroni, A., Bandelt, H.-J., 2008. The phylogeny of the four pan-
American MtDNA haplogroups: implications for evolutionary and diseasestudies. Plos One 3 e1764.
Achilli, A., Perego, U.A., Lancioni, H., Olivieri, A., Gandini, F., Hooshiar Kashani, B.,Battaglia, V., Grugni, V., Angerhofer, N., Rogers, M.P., Herrera, R.J.,Woodward, S.R., Labuda, D., Smith, D.G., Cybulski, J.S., Semino, O., Malhi, R.S.,Torroni, A., 2013. Reconciling migration models to the Americas with thevariation of North American native mitogenomes. Proc. Natl. Acad. Sci. 110,14308e14313.
Ardalan, A., Kluetsch, C.F.C., Zhang, A.-B., Erdogan, M., Uhl�en, M., Houshmand, M.,Tepeli, C., Ashtiani, S.R.M., Savolainen, P., 2011. Comprehensive study of mtDNAamong Southwest Asian dogs contradicts independent domestication of wolf,but implies dog-wolf hybridization. Ecol. Evol. 1, 373e385.
Axelsson, E., Ratnakumar, A., Arendt, M.-L., Maqbool, K., Webster, M.T., Perloski, M.,Liberg, O., Arnemo, J.M., Hedhammar, A., Lindblad-Toh, K., 2013. The genomicsignature of dog domestication reveals adaptation to a starch-rich diet. Nature 1e6.
Bandelt, H.-J., Forster, P., Rohl, A., 1999. Median-joining networks for inferringintraspecific phylogenies. Mol. Biol. Evol. 16, 37e48.
Barta, J.L., 2006. Addressing issues of domestication and cultural continuity. Ph.D.Dissertation, McMaster University.
Bielejec, F., Lemey, P., Carvalho, L., 2014. piBUSS: a parallel BEAST/BEAGLE utility forsequence simulation under complex evolutionary scenarios. BMC Bioinfor-matics 15.
Borgic, Q.L., Galloy, J.M., 2004. Domesticated dog remains from the Janey B. GoodeSite. Paper presented at the 2004 Joint Meetings of the Southeastern Archae-ological Conference and the Midwestern Archaeological Conference, St. Louis,Missouri, October 21e23, 2004.
Brown, S.K., Darwent, C.M., Sacks, B.N., 2013. Ancient DNA evidence for geneticcontinuity in Arctic dogs. J. Archaeol. Sci. 40, 1279e1288.
Byrd, B.F., Cornellas, A., Eerkens, J.W., Rosenthal, J.S., Carpenter, T.R., Leventhal, A.,Leonard, J.A., 2013. The role of canids in ritual and domestic contexts: newancient DNA insights from complex hunteregatherer sites in prehistoric CentralCalifornia. J. Archaeol. Sci. 40, 2176e2189.
Castroviejo-Fisher, S., Skoglund, P., Valadez, R., Vil�a, C., Leonard, J.A., 2011. VanishingNative American dog lineages. BMC Evol. Biol. 11, 73.
Clutton-Brock, J., 1995. Origins of the dog: domestication and early history. In:Serpell, J. (Ed.), The Domestic Dog: Its Evolution, Behaviour and Interactionswith People. Cambridge University Press, Cambridge, pp. 7e20.
Coppinger, R., Coppinger, L., 2001. Dogs: A New Understanding of Canine Origin,Behavior, and Evolution. University of Chicago Press, Chicago.
Cui, Y., Lindo, J., Hughes, C.E., Johnson, J.W., Hernandez, A.G., Kemp, B.M., Ma, J.,Cunningham, R., Petzelt, B., Mitchell, J., Archer, D., Cybulski, J.S., Malhi, R.S.,2013. Ancient DNA analysis of mid-Holocene individuals from the NorthwestCoast of North America reveals different evolutionary paths for mitogenomes.Plos One 8 e66948.
Dillehay, T., Collins, M., 1988. Early cultural evidence from Monte Verde in Chile.Nature 332, 150e152.
Ding, Z.-L., Oskarsson, M., Ardalan, A., Angleby, H., Dahlgren, L.-G., Tepeli, C.,Kirkness, E., Savolainen, P., Zhang, Y.-P., 2012. Origins of domestic dog insouthern East Asia is supported by analysis of Y-chromosome DNA. Heredity108, 507e514.
Drummond, A.J., Ho, S.Y.W., Phillips, M.J., Rambaut, A., 2006. Relaxed phylogeneticsand dating with confidence. Plos Biol. 4 e88.
Drummond, A.J., Suchard, M.A., Xie, D., Rambaut, A., 2012. Bayesian phylogeneticswith BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969e1973.
Druzhkova, A.S., Thalmann, O., Trifonov, V.A., Leonard, J.A., Vorobieva, N.V.,Ovodov, N.D., Graphodatsky, A.S., Wayne, R.K., 2013. Ancient DNA analysis af-firms the canid from Altai as a primitive dog. Plos One 8 e57754.
Erlandson, J., 2007. The Kelp Highway Hypothesis: Marine ecology, the coastal migra-tion theory, and the peopling of the Americas. J. Island Coast. Archaeol. 2, 161e174.
Excoffier, L., Lischer, H.E., 2010. Arlequin suite ver 3.5: A new series of programs toperform population genetics analyses under Linux andWindows. Mol. Ecol. Res.10, 564e567.
Fagundes, N.J.R., Kanitz, R., Eckert, R., Valls, A.C.S., Bogo, M.R., Salzano, F.M.,Smith, D.G., S Jr., W.A., Zago, M.A., Ribeiro-dos-Santos, A.K., Santos, S.E.B., Petzl-Erler, M.L., Bonatto, S.L., 2008. Mitochondrial population genomics supports asingle pre-Clovis origin with a coastal route for the peopling of the Americas.Am. J. Hum. Genet. 82, 583e592.
Ferreira, M.A.R., Suchard, M.A., 2008. Bayesian analysis of elapsed times incontinuous-time Markov chains. Can. J. Stat. 36, 355e368.
Forster, P., Harding, R., Torroni, A., Bandelt, H.J., 1996. Origin and evolution of NativeAmerican mtDNA variation: a reappraisal. Am. J. Hum. Genet. 59, 935e945.
Freedman, A.H., Gronau, I., Schweizer, R.M., Ortega-Del Vecchyo, D., Han, E.,Silva, P.M., Galaverni, M., Fan, Z., Marx, P., Lorente-Galdos, B., Beale, H.,Ramirez, O., Hormozdiari, F., Alkan, C., Vil�a, C., Squire, K., Geffen, E., Kusak, J.,Boyko, A.R., Parker, H.G., Lee, C., Tadigotla, V., Siepel, A., Bustamante, C.D.,Harkins, T.T., Nelson, S.F., Ostrander, E.A., Marques-Bonet, T., Wayne, R.K.,Novembre, J., 2014. Genome sequencing highlights the dynamic early history ofdogs. Plos Genet. 10 e1004016.
Fuller, T.K., 1995. Comparative population dynamics of North American wolves andAfrican Wild Dogs. In: Carbyn, L.N., Fritts, S.H., Seip, D.R. (Eds.), Ecology andConservation of Wolves in a Changing World. Canadian Circumpolar Institute,Edmonton, pp. 325e328.
Galloy, J.M., 2010. PART V: WOODLAND PERIOD The Janey B. Goode Site (11S1232):Highlights of investigations at a massive late prehistoric site in the Americanbottom. Illinois Archaeol. 22, 529e552.

K.E. Witt et al. / Journal of Human Evolution 79 (2015) 105e118 117
Germonpr�e, M., Sablin, M.V., Stevens, R.E., Hedges, R.E.M., Hofreiter, M., Stiller, M.,Despr�es, V.R., 2009. Fossil dogs and wolves from Palaeolithic sites in Belgium,the Ukraine and Russia: osteometry, ancient DNA and stable isotopes.J. Archaeol. Sci. 36, 473e490.
Gilbert, M.T.P., Hansen, A.J., Willerslev, E., Rudbeck, L., Barnes, I., Lynnerup, N.,Cooper, A., 2003. Characterization of genetic miscoding lesions caused bypostmortem damage. Am. J. Hum. Genet. 72, 48e61.
Grier, C., 2006. Temporality in Northwest Coast households. In: Sobel, E.A.,Gahr, D.A.T., Ames, K.M. (Eds.), Household Archaeology on the Northwest Coast.International Monographs in Prehistory, pp. 97e119. Ann Arbor.
Grier, C., Flanigan, K., Winters, M., Jordan, L.G., Lukowski, S., Kemp, B.M., 2013. Usingancient DNA identification and osteometric measures of archaeological Pacificsalmon vertebrae for reconstructing salmon fisheries and site seasonality atDionisio Point, British Columbia. J. Archaeol. Sci. 40, 544e555.
Heled, J., Drummond, A.J., 2008. Bayesian inference of population size history frommultiple loci. BMC Evol. Biol. 8, 289.
Hoffecker, J., Powers, W., Goebel, T., 1993. The colonization of Beringia and thepeopling of the New World. Science 259, 46e53.
Hooshiar Kashani, B., Perego, U.A., Olivieri, A., Angerhofer, N., Gandini, F., Carossa, V.,Lancioni, H., Semino, O., Woodward, S.R., Achilli, A., Torroni, A., 2012. Mito-chondrial haplogroup C4c: a rare lineage entering America through the ice-freecorridor? Am. J. Phys. Anthropol. 147, 35e39.
Kemp, B.M., Schurr, T.G., 2010. Ancient and modern genetic variation in theAmericas. In: Auerbach, B. (Ed.), Human Variation in the Americas: The Inte-gration of Archeology and Biological Anthropology. Southern Illinois UniversityPress, Carbondale, pp. 12e50.
Kemp, B.M., Malhi, R., McDonough, J., Bolnick, D.A., Eshleman, J.A., Rickards, O.,Martinez-Labarga, C., Johnson, J.R., Lorenz, J.G., Dixon, E.J., Fifield, T.E.,Heaton, T.H., Worl, R., Smith, D.G., 2007. Genetic analysis of early Holoceneskeletal remains from Alaska and its implications for the settlement of theAmericas. Am. J. Phys. Anthropol. 621, 605e621.
Kemp, B.M., Monroe, C., Judd, K.G., Reams, E., Grier, C., 2014. Evaluation of methodsthat subdue the effects of polymerase chain reaction inhibitors in the study ofancient and degraded DNA. J. Archaeol. Sci. 42, 373e380.
Kitchen, A., Miyamoto, M.M., Mulligan, C.J., 2008. A three-stage colonization modelfor the peopling of the Americas. Plos One 3 e1596.
Klütsch, C.F.C., de Caprona, M.D.C., 2010. The IGF1 small dog haplotype is derivedfrom Middle Eastern grey wolves: a closer look at statistics, sampling, and thealleged Middle Eastern origin of small dogs. BMC Biol. 8, 119.
Koop, B.F., Burbidge, M., Byun, A., Rink, U., Crockford, S., 2000. Ancient DNA evi-dence of a separate origin for North American indigenous dogs. In:Crockford, S.J. (Ed.), Dogs Through Time: An Archaeological Perspective. Pro-ceedings of the 1st ICAZ Symposium on the History of the Domestic Dog.August 23e29, 1998. Archaeopress, Oxford, pp. 271e286.
Kukekova, A.V., Temnykh, S.V., Johnson, J.L., Trut, L.N., Acland, G.M., 2012. Geneticsof behavior in the silver fox. Mamm. Genome 23, 164e177.
Leonard, J.A., Wayne, R.K., Wheeler, J., Valadez, R., Guill�en, S., Vil�a, C., 2002. AncientDNA evidence for Old World origin of NewWorld dogs. Science 298, 1613e1616.
Li, Y., Vonholdt, B.M., Reynolds, A., Boyko, A.R., Wayne, R.K., Wu, D.-D., Zhang, Y.-P.,2013. Artificial selection on brain expressed genes during the domestication ofdog. Mol. Biol. Evol. 30, 1867e1876.
Li, Y., Wu, D.-D., Boyko, A.R., Wang, G.-D., Wu, S.-F., Irwin, D.M., Zhang, Y.-P., 2014.Population variation revealed high-altitude adaptation of Tibetan Mastiffs. Mol.Biol. Evol. 31, 1200e1205.
Losey, R.J., Garvie-Lok, S., Leonard, J.A., Katzenberg, M.A., Germonpr�e, M.,Nomokonova, T., Sablin, M.V., Goriunova, O.I., Berdnikova, N.E., Savel'ev, N.A.,2013. Burying dogs in ancient Cis-Baikal, Siberia: temporal trends and re-lationships with human diet and subsistence practices. Plos One 8 e63740.
Malhi, R.S., Smith, D.G., 2002. Brief communication: Haplogroup X confirmed inprehistoric North America. Am. J. Phys. Anthropol. 119, 84e86.
Malhi, R.S., Eshleman, J.A., Greenberg, J.A., Weiss, D.A., Schultz Shook, B.A.,Kaestle, F.A., Lorenz, J.G., Kemp, B.M., Johnson, J.R., Smith, D.G., 2002. Thestructure of diversity within New World mitochondrial DNA haplogroups: im-plications for the prehistory of North America. Am. J. Hum. Genet. 70, 905e919.
Malhi, R.S., Kemp, B.M., Eshleman, J.A., Cybulski, J., Smith, D.G., Cousins, S., Harry, H.,2007. Mitochondrial haplogroup M discovered in prehistoric North Americans.J. Archaeol. Sci. 34, 642e648.
Malhi, R.S., Cybulski, J.S., Tito, R.Y., Johnson, J., Harry, H., Dan, C., 2010. Briefcommunication: mitochondrial haplotype C4c confirmed as a founding genomein the Americas. Am. J. Phys. Anthropol. 141, 494e497.
Matisoo-Smith, E., Robins, J.H., 2004. Origins and dispersals of Pacific peoples:evidence from mtDNA phylogenies of the Pacific rat. Proc. Natl. Acad. Sci. 101,9167e9172.
Meltzer, D., 2010. First Peoples in a New World: Colonizing Ice Age America. Uni-versity of California Press, Oakland.
Morey, D.F., Wiant, M.D., 1992. Early Holocene domestic dog burials from the NorthAmerican Midwest. Curr. Anthropol. 33, 224e230.
Mulligan, C.J., Kitchen, A., Miyamoto, M.M., 2008. Updated three-stage model forthe peopling of the Americas. Plos One 3 e3199.
O'Fallon, B.D., Fehren-Schmitz, L., 2011. Native Americans experienced a strongpopulation bottleneck coincident with European contact. Proc. Natl. Acad. Sci.108, 20444e20448.
Okumura, N., Ishiguro, N., Nakano, M., Matsui, A., Shigehara, N., Nishimoto, T.,Sahara, M., 1999. Variations in mitochondrial DNA of dogs isolated fromarch-aeological sites in Japan and neighboring islands. Anthropol. Sci. 107, 213e228.
Pang, J.-F., Kluetsch, C., Zou, X.-J., Zhang, A., Luo, L.-Y., Angleby, H., Ardalan, A.,Ekstr€om, C., Sk€ollermo, A., Lundeberg, J., Matsumura, S., Leitner, T., Zhang, Y.-P.,Savolainen, P., 2009. mtDNA data indicate a single origin for dogs south ofYangtze River, less than 16,300 years ago, from numerous wolves. Mol. Biol.Evol. 26, 2849e2864.
Perego, U.A., Achilli, A., Angerhofer, N., Accetturo, M., Pala, M., Olivieri, A., HooshiarKashani, B., Ritchie, K.H., Scozzari, R., Kong, Q.-P., Myres, N.M., Salas, A.,Semino, O., Bandelt, H.-J., Woodward, S.R., Torroni, A., 2009. Distinctive Paleo-Indian migration routes from Beringia marked by two rare mtDNA hap-logroups. Curr. Biol. 19, 1e8.
Perry, G.H., Dominy, N.J., Claw, K.G., Lee, A.S., Fiegler, H., Redon, R., Werner, J.,Villanea, F.A., Mountain, J.L., Misra, R., Carter, N.P., Lee, C., Stone, A.C., 2007. Dietand the evolution of human amylase gene copy number variation. Nat. Genet.39, 1256e1260.
Rambaut, A., Drummond, A.J., 2007. Tracer v. 1.4. Available from: http://beast.bio.ed.ac.uk/Tracer.
Rasmussen, M., Anzick, S.L., Waters, M.R., Skoglund, P., DeGiorgio, M., Stafford, T.W.,Rasmussen, S., Moltke, I., Albrechtsen, A., Doyle, S.M., Poznik, G.D.,Gudmundsdottir, V., Yadav, R., Malaspinas, A.-S., White, S.S., Allentoft, M.E.,Cornejo, O.E., Tambets, K., Eriksson, A., Heintzman, P.D., Karmin, M.,Korneliussen, T.S., Meltzer, D.J., Pierre, T.L., Stenderup, J., Saag, L.,Warmuth, V.M., Lopes, M.C., Malhi, R.S., Brunak, S., Sicheritz-Ponten, T.,Barnes, I., Collins, M., Orlando, L., Balloux, F., Manica, A., Gupta, R., Metspalu, M.,Bustamante, C.D., Jakobsson, M., Nielsen, R., Willerslev, E., 2014. The genome ofa Late Pleistocene human from a Clovis burial site in western Montana. Nature506, 225e229.
Ryan, S.C., 2004. Albert Porter Pueblo (Site 5MT123), Montezuma County, Colorado:Annual Report, 2004 Field Season.
Saetre, P., Lindberg, J., Leonard, J.A., Olsson, K., Pettersson, U., Ellegren, H.,Bergstr€om, T.F., Vil�a, C., Jazin, E., 2004. From wild wolf to domestic dog: geneexpression changes in the brain. Brain Res. Mol. Brain Res. 126, 198e206.
Savolainen, P., Zhang, Y., Luo, J., Lundeberg, J., Leitner, T., 2002. Genetic evidence foran East Asian origin of domestic dogs. Science 298, 1610e1613.
Scheinfeldt, L.B., Soi, S., Thompson, S., Ranciaro, A., Woldemeskel, D., Beggs, W.,Lambert, C., Jarvis, J.P., Abate, D., Belay, G., Tishkoff, S.A., 2012. Genetic adap-tation to high altitude in the Ethiopian highlands. Genome Biol. 13. R1.
Schroeder, K.B., Jakobsson, M., Crawford, M.H., Schurr, T.G., Boca, S.M., Conrad, D.F.,Tito, R.Y., Osipova, L.P., Tarskaia, L.A., Zhadanov, S.I., Wall, J.D., Pritchard, J.K.,Malhi, R.S., Smith, D.G., Rosenberg, N.A., 2009. Haplotypic background of aprivate allele at high frequency in the Americas. Mol. Biol. Evol. 26, 995e1016.
Schurr, T.G., Sherry, S.T., 2004. Mitochondrial DNA and Y chromosome diversity andthe peopling of the Americas: evolutionary and demographic evidence. Am. J.Hum. Biol. 16, 420e439.
Schwartz, M., 1997. A History of Dogs in the Early Americas. Yale University Press,New Haven.
Smith, D.G., Malhi, R.S., Eshleman, J., Lorenz, J.G., Kaestle, F.A., 1999. Distribution ofmtDNA haplogroup X among Native North Americans. Am. J. Phys. Anthropol.110, 271e284.
Soares, P., Ermini, L., Thomson, N., Mormina, M., Rito, T., R€ohl, A., Salas, A.,Oppenheimer, S., Macaulay, V., Richards, M.B., 2009. Correcting for purifyingselection: an improved human mitochondrial molecular clock. Am. J. Hum.Genet. 84, 740e759.
Takahasi, S., Miyahara, K., 2002. Lineage classification of canine inheritable disor-ders using mitochondrial DNA haplotypes. J. Vet. Med. Sci. 64, 255e259.
Tamm, E., Kivisild, T., Reidla, M., Metspalu, M., Smith, D.G., Mulligan, C.J., Bravi, C.M.,Rickards, O., Martinez-Labarga, C., Khusnutdinova, E.K., Fedorova, S.A.,Golubenko, M.V., Stepanov, V.A., Gubina, M.A., Zhadanov, S.I., Ossipova, L.P.,Damba, L., Voevoda, M.I., Dipierri, J.E., Villems, R., Malhi, R.S., 2007. Beringianstandstill and spread of Native American founders. Plos One 2 e829.
Tamura, K., Peterson, D., Peterson, N., 2011. MEGA5: molecular evolutionary ge-netics analysis using maximum likelihood, evolutionary distance, andmaximum parsimony methods. Mol. Biol. Evol. 28, 2731e2739.
Thalmann, O., Shapiro, B., Cui, P., Schuenemann, V.J., Sawyer, S.K., Greenfield, D.L.,Germonpre, M., Sablin, M.V., Lopez-Giraldez, F., Domingo-Roura, X.,Napierala, H., Uerpmann, H.-P., Loponte, D.M., Acosta, A., Giemsch, L.,Schmitz, R.W., Worthington, B., Buikstra, J.E., Druzhkova, A., Graphodatsky, A.S.,Ovodov, N.D., Wahlberg, N., Freedman, A.H., Schweizer, R.M., Koepfli, K.-P.,Leonard, J.A., Meyer, M., Krause, J., P€a€abo, S., Green, R.E., Wayne, R.K., 2013.Complete mitochondrial genomes of ancient canids suggest a European originof domestic dogs. Science 342, 871e874.
Torroni, A., Schurr, T.G., Cabell, M.F., Brown, M.D., Neel, J.V., Larsen, M., Smith, D.G.,Vullo, C.M., Wallace, D.C., 1993a. Asian affinities and continental radiation of thefour founding Native American mtDNAs. Am. J. Hum. Genet. 53, 563e590.
Torroni, A., Sukernik, R.I., Schurr, T.G., Starikorskaya, Y.B., Cabell, M.F.,Crawford, M.H., Comuzzie, A.G., Wallace, D.C., 1993b. mtDNA variation ofaboriginal Siberians reveals distinct genetic affinities with Native Americans.Am. J. Hum. Genet. 53, 591e608.
Tsuda, K., Kikkawa, Y., Yonekawa, H., 1997. Extensive interbreeding occurred amongmultiple matriarchal ancestors during the domestication of dogs: Evidencefrom inter- and intraspecies polymorphisms in the D-loop region of mito-chondrial DNA between dogs and wolves. Genes Genet. Syst. 72, 229e238.
Van Asch, B., Zhang, A.-B., Oskarsson, M.C.R., Klütsch, C.F.C., Amorim, A.,Savolainen, P., 2013. Pre-Columbian origins of Native American dog breeds,with only limited replacement by European dogs, confirmed by mtDNA anal-ysis. Proc. R. Soc. B 280, 20131142.

K.E. Witt et al. / Journal of Human Evolution 79 (2015) 105e118118
Vila, C., Savolainen, P., Maldonado, J.E., Amorim, I.R., Rice, J.E., Honeycutt, R.L.,Crandall, K.A., Lundeberg, J., Wayne, R.K., 1997. Multiple and ancient origins ofthe domestic dog. Science 276, 1687e1689.
Villanea, F.A., Bolnick, D.A., Monroe, C., Worl, R., Cambra, R., Leventhal, A.,Kemp, B.M., 2013. Brief communication: Evolution of a specific O allele(O1v(G542A)) supports unique ancestry of Native Americans. Am. J. Phys.Anthropol. 151, 649e657.
Von Valkenburgh, B., 1990. Skeletal and dental predictors of body mass in carni-vores. In: Damuth, J.D., MacFadden, B.J. (Eds.), Body Size in MammalianPaleobiology: Estimation and Biological Implications 1. Cambridge UniversityPress, Cambridge, pp. 181e205.
Vonholdt, B.M., Pollinger, J.P., Lohmueller, K.E., Han, E., Parker, H.G., Quignon, P.,Degenhardt, J.D., Boyko, A.R., Earl, D.A., Auton, A., Reynolds, A., Bryc, K.,Brisbin, A., Knowles, J.C., Mosher, D.S., Spady, T.C., Elkahloun, A., Geffen, E.,Pilot, M., Jedrzejewski, W., Greco, C., Randi, E., Bannasch, D., Wilton, A.,Shearman, J., Musiani, M., Cargill, M., Jones, P.G., Qian, Z., Huang, W., Ding, Z.-L.,Zhang, Y.-P., Bustamante, C.D., Ostrander, E.A., Novembre, J., Wayne, R.K., 2010.Genome-wide SNP and haplotype analyses reveal a rich history underlying dogdomestication. Nature 464, 898e902.
Walberg, M., Clayton, D., 1981. Sequence and properties of the human KB cell andmouse L cell D-loop regions of mitochondrial DNA. Nucleic Acids Res. 9,5411e5421.
Wang, S., Lewis, C.M., Jakobsson, M., Ramachandran, S., Ray, N., Bedoya, G.,Rojas, W., Parra, M.V., Molina, J.A., Gallo, C., Mazzotti, G., Poletti, G., Hill, K.,Hurtado, A.M., Labuda, D., Klitz, W., Barrantes, R., Bortolini, M.C., Salzano, F.M.,Petzl-Erler, M.L., Tsuneto, L.T., Llop, E., Rothhammer, F., Excoffier, L.,Feldman, M.W., Rosenberg, N.A., Ruiz-Linares, A., 2007. Genetic variation andpopulation structure in native Americans. Plos Genet. 3 e185.
Watterson, G., 1975. On the number of segregating sites in genetical models withoutrecombination. Theor. Popul. Biol. 276, 256e276.
Xu, S., Li, S., Yang, Y., Tan, J., Lou, H., Jin, W., Yang, L., Pan, X., Wang, J., Shen, Y., Wu, B.,Wang, H., Jin, L., 2011. A genome-wide search for signals of high-altitudeadaptation in Tibetans. Mol. Biol. Evol. 28, 1003e1011.
Yohe, R., Pavesic, M., 2000. Early archaic domestic dogs from Western Idaho, USA.In: Crockford, S.J. (Ed.), Dogs Through Time: An Archaeological Perspective.Proceedings of the 1st ICAZ Symposium on the History of the Domestic Dog.August 23e29, 1998, pp. 93e104.





























