Embed Size (px)
Citation preview

Postgraduate Medical Journal (August 1977) 53, 480-486.
Metaphyseal chondrodysplasia
J. W. SPRANGERM.D.
Children's Hospital, Johannes Gutenberg University, Mainz
SummaryAny review of the metaphyseal chondrodysplasias iscomplicated by their variety and mainly unknownpathogenesis. The more familiar types display con-siderable clinical and radiological diversity: evenmore so the rarer disorders which still require completedefinition, but differences in their mode of inheritancemake diagnostic precision mandatory. These dys-plasias present in infancy or in childhood, when thepatient, usually dwarfed, may be proportionate, sothat some forms may be confused with rickets or otherlesions. Mental retardation is unusual, but the skin,hair, nails and facies provide valuable diagnosticfeatures.
Radiological abnormalities mainly affect themetaphyses of the shortened limb bones, less often theskull, vertebrae, pelvis, ribs and. extremities, andsometimes their distribution may indicate the specifictype of dysplasia. In a further complex group multiplesystems are involved, notably the pancreas, intestineand lympho-reticular, causing malabsorption andhaematological or immunological disorders.
THE various metaphyseal chondrodysplasias areintrinsic disorders of skeletal development, charac-terized by abnormal radiographic appearances and,in most cases, by a disturbed histological structureof the metaphyses, the cause of which is unknown.The epiphyses and the vertebrae are only minimallyaffected. Their intrinsic nature implies that theseconditions are due to a defective growth potentialof cartilage and bone. They must be carefullydistinguished from acquired secondary metaphyseallesions such as those due to rickets, scurvy, trauma,etc., in which the growth potential of cartilage andbone is initially quite normal. These secondarymetaphyseal changes are not included in the groupunder review and, by convention, achondroplasiaand hypochondroplasia are excluded, although bothare radiologically and histologically somewhat
Correspondence: Professor J. W. Spranger, Children'sHospital, Johannes Gutenberg University, 6500 Mainz,Germany.
similar and might also be classified as metaphysealdisorders.
(1) Commoner forms of metaphyseal chondrodysplasias(Table 1)Hypophosphatasia
This is the only metaphyseal chondrodysplasia ofknown pathogenesis, being caused by a deficiency ofalkaline phosphatase, an enzyme which, in asimplistic description, behaves as a pyrophosphatase,destroying pyrophosphate which itself inhibitstissue mineralization. In the absence or deficiencyof the enzyme, pyrophosphate accumulates andinterferes with normal tissue calcification. Thisphosphatase deficiency is variable in degree.
TABLE 1. The metaphyseal chondrodysplasias
1. Well defined formsHypophosphatasiaJansen typeSchmid typeVaandrager-Pefia type2. Rarer forms (not well defined)Rimoin & McAlister (1971)Van Creveld et al. (1971)Wiedemann & Spranger (1970)Kozlowski (1964)Rosenblum & Smith (1965)Spahr & Spahr-Hartmann (1961)3. With multi-system involvementMetaphyseal chondrodysplasia with malabsorption and
neutropenia (Schwachman-Diamond syndrome)McKusick type (cartilage hair hypoplasia)Adenosine deaminase deficiencyMetaphyseal chondrodysplasia with humoral immuno-
deficiency
Severe forms of hypophosphatasia are incompat-ible with life, perinatal death occurring in days,less often months, from respiratory failure. Lessseverely affected patients may survive for varyingperiods, but hypercalcaemia gradually developsafter the first weeks of life with failure to thrive,episodic bouts of constipation, fever and nephro-calcinosis. If the infant survives the first year,
by copyright. on M
arch 21, 2020 by guest. Protected
http://pmj.bm
j.com/
Postgrad M
ed J: first published as 10.1136/pgmj.53.622.480 on 1 A
ugust 1977. Dow
nloaded from

Metaphyseal chondrodysplasia 481
spontaneous improvement takes place. Cranio-stenosis may occur with premature shedding ofteeth and these may be the only clinical symptoms inmild cases. Microscopically, the only changes notedare the tongues of unmineralized cartilage or osteoidextending into the shafts of the long bones.
It is important to note that the congenital lethaland the milder adult tarda forms have been observedin a single family and are hence expressions of asingle gene mutation (Macpherson, Kroeker andHouston, 1972). The range of genetic variability isquite remarkable for an autosomal recessive con-dition, simulating that reported by Siggers (1977)for cartilage hair hypoplasia.Both liver and bone alkaline phosphatase are
lowered, whilst the intestinal alkaline phosphatasemay be either normal or elevated. Clinically normalheterozygotes can be identified by a slightly reducedserum alkaline phosphatase and may also haveincreased urinary excretion ofphosphoethanolamine.It is important to note that the clinical and radio-graphic features of hypophosphatasia have beenobserved in patients with a normal level of bloodalkaline phosphatase. This has been described as'pseudo-hypophosphatasia', but there is at least onefamily described in which the various features ofhypophosphatasia occurred with normal or lowlevels of serum alkaline phosphatase (Mehes et al.,1972). Thus, so-called 'pseudo-hypophosphatasia'may not be a nosological entity.
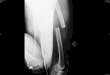
Metaphyseal chondrodysplasia-Jansen typeThis is well known and is inherited as an auto-
somal dominant. The original patient reported byJansen (1934) has been re-examined, and at 44 yearsof age is stated to be in a remarkably good state ofhealth and running a drug store (De Haas, De Boerand Griffioen, 1969).With advancing age the metaphyseal abnormalities
tend to disappear with fusion of the epiphyses, butthe bones remain short and deformed (Fig. 1). Theearly skull changes consist of a peculiar reticularpattern of the calvaria. Later there is a severedegree of sclerosis with hyperostosis of the skullbase, but the mandible may be small and there maybe persistent metaphyseal irregularities. One casereported by Ozonoff (1969) was described with thesechanges but did not have the Jansen type of meta-physeal chondrodysplasia, so that the early meta-physeal appearances should not be regarded asentirely diagnostic.
Metaphyseal chondrodysplasia-Schmid typeThis well-defined disorder is also transmitted as an
autosomal dominant, although in some patientswho are children of clinically and radiologicallynormal parents, a recessive inheritance must be
FIG. 1. Metaphyseal chondrodysplasia, Jansen type. Thetubular bones are severely shortened with striking meta-physeal irregularities, normal appearing epiphyses andvery wide physes. There are no major differentialdiagnostic problems.
considered possible. The Schmid type is commonerthan the Jansen type, and at least fifty-one caseshave been reported since the original paper bySchmid in 1949. In the Mainz Bone DysplasiaRegister there are nine cases of the Schmid type toonly three of the Jansen type.
This is a comparatively benign condition withoutmajor complications, with a normal life span and anadult height of 130-160 cm. Its main clinical signi-ficance is that it may be confused with, and some-times treated as vitamin D-resistant rickets.The most striking radiographic abnormalities
are well marked metaphyseal changes in the proximalrather than the distal femora (Fig. 2)-quite theopposite of the distribution found in cartilage hairhypoplasia (Fig. 3). Furthermore, in the Schmidtype there is much less coxa vara and osteotomiesare inadvisable as the spontaneous prognosis is veryfavourable.As mentioned above, the radiological changes of
metaphyseal chondrodysplasia may be closelysimulated by rickets (Fig. 4) and to a lesser degree byscurvy (Fig. 5) or by traumatic damage (Fig. 6)including the so-called 'battered child'.
by copyright. on M
arch 21, 2020 by guest. Protected
http://pmj.bm
j.com/
Postgrad M
ed J: first published as 10.1136/pgmj.53.622.480 on 1 A
ugust 1977. Dow
nloaded from

482 J. W. Spranger
1S~~~~~~~~~1~
A 4/
FIG. 2. Metaphyseal chondrodysplasia, Schmid type. Themetaphyscal changes are as severe in the proximal as inthe distal femora. The femoral necks are in varus posi-tion. Both fcaturcs help to differentiate the Schmid fromthe McKusick type (cartilage hair hypoplasia) of meta-physeal chondrodysplasias. The changes in this patientare rather marked; they maybe milder in other instances.
Metaphyseal chondrodysplasia-Vaandrager typeThis disorder is fairly well defined, although very
rare, and first described by Vaandrager (1960) whoreported monozygous twins with peculiar meta-physeal changes. Radiographically, there are longi-tudinal radiolucent areas extending deeply into thediaphyses, the ends of the more severely afflictedbones being expanded. The spine is normal, and theseverity of the long bone changes is again variable.Pefia (1965) described siblings with similar but moresevere changes, together with scoliosis and defectsof the cervical vertebrae. In a girl patient the authornoted that the bodies of the cervical vertebrae wereflattened, whilst those of her 14-year-old brotherwere uncalcified, these features being absent inVaandrager's patients. The knuckles were small, butradiographs of the hands were stated to be normal.Another patient with mild metaphyseal chondro-
dysplasia and slightly flattened vertebral bodies hasbeen described by Kozlowski and Sikorska (1970).With our present knowledge and information it isuncertain whether the differences between thesevarious cases are due to expression of a secondmutation, or whether there is an autosomal dominant
i. 'f.'W\:: . ',.g; d,}' ^w.'i._ E- k
f;'. ;.>.'.' _Bv
.?.
J..::
I KJFIG. 3. Metaphyseal chondrodysplasia, McKusick type(cartilage hair hypoplasia). The metaphyseal changes aremore pronounced in the distal than in the proximalfemora.
Vaandrager type with a normal spine and also anautosomal recessive or hump-backed variant withdistinct spinal changes.
(2) Metaphyseal chondrodysplasia-other rarer types(Table 1)These appear to be nosological entities, but as yet
not adequately described.
Rimoin and McAlister (1971)In 1971 Rimoin and McAlister described three
brothers, offsprings of a consanguineous mating,with short-limbed dwarfism, conductive hearingloss, mental retardation and a metaphyseal dys-plasia more severe in the middle and distal segmentsof the limbs than in the proximal. Similar bonechanges were described by Kozlowski and Rup-precht (1972) from a 7-year-old boy who wasmentally normal and no hearing defect was men-tioned.
Van Creveld et al. (1971)Van Creveld et al. (1971) de-scribed two unrelated
by copyright. on M
arch 21, 2020 by guest. Protected
http://pmj.bm
j.com/
Postgrad M
ed J: first published as 10.1136/pgmj.53.622.480 on 1 A
ugust 1977. Dow
nloaded from

Metaphyseal chondrodysplasia 483
U.'a..FIG. 4. Hypophosphataemic rickets. Note the strikingsimilarity of the bone changes to those in the meta-physeal chondrodysplasias. The major radiographicdifference is the abnormal bone structure in rickets. Vita-min D and phosphate therapy is required in rickets butcould be lethal in the chondrodysplasias.
patients with a severe disorder resembling theJansen type of chondrodysplasia. In contrast tothe Jansen type, however, there were large enchon-dromatous lesions extending into the ear bones.In the later stages of evolution, these defects werestudded with fine calcifications. There is someresemblance to multiple enchondromatosis, butthe condition is more symmetrical and moresevere than the usual case of Ollier disease.
Wiedemann and Spranger (1970)Another form of metaphyseal chondrodysplasia
was described by Wiedemann and Spranger (1970)with undoubtedly severe metaphyseal changes inthe long bones tending to disappear with advancingage and terminating, in contrast with other types ofmetaphyseal chondrodysplasia, with a normalstature. The vertebrae have separate ossificationcentres at birth; later they fuse, the changes regressand the spine eventually appears radiologicallynormal. There is one more case in the literature
FIG. 5. Scurvy. The metaphyseal changes could beconfused with those in the chondrodysplasias or those inbattered children. A major differential criterion is theabnormal bone structure in scurvy.
(Wiersbitzky, Weyrauch and Wiersbitzky, 1970)and it appears to be an entity.
Kozlowski (1964)Another condition was reported by Kozlowski
(1964) who described a family of five siblings andtwo unrelated children with short stature. Theradiographic abnormalities were so mild that Koz-lowski required to add arrows to his illustrations todemonstrate them. Possibly there are forms ofmetaphyseal chondrodysplasia that are extremelymild.
Rosenbloom and Smith (1965)One large family is often cited as a paradigm of
the Schmid type with autosomal dominant trans-mission; but the metaphyseal changes at the kneesare more severe than those in the proximal femora,a reversal of the usual distribution of the Schmidtype (Rosenbloom and Smith, 1965). In this family,on careful examination, epiphyseal changes may
by copyright. on M
arch 21, 2020 by guest. Protected
http://pmj.bm
j.com/
Postgrad M
ed J: first published as 10.1136/pgmj.53.622.480 on 1 A
ugust 1977. Dow
nloaded from

484 J. W. Spranger
I~~~~~~~~~~
mu.:
FIG. 6. Menkes syndrome. Metaphyseal irregularities areseen in the tibia and distal fibula. The left tibial shaft isexpanded owing to an old periosteal haematoma withsecondary bone formation. The appearance of thesebones is more likely to be confused with those of abattered child than with those of a metaphyseal chon-drodysplasia.
also be noted, but no coxa vara as might be ex-pected in the conventional Schmid type. Rather,there is coxa valga with flattening of the femoralheads. In this family the correctness of the diagnosisof Schmid type metaphyseal chondrodysplasiaseems questionable and possibly these patients mayhave arthro-ophthalmopathy described by Stickleret al (1965) as some had myopia and cleft palates.
Spahr and Spahr-Hartmann (1961)Two brothers were reported by Spahr and
Spahr-Hartmann (1961) in Switzerland who hadmetaphyseal abnormalities which were hard toclassify. The older sibling had severe constipation,probably caused by an aganglionic megacolon anddied from oesophageal haemorrhage due to severethrombocytopenia. The question arises that thesemay have been cases of cartilage hair hypoplasia.
(3) Metaphyseal chondrodysplasias with multi-systemdefects (Table 2)Metaphyseal chondrodysplasia, malabsorption, neu-tropenia (MMN-) syndromeTo date, about seventeen cases have been re-
ported. Since 1964 it has sometimes been referredto as the 'Schwachman-Diamond syndrome'(Schwachman et al., 1964). The metaphyseal defectswere first noted by Burke et al. (1967) and are mostmarked in the proximal femora. Unlike otherforms of metaphyseal chondrodysplasias, thesechanges are due to failure of the columnar cartilagecells to hypertrophy. Electron microscopic studieshave shown a peculiar accumulation of abnormalintracellular material with finger-print-like patternsin the rough endoplasmic reticulum of the chon-drocytes (Spycher et al., 1974). This may consistof a glycoprotein but is still undefined.The neutropenia in this disorder is unexplained;
some data point to a maturation arrest, others tohypoplasia of the neutrophil precursors. No definiteimmune defect has been demonstrated in thissyndrome, but recurrent respiratory infections arequite common although not related to the severity ofthe neutropenia.
Cartilage hair hypoplasiaThis has already been described in some detail in
the preceding paper by Dr Siggers et al. (1977). Thedisorder may be associated with defects of theintestine, the blood cells, the immune system andthe hair (Table 2). In the newborn the first sign isbowing of the femora which is diagnostic, with meta-physeal changes only apparent after 4 months of age.The original paper by McKusick et al. (1965)reported six of seventeen patients with intestinalmalabsorption and two with congenital megacolon.
TABLE 2. Metaphyseal chondrodysplasia with multi-systeminvolvement
MMN CHH ADAD MHD
Malabsorption + + +Hirschsprung disease - +Erythropenia + ±- +Neutropenia + + +Thrombocytopenia --Lymphopenia - + + -Cellular immunodeficiency - + +Humoral immunodeficiency - - + +Hair changes + + +Skin changes - - +
MMN, Metaphyseal chondrodysplasia, malabsorption,neutropenia; CHH, cartilage hair hypoplasia; ADAD,adenosine deaminase deficiency; MHD, metaphysealchondrodysplasia, humoral immunodeficiency; -1, reported;- not reported.
by copyright. on M
arch 21, 2020 by guest. Protected
http://pmj.bm
j.com/
Postgrad M
ed J: first published as 10.1136/pgmj.53.622.480 on 1 A
ugust 1977. Dow
nloaded from

Metaphyseal chondrodysplasia 485
This combination of metaphyseal chondrodysplasiaand intestinal abnormalities has been confirmed byothers.Haematological changes have often been re-
ported, e.g. gross neutropenia due to myeloid arrestat the metamyelocyte level with lymphopenia. Twocases were reported in the French literature bySacrez et al. (1965) and L'Hirondel et al. (1967)of congenital aplastic anaemia in cartilage hairhypoplasia. The marrow defect involves both thered and white cell systems.
In some cases, there is defective cellular immunitywhich explains the increased susceptibility to viralinfections, notably to chickenpox and smallpoxvaccination. Lymphoid cell transformation anddelayed hypersensitivity to Candida are normal insome patients, abnormal in others. No skin ab-normality has been reported in cartilage hairhypoplasia, but the hair is usually fine and sparse.
Adenosine deaminase deficiencyThis is the first of this group of dysplasias with a
pathogenetic explanation as to why so many systems-cartilage cells, immune and haematopoietictissues-are involved. The association of combinedB and T cell immunodeficiency with metaphysealchondrodysplasia was first observed by McKusickand Cross (1966), but at that time was thought to beco-incidental. Further reports, however, showedthat the association was pathogenetically relevant.It has recently been demonstrated that infants andyoung children with combined immunodeficiencyand metaphyseal chondrodysplasia had invariablya deficiency of the enzyme adenosine deaminase intheir erythrocytes (Meuwissen, Pollara and Pickering,1975). Bone changes, however, were only noted inpatients with severe immunodeficiency, but not inthose with some preservation of B or T cell function.The clinical picture of severe adenosine deaminasedeficiency is characterized by failure to thrive,chronic diarrhoea with malabsorption, chronic orrecurrent pneumonia, candidiasis and/or oppor-tunistic infections and short stature. The skin isthick and redundant, with scaling of the scalp andsparse body hair (Table 2).
In neonates and young infants, radiographs showan abnormally shaped pelvis, the wings of the iliabeing low and broad. There are metaphyseal changesand conspicuous flaring and cupping of the costo-chondral junctions.Adenosine deaminase which is normally present
in various tissues including liver, muscle and in-testinal mucosa, is involved in purine metabolism,converting adenosine to inosine. Possibly, if theenzyme is absent, adenosine may accumulate withincells and may suppress normal maturation or inhibit
the proliferation of lymphoid, cartilage and maybeother cells.
Metaphyseal chondrodysplasia and humoral immuno-deficiencyA case recently shown to the author by Professor
C. 0. Carter had metaphyseal chondrodysplasia anddeficient humoral immunity but normal cellularimmunity. This occurrence was first described byAmmann, Sutliff and Millinchick (1974) who re-ported a patient with a bone dysplasia, normalcellular and defective humoral immunity. Onefurther case was studied by Dr J. Herrmann inMadison, Wisconsin.Thus it appears that all three possibilities exist:
metaphyseal chondrodysplasia with cellular, humoralor combined immunodeficiency. Further studiesof the nucleic acid metabolism may be rewardingin these conditions.
ReferencesAMMANN, A.J., SUTLIFF, W. & MILLINCHICK, E. (1974)
Antibody-mediated immunodeficiency in short-limbeddwarfism. Journal of Paediatrics, 84, 200.
BURKE, V., COLEBATCH, J.H., ANDERSON, C.M. & SIMONS,M.J. (1967) Association of pancreatic insufficiency andchronic neutropenia in childhood. Archives of Disease inChildhood, 42, 147.
DE HAAS, W.H.D., DE BOER, W. & GRIFFIOEN, F. (1969)Metaphyseal dysostosis. Journal of Bone and Joint Surgery,S1B, 290.
JANSEN, M. (1934) (lber atypische Chondrodystrophie(Achondroplasie) und uber eine noch nicht beschriebeneangeborene Wachstumstorung des Knochensystems.Zeitschrift fur orthopadische Chirurgie, einschliesslich derHeilgymnastik und Massage, 61, 253.
KOZLOWSKI, K. (1964) Metaphyseal dysostosis. Report offive familial and two sporadic cases of a mild type. Ameri-can Journal of Roentgenology, Radiation Therapy andNuclear Medicine, 91, 602.
KOZLOWSKI, K. & RUPPRECHT, E. (1972) Metaphysealdysplasia with peripheral location. Helvetica paediatricaacta, 27, 85.
KOZLOWSKI, K. & SIKORSKA, B. (1970) Dysplasia meta-physaria, Typ Vaandrager-Pefia. Zeitschrift ftur Kinder-heilkunde, 108, 165.
L'HIRONDEL, J., CANE, DARIDON, & TILLET, (1967) Anemiede Blackfan-Diamond et dysostose metaphysaire re-cessive autosomique. Ouest M6dical, 20, 1152.
McKusIcK, V.A. & CRoss, H.E. (1966) Ataxia-telangiectasiaand Swiss type agammaglobulinemia. Journal of theAmerican Medical Association, 195, 739.
McKusIcK, V.A., ELDRIDGE, R., HOSTETLER, J.A., RUANG-WIT, 0. & EGELAND, J.A. (1965) Dwarfism in the Amish,II. Cartilage hair hypoplasia. Bulletin of the Johns HopkinsHospital, 116, 285.
MACPHERSON, R.I., KROEKER, M. & HOUSTON, C.S. (1972)Hypophosphatasia. Journal of the Canadian Association ofRadiologists, 23, 16.
MAROTEAUX, P., SAVART, P., LEFEBVRE, L. & ROYER, P. (1963)Les formes partielles de la dysostose metaphysaire.Presse Me'dicale, 71, 1523.
MtHES, K., KLUJBER, L., LASSu, G. & KAJTAR, P. (1972)Hypophosphatasia: screening and family investigationsin an endogamous Hungarian village. Clinical Genetics, 3,60.
by copyright. on M
arch 21, 2020 by guest. Protected
http://pmj.bm
j.com/
Postgrad M
ed J: first published as 10.1136/pgmj.53.622.480 on 1 A
ugust 1977. Dow
nloaded from

486 J. W. Spranger
MEUWISSEN, H.J., POLLARA, B. & PICKERING, R.J. (1975)Combined immunodeficiency disease associated withadenosine deaminase deficiency. Journal of Pediatrics, 80,1010.
OZONOFF, M.B. (1969) Metaphyseal dysostosis of Jansen.Radiology, 93, 1047.
PE&A, J. (1965) Disostosis metafisaria. Una revision conapertacion de una observacion familiar, una forma nuevade la enfermedad. Radiologica. Madrid, 47, 3.
RIMOIN, D.L. & MCALISTER, W.H. (1971) Metaphysealdysostosis, conductive hearing loss and mental retardation:a recessively inherited syndrome. Birth Defects, 7, 116.
RoSENBLOOM, A.L. & SMITH, D.W. (1965) The natural historyof metaphyseal dysostosis. Radiology, 83, 665.
SACREZ, R., LEVY, J.M., GODAR, G. & CASTANIER, J. (1965)An6mie de Blackfan-Diamond associ6 a des malformationsmultiples. Me'decine Infantile, 72, 493.
SCHMID, F. (1949) Beitrag zur Dysostosis EnchondralisMetaphysaria. Monatsschrift fur Kinderheilkunde, 97, 393.
SCHWACHMAN, H., DIAMOND, L.K., OSKI, F. & KON-T KHAW(1964) The syndrome of pancreatic insufficiency and bonemarrow dysfunction. Journal of Pediatrics, 65, 645.
SIGGERS, D.C., BURKE, J.B., MORRIS, B., NORMAND, I.C.S.,TANNER, J.M. & WILLIAMSON, D.A.J. (1977) Cartilage hairhypoplasia. Postgraduate Medical Journal, 53, 473.
SPAHR, A. & SPAHR-HARTMANN, 1. (1961) Dysostose m6ta-physaire familiale. Etude de 4 cas dans une famille.Helvetica paediatrica acta, 16, 834.
SPYCHER, M.A., GIEDION, A., SCHMERLING, D.H. & RUTT-NER, J.R. (1974) Electron microscopic examination ofcartilage in the syndrome of exocrine pancreatic in-sufficiency, neutropenia, multiple dysostosis and dwarfism.Helvetica paediatrica acta, 29, 471.
STICKLER, G.B., BELAU, P.G., FARRELL, F.J., JONES, J.D.,PUGH, D.G., STEINBERG, A.G. & WARD, L.M. (1965)Hereditary progressive arthro-ophthalmopathy. MayoClinic. Proceedings, 40, 433.
VAANDRAGER, G.J. (1960) Metafysaire dysostosis. Neder-lands Tijdschrift voor Geneeskunde, 104, 547.
VAN CREVELD, S., KOZLOWSKI, K., PIETRON, K. & VAN DERVALK, A. (1971) Metaphyseal chondrodysplasia cal-cificans. A report on two cases. British Journal ofRadiology,44, 773.
WIEDEMANN, H-R. & SPRANGER, J.W. (1970) Chondro-dysplasia metaphysaria (Dysostosis metaphysaria) einneuer typ. Zeitschrift fir Kinderheilkunde, 108, 171.
WIERSBITZKY, H., WEYRAUCH, P.C. & WIERSBITZKY, S.(1970) Dysostosis enchondralis metaphysaria (TypSchmid) bei einem Neugeborenen mit Morbus haemolyticusneonatorum (Anti-D). Deutsches Gesundheitswesen, 25,2225.
by copyright. on M
arch 21, 2020 by guest. Protected
http://pmj.bm
j.com/
Postgrad M
ed J: first published as 10.1136/pgmj.53.622.480 on 1 A
ugust 1977. Dow
nloaded from