Embed Size (px)
Citation preview

2009-05-28
1
KORELACJA IN VIVO-IN
VITRO – IVIVC DLA
PODANIA DOUSTNEGO
PREPARATY O NATYCHMIASTOWYM UWALNIANIU (IR)
I PREPARATY O PRZEDŁUŻONYM UWALNIANIU (ER)
KORELACJA IN VIVO - IN VITRO -
- IVIVC DLA PODANIA
DOUSTNEGO
IVIVC
Plan
prezentacjiCele badania IVIVC
FDA a IVIVC
Zastosowanie regulacyjne IVIVC dla postaci leków IR
Zastosowanie regulacyjne IVIVC dla postaci leków ER
Zastosowanie IVIVC dla rozwoju specyfikacji rozpadu in vitro
Koordynacja w czasie IVIVC w programie rozwoju preparatu
Podsumowanie
IVIVC

2009-05-28
2
Ważnym celem dla badaczy
farmaceutycznych jest uzyskanie danych
na temat korelacji między rozpadem
doustnej postaci leku badanej in vitro
(zwłaszcza tych z zastosowaniem
odpowiednich technologii nośnych), a
odpowiednimi wynikami in vivo.
IVIVC ma służyć jako narzędzie do
optymalizacji rozwoju nowych postaci leku
doustnego.
IVIVC A LEKI O PRZEDŁUŻONYM
UWALNIANIU
Koncepcja IVIVC dla postaci leku o
przedłużonym uwalnianiu pozwala
naukowcom przewidywać oczekiwaną
biodostępność substancji leczniczej
opartą o ich profil rozpadu in vitro.
IVIVC

2009-05-28
3
IVIV korelacja może być przydatna
m.in. w określaniu zmian w nowym
preparacie, stronie technicznej i w
samym procesie technologicznym.
Ustalono, że rozpad in vitro może być użyty
jako czuły, wiarygodny i powtarzalny
odpowiednik dla badań biorównoważności.
IVIVC
FOOD AND DRUG ADMINISTRATION
A KORELACJA IN VIVO IN VITRO FDA opisuje poziomy korelacji dostępne i wykorzystywane w
celach badawczych, jak i krytycznie ocenia zmieniające się
stopnie przydatności różnych poziomów IVIVC
IVIVC
Zarząd dostarcza informacji o zagadnieniach z zakresu
IVIVC oraz podpowiada, jak badać projektowanie i
zachowanie modeli IVIVC oraz jak ta korelacja powinna
zostać oceniona i praktycznie zastosowana
W 1997 roku instytucja FDA przedstawiła normy dotyczące
IVIVC również dla doustnych form dawkowania o
przedłużonym działaniu i nadal pozostaje jako jedyna
instytucja kompetentna w tej materiiJednakże zasady przedłożone w tym dokumencie powinny
być rozwijane i modyfikowane szczególnie w zakresie
technologii o kontrolowanym uwalnianiu substancji
leczniczej.

2009-05-28
4
IVIVC I FORMY POSTACI LEKU
UWALNIAJĄCE DAWKĘ
NATYCHMIASTOWO
Problemy z ustaleniem korelacji modelowej
spowodowane były:
brakiem liniowości rozkładu (tylko jedna trzecia
odpowiadających punktów czyniła rozwój danego
modelu prawie niemożliwy)
niemożnością ocenienia rozkładu (ograniczona
absorpcja)
Historycznie, usiłowało się szczęśliwie
rozwinąć korelację in vivo in vitro dla
modeli o natychmiastowym uwalnianiu
substancji leczniczej (IR - immediate
release), jednakże prawda okazała się
mniej pomyślna
Konsekwencja:
Korelacja nie została ustalona dla wielu
form postaci leku o natychmiastowym
uwalnianiu z powodu braku oczekiwanego
sukcesu, ponieważ
rezygnowano z dalszych badań.
Jednak okazał się, że rozwiązaniem
problemu będzie rozwój klasyfikacji BCS.

2009-05-28
5
STRUKTURY ORGANIZACYJNE
IVIVC
Food and Drug Administration (1997)- określona
jako instytucja specjalistyczna
SUPAC (Immediate Release Scale-up and Post
Approval Change) - dotycząca na dużą skalę
zakresu, zarówno IR dla stałych doustnych, form
dawkowania, jak i innych modyfikowanych postaci
leku
CPMP - The Committee for Proprietary Medicinal
Products (CPMP) within the European Agency for
the Evaluation of Medicinal Products (EMEA) -
organizacje te dostarczają informacji o
farmakokinetycznej i klinicznej ocenie
zmodyfikowanych doustnych postaci leku i tym
samym uczestniczą w rozwoju i ocenie IVIVC
-metod analitycznych
-procesów farmakokinetycznych
-wykwalifikowanego personelu o odpowiednim podejściu do rozwoju i wiedzy na temat stosowania modeli IVIVC
Struktury te powinny szybko informować naukowca,
iż obszar IVIVC wymaga współdziałania formulacji w
zakresie:

2009-05-28
6
ROZWÓJ I OCENA POZIOMÓW A
IVIVCWedług kryteriów FDA :
Wyznaczając AUC i C max absolutny błąd (%PE) nie
powinien być większy niż 15% dla pojedynczego
sformułowania jednocześnie należy zaznaczyć, że
absolutny błąd w przewidywaniu (MAPPE) powinien być
mniejszy niż 10% dla każdego parametru. Jeśli MAPPE
jest zawarte pomiędzy 10 – 20% dany model uważa się
za nieprzekonywujący i powinny zostać oznaczone dane
zewnętrzne (dane dodatkowe)
Ogólnie przyjmując dane użyte do badań IVIVC
określane jak dane wewnętrzne, dla doustnych postaci
posiadają dopuszczalny błąd do 15%
Dane dodatkowe, określane jako zewnętrzne, posiadają
dopuszczalny błąd do 10% i mogą wydawać się bardziej
zaostrzone czy też wygórowane
PODANIE IVIVC – PODSTAWOWE
PRAWO
IVIVC dla doustnych postaci leku pozwala
przewidzieć potencjalne nagłe zmiany zachodzące in
vivo w danej formulacji bez obowiązku prowadzenia
badań w tych warunkach
Jeśli zostaje opracowany nowy proces, bądź koryguje
się proces już istniejący poddany jest on wtedy
badaniom in vitro na testowanie rozkładu
Jeśli dwa produkty będą biorównoważne nowy
produkt zostaje zmieniony dla istnienia jednego w
programie rozwoju (po przeprowadzeniu badania BE
– bioequivalence)

2009-05-28
7
Jeśli badanie BE nie dało zadowalających wyników cykl zaczyna się od nowa i jest wtedy bardziej kosztowny
Proces, który spełnia warunki IVIVC jest podobny, ale decyzja o badaniu BE zostaje podjęta na podstawie wyników badań in vitro - takie postępowanie umożliwia otrzymanie korzyści finansowych wynikających z braku konieczności wykonywania badania BE
ZASTOSOWANIE IVIVC –
PODSTAWOWE PRAWO
Modele IVIVC pozwalają:
dokonywać zmian w procedurach invivo
unikać ryzyka, które istnieje podczas wykonywania badania BE
zyskać na czasie i podjąć decyzje o silnych zmianach w procesach in vivo dla danych form dawkowania.

2009-05-28
8
Przewidywany profil zależności stężenia
od czasu oraz odpowiednie parametry dla
nowego preparatu nie powinny być
porównywane z wcześniejszymi danymi
dotyczącymi istniejącego już preparatu,
ponieważ każdy model IVIVC jest
powiązany z (pewnym) błędem
Zatwierdzone modele IVIVC
mają błędy, które są ujęte
ilościowo i udowodnione,
aby spełnić kryteria
przedstawione w
wytycznych
IVIVC
PRZYKŁAD
Więc, dla identycznych profili rozpadu in vitro, 10%
różnica w AUC jest wkalkulowana. Stanowi ona
błąd związany z modelem IVIVC.
Założenie - Dopuszczalny błąd dla zatwierdzonego
modelu IVIVC przewidującego AUC dla preparatu
docelowego wynosi 10%. W pierwszym zakładzie wytwarzany jest dany
preparat. Drugi zakład wytwarza partię tego samego
preparatu, a testy rozpadu in vitro pokazują, że ma
on identyczny profil jak preparat wytworzony w
pierwszym miejscu.
Jednakże, porównanie modelu IVIVC- przewidywanego
AUC dla porcji z pierwszego miejsca ujawnia widoczną
10% różnicę.

2009-05-28
9
PRZYKŁAD CIĄG DALSZY
Użycie modelu IVIVC dla prognozowania działania
in vivo dla obydwu partii nie wskazałoby żadnej
różnicy w AUC, jak przewidywane jest z
identycznych profili in vitro.
IVIVC
Takie podejście jest również
zgodne z pojęciem, że model
IVIVC może zastąpić badania BE.
By można było przeprowadzić
takie badanie, porcja z każdego z
bieżących i nowo wytworzonych
miejsc powinna być oceniona w
tym samym badaniu.
ZASTOSOWANIE REGULACYJNE IVIVC DLA
POSTACI LEKÓW IR
Pomyślny rozwój modeli IVIVC dla postaci IR
może być ograniczony do substancji klasy II lub III
klasyfikacji BCS.
Mimo ograniczenia modele te mogą mieć jednak
zastosowanie dla postaci IR w celach
regulacyjnych.
W wytycznych SUPAC-IR z 1995r. omówione są
warunki uchylenia pełnego badania BE, gdzie
zmiana jest wymagana w dokumentacji BE in vivo.
Modele IVIVC mogą mieć także zastosowanie w
rozwoju specyfikacji rozpadu in vitro dla IR

2009-05-28
10
ZASTOSOWANIE REGULACYJNE IVIVC DLA
POSTACI LEKÓW O PRZEDŁUŻONYM
UWALNIANIU
Wytyczne IVIVC odnoszą się do zasad SUPAC-
MR dla przemysłu, gdzie konieczność
przeprowadzenia badania BE zmienia się w
zależności od poziomu skali.
Tam, gdzie wymagana jest taka dokumentacja,
potrzeba przeprowadzenia formalnych badań
może być uchylona w przypadku ustalonego
Wytyczne IVIVC dla doustnych postaci ER szczegółowo
omawiają zastosowanie IVIVC zwracając szczególną
uwagę na fakt, że przewidywalność modelu IVIVC musi
być ustalona, jeżeli ma on służyć jako substytut w
testach in vivo.
IVIVC NABIERA CORAZ WIĘKSZEGO
ZNACZENIA
5. zmiana w procesie wytwarzania
Obecnie, zgodnie z SUPAC-MR, IVIVC może być
użyty do podtrzymania rezygnacji z poniższych
zmian: 1. zmiana nieuwalnianej kontrolowanej substancji
pomocniczej leku
2. zmiana w uwalnianej kontrolowanej substancji
pomocniczej dla leku o wąskim indeksie
terapeutycznym3. zmiana w uwalnianej kontrolowanej
substancji pomocniczej leku
4. zmiana w miejscu wytwarzania

2009-05-28
11
Wytyczne FDA dla IVIVC omawiają także
zastosowanie IVIVC w uzasadnieniu uchylenia prośby
o zatwierdzenie niższych lub nowych stężeń.
W tych przypadkach, IVIVC powinien być opracowany
przy użyciu najwyższych stężeń, a nowe stężenia
powinny być kompozycyjnie proporcjonalne i
jakościowo takie same, mieć taki sam mechanizm
uwalniania oraz podobne profile rozpadu in vitro.
Omawiane wytyczne również kwalifikują zastosowanie
modeli IVIVC jako substytutu dla badań in vivo, na
podstawie liczby współczynnika uwalniania użytego w
rozwoju IVIVC oraz jeśli lek ma wąski indeks
terapeutyczny
Dla wszystkich form doustnych dawek
MR, w obecności ustalonego IVIVC,
jedynie zastosowanie testów lub ich
streszczenie powinno być
przeprowadzane (tylko dane dotyczące
uwalniania in vitro muszą być
potwierdzone przez korelującą
metodę).
Wytyczne dla przemysłu SUPAC-MR omawiają
zarówno możliwość użycia ustalonego IVIVC do
uchylenia wymagania przeprowadzenia badań BE
jak i to, jak IVIVC może zredukować ilość
dokumentacji dotyczącej rozpadu in vitro,
wymaganej dla wsparcia zmian preparatu lub
procesu.

2009-05-28
12
Zapewnienie maksymalnej różnicy na poziomie 20% w
przewidywanym Cmax i AUC dla wyższych i niższych
specyfikacji jest postrzegane jako optymalna sytuacja.
Wytyczne proponują, by ustalony IVIVC mógł pozwolić
na szersze specyfikacje, zależne od przewidywań
dotyczących IVIVC (tak długo, jak różnice w
przewidywanym Cmax i AUC dla niższych i wyższych
granic nie przekraczają 20%).
Wytyczne FDA IVIVC dla przemysłu przedstawiają w
zarysie, jak poziom A IVIVC może być zastosowany dla
ustawienia specyfikacji rozpadu.
Użycie modelu IVIVC, przewidziany Cmax oraz AUC są
uzyskane dla najszybszego i najwolniejszego
wskaźnika uwalniania dozwolonych przez specyfikację
rozpadu, tak że maksymalna istniejąca różnica to 20%.
ZASTOSOWANIE IVIVC DLA ROZWOJU
SPECYFIKACJI ROZPADU IN VITRO – ANALIZA
PRZYPADKU
Analiza ilustruje jak atestowany model A IVIVC
z dawki został zastosowany dla rozwoju
specyfikacji rozpadu in vitro.
Model IVIVC został rozwinięty przy użyciu
rozpadu in vitro danych próbek w 8 punktach
czasowych w ciągu 20 godzin. Ustanowiony
model najpierw miał zidentyfikować, który
punkt czasowy był krytyczny dla dokładnego
przewidywania Cmax dla preparatu
docelowego, którego dotyczyły specyfikacje.

2009-05-28
13
ANALIZA PRZYPADKU
Przez pominięcie każdego kolejnego
punktu czasowego oraz porównanie
prognozowanej wartości z wartością
uzyskaną przy użyciu pełnego profilu
rozpadu, model IVIVC szybko wskazał,
że jedynie punkt 6 godzinny był
krytycznym dla przewidywania Cmax.
ANALIZA PRZYPADKU
Model IVIVC został użyty by pokazać, że profil
stężenia i czasu dla preparatów docelowych
może być opisany przy użyciu tylko czterech
punktów czasowych. (rys. A)
2009-05-28
14
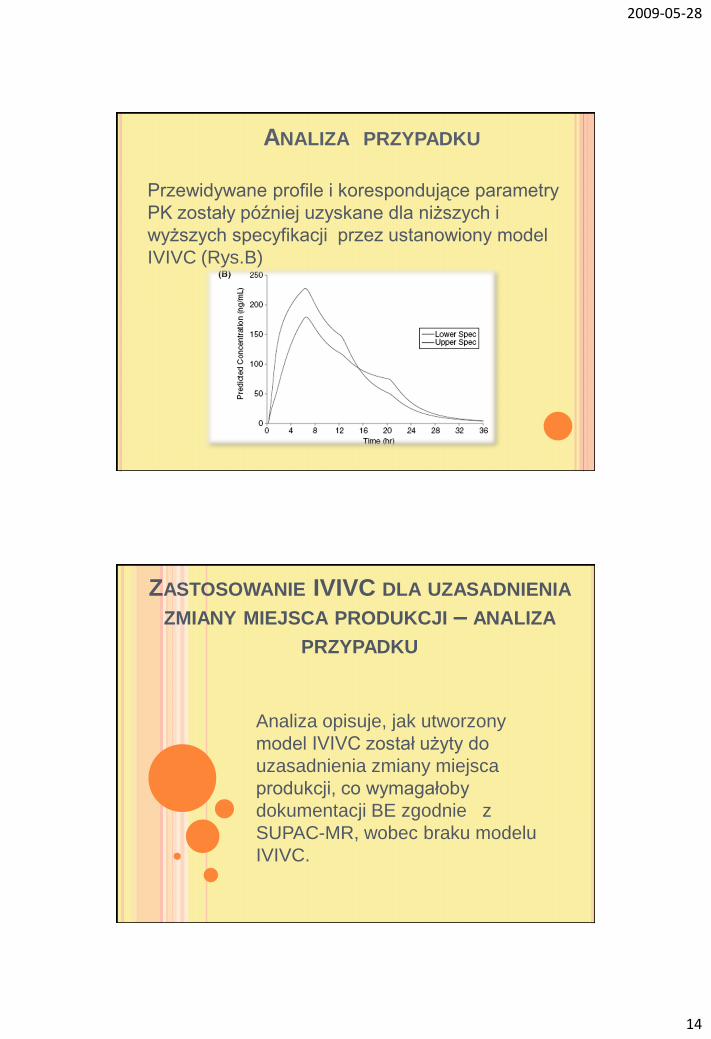
ANALIZA PRZYPADKU
Przewidywane profile i korespondujące parametry
PK zostały później uzyskane dla niższych i
wyższych specyfikacji przez ustanowiony model
IVIVC (Rys.B)
ZASTOSOWANIE IVIVC DLA UZASADNIENIA
ZMIANY MIEJSCA PRODUKCJI – ANALIZA
PRZYPADKU
Analiza opisuje, jak utworzony
model IVIVC został użyty do
uzasadnienia zmiany miejsca
produkcji, co wymagałoby
dokumentacji BE zgodnie z
SUPAC-MR, wobec braku modelu
IVIVC.
2009-05-28
15
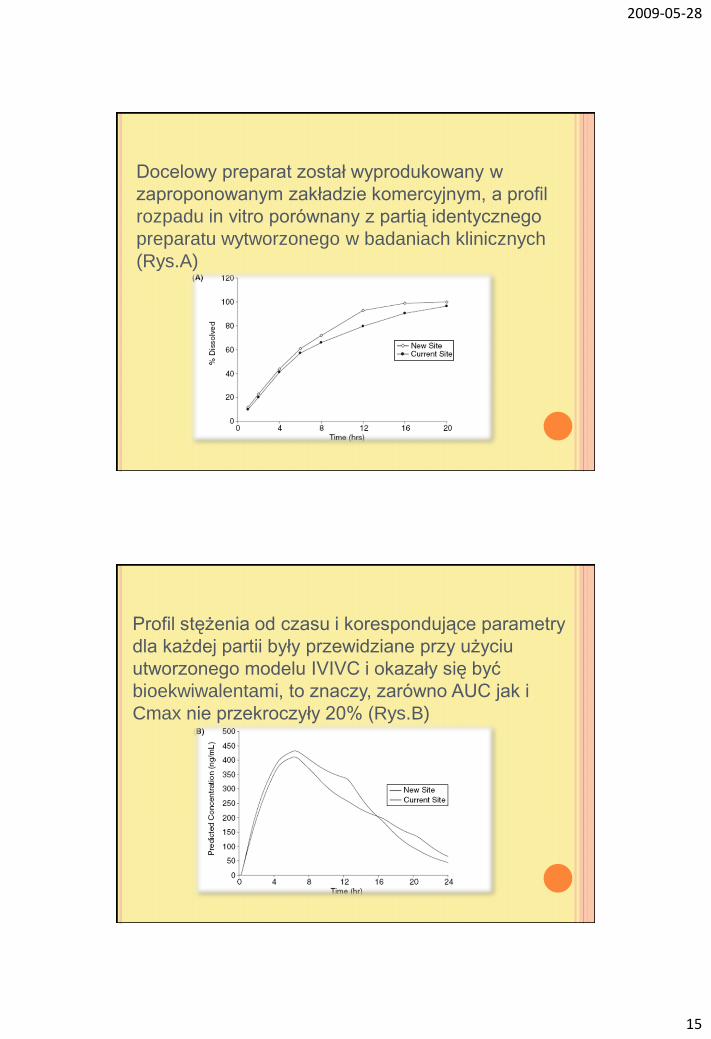
Docelowy preparat został wyprodukowany w
zaproponowanym zakładzie komercyjnym, a profil
rozpadu in vitro porównany z partią identycznego
preparatu wytworzonego w badaniach klinicznych
(Rys.A)
Profil stężenia od czasu i korespondujące parametry
dla każdej partii były przewidziane przy użyciu
utworzonego modelu IVIVC i okazały się być
bioekwiwalentami, to znaczy, zarówno AUC jak i
Cmax nie przekroczyły 20% (Rys.B)

2009-05-28
16
Obydwie analizy przypadku jasno
ilustrują zastosowanie
utworzonego modelu IVIVC
dopuszczając testy in vitro, jako
środek zastępczy badań BE, po to
żeby wpływ zmian w preparacie
lub zmian miejsca wytwarzania
mógł być szacowany szybko i
umożliwić kontynuację programu
rozwoju.
W ten sam sposób zmiany po
zatwierdzeniu mogą być poparte przy
użyciu IVIVC.
Zmiany wprowadzone w czasie
procesu przed zatwierdzeniem mogą
być również uzasadnione przy użyciu
danych dotyczących rozpadu in vitro i
utworzonego IVIVC.
Jednakże, firmy powinny wziąć pod
uwagę potencjalne ryzyko
użytkowania IVIVC, który ma dopiero
dostać akredytację od agencji
regulującej dla wsparcia takich zmian
przed jej otrzymaniem.

2009-05-28
17
KOORDYNACJA W CZASIE IVIVC W
PROGRAMIE ROZWOJU PREPARATU
Zakres preparatów ocenianych in vivo jest wystarczająco
szeroki, by umożliwić rozwój znaczącego modelu
IVIVC. Jeden z preparatów będzie reprezentować to, co
jak się sądzi spełni specyfikację produktu, ale preparaty
mające szybszy lub wolniejszy współczynnik uwalniania
też są zawarte w badaniu
Zastosowanie IVIVC dla rozwoju jakiejkolwiek
doustnej postaci leku jest zależne od tego kiedy
zostało przeprowadzone badanie zezwalające na
rozwój modelu. Koordynacja w czasie takich badań często odzwierciedla
stosunek firmy do IVIVC. W zakładach postrzegających
IVIVC, jako potężne narzędzie wspomagające rozwój
preparatu, badanie może być przeprowadzone w bardzo
wczesnej fazie programu.
IVIVC W BADANIACH
Włączenie rozwoju modelu IVIVC wcześnie do
programu preparatu umożliwia udoskonalanie i
rozszerzanie modelu w miarę zdobywania
dalszych danych.
Jeśli jeden z preparatów spełnia specyfikację
produktu, IVIVC może być użyty jako
wspomagający dalszą optymalizację. Jeśli żaden z
preparatów nie spełnia specyfikacji, IVIVC może
być użyty jako pomoc w rozwoju preparatu oraz
identyfikacji preparatów bliższych spełnienia
specyfikacji produktu.

2009-05-28
18
IVIVC W BADANIACH
Włączenie IVIVC w późnej fazie programu
rozwoju preparatu zazwyczaj wymaga
wytworzenia preparatów zaprojektowanych
tak, by były dostatecznie szybsze lub
wolniejsze niż docelowy, aby określić dla
projektowanych w przyszłości badań
znaczącej dla rozwoju korelacji IVIVC.
Inne firmy są mniej ofensywne w stosowaniu IVIVC i
mają tendencję postrzegania IVIVC jako narzędzia
wspierającego zmiany na już ustalonym docelowym
preparacie.
Takie podejście retrospektywne do IVIVC wymaga
odniesień zawieranych we wszystkich wczesnych
badaniach, by umożliwić odwrócenie skutków
dekonwolucji. Na przykład, wczesne badania
oceniające formy dawkowania ER wymagałyby
zawarcia odniesienia IR w każdym badaniu w celu
rozwoju IVIVC przy użyciu tradycyjnego podejścia
opartego na odwracaniu skutków dekonwolucji.
Omówienie programu rozwoju preparatu może
pomóc w zidentyfikowaniu odpowiednio
szerokiego zakresu preparatów, które zostały
ocenione w serii wczesnych badań i mogłyby być
włączone do pojedynczego IVIVC.

2009-05-28
19
Tak więc, mimo że IVIVC nie może
być pierwszym ani nawet drugim
celem wczesnych badań nad
rozwojem preparatu, należy
zwrócić uwagę na zaprojektowanie
takich badań umożliwiające
wykorzystanie uzyskanych wyników
do analizy IVIVC w późniejszym
etapie programu rozwoju
PODSUMOWANIE
Prawdopodobieństwo sukcesu IVIVC
i zastosowanie korelacji w
programach rozwoju doustnych form
leku może znacząco wzrosnąć przy
włączeniu strategii IVIVC do
wczesnej fazy badań.
Rola rozwoju IVIVC w doustnych postaciach leku
wzrosła w ostatnich latach, odzwierciedlając
użyteczność takich modeli, szczególnie Poziomu A
IVIVC,
- zarówno w pomocy rozwoju preparatu,
-regulacji istotnych specyfikacji rozpadu
- oraz we wspieraniu zmian przed i po zatwierdzeniu,
które w innych przypadkach wymagałyby
dokumentacji BE.

2009-05-28
20
Dziękujemy za uwagę
i owocną dyskusję ;)





























