Embed Size (px)
Citation preview

ADIS DRUG EVALUATION
Ivacaftor: A Review of Its Use in Patients with Cystic Fibrosis
Emma D. Deeks
Published online: 13 September 2013
� Springer International Publishing Switzerland 2013
Abstract Ivacaftor (KalydecoTM) is a potentiator of the
cystic fibrosis transmembrane conductance regulator
(CFTR) and is the first drug that treats an underlying cause
of cystic fibrosis to be licensed for use. Ivacaftor increases
the open probability (i.e. gating) of CFTR channels with
the G551D mutation, thus enhancing chloride transport,
and is indicated in a number of countries for the treatment
of cystic fibrosis in patients aged C6 years who carry this
mutation. This review focuses on pharmacological, clinical
efficacy and tolerability data relevant to the use of ivacaftor
in this indication. In two 48-week, double-blind, phase III
trials in patients aged C12 (STRIVE) or 6–11 (ENVISION)
years with cystic fibrosis and the G551D mutation, oral
ivacaftor 150 mg every 12 h significantly improved lung
function relative to placebo, when used in combination
with standard care. Significant improvements in pulmonary
exacerbation risk (in STRIVE) as well as bodyweight and
some aspects of health-related quality of life (both studies)
were also seen with the drug versus placebo. Moreover, the
beneficial effects of ivacaftor on parameters such as lung
function and bodyweight were maintained over up to
96 weeks of treatment in an ongoing open-label extension
of these studies. Ivacaftor was generally well tolerated,
with headache, oropharyngeal pain, upper respiratory tract
infection and nasal congestion being among the most
common adverse events. Thus, ivacaftor expands the
current treatment options for patients with cystic fibrosis
who have the G551D mutation. Its potential for use in
patients with other CFTR mutations is also of interest.
Ivacaftor in cystic fibrosis: a summary
First drug that treats an underlying cause of cystic
fibrosis to be licensed for use
Increases the open probability (i.e. gating) of cystic
fibrosis transmembrane conductance regulator
channels with the G551D mutation, thus augmenting
chloride transport
Convenient oral administration
Improves lung function and bodyweight parameters
when used in combination with standard care in adults,
adolescents and children (aged C6 years) with cystic
fibrosis and the G551D mutation
Generally well tolerated
1 Introduction
Cystic fibrosis is a complex, life-limiting, genetic disorder
that can affect multiple organs throughout the body, lead-
ing to pulmonary disease (most cystic fibrosis-related
deaths are due to pulmonary insufficiency), reproductive,
hepatic, pancreatic and gastrointestinal dysfunction and
malnutrition [1, 2]. Cystic fibrosis results from mutations in
the gene encoding the cystic fibrosis transmembrane con-
ductance regulator (CFTR), a glycoprotein present in the
apical membrane of epithelial cells, where it functions as a
chloride channel predominantly, but also regulates trans-
port of sodium (via epithelial sodium channels) as well as
various other processes [2, 3]. It is largely accepted that
The manuscript was reviewed by: F. Becq, Institut de Physiologie
et Biologie Cellulaires, Universite de Poitiers, Poitiers, France; R.S.Pettit, Riley Hospital for Children, Indiana University Health,
Indianapolis, IN, USA.
E. D. Deeks (&)
Adis, 41 Centorian Drive, Private Bag 65901, Mairangi Bay,
North Shore 0754, Auckland, New Zealand
e-mail: [email protected]
Drugs (2013) 73:1595–1604
DOI 10.1007/s40265-013-0115-2

defective ion transport caused by CFTR dysfunction leads
to depletion of airway surface liquid in the lungs of patients
with cystic fibrosis, which impairs ciliary function, result-
ing in mucus obstruction of the airways and consequently
infection and inflammation [4].
Cystic fibrosis is incurable at present [5]. Therapy can
involve a variety of different medications, including anti-
biotics (e.g. tobramycin) to treat lung infections, the DNase
dornase alfa to clear lung mucus, hypertonic saline to
improve mucociliary clearance, as well as anti-inflamma-
tory agents, bronchodilators and pancreatic enzymes [6, 7].
These therapies treat the downstream consequences of
CFTR dysfunction, rather than the underlying abnormality,
and although their aggressive use has helped to increase the
life expectancy of patients with cystic fibrosis [2], most
deaths still occur in early adulthood [8].
Understanding of the molecular biology of the CFTR
protein has increased over the last two decades and[1,500
CFTR mutations have now been identified [3]. Mutations
can be classified on the basis of their functional conse-
quences, which include CFTR protein that is truncated and
fails to reach the cell surface (class I) [e.g. R1162X]; is
misfolded, improperly processed and defective, little of
which reaches the cell surface (class II) [e.g. F508del]; is
unable to open and transport chloride properly (class III)
[e.g. G551D] or has reduced chloride conductance due to
channel narrowing (class IV) [e.g. R117H] but reaches the
cell surface; or transports chloride effectively but reaches
the cell surface in reduced amounts due to defective tran-
script splicing (class V) [e.g. 3120?1G?A] [1, 3]. As a
result of this knowledge, therapies specifically targeting
known CFTR defects have been a focus of drug develop-
ment for cystic fibrosis in recent years.
Ivacaftor (KalydecoTM) is the first drug to treat an
underlying cause of cystic fibrosis to be licensed for use in
the EU [9] and USA [10]. The drug potentiates the open
probability (i.e. gating) of the CFTR channel, thus
enhancing its transport of chloride, and is indicated for the
treatment of patients with cystic fibrosis aged C6 years
who carry the CFTR gating mutation G551D [9, 10]. This
narrative review focuses on pharmacological, clinical
efficacy and tolerability data relevant to the use of ivacaftor
in this indication.
2 Pharmacodynamic Properties
This section provides an overview of the pharmacody-
namic properties of ivacaftor. Data are from in vitro [11–
16] and ex vivo [17] studies, as well as from double-blind
[18–21] or single-blind [22], placebo-controlled trials in
patients with cystic fibrosis and the G551D mutation
(n = 8–167). Some data are available from the US
manufacturer’s prescribing information [10], the European
public assessment report (EPAR) [5] or abstracts [14, 17,
18, 22, 23]; further data from some clinical trials [19–21]
are discussed in Sect. 4.
2.1 In Vitro Studies
Ivacaftor (Fig. 1) is a CFTR potentiator that acts by
increasing the open probability of the CFTR channel, thus
enhancing its transport of chloride [10]. Ivacaftor binds
selectively to, and acts directly on, CFTR [5, 11, 12],
displaying little or no measurable activity at other sodium,
calcium or potassium channels tested, with the exception of
CaV1.2 and KV1.5, which it inhibited with moderate
potency (half maximal inhibitory concentrations of 1.3 and
3.4 lmol/L, respectively) [5]. Although CaV1.2 and KV1.5
are key cardiac ion channels, no clinically significant QT
interval prolongation was seen with ivacaftor in healthy
volunteers (Sect. 2.2).
In vitro, ivacaftor increased transepithelial current (a
measure of chloride secretion) by approximately fourfold
in rodent cells expressing human G551D CFTR (half
maximal effective concentration [EC50] 100 nmol/L) and
by approximately tenfold in human bronchial epithelial
(HBE) cells isolated from a cystic fibrosis patient with both
the G551D and F508del mutations (EC50 236 nmol/L) [10,
12]. This property of ivacaftor was shown in the rodent
cells to be dependent on prior treatment with a cyclic
adenosine monophosphate (cAMP) agonist [12], which is
consistent with the knowledge that phosphorylation of
CFTR by cAMP-dependent protein kinase (PKA), together
with adenosine triphosphate (ATP) binding, is needed for
the channel to be activated.
In membrane patches excised from recombinant rodent
cells exposed to both PKA and ATP, the open probability
of human G551D CFTR and wildtype CFTR increased
approximately sixfold and twofold at ivacaftor concentra-
tions of 10 and 1 lmol/L, respectively [12]. However,
recent in vitro data suggest ivacaftor may potentiate the
opening of phosphorylated CFTR in an ATP-independent
Fig. 1 Chemical structure of ivacaftor
1596 E. D. Deeks

manner [11, 15]. Such ATP-independent potentiation may
help to explain the drug’s benefit in cystic fibrosis patients
with the G551D-CFTR mutation (see Sect. 4), which is
known to cause disruption of the CFTR binding site usually
responsible for ATP-dependent gating [11, 15].
Of the two main circulating ivacaftor metabolites, only
one (M1; see Sect. 3) is considered to be pharmacologi-
cally active, potentiating CFTR-mediated chloride trans-
port with a potency approximately sixfold lower than that
of the parent drug in HBE cells expressing G551D/F508del
CFTR [5].
Ivacaftor may potentiate CFTR channels with gating
mutations other than G551D, according to additional
in vitro data [13]. For example, in rodent cells expressing
G551D/S, G178R, S549N/R, G970R, G1244E, S1251N,
S1255P or G1349D CFTR, ivacaftor increased channel
open probability from B5 % of normal at baseline to
30–118 % of normal and increased chloride transport C16-
fold (EC50 124–594 nmol/L). Further in vitro data suggest
that other CFTR proteins with residual function may also
be potentiated by ivacaftor, including those with mutations
that affect conductance (e.g. R117H, D110H), mildly affect
CFTR processing (e.g. E56K, P67L) or have uncharacter-
ized effects (e.g. D110E, S1235R) [5, 16].
The in vitro effect of ivacaftor on CFTR mutant proteins
with minimal chloride transport, such as those with muta-
tions affecting CFTR synthesis (e.g. G542X) [5] or
severely affecting CFTR conductance (e.g. R334W) or
processing (e.g. F508del [13]) [5, 16], was minimal [5, 13]
or less than its effect on CFTR proteins with mild con-
ductance or processing defects [16]. In another in vitro
study, transepithelial current was increased with ivacaftor
in HBE cultures from three of six patients homozygous for
the F508del mutation, although this effect was of lesser
magnitude than in HBE cells carrying both the F508del and
G551D mutation [12]. Notably, the affinity of the drug for
F508del CFTR may be higher than for G551D CFTR, as
the EC50 of ivacaftor was approximately tenfold lower in
homozygous than in heterozygous cells (22 vs. 236 nmol/
L) [12].
By potentiating CFTR-mediated chloride secretion,
ivacaftor may rescue the function of airway epithelial cells.
For example, the excessive absorption of sodium in HBE
cells expressing G551D/F508del CFTR was reduced with
ivacaftor, with a resultant increase in both apical surface
fluid and the beat frequency of cilia [12]. Augmentation of
cilia beat frequency also occurred with ivacaftor in primary
cultures of human sinonasal epithelia [14].
2.2 Studies in Humans
Improvements in CFTR function, as measured by the
concentration of chloride in sweat, were seen with oral
ivacaftor 150 mg every 12 h in patients with cystic fibrosis
and the G551D mutation. Ivacaftor significantly reduced
sweat chloride concentration relative to placebo through
week 24 of treatment in adults and adolescents aged
C12 years (mean adjusted change from baseline of -48.7
vs. -0.8 mmol/L; p \ 0.0001) [20] and children aged
6–11 years (-55.5 vs. -1.2 mmol/L; p \ 0.0001) [21] in
phase III trials, with this benefit being sustained through
48 weeks’ therapy in these populations [20, 21]; overall
mean values at baseline were 100 and &104 mmol/L in the
respective trials. Sweat chloride concentration was also
significantly (p B 0.02) reduced with this dosage of iva-
caftor versus placebo in phase II studies of up to 28 days’
duration in patients aged C18 [19] or C6 [18] years,
although the change from baseline in nasal potential dif-
ference (another measure of CFTR function) did not sig-
nificantly differ between the treatment groups [19].
Notably, reductions in sweat chloride did not correlate
directly with improvements in lung function (as measured
by forced expiratory volume in 1 s [FEV1]) in phase III
trials [10].
Treatment with ivacaftor 150 mg every 12 h for 28 days
generally led to significant (p \ 0.01) reductions in total
ventilation defects (assessed by hyperpolarized gas mag-
netic resonance imaging) [22] and ventilation inhomoge-
neity (measured by lung clearance index) [18] in patients
with cystic fibrosis and the G551D mutation in placebo-
controlled, phase II trials. Improvements in percent pre-
dicted FEV1 were also seen with ivacaftor in these studies
[18, 22]; at baseline, FEV1 was [90 % of predicted (i.e.
lung disease was mild) [18] or [40 % of predicted [22].
Neutrophil activity is dysregulated in cystic fibrosis and,
as a result, pathogens are not eliminated effectively, lead-
ing to lung infections [17, 24]. In an ex vivo study, the
impaired degranulation of secondary and tertiary neutro-
phil granules in patients with cystic fibrosis and the G551D
mutation was corrected following 1 year of treatment with
ivacaftor (dosage not specified), with levels of degranula-
tion significantly (p \ 0.05) increasing (by 130 %) to reach
those seen in healthy controls [17].
The Fridericia-corrected QT interval was not prolonged
to any clinically relevant extent with therapeutic (150 mg
every 12 h) or supratherapeutic (450 mg every 12 h) dos-
ages of ivacaftor relative to placebo over 5 days in a
moxifloxacin-controlled crossover study in 72 healthy
subjects [23].
3 Pharmacokinetic Properties
This section provides an overview of the pharmacokinetic
properties of ivacaftor. Data were obtained predominantly
from the US [10] and EU [9] prescribing information; some
Ivacaftor: A Review 1597

data are available from the EPAR [5] or as abstracts [25–
27].
Ivacaftor displays generally linear pharmacokinetics
with respect to time and across doses of 25–250 mg [9].
The mean maximum plasma concentration (Cmax) of the
drug (768 ng/mL) was reached within &4 h and the mean
area under the plasma concentration-time curve (AUC) was
10,600 ng � h/mL, following administration of a single oral
150 mg dose in healthy volunteers in a fed state [9, 10].
Steady-state ivacaftor plasma concentrations were attained
after 3–5 days of administering the drug every 12 h; the
accumulation ratio was 2.2–2.9 [9, 10]. The pharmacoki-
netic profile of ivacaftor in patients with cystic fibrosis is
similar to that in healthy adults [9, 10]; ivacaftor exposure
parameters in phase II and III trials are summarized in
Table 1 [9].
Administration of ivacaftor with fat-containing food
increased exposure to the drug approximately twofold to
fourfold; ivacaftor should be taken with fat-containing
foods, such as those prepared with oil or butter or con-
taining cheese, whole milk, eggs, nuts or meat [9, 10].
Exposure to ivacaftor appears to be related to FEV1 [9,
26] and sweat chloride [26] responses in patients with
cystic fibrosis and the G551D mutation, according to
population pharmacokinetic/pharmacodynamic modelling
of data from phase IIa and III trials. The concentration
leading to 90 % maximal response (EC90) was 405 ng/mL,
where specified, with the median minimal plasma drug
concentration at EC90 being the pharmacokinetic parameter
that was chosen as a target for efficacy [9].
Ivacaftor does not bind to human erythrocytes and is
highly plasma protein bound (&99 %), with albumin and
a1-acid glycoprotein contributing the most to binding [9,
10]. The mean apparent volume of distribution of ivacaftor
was 353 L in healthy volunteers who received oral iva-
caftor 150 mg every 12 h in the fed state for 7 days [9, 10].
Metabolism of ivacaftor in humans is extensive and
involves oxidation, reduction, dehydration, as well as sul-
fate and glucuronide conjugation [9, 10, 25]. Metabolism
occurs predominantly via the cytochrome P450 (CYP)
enzyme CYP3A4, with CYP3A5 probably contributing to
some extent [5], producing two major metabolites, M1 and
M6 [9, 10]. Of these metabolites, only M1 is considered to
be pharmacologically active, although is less potent than
the parent drug [9, 10] (see Sect. 2.1).
Elimination of ivacaftor after oral administration of a
radiolabelled dose in healthy volunteers was predominantly
via the faeces (87.8 %), with only 6.6 % being excreted via
the urine [25]. Of the drug that is eliminated, most
(&65 %) is the M1 or M6 metabolite; very little of an
administered dose is excreted unchanged (2.5 % via the
faeces; \0.01 % via the urine) [9, 10, 25]. The mean
apparent clearance of ivacaftor at steady state was 17.3 L/h
in healthy subjects receiving the 150 mg dose [9]. The
apparent terminal elimination half-life of the drug after a
single dose is &12 h in the fed state [9].
3.1 Special Patient Groups
The rate of ivacaftor absorption in children is similar to
that in adults, although the predicted total body clearance
of the drug in children is lower (e.g. 10 L/h in a 20 kg
child vs. 18.9 L/h in a 70 kg adult) according to a pop-
ulation pharmacokinetic analysis, which may explain why
exposure to ivacaftor was higher in children than in adults
in phase II and III studies (Table 1) [9]. The pharmaco-
kinetic profile of ivacaftor is consistent among adoles-
cents (aged [12 years) and adults [5]. The dosage of
ivacaftor does not require adjustment on the basis of
gender [9, 10].
In patients with moderate hepatic impairment (Child
Pugh class B), exposure to ivacaftor (as measured by the
AUC from time zero to infinity) was increased approxi-
mately twofold relative to healthy volunteers following a
single 150 mg dose, although the Cmax was similar; thus, a
reduction in dosage to 150 mg once daily is recommended
in these patients [9, 10]. What impact severe or mild
hepatic impairment may have on ivacaftor pharmacoki-
netics has not yet been studied. Exposure to ivacaftor is
expected to be increased substantially more by severe than
moderate impairment and, as a result, the drug is not rec-
ommended in patients with severe hepatic impairment in
the EU [9]; however, if the benefits outweigh the risks,
ivacaftor may be used (with caution [10]) at a reduced
dosage (150 mg once daily or less frequently in the USA
[10]; starting dosage of 150 mg every other day in the EU
[9]). Dosage adjustment is not considered necessary in
patients with mild hepatic impairment [9, 10].
The pharmacokinetics of ivacaftor have not been
studied in patients with renal impairment, although as
renal excretion of the drug is minimal, no dosage
adjustments are required in those with mild or moderate
impairment; however, ivacaftor should be used with
caution in patients with severe renal impairment (creati-
nine clearance B30 mL/min [B1.8 L/h]) or end-stage
renal disease [9, 10].
Table 1 Exposure parameters for ivacaftor 150 mg every 12 h;
values are means based on data from phase II and III trials [9]
Population Cmin (ng/mL) AUC (ng � h/mL)
Children (aged 6–11 years) 1,180 18,200
Adolescents (aged 12–17 years) 556 8,536
Adults 774 9,508
AUC area under the plasma drug concentration-time curve, Cmin
minimum plasma drug concentration
1598 E. D. Deeks

3.2 Potential Drug Interactions
Exposure to ivacaftor may be increased upon coadminis-
tration with agents that inhibit CYP3A enzymes either
strongly (e.g. ketoconazole, itraconazole, telithromycin,
clarithromycin) or moderately (e.g. fluconazole, erythro-
mycin) [9, 10, 27]; thus, the dosage of ivacaftor may need to
be reduced [9, 10]. Grapefruit juice may also increase iva-
caftor exposure via inhibition of CYP3A; grapefruit- and
Seville orange-containing foods should be avoided [9, 10].
Coadministering ivacaftor with agents that strongly
induce CYP3A (e.g. rifampin [rifampicin], rifabutin, car-
bamazepine, phenobarbital, phenytoin, hypericum [St
John’s Wort]) may reduce ivacaftor exposure (and thus
efficacy) [9, 10, 27], and is therefore not recommended [9,
10]. Exposure to ivacaftor may also be reduced by agents
that induce CYP3A weakly to moderately (e.g. dexa-
methasone, high-dose prednisone) [9].
Both CYP3A and permeability glycoprotein (p-gp) may
be inhibited by ivacaftor and its active metabolite M1 [9,
10]. Consequently, systemic exposure to drugs that are
CYP3A and/or p-gp substrates may be increased upon
coadministration with ivacaftor, which may increase/pro-
long not only their efficacy but also the adverse events
associated with their use. As a result, coadministration of
ivacaftor with midazolam, diazepam, alprazolam or tria-
zolam requires caution and monitoring for adverse events,
with similar recommendations applying to other substrates
of CYP3A and/or p-gp, including digoxin, tacrolimus and
cyclosporin [9, 10].
CYP2C9 may be inhibited by ivacaftor, necessitating
monitoring of the international normalized ratio if the drug
is coadministered with the CYP2C9 substrate warfarin [9,
10]. However, ivacaftor did not significantly affect expo-
sure to rosiglitazone (a CYP2C8 substrate), desipramine (a
CYP2D6 substrate) or an estrogen/progesterone oral con-
traceptive upon coadministration; thus, dosage adjustment
is not necessary for these [10] or other [9] CYP2C8 or
CYP2D6 substrates or for oral contraceptives [9, 10].
4 Therapeutic Efficacy
The potential for oral ivacaftor to be used in the treatment
of patients with cystic fibrosis who have a G551D mutation
in the CFTR gene was evaluated in a randomized, double-
blind, placebo-controlled, dose-ranging (25–250 mg every
12 h) trial (n = 39) [19]. On the basis of this multicentre
phase II study, an ivacaftor dosage of 150 mg every 12 h
was selected for subsequent trials.
This section focuses on the findings of two randomized,
double-blind, placebo-controlled trials, known as STRIVE
(n = 167 randomized) [20, 28] and ENVISION (n = 52)
[21, 29]. These multicentre, phase III studies were con-
ducted to evaluate the clinical efficacy of ivacaftor 150 mg
every 12 h in treating cystic fibrosis when used in addition
to existing therapy (the exception being inhaled hypertonic
saline) in adults and adolescents (aged C12 years; Sect.
4.1) [20, 28] and children (aged 6–11 years; Sect. 4.2) [21,
29] with the G551D mutation.
Patients eligible for these trials had confirmed cystic
fibrosis and the G551D mutation in at least one CFTR
allele [28, 29] and were required to have an FEV1 40–90 %
of predicted in STRIVE [28] and 40–105 % of predicted in
ENVISION [29]. Among the exclusion criteria of both
studies were pulmonary exacerbation, acute respiratory
infection, Mycobacterium abscessus, Burkholderia ceno-
cepacia or Burkholderia dolosa in sputum and changes in
pulmonary disease therapy in the last 4 weeks [10, 28, 29].
The primary endpoint of each trial was the absolute change
from baseline in percent predicted FEV1 through week 24
of treatment. Where specified, efficacy endpoint data were
adjusted for baseline parameters such as age, percent pre-
dicted FEV1, sweat chloride and/or Cystic Fibrosis Ques-
tionnaire-Revised (CFQ-R) domain score [28, 29].
Patients who completed STRIVE or ENVISION were
eligible to receive ivacaftor 150 mg every 12 h in addition
to their existing therapy (inhaled hypertonic saline per-
mitted [9]) in an open-label extension study, known as
PERSIST [30] (144 and 48 patients enrolled; the percent
predicted FEV1 was 29–127 % at extension baseline [9]);
data from a pre-specified interim analysis of PERSIST are
available. Some data in this section were obtained from
abstracts [30, 31] or the EU prescribing information [9].
4.1 In Adults and Adolescents
Most patients participating in STRIVE were aged
C18 years (78 % of patients) [range 12–53 years], had an
FEV1 \70 % predicted (58 % of patients) and had the
F508del mutation in the second CFTR allele (75.8 % of
patients); one patient randomized to placebo was found to
be homozygous for the F508del CFTR allele upon confir-
matory genotyping, although data from the patient were
still included [9, 20]. Of note, certain medicinal products,
including dornase alfa, tobramycin, salbutamol (albuterol)
and salmeterol/fluticasone, were being used by 8–13 %
fewer patients in the ivacaftor than in the placebo group at
the start of the study [9].
Oral ivacaftor 150 mg every 12 h improved lung func-
tion when used in combination with standard care in adults
and adolescents with cystic fibrosis and the G551D CFTR
mutation. Relative to placebo, ivacaftor significantly
increased the percent predicted FEV1 from baseline
through week 24 of treatment (primary endpoint), and
sustained this benefit through week 48 (Table 2) [20].
Ivacaftor: A Review 1599
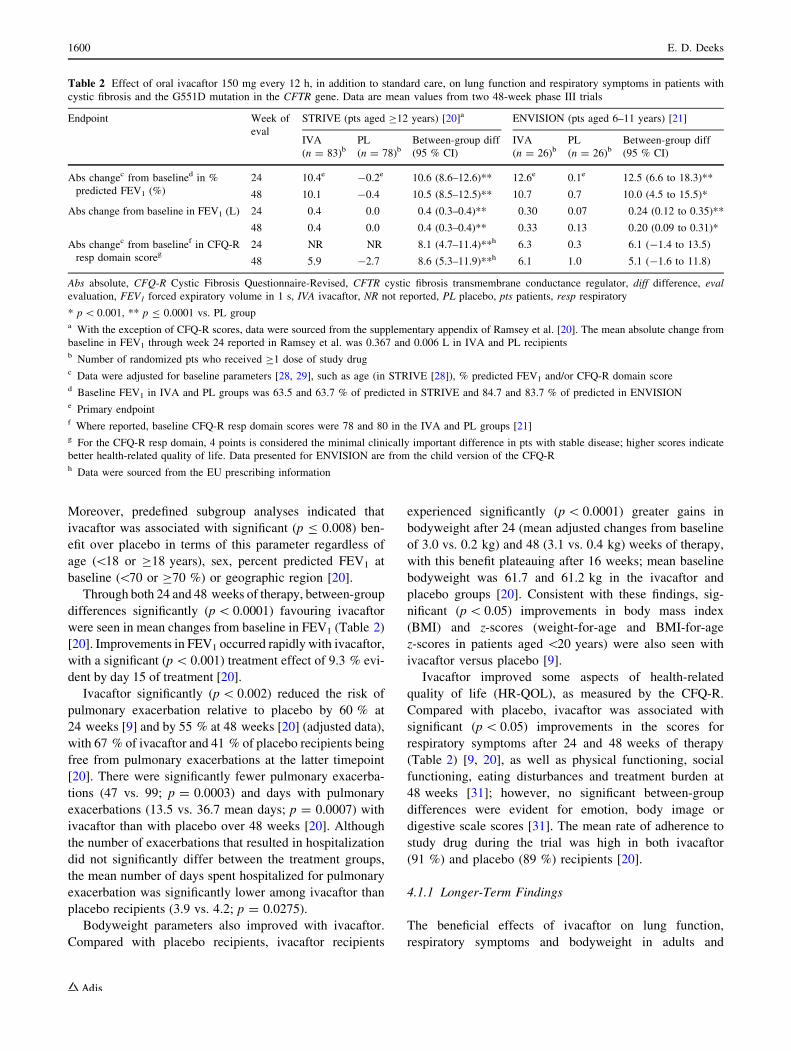
Moreover, predefined subgroup analyses indicated that
ivacaftor was associated with significant (p B 0.008) ben-
efit over placebo in terms of this parameter regardless of
age (\18 or C18 years), sex, percent predicted FEV1 at
baseline (\70 or C70 %) or geographic region [20].
Through both 24 and 48 weeks of therapy, between-group
differences significantly (p \ 0.0001) favouring ivacaftor
were seen in mean changes from baseline in FEV1 (Table 2)
[20]. Improvements in FEV1 occurred rapidly with ivacaftor,
with a significant (p \ 0.001) treatment effect of 9.3 % evi-
dent by day 15 of treatment [20].
Ivacaftor significantly (p \ 0.002) reduced the risk of
pulmonary exacerbation relative to placebo by 60 % at
24 weeks [9] and by 55 % at 48 weeks [20] (adjusted data),
with 67 % of ivacaftor and 41 % of placebo recipients being
free from pulmonary exacerbations at the latter timepoint
[20]. There were significantly fewer pulmonary exacerba-
tions (47 vs. 99; p = 0.0003) and days with pulmonary
exacerbations (13.5 vs. 36.7 mean days; p = 0.0007) with
ivacaftor than with placebo over 48 weeks [20]. Although
the number of exacerbations that resulted in hospitalization
did not significantly differ between the treatment groups,
the mean number of days spent hospitalized for pulmonary
exacerbation was significantly lower among ivacaftor than
placebo recipients (3.9 vs. 4.2; p = 0.0275).
Bodyweight parameters also improved with ivacaftor.
Compared with placebo recipients, ivacaftor recipients
experienced significantly (p \ 0.0001) greater gains in
bodyweight after 24 (mean adjusted changes from baseline
of 3.0 vs. 0.2 kg) and 48 (3.1 vs. 0.4 kg) weeks of therapy,
with this benefit plateauing after 16 weeks; mean baseline
bodyweight was 61.7 and 61.2 kg in the ivacaftor and
placebo groups [20]. Consistent with these findings, sig-
nificant (p \ 0.05) improvements in body mass index
(BMI) and z-scores (weight-for-age and BMI-for-age
z-scores in patients aged \20 years) were also seen with
ivacaftor versus placebo [9].
Ivacaftor improved some aspects of health-related
quality of life (HR-QOL), as measured by the CFQ-R.
Compared with placebo, ivacaftor was associated with
significant (p \ 0.05) improvements in the scores for
respiratory symptoms after 24 and 48 weeks of therapy
(Table 2) [9, 20], as well as physical functioning, social
functioning, eating disturbances and treatment burden at
48 weeks [31]; however, no significant between-group
differences were evident for emotion, body image or
digestive scale scores [31]. The mean rate of adherence to
study drug during the trial was high in both ivacaftor
(91 %) and placebo (89 %) recipients [20].
4.1.1 Longer-Term Findings
The beneficial effects of ivacaftor on lung function,
respiratory symptoms and bodyweight in adults and
Table 2 Effect of oral ivacaftor 150 mg every 12 h, in addition to standard care, on lung function and respiratory symptoms in patients with
cystic fibrosis and the G551D mutation in the CFTR gene. Data are mean values from two 48-week phase III trials
Endpoint Week of
eval
STRIVE (pts aged C12 years) [20]a ENVISION (pts aged 6–11 years) [21]
IVA
(n = 83)bPL
(n = 78)bBetween-group diff
(95 % CI)
IVA
(n = 26)bPL
(n = 26)bBetween-group diff
(95 % CI)
Abs changec from baselined in %
predicted FEV1 (%)
24 10.4e -0.2e 10.6 (8.6–12.6)** 12.6e 0.1e 12.5 (6.6 to 18.3)**
48 10.1 -0.4 10.5 (8.5–12.5)** 10.7 0.7 10.0 (4.5 to 15.5)*
Abs change from baseline in FEV1 (L) 24 0.4 0.0 0.4 (0.3–0.4)** 0.30 0.07 0.24 (0.12 to 0.35)**
48 0.4 0.0 0.4 (0.3–0.4)** 0.33 0.13 0.20 (0.09 to 0.31)*
Abs changec from baselinef in CFQ-R
resp domain scoreg24 NR NR 8.1 (4.7–11.4)**h 6.3 0.3 6.1 (-1.4 to 13.5)
48 5.9 -2.7 8.6 (5.3–11.9)**h 6.1 1.0 5.1 (-1.6 to 11.8)
Abs absolute, CFQ-R Cystic Fibrosis Questionnaire-Revised, CFTR cystic fibrosis transmembrane conductance regulator, diff difference, eval
evaluation, FEV1 forced expiratory volume in 1 s, IVA ivacaftor, NR not reported, PL placebo, pts patients, resp respiratory
* p \ 0.001, ** p B 0.0001 vs. PL groupa With the exception of CFQ-R scores, data were sourced from the supplementary appendix of Ramsey et al. [20]. The mean absolute change from
baseline in FEV1 through week 24 reported in Ramsey et al. was 0.367 and 0.006 L in IVA and PL recipientsb Number of randomized pts who received C1 dose of study drugc Data were adjusted for baseline parameters [28, 29], such as age (in STRIVE [28]), % predicted FEV1 and/or CFQ-R domain scored Baseline FEV1 in IVA and PL groups was 63.5 and 63.7 % of predicted in STRIVE and 84.7 and 83.7 % of predicted in ENVISIONe Primary endpointf Where reported, baseline CFQ-R resp domain scores were 78 and 80 in the IVA and PL groups [21]g For the CFQ-R resp domain, 4 points is considered the minimal clinically important difference in pts with stable disease; higher scores indicate
better health-related quality of life. Data presented for ENVISION are from the child version of the CFQ-Rh Data were sourced from the EU prescribing information
1600 E. D. Deeks

adolescents were maintained during longer-term treatment,
according to a 48-week interim analysis of the extension
study PERSIST [30]. For example, among patients who
received 48 weeks’ treatment with ivacaftor in STRIVE
and continued to receive the drug in PERSIST (n = 73),
the mean absolute change from STRIVE baseline in per-
cent predicted FEV1 was 9.5 % after a total of 96 weeks’
therapy versus 9.4 % at the start of the extension [9].
Patients who had received placebo in STRIVE and were
switched to ivacaftor for the extension (n = 63) had a
corresponding improvement in this parameter of 9.4 %
48 weeks after switching [9].
4.2 In Children
At baseline, patients participating in ENVISION had a
mean age of 9 years, most had the F508del mutation in the
second CFTR allele (80.8 %) and 15 % of those in the
ivacaftor group versus 31 % of those in the placebo group
had an FEV1 \70 % of predicted, although the between-
group difference was not significant [9, 21].
Oral ivacaftor 150 mg every 12 h, in combination with
standard care, was effective in improving lung function in
children aged 6–11 years with cystic fibrosis and the
G551D CFTR mutation, according to primary endpoint
analysis. Percent predicted FEV1 increased from baseline
to a significantly greater extent with ivacaftor than with
placebo through week 24 of treatment (primary endpoint;
Table 2), with the between-group difference favouring
ivacaftor from day 15 of therapy and remaining significant
through week 48 (Table 2) [21]. Predefined subgroup
analyses suggested this measure was significantly
improved with ivacaftor relative to placebo among patients
who had an FEV1 B90 % of predicted at baseline, were
female or were participating at a European centre [21].
Ivacaftor also provided benefit over placebo in terms of
other lung function endpoints, including the mean absolute
change from baseline in FEV1 through 24 and 48 weeks’
treatment (Table 2) [21]. Pulmonary exacerbations, as
defined in the trial protocol, were uncommon (four
occurred with ivacaftor, three with placebo) [21].
Ivacaftor had beneficial effects on bodyweight parame-
ters. Significant bodyweight gain occurred with ivacaftor
relative to placebo after 24 weeks of therapy (mean adjus-
ted changes from baseline of 3.7 vs. 1.8 kg; p = 0.0004)
and was maintained through week 48 (treatment difference
of 2.8 kg; p = 0.0002); mean baseline bodyweight was
31.8 and 30.0 kg in the respective groups [21]. Similarly,
mean changes from baseline in BMI and both BMI-for-age
and weight-for-age z-scores also significantly (p \ 0.001)
favoured ivacaftor over placebo at 48 weeks [9].
Unlike placebo recipients, ivacaftor recipients had
clinically relevant improvements from baseline in
respiratory symptoms, as measured by the child version of
the CFQ-R respiratory domain, although the difference
between the treatment groups was not statistically signifi-
cant through 24 or 48 weeks’ therapy (Table 2) [21]. There
were also no significant between-group differences for
other CFQ-R domain scores [31]. However, in the parent/
caregiver CFQ-R, scores for respiratory symptoms
(through week 24) [21] as well as body image and body-
weight (through week 48) [31] significantly (p = 0.033
where reported [21]) favoured ivacaftor over placebo.
Compliance with study drug was high (mean rate C94 %)
in both treatment groups [21].
4.2.1 Longer-Term Findings
Ivacaftor provided beneficial effects on parameters such as
lung function and bodyweight for up to 72 weeks,
according to an interim analysis of the extension study
PERSIST [30]. For instance, among patients who received
ivacaftor for 48 weeks in ENVISION and received the drug
for a further 24 weeks in PERSIST (n = 26), the mean
absolute change from ENVISION baseline in percent pre-
dicted FEV1 was 10.1 % after a total of 72 weeks’ therapy
versus 10.2 % at the start of the extension [9]. Patients who
had received placebo in ENVISION experienced a corre-
sponding mean improvement in this measure of 8.1 %
24 weeks after switching to ivacaftor for the extension
(n = 22) [9].
5 Tolerability
Tolerability data concerning the use of oral ivacaftor in
patients aged C6 years with cystic fibrosis who have the
G551D CFTR mutation are available from the clinical
studies discussed in Sect. 4. Some data, including a pooled
analysis of the two pivotal phase III trials that compared
ivacaftor 150 mg every 12 h with placebo in adults and
adolescents (STRIVE) or children (ENVISION), are
available from the US [10] and EU [9] prescribing
information.
Treatment with ivacaftor 150 mg every 12 h for up to
48 weeks was generally well tolerated in adults [20],
adolescents [20] and children [21] with cystic fibrosis and
the G551D CFTR mutation participating in the STRIVE
[20] and ENVISION [21] trials. In each of these studies,
adverse events occurred in the vast majority (C96 %) of
ivacaftor and placebo recipients, although the incidence of
those considered to be serious was numerically lower with
ivacaftor than with placebo (19 vs. 23 % [21] and 24 vs.
42 % [20]).
Serious adverse events reported (more than once [21])
with ivacaftor or placebo included pulmonary exacerbation
Ivacaftor: A Review 1601
(two vs. three patients) and productive cough (one patient
per group) in ENVISION [21] and pulmonary exacerba-
tions (13 vs. 33 % of patients), haemoptysis (1 vs. 5 %)
and hypoglycaemia (2 vs. 0 %) in STRIVE [20]. Among
the two ivacaftor recipients who had hypoglycaemia, one
was receiving insulin for diabetes mellitus and the other
had had symptoms suggestive of hypoglycaemia previously
[20]. No patients died in either study [20, 21].
In the trial in adults and adolescents, adverse events led
to interruption of study drug in 13 % of ivacaftor and 6 %
of placebo recipients and discontinuation of study drug in 1
and 5 %, with the reason for discontinuation in the iva-
caftor group being increased levels of hepatic enzymes
[20]. In ENVISION, threefold fewer ivacaftor than placebo
recipients had their study drug interrupted because of
adverse events (3.8 vs. 11.5 %) and no children receiving
ivacaftor discontinued therapy permanently because of
adverse events (vs. 3.8 % of placebo recipients) [21].
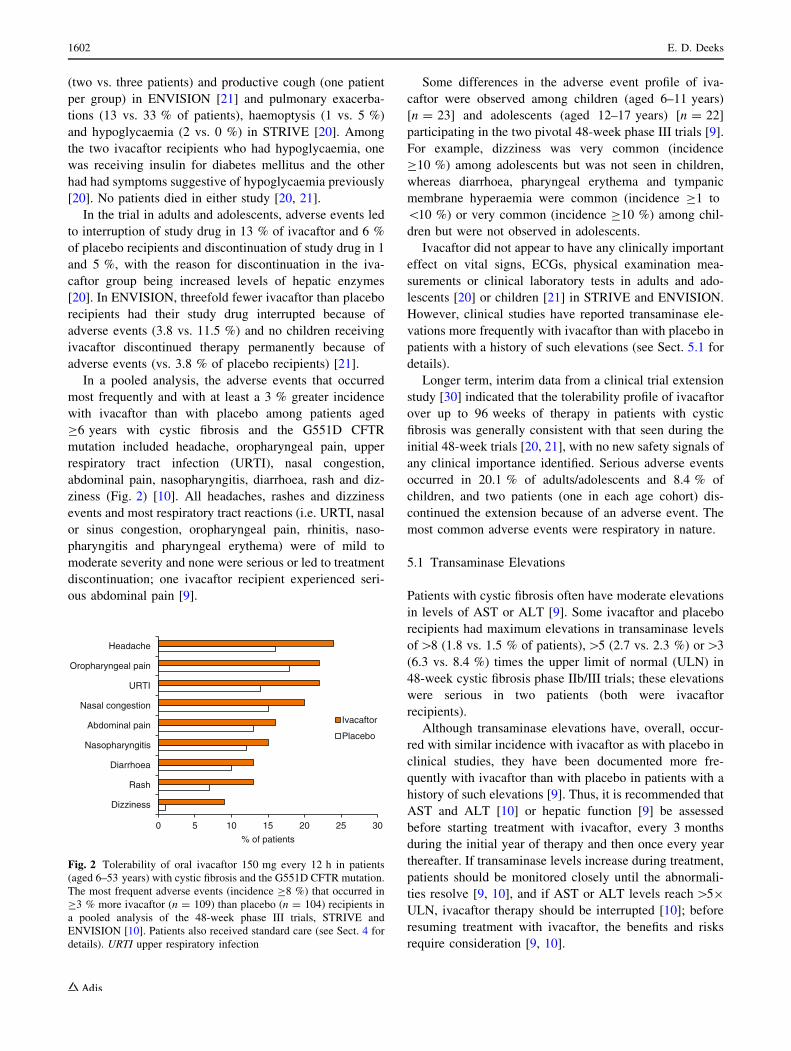
In a pooled analysis, the adverse events that occurred
most frequently and with at least a 3 % greater incidence
with ivacaftor than with placebo among patients aged
C6 years with cystic fibrosis and the G551D CFTR
mutation included headache, oropharyngeal pain, upper
respiratory tract infection (URTI), nasal congestion,
abdominal pain, nasopharyngitis, diarrhoea, rash and diz-
ziness (Fig. 2) [10]. All headaches, rashes and dizziness
events and most respiratory tract reactions (i.e. URTI, nasal
or sinus congestion, oropharyngeal pain, rhinitis, naso-
pharyngitis and pharyngeal erythema) were of mild to
moderate severity and none were serious or led to treatment
discontinuation; one ivacaftor recipient experienced seri-
ous abdominal pain [9].
Some differences in the adverse event profile of iva-
caftor were observed among children (aged 6–11 years)
[n = 23] and adolescents (aged 12–17 years) [n = 22]
participating in the two pivotal 48-week phase III trials [9].
For example, dizziness was very common (incidence
C10 %) among adolescents but was not seen in children,
whereas diarrhoea, pharyngeal erythema and tympanic
membrane hyperaemia were common (incidence C1 to
\10 %) or very common (incidence C10 %) among chil-
dren but were not observed in adolescents.
Ivacaftor did not appear to have any clinically important
effect on vital signs, ECGs, physical examination mea-
surements or clinical laboratory tests in adults and ado-
lescents [20] or children [21] in STRIVE and ENVISION.
However, clinical studies have reported transaminase ele-
vations more frequently with ivacaftor than with placebo in
patients with a history of such elevations (see Sect. 5.1 for
details).
Longer term, interim data from a clinical trial extension
study [30] indicated that the tolerability profile of ivacaftor
over up to 96 weeks of therapy in patients with cystic
fibrosis was generally consistent with that seen during the
initial 48-week trials [20, 21], with no new safety signals of
any clinical importance identified. Serious adverse events
occurred in 20.1 % of adults/adolescents and 8.4 % of
children, and two patients (one in each age cohort) dis-
continued the extension because of an adverse event. The
most common adverse events were respiratory in nature.
5.1 Transaminase Elevations
Patients with cystic fibrosis often have moderate elevations
in levels of AST or ALT [9]. Some ivacaftor and placebo
recipients had maximum elevations in transaminase levels
of[8 (1.8 vs. 1.5 % of patients),[5 (2.7 vs. 2.3 %) or[3
(6.3 vs. 8.4 %) times the upper limit of normal (ULN) in
48-week cystic fibrosis phase IIb/III trials; these elevations
were serious in two patients (both were ivacaftor
recipients).
Although transaminase elevations have, overall, occur-
red with similar incidence with ivacaftor as with placebo in
clinical studies, they have been documented more fre-
quently with ivacaftor than with placebo in patients with a
history of such elevations [9]. Thus, it is recommended that
AST and ALT [10] or hepatic function [9] be assessed
before starting treatment with ivacaftor, every 3 months
during the initial year of therapy and then once every year
thereafter. If transaminase levels increase during treatment,
patients should be monitored closely until the abnormali-
ties resolve [9, 10], and if AST or ALT levels reach [59
ULN, ivacaftor therapy should be interrupted [10]; before
resuming treatment with ivacaftor, the benefits and risks
require consideration [9, 10].
0 5 10 15 20 25 30
Dizziness
Rash
Diarrhoea
Nasopharyngitis
Abdominal pain
Nasal congestion
URTI
Oropharyngeal pain
Headache
% of patients
Ivacaftor
Placebo
Fig. 2 Tolerability of oral ivacaftor 150 mg every 12 h in patients
(aged 6–53 years) with cystic fibrosis and the G551D CFTR mutation.
The most frequent adverse events (incidence C8 %) that occurred in
C3 % more ivacaftor (n = 109) than placebo (n = 104) recipients in
a pooled analysis of the 48-week phase III trials, STRIVE and
ENVISION [10]. Patients also received standard care (see Sect. 4 for
details). URTI upper respiratory infection
1602 E. D. Deeks

6 Dosage and Administration
For the treatment of patients with cystic fibrosis and the
G551D CFTR mutation, the recommended dosage of iva-
caftor in the USA [10] and EU [9] is 150 mg every 12 h, taken
orally with fat-containing food. Patients with an unknown
genotype should have the presence of the G551D mutation
confirmed before being treated with ivacaftor. Local pre-
scribing information should be consulted for suggested dos-
age modifications in special patient populations and patients
receiving concomitant CYP3A4 inhibitor therapy and for
information regarding contraindications, drug interactions,
and other warnings and precautions.
7 Current Status of Ivacaftor in Cystic Fibrosis
Oral ivacaftor is indicated in a number of countries, including
the USA [10] and those of the EU [9], for the treatment of
cystic fibrosis in patients aged C6 years who have the G551D
CFTR mutation and its use is strongly recommended in this
patient population in the most recent guidelines of the Pul-
monary Clinical Practice Guidelines Committee (established
by the Cystic Fibrosis Foundation) [7]. In two well-designed,
48-week trials in patients aged 6–11 years (ENVISION) or
C12 years (STRIVE) with cystic fibrosis and this mutation,
ivacaftor 150 mg every 12 h significantly improved lung
function relative to placebo when used in combination with
standard care. Significant improvements in other measures
such as pulmonary exacerbation risk (in STRIVE) and
bodyweight (in both studies) were also seen with the drug
versus placebo. In an ongoing extension of these trials, ben-
eficial effects of ivacaftor were maintained for up to 96 weeks
of therapy, although longer-term data in this setting would be
beneficial given that cystic fibrosis is a life-long condition.
Ivacaftor was generally well tolerated and adherence/com-
pliance to the drug was high at [90 %, which compares
favourably with adherence rates reported for some other cystic
fibrosis therapies, such as inhaled antibiotics (31–53 % [32]).
Whether use of ivacaftor could enable some standard care
medications to be discontinued remains to be determined [1].
The G551D mutation is present in &3 % of patients with
cystic fibrosis, making it the third most common mutation
of the CFTR gene [3]. The efficacy demonstrated by iva-
caftor in patients with this gating (i.e. class III) mutation has
prompted investigation into the potential use of the drug in
patients with other CFTR gating mutations, and such trials,
including one in younger patients aged 2–5 years, are cur-
rently ongoing or recruiting participants [33].
In addition to gating mutations, the efficacy and safety of
ivacaftor has also been evaluated in patients homozygous for
the class II mutation F508del [34], the most common CFTR
mutation in those with cystic fibrosis (accounting for around
two-thirds of alleles) [35]. In a randomized, double-blind,
phase II trial in this patient population, which was powered to
assess safety rather than efficacy [34], treatment with ivacaftor
150 mg every 12 h (n = 112) was generally well tolerated but
did not significantly improve lung function relative to placebo
(n = 28), as measured by the change from baseline in percent
predicted FEV1 through week 16 (primary efficacy endpoint).
As discussed in Sect. 1, class II mutations result in CFTR
that is misfolded and improperly processed, and only a small
amount of the defective protein (if any) reaches the cell sur-
face [1], which may in part explain the lack of benefit
observed with ivacaftor in the trial discussed above. One
potential solution may be to use a CFTR potentiator, such as
ivacaftor, in combination with a CFTR corrector, i.e. a
compound that enhances the processing and maturation of
class II mutant CFTR proteins such as F508del, with the idea
being that the amount of CFTR reaching the cell surface will
be increased by the corrector and the function of the channels
reaching the membrane will be increased by the potentiator
[1]. Indeed, small (n B 82) phase II studies assessing iva-
caftor in combination with the investigational corrector lu-
macaftor in patients homozygous for the F508del mutation
have been promising [36, 37], and phase III trials in this
setting have been initiated [33]. A phase III ivacaftor trial in
patients with the class IV CFTR mutation, R117H, is also
recruiting participants [33]. Results of these studies are
awaited with interest.
Data selection sources: Relevant medical literature (including
published and unpublished data) on ivacaftor was identified by
searching databases including MEDLINE (from 1946) and EM-
BASE (from 1996) [searches last updated 23 August 2013],
bibliographies from published literature, clinical trial registries/
databases and websites. Additional information was also
requested from the company developing the drug.
Search terms: Ivacaftor, kalydeco, vx-770, vx770, vrt-813077,
vrt813077.
Study selection: Studies in patients with cystic fibrosis who
received ivacaftor. When available, large, well-designed, com-
parative trials with appropriate statistical methodology were
preferred. Relevant pharmacodynamic and pharmacokinetic data
are also included. All data included are publically available.
Disclosure The preparation of this review was not supported by any
external funding. During the peer review process, the manufacturer of
the agent under review was offered an opportunity to comment on this
article. Changes resulting from comments received were made by the
author on the basis of scientific and editorial merit.
References
1. Pettit RS. Cystic fibrosis transmembrane conductance regulator-
modifying medications: the future of cystic fibrosis treatment.
Ann Pharmacother. 2012;46(7–8):1065–75.
2. O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet.
2009;373(9678):1891–904.
Ivacaftor: A Review 1603

3. Rogan MP, Stoltz DA, Hornick DB. Cystic fibrosis transmem-
brane conductance regulator intracellular processing, trafficking,
and opportunities for mutation-specific treatment. Chest.
2011;139(6):1480–90.
4. Ratjen FA. Cystic fibrosis: pathogenesis and future treatment
strategies. Respir Care. 2009;54(5):595–605.
5. European Medicines Agency. Assessment report: Kalydeco (iva-
caftor). 2012. http://www.ema.europa.eu/docs/en_GB/document_
library/EPAR_-_Public_assessment_report/human/002494/WC5001
30766.pdf (Accessed 21 Aug 2013).
6. Borowitz D, Baker RD, Stallings V. Consensus report on nutri-
tion for pediatric patients with cystic fibrosis. J Pediatr Gastro-
enterol Nutr. 2002;35(3):246–59.
7. Mogayzel PJ Jr, Naureckas ET, Robinson KA, et al. Cystic
fibrosis pulmonary guidelines. Chronic medications for mainte-
nance of lung health. Am J Respir Crit Care Med.
2013;187(7):680–9.
8. Cystic Fibrosis Trust. Standards for the clinical care of children
and adults with cystic fibrosis in the UK; 2011. http://www.
cftrust.org.uk/aboutcf/publications/consensusdoc/CF_Trust_Stan
dards_of_Care_2011_%28website_Apr_12%29.pdf (Accessed 21
Aug 2013).
9. European Medicines Agency. Kalydeco 150 mg film-coated tablets:
summary of product characteristics; 2013. http://www.ema.europa.
eu/docs/en_GB/document_library/EPAR_-_Product_Information/
human/002494/WC500130696.pdf (Accessed 21 Aug 2013).
10. US Food and Drug Administration. Kalydeco (ivacaftor) tablets: US
prescribing information; 2012. http://www.accessdata.fda.gov/
drugsatfda_docs/label/2012/203188s001lbl.pdf (Accessed 21 Aug
2013).
11. Eckford PDW, Li C, Ramjeesingh M, et al. Cystic fibrosis
transmembrane conductance regulator (CFTR) potentiator VX-
770 (ivacaftor) opens the defective channel gate of mutant CFTR
in a phosphorylation-dependent but ATP-independent manner.
J Biol Chem. 2012;287(44):36639–49.
12. Van Goor F, Hadida S, Grootenhuis PDJ, et al. Rescue of CF
airway epithelial cell function in vitro by a CFTR potentiator,
VX-770. Proc Natl Acad Sci USA. 2009;106(44):18825–30.
13. Yu H, Burton B, Huang C-J, et al. Ivacaftor potentiation of
multiple CFTR channels with gating mutations. J Cyst Fibros.
2012;11(3):237–45.
14. Woodworth BA, Zhang S, Skinner D, et al. Comparison of
CFTR and ciliary beat frequency activation by the CFTR
modulators VX-770, VRT532, and UCCF-152 in primary sin-
onasal epithelial cultures [abstract no. 114]. Pediatr Pulmonol.
2011;46:251–2.
15. Jih KY, Hwang TC. Vx-770 potentiates CFTR function by pro-
moting decoupling between the gating cycle and ATP hydrolysis
cycle. Proc Natl Acad Sci USA. 2013;110(11):4404–9.
16. Van Goor F, Yu H, Burton B, et al. Effect of ivacaftor on CFTR forms
with missense mutations associated with defects in protein processing
or function. J Cyst Fibros. 2013. doi:10.1016/j.jcf.2013.06.008.
17. Pohl K, Reeves EP, McElvaney NG. The CFTR potentiator i-
vacaftor corrects defective degranulation of secondary and ter-
tiary granules by cystic fibrosis neutrophils [abstract no. WS9.9].
J Cyst Fibros. 2012;11(Suppl. 1):S21.
18. Davies JC, Sheridan H, Lee P, et al. Lung clearance index to
evaluate the effect of ivacaftor on lung function in subjects with
CF who have the G551D-CFTR mutation and mild lung disease
[abstract no. 249]. Pediatr Pulmonol. 2012;47:311.
19. Accurso FJ, Rowe SM, Clancy JP, et al. Effect of VX-770 in
persons with cystic fibrosis and the G551D-CFTR mutation.
N Engl J Med. 2010;363(21):1991–2003.
20. Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator
in patients with cystic fibrosis and the G551D mutation. N Engl J
Med. 2011;365(18):1663–72 (plus supplementary appendix).
21. Davies JC, Wainwright CE, Canny GJ, et al. Efficacy and safety
of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with
a G551D mutation. Am J Respir Crit Care Med.
2013;187(11):1219–25 (plus supplementary appendix).
22. Altes T, Johnson MA, Miller GW, et al. Hyperpolarized gas MRI
of ivacaftor therapy in persons with cystic fibrosis and the
G551D-CFTR mutation [abstract no. 196]. Pediatr Pulmonol.
2012;47:291.
23. Li C, Song T, Ordonez C, et al. No effect of therapeutic and
supratherapeutic doses of ivacaftor on QTc interval in healthy
subjects [abstract no. PII-66]. Clin Pharmacol Ther.
2013;93(S1):S76.
24. Hayes E, Pohl K, McElvaney NG, et al. The cystic fibrosis
neutrophil: a specialized yet potentially defective cell. Arch
Immunol Ther Exp (Warsz). 2011;59(2):97–112.
25. Zha J, Zhang J, Ordonez C. Pharmacokinetics (PK), metabolism
and excretion of [14C] VX-770, a potentiator of cystic fibrosis
transmembrane conductance regulator (CFTR), in healthy subjects
[abstract no. 1124131]. J Clin Pharmacol. 2011;51(9):1358–9.
26. Zha J, Kumor K, Zhang J, et al. Exposure–response relationship
for FEV1 and sweat chloride in patients with cystic fibrosis
treated with ivacaftor, a CFTR potentiator [abstract no. 235].
Pediatr Pulmonol. 2012;47:306.
27. Zha J, Zhang J, Ordonez C, et al. Clinical pharmacology profile
of ivacaftor, a CFTR potentiator [abstract no. 236]. Pediatr Pul-
monol. 2012;47:306–7.
28. Vertex Pharmaceuticals Incorporated. Study of ivacaftor in cystic
fibrosis subjects aged 12 years and older with the G551D muta-
tion (STRIVE) [ClinicalTrials.gov identifier NCT00909532]. US
National Institutes of Health, ClinicalTrials.gov; 2013. http://
www.clinicaltrials.gov (Accessed 21 Aug 2013).
29. Vertex Pharmaceuticals Incorporated. Study of ivacaftor in cystic
fibrosis subjects aged 6 to 11 years with the G551D mutation
(ENVISION) [ClinicalTrials.gov identifier NCT00909727]. US
National Institutes of Health, ClinicalTrials.gov; 2012. http://
www.clinicaltrials.gov (Accessed 21 Aug 2013).
30. McKone E, Li H, Gilmartin G, et al. Long-term safety and effi-
cacy of ivacaftor in persons with cystic fibrosis who have the
G551D-CFTR mutation [abstract no. 211]. Pediatr Pulmonol.
2012;47(S35):296–7.
31. Quittner A, Ramsey B, Rodriguez S, et al. Patient-reported out-
comes in phase 3 trials of ivacaftor in subjects with CF who have
the G551D-CFTR mutation [abstract no. 212]. Pediatr Pulmonol.
2012;47:297.
32. Eakin MN, Bilderback A, Boyle MP, et al. Longitudinal associ-
ation between medication adherence and lung health in people
with cystic fibrosis. J Cyst Fibros. 2011;10(4):258–64.
33. US National Institutes of Health. ClinicalTrials.gov; 2013. http://
www.clinicaltrials.gov (Accessed 21 Aug 2013).
34. Flume PA, Liou TG, Borowitz DS, et al. Ivacaftor in subjects
with cystic fibrosis who are homozygous for the F508del-CFTR
mutation. Chest. 2012;142(3):718–24.
35. Castellani C, Cuppens H, Macek M Jr, et al. Consensus on the use
and interpretation of cystic fibrosis mutation analysis in clinical
practice. J Cyst Fibros. 2008;7(3):179–96.
36. Boyle MP, Bell S, Konstan M, et al. The investigational CFTR
corrector, VX-809 (lumacaftor) co-administered with the oral
potentiator ivacaftor improved CFTR and lung function in
F508DEL homozygous patients: phase II study results [abstract
no. 260]. Pediatr Pulmonol. 2012;47:315.
37. Boyle MP, Bell S, Konstan MW, et al. VX-809, an investiga-
tional CFTR corrector, in combination with VX-770, an inves-
tigational CFTR potentiator, in subjects with CF and homozygous
for the F508del-CFTR mutation [abstract no. 212]. Pediatr Pul-
monol. 2011;46:287.
1604 E. D. Deeks











