Embed Size (px)
Citation preview

Ž .Mutation Research 422 1998 43–53
Involvement of p16CDKN2A in cell cycle delays after low doseUV irradiation
A. Milligan, B.G. Gabrielli ), J.M. Clark, N.K. Hayward, K.A.O. EllemQueensland Cancer Fund Cancer Unit, Joint Experimental Oncology Program, Queensland Institute of Medical Research,
P.O. Royal Brisbane Hospital, Herston, Queensland 4029, Australia
Received 17 March 1997; revised 24 July 1997; accepted 31 July 1997
Abstract
Ž .Ultraviolet UV radiation contributes to the aetiology of melanoma, but the precise mechanistic details are still unclear.The CDKN2A gene which is associated with familial and sporadic melanoma, encodes a tumour suppressor, p16. We havepreviously shown that in response to low doses of UV radiation the level of p16 increases, and that this correlates with a G2delay. Here we report that in melanoma cell lines which do not express p16, or express a mutant p16, no G2 delay isobserved in response to UV. The loss of functional p16 also correlates with an increase in DNA damage as judged byincreased numbers of bi- and multinuclear cells and cells containing 1–2 micronuclei following UV irradiation. This workprovides a further link between UV radiation, CDKN2A and melanoma, suggesting that the functional inactivation ofCDKN2A disrupts a p16-dependent G2 cell cycle checkpoint, thus contributing to the development of this neoplasm. q 1998Elsevier Science B.V. All rights reserved.
Keywords: UV; p16; Melanoma; Cell cycle; CDKN2A
1. Introduction
Skin cancer is the most common human malig-nancy and is strongly associated with exposure to
Ž .ultraviolet UV radiation. The UV radiation in sun-light is subdivided into three wavebands, UVCŽ .190–280 nm which is absorbed by the earth’s
Ž .atmosphere, while UVB 280–320 nm and UVAŽ .320–400 nm both penetrate the earth’s ozone layer.UVB is the major waveband involved in the devel-opment of skin cancer, primarily targeting DNA,
) Corresponding author. Tel.: q61-7-3362-0304; Fax: q61-7-3362-0107; E-mail: [email protected]
producing 6–4 photoproducts and cyclobutanepyrimidine dimers, which are also produced by UVC
Ž w x.exposure in vitro reviewed in Ref. 1 . The deple-tion of the ozone layer is expected to contribute toincreased penetration of UV radiation with a conse-quently increased rate of UV-induced DNA damage
w xand higher incidence of skin cancer 2 .Exposure to UV radiation induces a variety of
cellular responses including transcriptional inductionof a number of genes including c-jun, c-fos and
Ž w x .TP53 Ref. 3 and references therein and cell cyclew xeffects 4–7 . Little is known regarding the mecha-
nisms by which UV light affects the cell cyclemachinery. In mammalian cells, cell cycle progres-
0027-5107r98r$ - see front matter q 1998 Elsevier Science B.V. All rights reserved.Ž .PII: S0027-5107 98 00174-2

( )A. Milligan et al.rMutation Research 422 1998 43–5344
sion is controlled by the sequential formation andŽ .activation of cyclinrcyclin-dependent kinase cdk
Ž w x.complexes reviewed in Ref. 8 . The recent identifi-cation of low molecular weight inhibitors ofcyclinrcdk complexes, the p16 and p21 families ofproteins, has introduced a new mechanism for regu-
Ž w x.lating cell cycle progression reviewed in Ref. 9 .The expression of some of these proteins has beenshown to be upregulated in response to cellularstress. For example, the p53 tumour suppressor pro-tein has been shown to participate in DNA damage-
w xactivated cell cycle checkpoints 4,10 , in part by upw xregulating the expression of p21 7,11–13 .
p16 is the founding member of a rapidly growingfamily of inhibitors of cyclinrcdk complexes whichspecifically bind and inhibit the activity of cdk4 and
w xcdk6 14,15 , which are in turn required for thetransition from G1 in to S phase in the eukaryotic
w xcell cycle 16 . Deletion or mutational inactivation ofCDKN2A, the gene encoding p16, could lead todisruption of the cell cycle and as a consequence,contribute to tumorigenesis.
CDKN2A maps to the chromosomal region 9p21–22 and has been identified as a melanoma suscepti-
Ž w x.bility gene reviewed in Ref. 17 . Analysis of anumber of CDKN2A mutations identified in affectedmembers of melanoma kindreds showing linkage to9p21–22, has confirmed that they encode function-
w xally compromised p16 proteins 18 . This suggeststhat p16 is a tumour suppressor and that loss of p16function may be a predisposing event in familialmelanoma. The CDKN2A gene is also mutated ordeleted in cell lines and primary tumour cells from a
w xwide variety of cancer types 19–21 , indicating thatit may have a more general role in tumour suppres-sion.
Previous work in this laboratory has demonstratedthat in response to low doses of UVC radiation,HeLa cells were delayed in the S and G2rM phases
w xof the cell cycle 5 . Similar UV-induced delays havew xbeen observed by others 4,6,7 . In HeLa cells, the
G2 delay correlated with elevated p16 levels, andloss of p16 expression resulted in the loss of the
w xUVC-induced cell cycle delay 22 . We were inter-ested to see whether this UV response was abrogatedin melanoma cell lines which express mutated p16 orelse carry homozygous deletions of CDKN2A. Wehave therefore investigated the cell cycle and growth
effects of both UVC and UVB radiation on a numberof different melanoma cell lines. We now report thatinduction of p16 following UV irradiation results ina G2 cell cycle delay in those cells expressingfunctional p16, but that melanoma cell lines whichhave lost functional p16 expression do not delay inG2 following irradiation, and accumulate signifi-cantly more DNA damage.
2. Materials and methods
2.1. Cell lines and culture conditions
Melanoma cell lines, MM96L, MM384, MM473,SK-MEL-13, SK-MEL-28 and A2058, 18-11-TiŽ .HPV-transformed human keratinocytes and HeLaŽ .human cervical carcinoma were cultured as mono-layers in RPMI-1640 medium supplemented with 0.1mgrml streptomycin, 100 Urml penicillin and 10%fetal bovine serum or 5–10% fetal bovine serum. Allcell lines used were shown to be free of mycoplasmainfection.
2.2. UltraÕiolet radiation
Irradiation with UVC was performed using aŽUVS-52 Mineralite lamp Ultraviolet Products, San
.Gabriel, CA with maximal output at 254 nm asw xdescribed previously 5 . For UVB irradiation, a
Ž .single FS2OT12 UVB lamp Light Sources withmaximal output at 313 nm was used. Asynchronouscultures were seeded in 100 mm diameter dishes, and
Ž .on the following day day 0 , cells were overlaidŽ .with 2 ml phosphate buffered saline PBS and irra-
y2 Ždiated with 100 J m UVB according to cellsurvival studies 100 J my2 UVB is approximatelyequitoxic to 13 J my2 UVC; P. Parsons, personal
.communication . Immediately after irradiation, thecells were supplemented with fresh medium andreturned to the incubator. Controls were handledsimilarly, but were not exposed to UV.
Duplicate cultures were removed 0, 1, 2, 3 and 6days after irradiation for analysis. The cells wereharvested, resuspended in PBS and counted using ahaemocytometer. The cells were then pooled and analiquot removed, fixed in 1 ml of cold 70% ethanoland stored at y208C for flow cytometric analysis.
( )A. Milligan et al.rMutation Research 422 1998 43–53 45
The remainder of the sample was pelleted and storedat y208C.
2.3. Flow cytometric analysis
Cell cycle phase distributions were determined byŽ .flow cytometry of propidium iodide PI stained
cells. Fixed cells were washed twice in PBS thenresuspended in 0.5 ml PBS containing 0.5 mgrmlRNase A and 5 mgrml PI, incubated at room tem-perature for 10 min, filtered through a 4 mm nylongauze, and analysed for DNA content by flow cy-tometry using a Becton-Dickinson FACScan. Quanti-
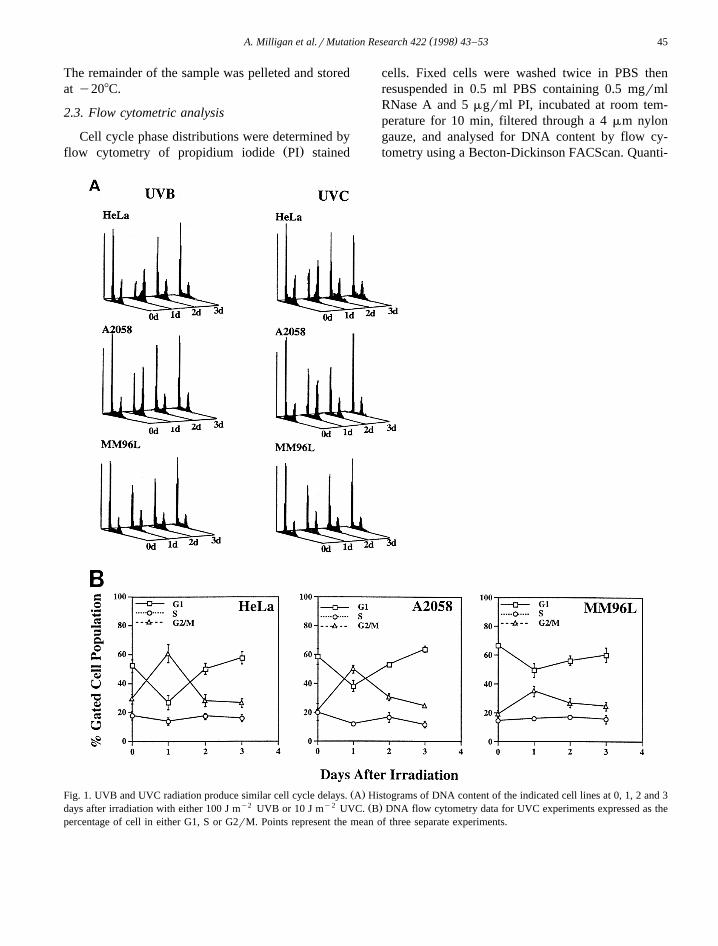
Ž .Fig. 1. UVB and UVC radiation produce similar cell cycle delays. A Histograms of DNA content of the indicated cell lines at 0, 1, 2 and 3y2 y2 Ž .days after irradiation with either 100 J m UVB or 10 J m UVC. B DNA flow cytometry data for UVC experiments expressed as the
percentage of cell in either G1, S or G2rM. Points represent the mean of three separate experiments.

( )A. Milligan et al.rMutation Research 422 1998 43–5346
tation of the cell cycle phase distribution was gener-ated from the flow cytometric data by using Lysis II
w xanalysis software 5 .
2.4. Immunoblotting analysis
Thawed cell pellets were lysed in NETN bufferŽ20 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA,
.0.5% Nonidet P-40 supplemented with 0.3 M NaCl,5 mgrml each of leupeptin, apoprotin, pepstatin and0.5 mM PMSF, 10 mM NaF and 0.1 mM sodiumvanadate, at a concentration of 5=106 cellsrml.The lysate supernatants were diluted 1 in 2 with 2=
SDS sample buffer then boiled. Aliquots equivalentto 5=104 cells were loaded onto 12% or 15%SDS-PAGE gels, then blotted onto polyvinylidine
Ž .difluoridine PVDF using semi dry electrotransfer.After transfer the membranes were blocked in 5%
Žblotto 0.05% Tween 20 in PBS containing 5% skim.milk powder for 2 h and then incubated in primary
antibody overnight at 48C, diluted in 1% blotto.Anti-p16 antibody was used as described previouslyw x22 and visualised using enhanced chemilumines-
Ž .cence detection ECL, NEN .
2.5. Immunofluorescent studies
Cells were grown on coverslips and irradiatedwith 10 J my2 UVC or mock irradiated, and col-lected at times after, washed once with PBS andfixed with y208C 100% methanol. The cells wererehydrated and stained with a-tubulin monoclonalantibody and Hoechst H33342 as described previ-
w xously 22 .
3. Results
3.1. UVC and UVB irradiation produce similar cellcycle and growth effects
ŽEquitoxic doses of UVC and UVB radiation 10 Jy2 y2 .m and 100 J m , respectively produced similar
cell cycle delays in G2rM at 24 h in both HeLa cellsŽ .and the melanoma line A2058 Fig. 1A . Another
melanoma line, MM96L, showed little change in itsŽcell cycle distribution, even with higher doses 30 J
y2 .m UVC; data not shown , although a small in-crease in the G2rM population was consistently
Ž .observed at 24 h Fig. 1B . Both cell lines overcomethe G2rM delay by 48 h and return to a normal cellcycle distribution. Our earlier work had shown astrong correlation between the UV-induced G2rM
w xdelay and increased levels of p16 in HeLa cells 22 .Increased p16 levels were observed in HeLa andA2058 cells by 24 h after UV irradiation, and thiscorrelated with both UVB and UVC induced G2rM
Ž .delay in these cells at 24 h Fig. 2 . The levels andpatterns of p16 expression varied between the twolines and with the UV band used. As shown previ-ously, HeLa p16 increased by 24 h after UVC irradi-ation but had decreased by 48 h, although it re-mained above control levels. Similar p16 accumula-tion and G2rM delay at 24 h after irradiation with10 J my2 UVC was seen in the HPV-transformed
Ž .keratinocyte line, 18-11-Ti data not shown . WithUVB, the levels of p16 also increased at 24 h butthis level was maintained for at least 72 h after
Ž .irradiation Fig. 2 . In A2058 cells, a similar increasein p16 was detected following UVC exposure asseen in HeLa, but the maximal p16 levels were seenat 48 h, when the cells displayed a normal cell cycledistribution. With UVB, the maximal level of p16accumulated was not as high as with UVC, but thislevel was sustained for the 72 h of this experiment.Surprisingly, the cell cycle profile of both HeLa andA2058 cells after the initial G2rM delay at 24 h was
Fig. 2. Increased p16 correlates with cell cycle delays. Equalnumbers of HeLa and A2058 cells, from either control or UVBand UVC irradiated samples harvested at the indicated times afterirradiation were immunoblotted for p16.

( )A. Milligan et al.rMutation Research 422 1998 43–53 47
Ž .Fig. 3. Cell proliferation following UV irradiation. The averages of duplicate cell counts for either mock irradiated control or cellsirradiated with either 100 J my2 UVB or 10 J my2 UVC and harvested at the indicated times following irradiation are shown. These dataare from the same experiment shown in Fig. 2 expressed as the log of the fold increase over the day zero cell numbers. Similar results were
Ž .obtained in two other separate experiments not shown .
similar to that of exponentially growing cells. Nop16 was detected in MM96L either before or afterirradiation.
The flow cytometry data for the three cell lines,and different p16 expression patterns in response tothe different UV bands in HeLa and A2058, sug-gested different growth consequences for these cellsin response to UV. This was measured by increase incell number following irradiation. Cell viability mea-sured for the first 3 days of the experiment wasgreater than 90% for each of the cell lines and didnot vary significantly with either UVB or UVC
Ž .irradiation data not shown . Both HeLa and A2058displayed an initial block in cell proliferation duringthe first 24 h following irradiation with either waveband, corresponding to the cell cycle delay observedŽ .Fig. 1B , after which time the cells recommenced
Ž .logarithmic growth at the control rate Fig. 3 . Incontrast, irradiated MM96L did not have a dis-cernible delay in proliferation, and although the UVirradiated cultures had a slightly lower cell number,the cells proliferated at a similar rate to the controlsŽ .Fig. 3 .
3.2. Loss of functional p16 response to UVirradiation correlates with absence of a G2rMdelay
This study was extended to investigate the UVresponses of a number of other melanoma cell lines
Žwhich were either deleted for CDKN2A SK-MEL-
. Ž13 , contained mutant CDKN2A MM384 and. ŽMM473 , or expressed the CDK4 R24C mutant SK-
.Mel-28; Castellano et al., submitted which does notw xbind p16 24 , to test whether loss or mutation of
CDKN2A or CDK4 abrogated the UV-induced cellcycle and growth effects seen with HeLa and A2058lines. Since irradiation with both UVB and UVCresulted in p16 accumulation and G2rM delays at 24h after irradiation in the p16 expressing cell linestested above, only UVC radiation was used in thesefurther studies. No cell cycle delays were observedby flow cytometry following UVC irradiation of
Žeither the SK-MEL-13 or SK-MEL-28 lines Fig..4A . MM473 were a mixed population of diploid and
tetraploid cells, and the relative percentages of G1and G2rM phase cells could not be accurately calcu-lated from single parameter DNA flow cytometrydata. The relative abundance of the 2N, 4N and 8NDNA peaks did not change following UVC irradia-tion, however, indicating that no cell cycle phase
Ž .specific delays occurred in these cells Fig. 4B .MM384 appeared to display small, reproducible Sthen G2rM accumulations as seen with HeLa, al-
Ž .though with much slower kinetics Fig. 4A . Im-munoblotting for p16 in the cell lines revealed that inSK-MEL-28 and MM384, the level of the p16 pro-tein expressed was unaffected by UVC irradiation,whereas MM473 p16 showed a UVC-induced in-crease at 24 h, similar to that seen in HeLa cellsŽ .Fig. 4C .

( )A. Milligan et al.rMutation Research 422 1998 43–5348
Ž .Fig. 4. Loss of functional p16 response to UV results in absence of cell cycle delays. A DNA flow cytometry data from three separatey2 Ž .experiments expressed as percentage of cells in G1, S or G2rM at the indicated times following irradiation with 10 J m UVC. B DNA
y2 Ž .flow cytometry histogram of control and irradiated MM473 cells at 24 h after exposure to 10 J m UVC. C Equal cell numbers fromŽ . y2 Ž .either control 0 or samples irradiated with 10 J m UVC and harvested at the indicated times in hours after irradiation, were
immunoblotted for p16.
The growth consequences of the UVC irradiation,measured by increase in cell number, showed thatthe growth rate of SK-MEL-13 and SK-MEL-28 was
unaffected by UVC irradiation, although the increasein cell number was a little reduced at 24 h afterirradiation for both lines compared to controls

( )A. Milligan et al.rMutation Research 422 1998 43–53 49
Fig. 5. Cell proliferation following UVC irradiation. The mean values of duplicate cell counts for mock irradiated and cells irradiated with10 J my2 UVC and harvested at the indicated times after irradiation are shown. Similar data were obtained in two other separate
Ž .experiments for SK-MEL-13 and SK-MEl-28 lines and one other experiment for the MM384 and MM473 lines not shown .
Ž .Fig. 5 . MM473 and MM384 had a reduced cellnumber at 24 h compared to the number at day 0,presumably due to detachment of dying cells fromthe plates following irradiation in these lines. MM473recovered to normal logarithmic growth at the con-trol rate by 48 h, whereas the MM384 cell numbercontinued to decrease during the first 3 days follow-
Ž .ing irradiation Fig. 5 .
3.3. UV irradiation induces an increase in aberrantnuclear DNA structures
The major cellular target of the short wavelengthw xUV radiation used in these experiments is DNA 1 .
Our working hypothesis was that the G2rM cellcycle delay following UV irradiation was used by
the cell to repair the DNA damage. The absence of acell cycle delay could lead to cells attempting toprogress through cell division with damaged DNA,possibly resulting in macroscopic changes in thenucleus due to an inability of damaged chromosomesto undergo normal disjunction at mitosis. Inspectionof nuclei of fixed cells, stained with Hoechst 33342and counterstained for microtubules with an a-tubu-lin antibody, showed that even unirradiated controlcultures of the cell lines tested contained a range ofaberrant nuclear structures. These included binuclearcells, cells containing 1–2 small micronuclei, and ina very small percentage of cases, enlarged cells with
Ž .multiple nuclei of various sizes Fig. 6B–D . Each ofthe multiple nuclei appear to have intact nuclearmembranes, and the microtubules can be seen to

( )A. Milligan et al.rMutation Research 422 1998 43–5350
have formed a network around them as they doaround normal nuclei. The DNA appeared to bedecondensed in these cells, distinguishing them from
apoptotic cells. Indeed few apoptotic cells were de-tected in these experiments, probably due to theirloss by detachment from the substrate surface. Quan-

( )A. Milligan et al.rMutation Research 422 1998 43–53 51
titation of the number of cells containing these ab-normal nuclei showed that the SK-MEL-13 andMM384 lines normally contained a significant per-
Ž .centage 10–30% of cells with aberrant nuclearŽ .morphologies Fig. 6A . The SK-MEL-28, MM473
and MM96L cells had a lower proportion of cellsŽ .with aberrant nuclei 10–15% , but always more
than HeLa and A2058 cells which normally had only5% of cells with deformed or multiple nuclei. Fol-lowing UV irradiation, the numbers of cells contain-
Žing aberrant nuclei increased in all cell lines Fig..6A . In contrast, no abnormal nuclei were observed
in normal human foreskin fibroblasts in the sameexperiment. The two cell lines with UV-induced wildtype p16 accumulation and a G2rM delay, HeLaand A2058, had the lowest number of cells with
Ž .aberrant nuclei by 72 h after irradiation 15–20% .MM96L and MM473, which did not express wildtype p16 had more cells with aberrant nuclei by 72 hŽ .20–30% . The remaining three cell lines, SK-MEL-13, SK-MEL-28, and MM384, which did not havedetectable UV-induced p16 responses, had the high-est numbers of cells with abnormal nuclei at 72 h,with up to 60% of the attached MM384 cells con-
Ž .taining abnormal nuclear structures Fig. 6A .
4. Discussion
The work described here provides further supportfor a role for elevated levels of p16 in the initialG2rM delay seen in response to both UVC and thephysiologically relevant UVB radiation. This data is
Žsummarised in Table 1. In 3r3 cell lines HeLa,.A2058 and 18-11-Ti expressing wild type p16 which
accumulated to greater than 4-fold following UVirradiation, an initial G2rM delay was observed. Thedelay in HeLa and A2058 cells is due to a block inprogression through G2 phase, as the activation of
Table 1Summary of p16 mutation and expression status, and UV re-sponses in the cell lines tested
p16 G2 delay
DNA Protein
Normal qUV
HeLa wt qq qqqqqqq yesaA2058 wt q qqqqqqq yes
18-11-Ti wt qq qqqqqq yesbMM96L wt y y slight?b,cSK-Mel-28 wt q q no
bSK-Mel-13 deleted y y nobMM473 mutant qq qqqq nobMM384 mutant qq qq No
ysnot detected.qqs indicates level of p16 expression.n.d.snot determined.aUnpublished data.b w xRef. 28 .c ŽOne CDK4 allele has R24C mutation Castellano et al., submit-
.ted .
both cyclin Arcdk2 and cyclin B1rcdc2 is inhibitedw x24 . A G2 delay has also been reported for keratino-
w xcytes irradiated with low doses of UVB 6 . Thereare two further cell lines with wild type CDKN2A.Of these, MM96L does not express p16 and showslittle cell cycle delay, while SK-MEL-28 expresses afunctional p16 protein but also the CDK4 mutant
Ž . Ž .protein R24C Castellano et al., submitted whichw xdoes not bind p16 23 —and hence is effectively p16
null—shows no G2rM delay. The remainingmelanoma lines are either deleted for CDKN2A, orexpress mutant p16. Two of the three show no G2delay in response to UV treatment. The exception isthe MM384 line, in which a small proportion of cellsappears to slowly accumulate in G2rM followingUV irradiation. However, inspection of the nuclei ofthese cells showed that at 2 and 3 days after irradia-tion, 40–50% of the attached cells were either binu-
Ž . Ž . y2Fig. 6. A Coverslip cultures of the indicated cell lines were either mock irradiated 0 or irradiated with 10 J m UVC and harvested atthe indicated times afterwards. The cells were fixed, then stained for DNA and tubulin. Cells were inspected by immunofluorescent
Ž . Ž . Ž .microscopy and the numbers of binuclear cells dots , micronuclei solid and multinuclei crosshatching containing cells were quantitated.Ž .In each case between 100 and 300 cells were counted. The numbers of each are expressed as a percentage of the total cells counted. B–D
Ž . Ž . Ž .Immunofluorescent stains of DNA and tubulin staining the microtubules; Mt of control MM384 cells B showing a binuclear cell rightŽ . Ž . Ž .and a micronucleus left, arrow . C Multiple nuclei are seen arrows in the lower A2058 cell at 72 h following irradiation. These small
Ž . Ž .nuclei are clearly defined by the microtubules surrounding them arrows . D A more extreme example of a multinuclear cell. This MM384cell at 72 h following irradiation clearly contains many small nuclei and a DNA content )4N.

( )A. Milligan et al.rMutation Research 422 1998 43–5352
clear or contained multiple fractured nuclei. Thus theapparent accumulation of cells with normal G2 DNAcontent at 3 days represents this high proportion ofcells with 2 or more nuclei, detected as 4N DNAcontent by flow cytometry. The appearance of thesebi- and multinuclear cells indicates a block in cytoki-nesis, but not a G2 delay. Thus all three of thedeleted or mutant CDKN2A lines have lost the G2rMcell cycle delay response following UVC irradiation.
Our findings suggest that p16 acts specificallythrough its binding to cdk4 to produce the G2 delayresponses. Two of the melanoma lines which did nothave a UV-induced G2 delay have impairedp16rcdk4 but not p16rcdk6 association; SK-MEL-28, which expresses the R24C mutant of CDK4
w xwhich does not bind p16 23 , and MM384, whichhas a mutant p16 that binds cdk6 but not cdk4Ž .Castellano et al., submitted . Furthermore, a cdk4rcyclin D kinase activity has been identified in late S,
w xearly G2 phase of the cell cycle 25 , supporting thenotion that p16 acts through cdk4 in a DNA damageinitiated G2 checkpoint.
One apparent inconsistency in the findings pre-sented here relates to the high levels of p16 that weremaintained in HeLa and A2058 lines for at least 3days following UVC treatment, by which time theyhad recommenced normal logarithmic growth. Wehave stably transfected HeLa and A2058 cells withp16 cDNA and produced clones which express levelsof p16 equivalent to the elevated, UV-induced levels,and these clones also appear to proliferate normallyŽ .unpublished data . This suggests that although ele-vated levels of p16 are not sufficient to produce theG2 delay, the lack of a G2 delay in cells notexpressing a functional p16 response to UV wouldargue that elevated p16 levels are necessary for thisdelay. The p16 may co-operate with a second factorwhich is induced by UV to cause the cell cycledelay, or alternatively, p16 may be involved in eithersensing DNA damage andror recruiting repair ma-chinery onto the DNA. Thus loss of p16 would resultin cells not recognising the need to stop and repairDNA damage, while high levels of p16 in the ab-sence of DNA damage would have no consequenceson cell cycle progression. These models are currentlyunder investigation.
A large body of literature is accumulating on theinvolvement of the tumour suppressor protein p53 in
UV-induced DNA damage repair and cell cycle de-lays. Increased expression of the cdk inhibitor pro-tein p21 mediates some of the p53-dependent G1 andS phase cell cycle effects following UV irradiationw x7,26 . It is unlikely that p53rp21 are involved in theG2 delay being investigated here, as cell lines with
Žnon-functional p53 including the HeLa and A2058.cells used in this study retain the UV-induced G2
w xdelay 6,22,24 . However, p53 may also be involvedw xin a mitotic spindle assembly checkpoint 27 , loss of
which may account for the aberrant nuclear struc-tures observed in the irradiated HeLa and A2058cells and may possibly account for a proportion ofthose in the other melanoma lines.
The data presented here support a role for p16 inthe UV-induced G2 delay. The p16 response appearsto be independent from the p53-mediated UV re-sponses. Precisely why there are two independentcell cycle checkpoints for DNA damage responseremains unclear. The loss of a functional p16 re-sponse to UV irradiation has clear implications forthe development of melanoma. An inability to detectand stop to repair DNA damage could result in anincrease in the mutation load in cells; in appropriatecircumstances this would lead to neoplastic transfor-mation, an initial step in tumour development.
Acknowledgements
The authors are grateful to Dr. Peter Parsons forthe cell lines and Dr. Marina Castellano for helpfuldiscussions. This work was supported by the Queens-land Cancer Fund. BG is a QIMR Fuiczek Fellow.
References
w x1 E. Sage, Distribution and repair of photolesions in DNA:genetic consequences and the role of sequence context, Pho-
Ž .tochem. Photobiol. 57 1993 163–174.w x2 S.E. Freeman, H. Hacham, R.W. Gange, D.J. Maytum, J.C.
Sutherland, B.M. Sutherland, Wavelength dependence ofpyrimidine dimer formation in DNA of human skin irradiatedin situ with ultraviolet light, Proc. Natl. Acad. Sci. USA 86Ž .1989 5605–5609.
w x3 C. Campbell, A.G. Quinn, B. Angus, P.M. Farr, J.L. Ress,Wavelength specific patterns of p53 induction in human skin
Ž .following exposure to UV radiation, Cancer Res. 53 19932697–2699.

( )A. Milligan et al.rMutation Research 422 1998 43–53 53
w x4 X. Lu, D.P. Lane, Differential induction of transcriptionallyactive p53 following UV or ionising radiation: defects in
Ž .chromosome instability syndromes?, Cell 75 1993 765–778.w x5 X.-Q. Wang, K.A.O. Ellem, Heterogeneity in the HeLa cell
cycle response to UVC analysed by the BrdUrd two-parame-Ž .ter method, Exp. Cell Res. 212 1994 176–189.
w x6 T. Herzinger, J.O. Funk, K. Hillmer, D. Eick, D.A. Wolf, P.Kind, Ultraviolet B irradiation-induced G2 cell cycle arrest inhuman keratinoctyes by inhibitory phosphorylation of the
Ž .cdc2 cell cycle kinase, Oncogene 11 1995 2151–2156.w x7 T. Petrocelli, R. Poon, D.J. Drucker, J.M. Slingerland, C.F.
Rosen, UVB radiation induces p21 and mediates G1 and SŽ .phase checkpoints, Oncogene 12 1996 1387–1396.
w x8 D.O. Morgan, Principles of CDK regulation, Nature 374Ž .1995 131–134.
w x9 C.J. Sherr, J.M. Roberts, Inhibitors of mammalian G1Ž .cyclin-dependent kinases, Genes Dev. 9 1995 1149–1163.
w x10 M.B. Kastan, Q. Zhan, W.S. El Deiry, F. Carrier, T. Jacks,W.V. Walsh, B.S. Plunlett, B. Vogelstein, A.J. Fornace, Amammalian cell cycle checkpoint utilising p53 and GADD45
Ž .is defective in Ataxia-Telangiectasia, Cell 71 1992 587–597.
w x11 A. Di Leonardi, S.P. Linke, K. Clarkin, G.M. Wahl, DNAdamage triggers a prolonged p53-dependent G1 arrest andlong-term induction of Cip1 in normal human fibroblasts,
Ž .Genes Dev. 8 1994 2540–2551.w x12 V. Dulic, W.K. Kaufamann, S.J. Wilson, T.D. Tlsty, E. Lees,
J.W. Harper, S.J. Elledge, S.I. Reed, p53-Dependent inhibi-tion of cyclin-dependent kinase activities in human fibrob-
Ž .lasts during radiation-induced G1 arrest, Cell 76 19941013–1023.
w x13 E.E. Medrano, S. Im, F. Yang, Z.A. Abdel-Malek, Ultravio-let B light induces G1 arrest in human melanocytes byprolonged inhibition of retinoblastoma protein phosphoryla-tion associated with long-term expression of the p21 protein,
Ž .Cancer Res. 55 1995 4047–4052.w x14 M. Serrano, G.J. Hannon, D. Beach, A new regulatory motif
in cell cycle control causing specific inhibition of cyclinŽ .DrCDK4, Nature 366 1993 704–707.
w x15 K. Guan, C.W. Jenkins, Y. Li, M.A. Nichols, X. Wu, C.L.O’Keefe, A.G. Matera, Y. Xiong, Growth suppression byp18, a p16INK4rMTS1 and p14INK4BrMTS2-relatedCDK6 inhibitor, correlates with wild-type pRb function,
Ž .Genes Dev. 8 1994 2939–2952.w x16 M.E. Ewen, H.K. Sluss, C.J. Sherr, H. Matsushime, J. Kato,
D.M. Livingston, Functional interactions of the retinoblas-Ž .toma protein with mammalian D-type cyclins, Cell 73 1993
487–497.
w x17 N.K. Hayward, The current situation with regard to humanmelanoma and genetic inferences, Curr. Opin. Oncol. 8Ž .1996 136–142.
w x18 K. Ranade, C.J. Hussussian, R.S. Sikorski, H.E. Varmus,A.M. Goldstein, M.A. Tucker, M. Serrano, G.J. Hannon, D.Beach, N.C. Dracopoli, Mutations associated with familial
Ž .melanoma impair p16INK4 function, Nature Genet. 10 1995114–116.
w x19 A. Kamb, N.A. Gruis, J. Weaver-Feldhaus, Q. Liu, K. Harsh-man, S.V. Tavtigian, E. Stockert, R.S. Day, B.E. Johnson,M.H. Skolnick, A cell cycle regulator potentially involved in
Ž .genesis of many tumor types, Science 264 1994 436–440.w x20 T. Nobori, K. Miura, D.J. Wu, A. Lois, K. Takabayashi,
D.A. Carson, Deletions of the cyclin-dependent kinase-4Ž .inhibitor gene in multiple human cancers, Nature 368 1994
753–756.w x21 P.M. Pollock, J.V. Pearson, N.K. Hayward, Compilation of
somatic mutations of the CDKN2 gene in human cancers:non-random distribution of base substitutions, Genes Chro-
Ž .mosomes Cancer 15 1996 77–88.w x22 X.Q. Wang, B.G. Gabrielli, A. Milligan, J.L. Dickinson,
T.M. Antalis, K.A.O. Ellem, Accumulation of p16CDKN2A inresponse to ultraviolet irradiation correlates with a late S–G2
Ž .phase cell cycle delay, Cancer Res. 56 1996 2510–2514.w x23 T. Wolfel, M. Hauer, J. Schneider, M. Serrano, C. Wolfel, E.
Klehmann-Hieb, E. De Plaen, T. Hankeln, K.-H. Mayer zumBuschenfelde, D. Beach, A p16INK4a-insensitive CDK4 mu-tant targeted by cytolytic T lymphocytes in a human
Ž .melanoma, Science 269 1995 1281–1284.w x24 B.G. Gabrielli, J.M. Clark, A.K. McCormack, K.A.O. Ellem,
Ultraviolet light-induced G2 phase cell cycle checkpointblocks cdc25-dependent progression into mitosis, OncogeneŽ .1997 in press.
w x25 H. Matsushime, D.E. Quelle, S.A. Shurtleff, M. Shibuya,C.J. Sherr, J.-Y. Kato, D-type cyclin-dependent kinase activ-
Ž .ity in mammalian cells, Mol. Cell. Biol. 14 1994 2066–2076.
w x26 R.Y.C. Poon, W. Jiang, H. Toyoshima, T. Hunter, Cyclin-de-pendent kinases are inactivated by a combination of p21 andThr14rTyr15 phosphorylation after UV-induced DNA dam-
Ž .age, J. Biol. Chem. 271 1996 13283–13291.w x27 S.M. Cross, C.A. Sanchez, C.A. Morgan, M.K. Sckimke, S.
Ramel, R.L. Idzerda, W.H. Raskind, B.J. Reid, A p53-depen-Ž .dent mouse spindle checkpoint, Science 267 1995 1353–
1356.w x28 P.M. Pollock, F. Yu, L. Qiu, P.G. Parsons, N.K. Hayward,
Evidence for UV induction of CDKN2 mutations inŽ .melanoma cell lines, Oncogene 11 1995 663–668.





























