Embed Size (px)
Citation preview

ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
ISSN 1651-6206ISBN 978-91-554-8586-3urn:nbn:se:uu:diva-1925312013
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 862
Intensive care Muscle Wasting and Weakness
Underlying Mechanisms, Muscle Specific Differences and a Specific Intervention Strategy
GUIllaUMe ReNaUD

Dissertation presented at Uppsala University to be publicly examined in Hedstrandsalen,Ingång 70, bv, Akademiska sjukhuset, Uppsala, Friday, March 8, 2013 at 13:15 for thedegree of Doctor of Philosophy (Faculty of Medicine). The examination will be conductedin English.
AbstractRenaud, G. 2013. Intensive care Muscle Wasting and Weakness: Underlying Mechanisms,Muscle Specific Differences and a Specific Intervention Strategy. Acta UniversitatisUpsaliensis. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty ofMedicine 862. 57 pp. Uppsala. ISBN 978-91-554-8586-3.
The intensive care unit (ICU) condition, i.e., immobilisation, sedation and mechanicalventilation, often results in severe muscle wasting and weakness as well as a specific acquiredmyopathy, i.e., Acute Quadriplegic Myopathy (AQM). The exact mechanisms underlying AQMremain incomplete, but this myopathy is characterised a preferential myosin loss and a decreasedmuscle membrane leading to a delayed recovery from the primary disease, increased mortalityand morbidity and altered quality of life of survivors. This project aims at improving ourunderstanding of the mechanisms underlying the muscle wasting and weakness associated withAQM and explore the effects of a specific intervention strategy. Time-resolved analyses havebeen undertaken using a unique experimental rodent ICU model and specifically studying themuscle wasting and weakness in limb and diaphragm muscles over a two week period. Further,we used passive mechanical loading in an attempt to alleviate the impaired muscle function andwasting associated with the ICU condition. Subsequently, the knowledge gained from the animalmodel was translated into a clinical study. Mechanical silencing (absence of external and internalstrain) due to immobilisation, pharmacological neuromuscular blockade and sedation, wasidentified as a key factor triggering the muscle wasting and weakness associated with AQM inlimb muscles. In addition, MuRF1, a member of the ubiquitin proteasome degradation pathwayis playing a major role in the contractile protein degradation observed in both the diaphragm andlimb muscles offering a potential candidate for future therapeutic approaches. Moreover, passivemechanical loading resulted in significant positive effects on muscle structure and function inthe rodent ICU model, decreasing muscle atrophy and the loss of force generating capacity. InICU patients passive mechanical loading improved the muscle fibre force generating capacitybut did not affect muscle wasting. Nevertheless, this work strongly supports the importanceof early physical therapy and mobilization in deeply sedated and mechanically ventilated ICUpatients.
Furthermore, we observed significant differences in the phenotype and mechanism underlyingthe loss of force generating capacity between the diaphragm and limb muscles in response tocontrolled mechanical ventilation (CMV) and immobilisation. This knowledge will have to betaken into account when designing intervention strategies to alleviate the muscle wasting andweakness that occurs in mechanically ventilated and immobilized ICU patients.
Guillaume Renaud, Uppsala University, Department of Neuroscience, ClinicalNeurophysiology, Akademiska sjukhuset, SE-751 85 Uppsala, Sweden.
© Guillaume Renaud 2013
ISSN 1651-6206ISBN 978-91-554-8586-3urn:nbn:se:uu:diva-192531 (http://urn.kb.se/resolve?urn=urn:nbn:se:uu:diva-192531)

A mes Neveux et Nièces


List of Papers
This thesis is based on the following papers, which are referred to in the text by their Roman numerals.
I Ochala J, Gustafson AM, Diez ML, Renaud G, Li M, Aare S,
Qaisar R, Banduseela VC, Hedstrom Y, Tang X, Dworkin B, Ford GC, Nair KS, Perera S, Gautel M, and Larsson L. Prefer-ential skeletal muscle myosin loss in response to mechanical si-lencing in a novel rat intensive care unit model: Underlying mechanisms. J Physiol 589: 2007-2026, 2011.
II Renaud† G, Diez† ML, Ravar B, Gorza L, Feng HZ, Jin JP, Cacciani N, Gustafson AM, Ochala J, Corpeño R, Li M, Hed-ström Y, Ford GC, Nair KS, Larsson L. Sparing of muscle mass and function by passive loading in an experimental intensive care unit model. J physiol. In press.
III Diez† ML, Renaud† G, Andersson M, Gonzales Marrero H, Cacciani N, Engquist H, Corpeño R, Artemenko K, Bergquist J, and Larsson L. Intensive care unit muscle wasting: mechanisms and intervention strategies. Crit Care, 16:R209, 2012
IV Renaud G, Corpeño R, Gorza L, Jin JP, Gustafson AM, Iwa-moto H, Yagi N, and Larsson L. Time-course analysis of me-chanical ventilation-induced diaphragm contractile muscle dys-function. In manuscript.
† Contributed equally to the study
Reprints were made with permission from the respective publishers.


Contents
Introduction ................................................................................................... 11 Skeletal Muscle ........................................................................................ 11 Contractile proteins .................................................................................. 13
Actin .................................................................................................... 13 Myosin ................................................................................................. 13
Muscle Contraction .................................................................................. 15 Regulation of muscle mass ....................................................................... 17 Oxidative stress ........................................................................................ 18 Acute Quadriplegic Myopathy ................................................................. 21
Characteristics ..................................................................................... 22 Diagnosis ............................................................................................. 22 Risk factors .......................................................................................... 23 Interventions ........................................................................................ 24
Aims of the present investigation.................................................................. 25
Materials and methods .................................................................................. 26 Animals (I, II and IV) .......................................................................... 26 Human patients (III) ............................................................................ 26 Muscle biopsy and muscle fibre membrane permeabilization (I, II, III and IV) ............................................................................................ 27 Contractile measurements of single muscle fibres (I, II, III and IV) ... 27 Enzyme-histochemistry and immunocytochemistry (I and III) ........... 28 Myosin heavy chain isoform expression (I, II, III and IV) .................. 28 Actin, myosin and total protein quantification (I, II, III and IV) ......... 28 Western blots (I, II and IV) .................................................................. 28 Total RNA isolation and quantification (I, II, III and IV) ................... 29 Quantitative RT-PCR (I, II, III and IV) ............................................... 29 Protein oxidation detection (II and IV) ................................................ 29 Fractional synthesis rate (I, II) ............................................................. 29 Ultrasound measurements (III) ............................................................ 29 Electrophysiological measurements (III) ............................................. 30 Gene expression profiling (II) ............................................................. 30 Post-translational modifications (III) ................................................... 30 X-ray diffraction (IV) .......................................................................... 30 Statistics (I, II, III and IV) ................................................................... 31

Results and Discussion ................................................................................. 32 A rodent model that mimics ICU conditions (I) ....................................... 32 Mechanical silencing is key for triggering AQM (I) ................................ 33 MuRF1 is a key factor underlying muscle proteolysis (I and IV) ............ 33 Passive mechanical loading is beneficial on skeletal muscle (II and III) . 34 Passive mechanical loading reduces the oxidative stress associated with mechanical silencing (II) .................................................................. 35 ICU condition induces specific myosin post-translational modifications (III) ........................................................................................................... 36 Controlled mechanical ventilation causes early contractile dysfunction in the diaphragm (IV) ............................................................................... 37 Diaphragm atrophy differs from the one observed in limb muscles (IV) 37 Mechanical ventilation induces a severe oxidative stress (IV) ................ 38
Conclusions ................................................................................................... 39
Acknowledgments......................................................................................... 41
References ..................................................................................................... 43

Abbreviations
AQM AKT CIM CIP CMAP CMV CS CSA DMS DNPH EDL EMG eNOS FoxO FSR GC GRP94 HMM ICU IGF-1 iNOS LC/MS MAPKs mTOR MUP MuRF MyHC MyLC NF-кB NMBA NO nNOS ONO2
- PTM ROS RNS
Acute quadriplegic myopathy Protein kinase B Critical illness myopathy Critical illness polyneuropathy Compound muscle action potential Controlled mechanical ventilation Costicosteroids Cross-sectional area Direct muscle stimulation Dinitrophenylhydrazine Extensor digitorum longus Electromyography Endothelial nitric oxide synthase Forkhead-box-O Fractional synthesis rate Glucocorticosteroid Glucose regulated protein 94 Heavy meromyosin Intensive care unit Insulin growth factor 1 Inducible nitric oxide synthase Liquid chromatography-mass spectrometry Mitogen activated protein kinase Mammalian target of rapamycin Motor unit potential Muscle specific ring finger Myosin heavy chain Myosin light chain Nuclear factor kappa B Neuromuscular blocking agent Nitric oxide Neuronal nitric oxide synthase Peroxynitrite Post-translational modification Reactive oxygen species Reactive nitrogen species

RyR SERCA SNAP SR TA V0
Ryanodine receptors Sarcoplasmic reticulum Ca2+ pumps Sensory nerve action potential Sarcoplasmic reticulum Tibialis anterior Shortening velocity
11
Introduction
Skeletal Muscle Skeletal muscle, also known as striated muscle, is essential for performing all voluntary movement. It is the largest tissue in the body, comprising 40-45% of the total body weight. Each muscle consists of thousands of muscle fibres.
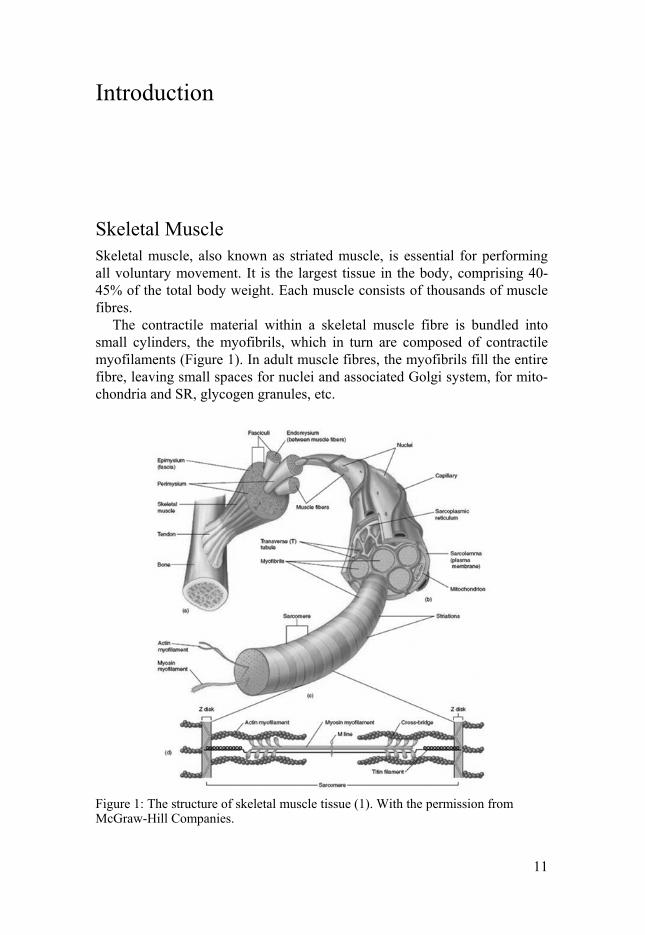
The contractile material within a skeletal muscle fibre is bundled into small cylinders, the myofibrils, which in turn are composed of contractile myofilaments (Figure 1). In adult muscle fibres, the myofibrils fill the entire fibre, leaving small spaces for nuclei and associated Golgi system, for mito-chondria and SR, glycogen granules, etc.
Figure 1: The structure of skeletal muscle tissue (1). With the permission from McGraw-Hill Companies.

12
The myofibrils are composed of repeating units, or segments a few mi-crons in length, called sarcomeres. By convention, the sarcomere starts and ends at the Z-lines or Z-bands: thins bands containing a high density of pro-tein, with a high refractive index and a high electron density in the electron microscope. Between Z-lines are a series of bands with variable light optical and electrical properties that are determined by their structural components (Figure 2).
The centre of the sarcomere is occupied by the A-band, which is optically anisotropic or birefringent. This is due to the presence and parallel aligne-ment of “thick” filaments composed mostly of myosin. The A-band length is the same (1.6 µm) in all vertebrate skeletal muscles, and the edges of the A-band are sharp because the thick filaments have uniform length. The I-band, on either side of the A-band and bisected by the Z-line, is composed of actin and associated proteins. Thin actin filaments are thinner, more flexible, and mostly less well aligned than the thick myosin filament (2). Thin filaments run from the Z-line across the I-band into the A-band, where they overlap with the thick filaments and reach up to the edges of the H-zone. The very central region of the H-zone, called the M-band is very dense due to the presence of cross-links connecting thin filaments into a network. Immedi-ately adjacent to the M-band, on either side of it, is a narrow lighter band, the bridge-free zone where no myosin cross-bridges project from the surface of the thick filament. Sarcomere shortening results in the sliding of thin filaments past the thick filaments into the A-band, so that the I-band and H-zone shorten, while the widths of Z-line, A-band M-line and the bridge-free region do not change. (3, 4).
Figure 2: Structure of a sarcomere (5). With permission from John Wiley & Sons.

13
Contractile proteins Actin At physiological ionic strength, and in the presence of Mg++ and K+, actin monomers (G-actin) polymerize into long thin filaments (F-actin), in which the molecules form a tightly wound “genetic” helix, with a pitch of 5.9 nm. A further twist of the helix gives the filament the appearance of two twisted strands of beads, with crossover points at ~37 nm. The length of actin fila-ments is not stable and thus not uniquely determined, particularly for “cyto-plasmic” actin, but it can be stabilized by capping proteins that block loss and/or addition of monomers at the ends of the filaments. In skeletal muscle, the length of actin filaments depends on the species and muscles in question.
Actin filaments polymerize from the edges of the Z-line, with the “barbed” end at the Z-line end. Anchoring of thin filaments to the Z-line and/or nucleation, a necessary step in the initiation of actin polymerization are mediated by Cap Z, also called beta actinin (6, 7). A second set of pro-teins, part of the formin family, also regulates actin assembly and/or mainte-nance in striated muscle, perhaps acting as elongating factors (8). Capping of the pointed (slow growing) end of the filaments (away from the Z-lines) is important in maintaining the thin filaments’ lengths (9, 10). However, the thin filament lengths are precise in different muscles, therefore, in addition to capping, a guide is necessary; a role nebulin is thought to play.
Skeletal muscle actin is a 5-10 nm wide, 42kDa protein composed of a double helical strand of individual actin molecules (11). Actin has a 2-fold slower turnover rate compared to myosin heavy chain (12). During muscle contraction, actin filament is exposed to myosin heads which then appear as arrowhead-like structures along the filament length.
Myosin Myosin is the force generator of skeletal, cardiac and smooth muscle. The myosin found in muscle was the first protein discovered in what is now a large superfamily of motor proteins (13). Muscle myosin is now referred to as myosin II, or conventional myosin, it is a hexameric protein consisting of four light chains and two heavy chains. Each of the two heavy chains is ap-proximately 200 kDa (14). Each heavy chain is associated with two light chains of approximately 24 kDa each. The heavy chains consist of two dis-tinct regions: the head and the tail. The tail, also named rod, is an alpha-helical coiled coil structure approximately 1500 Å long and 20 Å in diameter (15). The head is the enzymatic region, containing ATP hydrolysis and actin binding sites (16-18). Biochemical studies (16-18) and the first high resolu-tion crystal structure (19) identified regions in the myosin heavy chain head
14
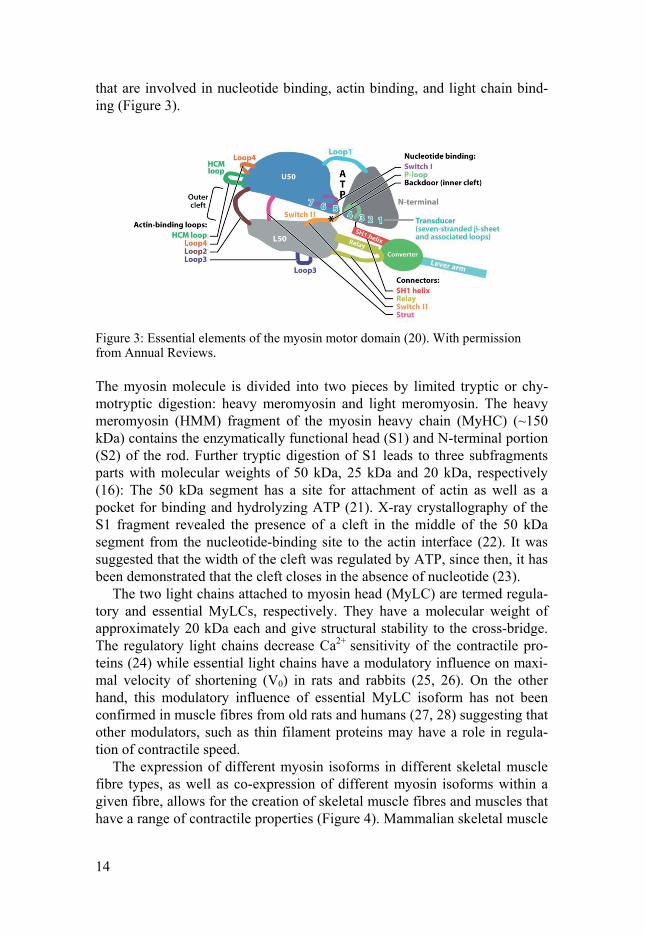
that are involved in nucleotide binding, actin binding, and light chain bind-ing (Figure 3).
Figure 3: Essential elements of the myosin motor domain (20). With permission from Annual Reviews.
The myosin molecule is divided into two pieces by limited tryptic or chy-motryptic digestion: heavy meromyosin and light meromyosin. The heavy meromyosin (HMM) fragment of the myosin heavy chain (MyHC) (~150 kDa) contains the enzymatically functional head (S1) and N-terminal portion (S2) of the rod. Further tryptic digestion of S1 leads to three subfragments parts with molecular weights of 50 kDa, 25 kDa and 20 kDa, respectively (16): The 50 kDa segment has a site for attachment of actin as well as a pocket for binding and hydrolyzing ATP (21). X-ray crystallography of the S1 fragment revealed the presence of a cleft in the middle of the 50 kDa segment from the nucleotide-binding site to the actin interface (22). It was suggested that the width of the cleft was regulated by ATP, since then, it has been demonstrated that the cleft closes in the absence of nucleotide (23).
The two light chains attached to myosin head (MyLC) are termed regula-tory and essential MyLCs, respectively. They have a molecular weight of approximately 20 kDa each and give structural stability to the cross-bridge. The regulatory light chains decrease Ca2+ sensitivity of the contractile pro-teins (24) while essential light chains have a modulatory influence on maxi-mal velocity of shortening (V0) in rats and rabbits (25, 26). On the other hand, this modulatory influence of essential MyLC isoform has not been confirmed in muscle fibres from old rats and humans (27, 28) suggesting that other modulators, such as thin filament proteins may have a role in regula-tion of contractile speed.
The expression of different myosin isoforms in different skeletal muscle fibre types, as well as co-expression of different myosin isoforms within a given fibre, allows for the creation of skeletal muscle fibres and muscles that have a range of contractile properties (Figure 4). Mammalian skeletal muscle

15
fibres express members of three gene families of myosin II heavy chains: fast skeletal, slow skeletal/cardiac, and non muscle, and potentially three additional sarcomeric myosin heavy chain genes: superfast, slow A and and slow B (29). The fast skeletal muscle locus is comprised of six distinct heavy chain genes. These are the embryonic, neonatal, fast type IIa, fast type IIx, fast type IIb, and extraocular. These myosin heavy chain forms are expressed only in skeletal muscle. However, in human muscles, MyHC-IIB is not de-tectable, although the corresponding MYH4 gene is present in the genome, and fibres typed as IIB based on ATPase staining are in fact IIX fibres based on the MyHC composition (30). It has been demonstrated that in addition to pure fibre types, skeletal muscles contain hybrid fibres with a mixed MyHC composition (31-33), this supports the evidence of a spectrum of fibre types: I ↔ I/IIa ↔ IIa ↔ IIa/IIx ↔ IIx ↔ IIx/IIb ↔ IIb. MyHC composition gov-erns fibres oxidative vs. glycolytic metabolic properties. Type I fibres have a high amount of oxidative enzyme and myoglobin complement with lower levels of glyocolytic enzymes whereas type IIb fibres have an opposite metabolic profile.
Figure 4 Panel of sarcomeric MYH genes in mammals with the corresponding pro-tein products and their expression pattern (34). With permission from American Physiological Society.
Muscle Contraction Individual myofibrils are surrounded by an elaborated meshwork of channels called sarcoplasmic reticulum (SR) which release Ca2+ around the myofibrils through ryanodine receptors (RyR). These channels appear anchored to a set of sarcoplasmic invaginations called T-tubules which conduct action poten-tial leading to release of Ca2+ from SR. In the relaxed state, myosin-binding

16
site on actin is covered by tropomyosin. When Ca2+ is released into the cyto-sol, it binds to troponin C which then undergoes conformational change ex-posing the binding sites on actin by displacing tropomyosin which initiates contraction and actomyosin interactions (35).
Figure 5: General kinetic scheme of the actomyosin ATPase cycle (20). With per-mission from Annual Reviews.
Many years of kinetics studies of the actin-myosin interaction have defined an ATP consuming cycle that is catalyzed by actin association (Figure 5). Structural studies have shown that more states exists (20). The ATPase ac-tivity of myosin resides in its conserved motor domain, which interacts with actin, hydrolyses ATP, and produces the force necessary for movement along actin filaments. During the actomyosin ATPase cycle, weak actin-binding states (ATP and ADP-Pi states) alternate with string actin-binding states (ADP states and nucleotide-free or rigor state). ATP binding dissoci-ates the actomyosin complex, and ATP hydrolysis is rapid when myosin is not associated with actin. Phosphate release precedes ADP release and both product release steps are accelerated considerably upon actin binding. Force development occurs when myosin bind strongly to actin and is associated with actin-induced acceleration of phosphate release. We have little insight into the structural intermediates between the initial weak interaction of the pre-powerstroke state with actin, and the release of inorganic phosphate and formation of strong acting binding. However, muscle fibres studies suggest

17
that force generation, and thus strong actin binding, occurs prior to phos-phate release (36). The myosin filament remains stationary during the con-traction with each cross-bridge acting as a force generator. The number of cross-bridges and the force generated per cross bridge determine the amount of force generated by a muscle fibre (37, 38). After the contraction phase is over, Ca2+ is removed from the cytosol by ATP-driven sarcoplasmic reticu-lum Ca2+ pumps (SERCA) (39).
Regulation of muscle mass Recent evidence shows the presence of a well-regulated series of events in skeletal muscle concerning the control of net protein balance that are medi-ated by the relative rates controlling protein synthesis vs. protein degradation (40). For example, IGF-1 stimulates muscle protein synthesis and hypertro-phy via the Akt/mammalian target of rapamycin (mTOR) pathway that leads to the concomitant activation of initiation and elongation factors resulting in the elevation of protein translation and the downregulation of ubiquitin pro-teasome components through Forkhead-box O (FoxO) transcription factors, e.g., atrogin-1 and muscle-specific ring finger 1 (MuRF1) (41-43). In con-trast, downregulation of Akt signalling leads to net protein degradation and subsequent atrophy due to the upregulation of atrogin-1 and MuRF1 (44, 45). Together, this data demonstrate a well-coordinated balance between catabolic and anabolic pathways of multiple protein systems that are regu-lated and maintained to sustain homeostatic and adaptive events in skeletal muscle.
Stretch has been shown to be a powerful stimulation of muscle protein synthesis and growth (46). Numerous studies show that a critical variable in the maintenance of muscle mass, particularly for the muscle to be able to generate force, is that it must generate strain that, in turn, triggers a series of tissue adaptive events. For example, chronically stretching the muscle can increase protein synthesis by adding sarcomeres in parallel and in series so that the sarcomere length is adjusted back to the optimum for force genera-tion, velocity, and hence power output (47-49). Furthermore, when chronic muscle stretch is combined with electrical stimulation, there is a pronounced additive effect on the enhancement of protein synthesis (50).
In response to functional overload of a muscle an increase in mass di-rectly proportional to the increase in amino acid incorporation is observed (51). Within a few hours after an acute bout of resistance exercise or after 2 weeks of resistance training there is a marked increase in protein synthesis as well as myofibrillar protein synthesis in both fast and slow muscles (52-55). Despite an elevated rate of protein degradation after a single bout of resis-tance exercise, there is a greater net increase in protein synthesis, shifting the balance toward greater protein synthesis (55).

18
Similar adaptive phenomena in protein synthesis and degradation occur with models of disuse and injury, i.e., protein synthesis is decreased and protein degradation is increased, leading to net protein loss as observed with denervation (56), hindlimb unloading (57), spinal cord injury (58, 59), bed rest (60), immobilisation (61) and in elderly human subjects (52). Neverthe-less, considerable progress has been made in blunting the deleterious effects observed in skeletal muscle after disuse, or injury using a variety of pharma-cological, electromechanical, and rehabilitative interventions (62, 63).
Oxidative stress Reactive oxygen species (ROS) can be produced by many different chemical reactions originating from cellular events, e.g., metal-catalyzed reactions, mitochondrial electron transport reactions, and neutrophil or macrophage activation during inflammation (64), or external events, e.g., UV light, X-rays, or pollutants. ROS, a natural byproduct of the oxygen metabolism, plays important roles in cellular signaling (65, 66), similar to reactive nitro-gen species (RNS) (64).
Excessive production of ROS within the cells, caused, for example, in muscles by extensive exercise, or reduced capture capabilities of the antioxi-dative systems, can lead to oxidative stress (67). This deleterious condition is defined as an imbalance between ROS production and antioxidant defense systems. Proteins can, therefore, be severely damaged, as they capture 50% to 75% of all highly reactive species with functional groups in their side chains (68). These reactions, among the diverse ROS and different func-tional protein groups, as well as the consecutive reactions among the result-ing primary oxidation products, are not well understood, especially as the primary oxidation products can undergo consecutive reactions with other functional groups of the same protein or other molecules (69). One group of reactions is protein carbonylation, which refers to the production of highly reactive carbonyl groups in proteins that are reactive to hydrazine com-pounds (70). This irreversible modification includes aldehydes and ketones formed via different mechanisms: (i) direct oxidation of the polypeptide backbone leading to truncated peptides with N-terminal α-ketoacyl amino acid residues; (ii) oxidation of the side chains of lysine, arginine, proline, and threonine residues; (iii) reaction of histidine, cysteine, and lysine amino acid residues with aldehydes, e.g., produced by lipid peroxidation; and (iv) glycation (nonenzymatic glycosylation) of lysine residues forming Amadori and Heyns products. Protein carbonylation is used in many studies as a pri-mary marker for oxidative stress. It can be easily quantified with 2,4-dinitrophenylhydrazine (DNPH), which forms 2,4-dinitrophenyl (DNP) de-rivatives with an absorption maximum at 370 nm. DNP-modified proteins can also be quantified with specific anti-DNP-antibodies (71-73).

19
Cellular function is controlled by a myriad of signaling events and ROS and reactive nitrogen species (RNS) are involved in many aspects of this signal-ing. ROS/RNS have classically been associated with “oxidative/nitrosative stress” resulting in cellular dysfunction and disease. However, it is becoming increasingly clear that ROS/RNS are also important components in normal cellular signaling. For instance, ROS can affect muscle cell function via altering the activity of kinases and phosphatases and hence the cellular phos-phorylation status (74, 75). ROS/RNS can induce both acute reversible ef-fects and long-term effects, which are mediated via altered gene and protein expression or via partially irreversible changes in existing proteins and lip-ids. Positive acute effects of ROS/RNS in skeletal muscle include a role in contraction-mediated glucose uptake (76-78) and a long-term effect is the involvement in induction of beneficial adaptations in response to endurance training (79). On the other hand, an excessive ROS production can cause an acute decrease in force production during repeated contractions (80), and prolonged increases in ROS/RNS appear to have pivotal roles in the skeletal muscle dysfunction observed in diseases such as muscle dystrophies and generalized inflammatory diseases (81, 82).
Superoxide anion (O2-•) is the primary oxygen free radical. In skeletal
muscle, O2-• is considered to be mainly produced by mitochondria and by
NADPH oxidases (83). In mitochondria O2-• is produced as a by-product of
oxidative phosphorylation and hence the rate of production would be ex-pected to increase during physical exercise and in other situations with in-creased mitochondrial respiration. In mitochondria, O2
-• is mainly produced by complexes I and III, which release O2
-• to the matrix, and complex III, which releases O2
-• to the intermembrane space (83, 84). Early data indicated that the rate of mitochondrial O2
-• production amounts to 1%–2% of the O2 consumption (85). However, this cannot be the situation in exercising skele-tal muscle, because prolonged physical exercise and endurance training would then result in dangerously high ROS levels, severe oxidative stress, and muscle damage. Accordingly, more recent measurements of mitochon-drial O2
-• production show that the O2-• production is only 0.15% of the O2
consumption (86). It should also be noted that there is no fixed relation be-tween the rate of mitochondrial O2 consumption and ROS production; for instance, uncoupling of oxidative phosphorylation with 2,4-dinitrophenol results in increased O2 consumption accompanied by decreased ROS levels (87) and mitochondrial ROS production appears to be higher during state 4 (basal) as compared to state 3 (maximal ADP-stimulated) respiration, and the latter dominates during aerobic exercise (83).
Nitric oxide (NO•) is the primary nitrogen free radical. NO• is synthesized from the amino acid L-arginine by nitric oxide synthases (NOS) (88). In addition, it was recently shown that NO• can be formed from the inorganic anions nitrate (NO3
-) and nitrite (NO2-) (89). Adult skeletal muscle constitu-
tively coexpresses two NOS isoforms: the neuronal (nNOS or type I) and the

20
endothelial (eNOS or type III) isoform (90). A third isoform, inducible NOS (iNOS or type II), is expressed in skeletal muscle in some inflammatory conditions (90). The release of NO• from isolated rat fast-twitch extensor digitorum longus (EDL) muscles and mouse diaphragm and slow-twitch soleus muscles was increased by electrically stimulated contractions (91, 92). The NO• release both under resting conditions and during repeated con-tractions was similar in eNOS-deficient and wild-type mouse muscles, which indicates that nNOS is the dominating constitutively expressed isoform (92). It should be noted that, with the use of intact whole muscles, it cannot be excluded that an increased NO• production occurred in cell types other than the skeletal muscle fibres. However, a more recent study showed an in-creased rate of intracellular NO• production with electrically induced con-tractions in isolated adult mouse FDB fibres (93). Thus, although less stud-ied than ROS, there is experimental support also for increased RNS produc-tion in muscle fibres during repeated contractions.
Oxidative stress has been shown to occur in disuse and in many patho-logical conditions, and is now widely considered a major trigger of the im-balance between protein synthesis and degradation leading to muscle atrophy (67, 94, 95). However, the causal role of oxidative stress in determining dis-use atrophy has not yet been definitely established. Some contradictory re-sults have been reported, suggesting that care should be taken and avoid over generalising conclusions based on observations from different species, dis-use models and muscles. We will focus on the most widely studied models of disuse in animals (mechanical ventilation, limb immobilisation and hindlimb unloading), and in humans (bed rest, unilateral lower limb suspen-sion, limb immobilisation), and we will consider how conclusive the evi-dence for a causal role of oxidative stress is.
The mechanisms underlying the opposite effects of ROS on muscle ho-meostasis in different conditions are still unclear. It could be that small, compartmentalised, or transient (minutes) increases in ROS mostly modulate intracellular signals by reversible oxidation of specific protein residues (96, 97) and consequently affect gene expression (94, 95, 98, 99). The latter phe-nomenon could occur in response to moderate exercise. In heavy exercise, disuse and pathological conditions, sustained (hours, days), large increases in ROS could (i) have a direct, non-specific, large scale oxidative effect on proteins, which would become more susceptible to proteolysis; (ii) damage plasma membrane and sarcoplasmic reticulum altering calcium homeostasis and activating proteases (e.g. calpains), enhancing proteolysis, (iii) damage lysosome and cause a leakage of catabolic enzymes in the cytosol. Oxidized proteins could be more susceptible to proteolysis because they are more eas-ily targeted by the ubiquitin–proteasome system, which is up-regulated by ROS (100, 101), because their recognition by calpain and caspase is en-hanced (102), or because they could be directly degraded by proteasome without being ubiquitinated (103), or for all the above causes.

21
Figure 6: Major ROS/RNS in muscle and tentative targets (104). With permission from Mary Ann Liebert.
As ROS are short lived and the direct determination of their concentration is complex and exposed to error (105, 106), in most studies on disuse atrophy, ROS activity has been studied ‘indirectly', namely by measuring protein oxidation and lipid peroxidation (107, 108). The latter approaches have been mostly combined with another indirect index of ROS activity, namely the activity or content of antioxidant defense systems among them superoxide dismutase, catalase and glutathione peroxidase. The correlation between muscle atrophy, increase in protein and lipid peroxidation and impairment of antioxidant defense systems has been taken as a strong indication that oxida-tive stress occurred and was involved in muscle wasting (95, 107). The ad-ministration of antioxidants has been used to counteract oxidative stress with the goal of confirming the role of ROS through amelioration of muscle atro-phy (109-111).
Acute Quadriplegic Myopathy Found in mechanical ventilated intensive care unit (ICU) patients, acute quadriplegic myopathy (AQM) is characterised by severe skeletal muscle wasting and persistent weakness leading to impairment of muscle function (112). AQM was first described by MacFarlane and Rosenthal (1977) in a 24-year-old woman with status asthmaticus (113). The patient was mechani-cally ventilated and treated with high doses of costicosteroids (CS) as well as non-depolarizing neuromuscular blocking agents (NMBAs). Three weeks post-ICU the patient was able to walk, but weakness remained 2 months after ICU discharge. This myopathy has been given many different names such as critical illness myopathy (CIM) and thick filament myopathy (112).

22
AQM is a common acquired neuromuscular disorder that affects more than a third of the ICU population (114, 115). The exact cause and underly-ing mechanisms of the disease remain unknown, although mechanical venti-lation, immobilisation, CS, NMBAs have been proposed as triggering fac-tors. The effects of AQM are not limited to muscle weakness, it delays the recovery, increases morbidity and mortality rates. It also has a financial im-pact, partly due to the fact that the average weaning time from mechanical ventilation is doubled in critically ill patients suffering from severe weakness (116).
Characteristics AQM is anticipated in patients with weaning difficulties 7-10 days after mechanical ventilation (117) and is characterised by diffuse flaccid weak-ness of limb muscles as well as severe skeletal muscle atrophy. Abnormal electrophysiology has been demonstrated in AQM patients: a reduced com-pound muscle action potential (CMAP), short duration and low motor unit potentials (MUP) but intact sensory nerve action potentials (SNAP). Muscle membrane excitability is decreased resulting in decreased CMAP amplitude independent on if the muscle is stimulated directly or indirectly via the mo-tor nerve (118). A preferential myosin loss and myosin associated proteins is pathognomonic of AQM (119, 120). The ratio between actin and myosin has been used as a tool for diagnosing AQM (119, 120). The progression of the onset of weakness is unknown, but weakness has been detected as early as 4 days after ICU treatment (121).
Diagnosis AQM diagnosis is difficult for several reasons. The main reason is that col-laboration of the patient is necessary for major electromyography criteria (EMG) and ICU patients are usually unable to respond due to deep sedation, NMBAs or encephalopathy (122). In addition, it is not possible to differenti-ate between polyneuropathy and myopathy using conventional EMG. An-other reason is the co-existence of critical illness polyneuropathy (CIP) which can lead to a mis-diagnosis. Furthermore, other factors can interfere with AQM diagnosis such as pharmacological treatments and underlying diseases.
Nevertheless, several criteria (112, 123, 124) has been proposed for AQM diagnosis which include SNAP amplitudes >80% of the lower limit for two or more nerves, needle EMG with short-duration, reduced muscle membrane excitability using direct muscle stimulation (DMS), low amplitude MUPs with an early or normal full recruitment with or without fibrillation poten-tials, absence of a decremental response on repetitive nerve stimulation, muscle histopathologic findings of myopathy with myosin loss. Despite not

23
being specific to AQM, serum CK level increase has also been suggested as criteria for an evolving myopathy (115).
At the moment, a clear diagnosis of AQM can only be made through muscle biopsy, an invasive method. A muscle biopsy allows us to look for AQM typical features, which are not usually observed in neuropathic disor-ders such as selective loss of myosin and myosin associated proteins and decreased myosin:actin ratio determined by gel electrophoresis (125).
Risk factors Corticosteroids Glucocorticosteroids (GC) are highly lipid soluble and easily partition into the cell membrane. Inside the cell, these hormones act at the transcriptional level, inhibiting protein synthesis and also increasing mRNAs encoding for ubiquitin and proteasome subunits (126).
Administration of GC in ICU patients is still debated, weighting the bene-fits during sepsis or autoimmune disorders against negative effects. Nerver-theless, there is conflicting data concerning the association between CS treatment and the occurrence of AQM. In a study, CS were suggested as a key factor in developing muscle weakness in ICU (127), however, several studies failed to show a consistent relationship between CS treatment and the development of AQM (128, 129).
Immobilisation Immobilisation induces muscles changes and muscle weakness, although the time required for atrophy to occur in healthy individuals is much longer (around 30 days) (130) than in AQM patients in whom a severe atrophy is seen after a few days of immobilisation. In addition, in healthy controls, bed rest induces a 3% loss of muscle mass per week (131) but never reaches the severity observed in AQM patients, even after extensive time periods (130, 132). Inactivity is associated with muscle atrophy, characterised by loss of myonuclei, myosin filament defects, an increase in proteolytic enzymes as well as an increase in pro-inflammatory cytokines (133).
Neuromuscular blocking agents NMBAs are large molecules that contain regions similar to acetylcholine. They are given to facilitate mechanical ventilation in ICU patients and im-prove arterial partial pressure of oxygen. There are different non-depolarizing muscle relaxants exists, such as aminosteroid and histamine non-depolarizing muscle relaxants. Which NMBA to use is patient depend-ent, as they have different effects based on the excretion method as well as secondary effects. A positive correlation between the dose and duration of NMBA treatment and the occurrence of myopathy has been described (115,

24
134). Nowadays, the use of NMBAs in theICU is decreasing as they are associated with an increased ICU stay, delayed weaning and higher mortality and morbidity rates (135). The relationship between NMBA administration and the development of AQM is not clear and contradictory results have been reported, i.e., a link between prolonged weakness after long term or high NMBA doses have been reported by some (129, 136) but not by others (127, 137).
Other risk factors Other factors have been speculated to be implicated in the development of AQM such as sepsis and mitochondrial dysfunction (138, 139). Mechanical ventilation duration has also been hypothesised to be strongly related to nerve disfunction (140).
Interventions To date there is no specific AQM treatment, but different therapies have been suggested to reduce the severity of the myopathy such as reducing the use of CS/NMBAs and nutritional interventions. Recently, early mobilisa-tion has been proposed as a best candidate to slow the development of myopathy in ICU patients. Early physical therapy has proven beneficial in ICU patients, reducing the length of the stay, decreasing muscle atrophy and loss in muscle strength, resulting in a quicker recovery time (141-144). Due to immobilisation and deep sedation of ICU patients, active movement has proven to be difficult to use, therefore passive movement has been suggested as an alternative. While passive movement does not prevent AQM (145), it slows the progression of skeletal muscle atrophy. Several studies have shown that passive movement reduces protein loss and muscle fibre size (146). The mechanism behind such changes might be related to the spring-like protein titin, which is an important mechanosensor in the sarcomere and is implicated in hypertrophy and protein turnover (147).

25
Aims of the present investigation
The general objective of this thesis was to achieve a better understanding about the cellular and molecular events occurring during the muscle atrophy and weakness in Acute Quadriplegic Myopathy patients and to assess thera-peutic strategies.
This thesis can be divided into four parts. The first part (Paper I) identifies mechanical silencing as a critical triggering factor for developing AQM in a rodent ICU model. The second part (Paper II) assesses the efficacy of pas-sive mechanical loading in the ICU rodent model. The third part (Paper III) involves the translation of the knowledge obtain from the rodent model into a clinical intervention study. The fourth part (Paper IV) investigates the spe-cific response of diaphragm muscle to long-term controlled mechanical ven-tilation.
Specific aims: 1) To follow the time course changes of skeletal muscle structure and func-
tion, as well as mRNA and protein expression during AQM (Paper I). 2) To determine the mechanisms underlying the preferential myosin loss
and impaired skeletal muscle structure and function response to immobi-lisation and controlled mechanical ventilation. (Paper I).
3) To determine the effects of passive mechanical loading on muscle wast-ing and function in an experimental rat ICU model (Paper II).
4) To identify the time course of gene expression pattern in response to passive mechanical loading in an experimental rat ICU model (Paper II).
5) To determine the effects of passive mechanical loading on muscle wast-ing and function in ICU patients (Paper III).
6) To follow the time course changes of diaphragm muscle structure and function, as well as mRNA and protein expression during the ICU con-dition (Paper IV).

26
Materials and methods
In this section, methods from the work are summarized; the reader is referred to the individual paper (I-IV) for further details.
Animals (I, II and IV) Female Sprague-Dawley rats were anaesthetized (isoflurane), mechanically ventilated, monitored and pharmacologically paralyzed with α-cobratoxin for durations varying from 6h to 14 days. The sham-operated control animals were anesthetized (isoflurane), spontaneously breathing, given intra-arterial and intra-venous solutions, and sacrificed within two hours after the initial anaesthesia and surgery, but they were not pharmacologically paralyzed with α –cobratoxin. The left leg of the animal (II) was activated for 6 hours at the shortest duration and 12 hours per day at durations 12 hours and longer throughout the experiment, using a mechanical lever arm that produced con-tinous passive mechanical ankle joint flexions-extensions at a speed of 13.3 cycles per minute.
Human patients (III) A total of seven (four females and three males, aged 56-67 years) mechani-cally ventilated ICU patients numbered M1–M7 were included in this study. Patients anticipated to require mechanical ventilation for 10 consecutive days or longer were recruited. Patients had typically been exposed to me-chanical ventilation and immobilisation for 0-3 days (1.7 ± 0.9 days) prior to initiating the intervention and monitoring due to delays related to obtaining signed informed consents. Patients with a previous history of neuromuscular disease were not included in the study. No evidence based guidelines for severe sepsis were found in these patients. None of them received systemic administration of neuromuscular blocking agents (NMBA) and only one patient (M1) received administration of inhaled corticosteroids (daily dose: 1 mg) due to asthma. Propofol was intravenously administered in all patients. The patients left leg was subjected to passive mechanical loading for 2.5 hours, 4 times per day during 7-11 days, i.e., continuous passive anatomi-cally correct motion from 30° plantar flexion to 25° dorsiflexion were gener-ated at a speed corresponding to 150° per minute.

27
Muscle biopsies were obtained from the tibialis anterior (TA) muscle on both the unloaded and loaded side using the percutaneous conchotome method at the final day of the observation period. Muscle biopsies were not taken at the start of the intervention in order to eliminate the risk of the first biopsy sampling procedure interfering with the results from the biopsy ob-tained at the end of the observation period.
Muscle biopsy and muscle fibre membrane permeabilization (I, II, III and IV) In paper III, muscle samples were obtained from both the loaded and unloaded TA muscle and when specified, from the vastus lateralis, using percutaneous conchotome method at the final day of the observation period.
In papers I, II and IV, the diaphragm, TA, plantaris, EDL, gastrocnemius and soleus muscles were dissected from the loaded left leg and the unloaded right leg immediately after euthanasia. One half of the diaphragm, soleus, TA and EDL muscles together with plantaris and gastrocnemius muscles were quickly frozen in liquid propane cooled by liquid nitrogen and stored at -160 °C for further analyses. The other halves of the diaphragm, soleus, EDL and TA muscles were immediately placed in ice-cold relaxing solution (in mmol/l: 100KCL, 20 imidazole, 7 MgCL2, 2 EGTA, 4ATP, pH 7.0: 4 °C). Small bundles of ~25-50 fibres were dissected free from the muscle and tied with surgical silk to glass capillary tubes at slightly stretched lengths. The bundles were then placed in skinning solution (relaxing solution containing glycerol, 50:50 vol/vol) at 4 °C for 24h, after which they were transferred to -20 °C. All the bundles were cryo-protected within one-two weeks after skinning and subsequently snap frozen in liquid propane cooled by liquid nitrogen and stored at -160 °C for further studies in single muscle fibre.
Contractile measurements of single muscle fibres (I, II, III and IV) To assess fibre force generating capacity and structure, skinned single mus-cle fibre experiments were conducted in EDL and soleus rat muscles (paper I and II), TA from ICU patients (paper III) and diaphragm rat muscle (paper IV). A fibre segment 1 to 2 mm long was attached to a force transducer (model 400A, Aurora Scientific) and a lever arm system (model 308B, Aurora Scientific). While the fibre segments were in relaxing solution, the sarcomere length was set to 2.65-2.75 µm by adjusting the overall segment length and resting tension was assessed. The fibre was then moved to acti-vating solution (pCa 4.5) in which all the recordings were done. The focus-ing control of the microscope was used as a micrometer. Fibre cross-sectional area (CSA) was calculated from the diameter and depth, assuming

28
an elliptical circumference, and was corrected for the 20% swelling that is known to occur during skinning.
Enzyme-histochemistry and immunocytochemistry (I and III) CSA measurement was performed using enzyme histochemistry. Cross-sections (10µm) were cut perpendicular to the longitudinal axis of muscle fibres with a cryostat at -20 °C. The sections were stained for myofibrillar ATPase after alkaline and acid pre-incubations (paper I) and NADH (paper III). CSAs, roundness and the smaller diameter were measured in a total of 50-100 muscle fibres in the central region of the biopsy cross-section using an inverted microscope and imaging software.
To detect the presence of a specific protein and its localization in the muscle fibre, immunocytochemistry was performed. Cross-sections (8µm) were cut perpendicular to the longitudinal axis of muscle fibres with a cry-ostat at -20 °C. Expression and subcellular localisation of MuRF1/2, p62, serum response factor (SRF) (paper I), nNOS (paper III) and laminin (paper III) were performed. All samples were stained with identical primary and secondary antibody dilutions and immunofluorescence was analysed by con-focal microscopy.
Myosin heavy chain isoform expression (I, II, III and IV) MyHC isoform expression was detected by 6% SDS-PAGE. The gels were silver stained, scanned and the volume integration function was used to de-termine the fibre type proportion.
Actin, myosin and total protein quantification (I, II, III and IV) Actin and myosin content was determined by 12% SDS-PAGE. The gels were stained with coomassie blue, scanned and the volume integration func-tion was used to quantify the amount of myosin and actin which were nor-malized to total protein content. Total protein content was determined using the NanoOrange Protein Quanti-fication Kit (Invitrogen, Carlsbad, CA, USA) in papers I and II, or Pierce 660 Protein assay (Thermo Fisher Scientific Inc, Rockford, IL, USA) in pa-per III, according to manufacturer’s instructions. The fluorescence of the samples was measured and related to a standard curve.
Western blots (I, II and IV) Identification and quantification specific proteins were performed using western blots. In paper I, HSP-70, aB-crystallin, atrogin-1, calpain-1, LC3B and MuRF1 content were measured in the EDL. In paper II, Troponin (Tn)

29
isoform expression was measured in the TA, Grp94 protein levels were quantified in the plantaris muscle and atrogin-1, calpain-1 and MuRF1 were measured in the soleus. In paper IV, calpain-1, LC3B and MuRF1 content were measured in the diaphragm. The immunoblots were scanned in a soft laser densitometer and the signal intensities were quantified using the vol-ume integration function and normalized to actin content or total protein.
Total RNA isolation and quantification (I, II, III and IV) Total RNA was extracted from frozen tissue (10-30mg) using Qiagen RNeasy Mini Kit (Quigen, Inc., Valencia, CA). RNA-concentration were then quantified using the fluorescent nucleic acid stain, Ribogreen (Molecu-lar Probes, Eugene, OR), on a fluorescence spectrophotometer.
Quantitative RT-PCR (I, II, III and IV) qRT-PCR was used to quantify mRNA levels for MyHCs isoforms, skeletal a-actin, myosin binding proteins, as well as MAFbx/atrogin-1, MRF1, cal-pain-1 and Map11c3b.
Protein oxidation detection (II and IV) The levels of protein carbonyl groups were assessed in the diaphragm ( pa-per IV) and the plantaris muscle (paper II) using the OxyBlot protein oxida-tion detection kit (Chemicon, Hampshire, UK) according to manufacturer’s instructions. Levels of oxidized proteins were quantified using the NIH Im-ageJ analysis software and normalized to the densitometric value of the Pon-ceau red staining of the corresponding albumin band.
Fractional synthesis rate (I, II) Fractional protein synthesis rate (FSR) was used to measure the rate of mus-cle protein synthesis in the superficial and deep part of the grastrocnemius using [ring-13C6]phenylalanine (15µg/g body weight) as tracer.
Ultrasound measurements (III) The tibialis anterior (TA) CSAs of the ICU patients were measured every day during the intervention period (7-11days) using real-time ultrasound scanner. Scans were taken transversally on relaxed muscles at three loca-tions: 50, 40 and 30% of the distance from the proximal part of the fibula head to the distal tip of the lateral malleolus. The captured muscle images were stored, the region of interest (TA muscle mass) was manually selected

30
and CSA was measured. CSA mean was calculated as the mean of the three consecutive measurements at the three different locations (50, 40 and 30%).
Electrophysiological measurements (III) To investigate the properties of the nerve-muscle interaction, electrophysiol-ogy was performed. Motor (n. fibularis, and tibialis) and sensory nerve (n. suralis and fibularis superficialis) were measured bilaterally using surface electrode both for stimulation and recording. Studies were performed on the first and final day of the observation period in all patients. At the final day of the period, concentric needle EMG was performed in the m. vastus lateralis and tibialis anterior bilaterally.
Gene expression profiling (II) A micro-array was used to examine the gene expression in response to pas-sive mechanical loading in the rodent ICU model in a time-resolved manner. Three micrograms of total RNA from the proximal gastrocnemius muscle samples were extracted and processed to generate biotin-labeled cRNA. Samples were then hybridized to an Affimetrix Rat Gene 1.0 ST Array (Af-fimetrix,). Microarray data were background-adjusted, normalized and log-transformed summarized values. In order to search for the differentially ex-pressed genes between samples from different days, an empirical Bayes moderated t-test was applied. A linear model was fitted to the data unloaded vs. loaded at the following time durations: 6h-4 days, 5-8 days, and 9-14 days. To address the problem with multiple testing, the p-values were ad-justed according to Benjamini and Hochberg. Probe sets with a minimum fold change of ± 1.5 and adjusted p < 0.05 in at least one time point were included in further analyses.
Post-translational modifications (III) Liquid chromatography–mass spectrometry (LC/MS) analysis of myosin heavy chain isoforms was performed to investigate the post-translational modifications of myosin.
X-ray diffraction (IV) Structural analysis of diaphragm fibre bundles was assessed by x-ray diffrac-tion. On the day of x-ray recordings, fibre bundles were placed in a plastic dish containing a relaxing solution and washed thoroughly to remove the glycerol. They were then transferred to a specimen chamber filled with the same relaxing solution. Each fibre bundle was then fixed (clamped ends)

31
and slightly stretched. Subsequently, x-ray diffraction patterns were re-corded at 15 °C.´
Statistics (I, II, III and IV) SigmaPlot software (Systat Software, Inc., CA, USA) was used to generate descriptive statistics. Means, standard errors of the means (SEM) and linear regression analysis were calculated according to standard procedures. Paired t-test was used in pairwise comparisons between unloaded and loaded leg. One way analysis of variance (ANOVA) and the Tukey post-hoc test were used when comparing multiple groups. When the normality test failed, a one way ANOVA on ranks, i.e., Kruskal–Wallis, and the Dunn's post-hoc were performed. Differences were considered significant at p < 0.05.

32
Results and Discussion
A rodent model that mimics ICU conditions (I) The knowledge of etiological factors underlying AQM remains insufficient, e.g. relative importance of different agents linked to the disease, the mecha-nisms underlying the reduced muscle membrane excitability, the signaling pathways regulating protein synthesis and degradation and reassembly of the contractile apparatus during recovery. Typically, AQM is not diagnosed until 2–3 weeks after admission to the ICU. This delayed diagnosis has signifi-cantly hampered our understanding of the mechanisms underlying AQM. Accordingly, there is a need for an experimental animal model where the possible causative agents and the mechanisms underlying AQM can be stud-ied in detail. One accepted animal model for AQM involves peripheral den-ervation of the hind limb and administration of corticosteroids in the rat (148, 149). This model is insufficient to study this ICU-related disease, as patients are immobilized, mechanically ventilated, have an underlying sys-temic illness, and receive a number of pharmacological agents, apart from corticosteroids. Previous studies have also used a porcine ICU model with significant advantages such as similarities with humans, regarding size and metabolism as well as being an established model to study mechanical venti-lation (150-152). Nevertheless, due to the size and cost of the animal, it is not practical for long duration experiments.
In this project, we have used a unique experimental rat ICU model where animals were mechanically ventilated, sedated, pharmacologically paralysed with NMBAs, and extensively monitored between 6 hours and 14 days. This model allows for muscle unloading, mechanical ventilation, sedation and blocked neuromuscular transmission during an extended period of time simi-lar to modern intensive care treatment. Advantages of this model include the absence of confounding factors such as pharmacological treatment and sys-temic diseases. Moreover, results showed that, 1) muscles fibre size and contractibility were preserved during the first 4 days followed by a severe muscle atrophy and contractile dysfunction and 2) preferential myosin loss was observed, which is considered pathognomonic of AQM (119). This is in accordance with a study in ICU patients, where no signs of atrophy were detected before the first four days in the ICU (121) as well as previous stud-ies using a porcine experimental model in which muscle fibre mass, contrac-tility and myofibrillar proteins were preserved during 5 days of ICU condi-

33
tions (153, 154). Therefore, this rodent ICU model mimics the ICU condi-tions of long-term mechanical ventilation, sedation and muscle unloading.
Mechanical silencing is key for triggering AQM (I) To date, multiple risk factors have been proposed for the development of AQM such as: immobilisation, prolonged mechanical ventilation, NMBAs, and CS. A number of other factors have been suggested as causative agents: underlying systemic illness, severe asthma, sepsis, multi-organ failure, hy-perglycemia and malnutrition (112, 155, 156). However, studies have shown that AQM could be acquired in the absence of CS and/or NMBAs, and inde-pendent of underlying disease (127, 129, 137, 157), thus, a discrepancy ex-ists in the literature in regard to causative agents and the mechanisms under-lying AQM remains to be deciphered. Nevertheless, ICU intervention per se, i.e., immobilisation, prolonged mechanical ventilation and sedation, appears to play a major role in the development of AQM.
Using the rodent experimental ICU model we showed that immobilisa-tion, prolonged mechanical ventilation, and sedation, alone induced muscle wasting, impaired muscle function and a preferential loss of myosin, a fea-ture pathognomonic of AQM (112, 119). Therefore, mechanical silencing (absence of external strain and internal strain) that occurs during the ICU condition is forwarded as a major factor triggering AQM, while other factors such as CS, NMBas contribute to aggravate its prognosis (119, 158, 159).
MuRF1 is a key factor underlying muscle proteolysis (I and IV) We investigated in a time course manner which proteolysis pathways were involved in the muscle wasting associated with ICU intervention (I). The three main proteolysis pathways were studied: ubiquitin proteasome, ly-sosomal and calcium activated calpain systems. The ubiquitin proteasome is the main degradation pathway in cells (160). In addition, it has been shown that MuRF1, a muscle specific ubiquitin ligase, is capable of degrading the thick filament protein myosin without the activation of other pathways (161). Nevertheless, some studies have shown that calpains and caspases play an important role in protein degradation under disuse conditions. In an attempt to improve our understanding of the mechanisms underlying this particular muscle atrophy, the rat ICU model was used.
A specific temporal pattern of proteolytic pathways was found during 14 days of ICU intervention. At the gene level, an early up-regulation of the atrogenes, atrogin-1 and MuRF1 and Lc3b, a marker of autophagy lysosome

34
from 6 hours to 4 days was observed, atrogenes up-regulation was main-tained until 8 days while Lc3b mRNA level returned to normal after 4 days. Calpain-1 expression, a marker of the calpain system remained unchanged throughout the entire duration. This was paralleled by a severe down-regulation of actin and MyHC isoforms at the transcriptional level from 5 to 14 days. At the protein level, MuRF1 was up-regulated at early durations (6h to 14 days), while Atrogin-1 content remained the same during the observa-tion period. LC3b and calpain-1, taken as markers of the autophy-lyosome and calpain systems respectively, increased their expression from 9 to 14 days.
In addition to protein degradation, we suspected RING finger proteins MuRF1/2 to play a role at the transcription level. MuRF1/2 are both associ-ated with the giant sarcomeric protein titin (162), under stress they can trans-locate to the nucleus and alter the transcription of a number of genes, MyHC and actin in particular (162) (163). In addition, nuclear translocation of MuRF2 represses serum response factor transcriptional activity (147). In the rat ICU model, 4 days of the ICU condition induced a strong nuclear translo-cation of MuRF1/2 which persisted up to 9 days, furthermore, a perinuclear accumulation of both MuRFs was observed. Moreover, after 4 days, MuRF2 and its ligand p62 colocalized with SRF in the nucleus. After 9 days, a peri-nuclear colocalization of MuRF2, p62, and SRF was observed. Therefore, MuRFs do not only control the degradation of the thick filaments proteins, but are likely to play a role in the transcriptional downregulation of contrac-tile proteins observed under ICU conditions.
Similar results were observed in the diaphragm in response to controlled mechanical ventilation (CMV) (IV), i.e., an early increase in MuRF1 protein content while Calpain-1 and LC3B contents did not change significantly until 5 days of exposure. Thus, early loss of myosin is suggested to be caused by activation of MuRF1 and activation of the ubiquitin-proteasome pathway. This is in accordance with observations by Cohen and colleagues showing that myosin is primarily degraded by a MuRF1 dependent ubiquity-lation without the activation of calpains or caspases (164). The subsequent activation of the autophagy-lysosomal pathway and calpains system may be responsible for the muscle atrophy as suggested by studies in other animal models (165).
Passive mechanical loading is beneficial on skeletal muscle (II and III) Mechanical silencing was postulated as being a key factor underlying muscle wasting associated with ICU conditions. Therefore, a physical therapy inter-vention was tested in order to test this hypothesis. Using the rat ICU model

35
(II), animals were subjected to unilateral passive mechanical loading for 12 hours per day for up to 14 days. Results showed a significant decrease in muscle atrophy associated with the ICU condition, independent of muscle type. At the single fibre level, a 44% higher CSA and a doubling of the force generating capacity was observed. This was paralleled by a sparing of my-osin protein as well as transcriptional up-regulation of contractile proteins and muscle growth factors. This is in accordance with other studies that have shown that static passive stretching suppresses muscle wasting associated with disuse (166, 167).
In addition, a reduced MuRF1 protein content was observed in response to passive mechanical loading. Coupled with the higher myosin content, this indicates that passive mechanical loading has a beneficial effects on prote-olysis.
As passive mechanical loading had shown beneficial effects in the rodent ICU model, a similar approach was taken in ICU patients. Seven ICU pa-tients were subjected to unilateral passive mechanical loading for 2.5 hours, 4 times per day during 9 ± 1 days. Although it was not sufficient to counter-act muscle atrophy, muscle fibre function was increased by 35%. This dis-crepancy between the experimental model and the clinical study may be explained by background heterogeneities in patients compared with animal models, the influence of underlying disease in the ICU patients, species- and age-related differences, the delayed monitoring of muscle mass in ICU patients due to delays in obtaining informed signed consents.
The beneficial effect of passive mechanical loading on alleviating the muscle atrophy in sedated, pharmacologically paralyzed and mechanically ventilated rats together with the improvement in muscle function seen in both the experimental rat model and the ICU patients support the importance of early physical mobility therapy in deeply sedated and mechanically venti-lated ICU patients, resulting in shortening of ventilator days and ICU stay, as well as reducing the financial costs and enhancing patients prognosis and quality of life of survivors (143, 168, 169).
Passive mechanical loading reduces the oxidative stress associated with mechanical silencing (II) During disuse conditions, an increase in ROS and RNS has been reported and evidences suggest that they play a role in muscle atrophy. Under a physiological conditions, ROS and RNS control several signalling events such as NF-KB, MAPKs and FoxO, and are needed for muscle contractions. However, an imbalance between oxidant and antioxidants lead to an oxida-tive stress which can be detrimental to cells and results in muscle atrophy (170). We investigated the impact of passive mechanical loading on carbonyl

36
levels in the rat ICU model. Carbonylation is a common marker of oxidative stress since it is an irreversible reaction and carbonyls are quite stable (70, 171). Results showed that protein carbonyl levels increase due to the ICU intervention after 9 to 14 days. However, on the loaded side, protein carbon-yls levels remained unchanged coupled with a decrease of genes involved in the oxidative stress response. In addition, the oxidative stress response was also examined by quantifying the calcium sensitive chaperone GRP94 pro-tein content. GRP94 over expression has been shown to protect against oxi-dative stress as well as apoptosis induced by ischemia and ER-stress (172, 173). GRP94 protein content decreased on both sides during 6h to 4 days but was followed by a high increase on the loaded side from 5 to 8 days com-pared with the unloaded side. Taken together, these results indicate that 1) passive loading has a protective role against the oxidative stress associated with the ICU condition; 2) it remains unknown whether oxidative stress plays a key role in the early degradation of myosin protein and thus it should be addressed in future studies.
ICU condition induces specific myosin post-translational modifications (III) As passive mechanical loading resulted in an increase in fibre contractility but did not affect myosin loss, it was hypothesized that passive loading al-tered protein quality and thus, induced MyHC post-translational modifica-tions (PTMs). However, mass-spectrometry analysis did not detect any spe-cific MyHC PTMs associated with passive loading in ICU patients. Other sarcomeric proteins could be affected by loading and explained the differ-ence in contractibility as it has been shown that modifications of actin, MyLC or tropomyosin could result in an impaired muscle function (174, 175).
Mass-spectrometry analysis revealed specific PTMs in immobilized, se-dated and mechanically ventilated ICU patients, which were absent in healthy controls. Almost all the new modifications were located deep in the motor domain of the myosin, a region less prone to oxidative modifications due to its structure and the presence of hydrophobic domains (176); there-fore, PTMs in the myosin motor domain may be a sign of acute oxidative stress. We speculated that nNOS plays a role in these modifications as three out of the five new PTMs were deamidations and it is known that nitrite anions can induce the deamidation of proteins (177). nNOS generates NO that produces peroxynitrite (ONO2
-), a reactive oxidant and nitrating agent that is tightly regulated under physiological conditions but under acute oxi-dative stress has detrimental effects (178). Results showed an increase of nNOS levels (an indicator of NO activity) and a shift from the sarcolemma

37
to the cytoplasm. This dislocation has also been reported during muscle at-rophy caused by hind-limb suspension or amyotrophic lateral sclerosis (179, 180), demonstrating that cytoplasmic nNOS produces NO and up-regulates MuRF1 and atrogin1 via FoxO3 (180). In conclusion, nNOS has arisen as a key factor underlying the specific myosin PTMs induced by mechanical silencing. These new PTMs may affect the regulation of contraction and lead to muscle proteolysis (103, 181).
Controlled mechanical ventilation causes early contractile dysfunction in the diaphragm (IV) An increasing number of studies indicate that prolonged CMV is a key factor triggering the impairment in diaphragm muscle function (152, 182-187). To date, however, clinical and experimental studies have primarily focused on the early effects of CMV (182-184), or at specific time points (152, 186, 187). In an attempt to unravel the underlying mechanisms, a temporal analy-sis of diaphragm muscle was performed using the rat ICU model.
In accordance with previous studies (174, 182-184), the force generation capacity of individual diaphragm muscle fibres was impaired after early exposure to CMV. However, during the initial 4 days of mechanical ventila-tion, single fibre size was maintained in accordance with our previous obser-vations in the diaphragm muscle fibres in a porcine experimental ICU model, i.e., pigs exposed to sedation and mechanical ventilation for five days alone or in combination with neuromuscular blockade, sepsis and/or sepsis (174). In other studies, on the other hand, severe atrophy has been reported in response to a very short duration of mechanical ventilation in both ex-perimental and clinical studies (184, 188, 189). The discrepancy between the results may be due to other confounding factors beside mechanical ventila-tions when commercially available ventilators are used even at durations shorter than 24 hours.
Diaphragm atrophy differs from the one observed in limb muscles (IV) In contrast to our previous observations in limb muscles where both fibre size and specific force was maintained in the 0.25 to 4 day group (190), dia-phragm fibre specific force was impaired after early exposure to CMV. Sur-prisingly, myosin:actin ratio remained unchanged during the whole duration of the experiment and myosin protein content decreased slightly in a linear manner over time indicating that actin proteolysis occurs and is not specific to myosin in the diaphragm muscle in contrast to limb muscles (190). Those

38
results are in accordance with a recent study which reported MyHC and actin degradation in brain dead organ donors following 18 to 72 hours of CMV (191). Nevertheless, the contractile protein proteolysis does not correlate with the dramatic decrease in force generating capacity suggesting that pro-tein degradation alone is not responsible for the impaired force production. One possible mechanism behind the significant loss of muscle function could be sarcomeric protein PTMs induced by CMV as it has been shown that PTMs could affect muscle fibre function (192, 193).
Moreover, at the gene level, myosin and actin expression remained un-changed until 9 to 14 days of exposure to CMV contrary to our previous observations in limb muscles where dramatic decreases of myosin and actin mRNA expression were observed early during exposure and remained de-pressed at long durations (190) demonstrating a significant difference in the transcriptional regulation of contractile proteins between diaphragm and limb muscles.
Mechanical ventilation induces a severe oxidative stress (IV) CMV has been repeatedly shown to induce oxidative stress and ROS produc-tion (184, 194, 195). In accordance with those studies, we observed an in-creased amount of protein carbonyls after early exposure to CMV and re-mained elevated until 8 days. It is suggested that the myosin degradation is a response to an early oxidative stress via a MuRF1 dependent ubiquitylation process which in part explains the impaired force producing capacity, while the activation of the calpains and lysosomal pathways are caused by a maintained oxidative stress and accumulation of ROS leading to a reduced CSA. Abnormal ROS levels result in mitochondrial dysfunction (196, 197) that may lead to intrinsic apoptosis and activation of the proteasome pathway (198). A recent study showed that treating rats with the antioxidant Trolox before and during exposure to 12 hours of CMV prevented oxidative damage to the diaphragm as well as the activation of caspases-3 and calpains which supports such hypothesis (195).

39
Conclusions
This study has investigated the mechanisms of AQM and shown that the ICU condition per se, i.e., immobilisation, sedation and mechanical ventila-tion induces severe muscle atrophy, a dramatic decrease in muscle fibre force generating capacity and preferential myosin loss, features which are similar to what is observed in ICU patients with AQM. In addition, complete mechanical silencing of the muscle (absence of external and internal strain), due to pharmacological paralysis and sedation is forwarded as a major player in triggering AQM. Time-resolved analysis showed an early and maintained up-regulation (6h-14d) of MuRF1 at the protein level, identifying the ubiq-uitin proteasome pathway as a major player in the specific myosin degrada-tion associated with AQM.
Further investigation of the localization of MuRFs revealed that in addition to the up-regulation at the protein level, a translocation from the cytoplasm to the nucleus occurred, indicating their role in the transcriptional down-regulation of sarcomeric proteins. Thus, MuRFs are good candidates for pharmacological treatment and blocking either their activation or transloca-tion could delay the development of AQM.
In ICU patients we observed an increased expression of nNOS coupled with a cytoplasmic translocation. While nNOS cytoplasmic translocation has been reported to trigger atrophy through MuRFs activation (180), in our study we could not determine whether it was the cause of the muscle atrophy or a con-sequence and thus further studies are still needed.
Passive mechanical loading resulted in a significant positive effect on mus-cle wasting and function in the rodent ICU model. It also decreased the amount of myosin protein lost in the slow twitch soleus muscle as well as MuRF1 protein content. On the other hand, in immobilized, sedated and mechanically ventilated ICU patients, while we also observed a positive effect on muscle function, passive mechanical loading did not affect muscle wasting. Nevertheless, those results support the use of early physical therapy among immobilized ICU patients.
The lack of preferential myosin loss and early oxidative stress observed in the diaphragm in response to CMV and immobilisation differs dramatically to what is occurring in limb muscles. This underlines muscle type differ-

40
ences in the mechanism behind the loss of muscle function associated with the ICU condition. This knowledge will have to be taken into account when designing intervention strategies to alleviate the muscle wasting and weak-ness that is found in mechanically ventilated and immobilized ICU patients.

41
Acknowledgments
This work was carried out at the Clinical Neurophysiology Department, Uppsala University, Uppsala, Sweden, with collaborations at the Department of Chemistry, BMC, Uppsala University, Uppsala, Sweden; Hershey Medi-cal Center, Hershey, USA; the Mayo Clinic College of Medicine, Rochester, USA; King´s College London BHF Centre for Research Excellence, London, UK; the Division of Endocrinology, Wayne State University, Detroit, USA; the Departement of Sciences, Padova University, Padova, Italy, and at Japan Synchrotron Radiation Research Institute, Sayo-cho, Sayo-gun, Hyogo, Ja-pan. I would like to thank all staff, colleagues and friends for their direct or indi-rect contribution to this work. I would like to express my sincere gratitude to: Professor Lars Larsson, my supervisor for giving me fantastic opportunities, being always honest and for pushing me to strive harder. Dr Julien Ochala, my co-supervisor, for all the scientific or non-scientific talks and for all the help you have provided me with during all these years. Professor Naoto Yagi and Dr Hiroyuki Iwamoto, for their kindness and for granting me the opportunity to discover a new part of Japan. Professor Jonas Berquist and Dr Konstantin Artemenko, for their invaluable support. My old office mate: Rizwan, for all the good times and cooking sessions. All the current and previous lab members for their help and support as well as the enjoyment of ¨fika¨. Inger Hedlund and Gun Schönnings, for taking care of all the administrative duties. All the people at Ängström, without you all the last year and a half would not have been the same. Especially Seif, for dragging me back to the gym

42
and for our endless talks. Girma, for outrunning Seif. Mir Mahadi, for our cooking sessions late at night. My friends in France and Switzerland, for their support and for all the crazy events. A Flo, pour être aussi charmante que casse-pieds! A mes parents, vous m’avez aidé toutes ces années durant et pour cela je vous serait toujours reconnaissant. Ainsi qu’au reste de ma famille, pour leur soutien. Tout spécialement mon neveu Matthieu, ce message est pour toi . A Sandrine, en dépit de la distance tu m’as toujours soutenu quelque soit mes decisions. And to you reader for taking the time to read this thesis. This doctoral thesis was financially supported by the Swedish Research Coundil (8651), the European Commission (MyoAge, EC Fp7 CT-223756 and COST CM 1001), King Gustaf V and Queen Victoria’s Foundation, STINT, Association Française contre les Myopathies, Uppsala University and Uppsala University Hospital.
Guillaume Renaud 22nd January 2013

43
References
1. Seeley, R., Stephens, T., and Tate, P. (2008) Anatomy and Physiology, McGraw-Hill Companies
2. Craig, R. (2004) Molecular structure of the sarcomere, McGraw-Hill, New York
3. Huxley, H., and Hanson, J. (1954) Changes in the cross-striations of muscle during contraction and stretch and their structural interpretation. Nature 173, 973-976
4. Huxley, A. F., and Niedergerke, R. (1954) Structural changes in muscle during contraction; interference microscopy of living muscle fibres. Nature 173, 971-973
5. Tortora, G. J., and Derrickson, B. H. (2008) Principles of Anatomy and Physiology, John Wiley & Sons
6. Casella, J. F., Casella, S. J., Hollands, J. A., Caldwell, J. E., and Cooper, J. A. (1989) Isolation and characterization of cDNA encoding the alpha subunit of Cap Z(36/32), an actin-capping protein from the Z line of skeletal muscle. Proc Natl Acad Sci U S A 86, 5800-5804
7. Maruyama, K. (2002) beta-Actinin, Cap Z, connectin and titin: what's in a name? Trends Biochem Sci 27, 264-266
8. Taniguchi, K., Takeya, R., Suetsugu, S., Kan, O. M., Narusawa, M., Shiose, A., Tominaga, R., and Sumimoto, H. (2009) Mammalian formin fhod3 regulates actin assembly and sarcomere organization in striated muscles. J Biol Chem 284, 29873-29881
9. Gregorio, C. C., Weber, A., Bondad, M., Pennise, C. R., and Fowler, V. M. (1995) Requirement of pointed-end capping by tropomodulin to maintain actin filament length in embryonic chick cardiac myocytes. Nature 377, 83-86
10. Almenar-Queralt, A., Lee, A., Conley, C. A., Ribas de Pouplana, L., and Fowler, V. M. (1999) Identification of a novel tropomodulin isoform, skeletal tropomodulin, that caps actin filament pointed ends in fast skeletal muscle. J Biol Chem 274, 28466-28475
11. Au, Y. (2004) The muscle ultrastructure: a structural perspective of the sarcomere. Cellular and molecular life sciences : CMLS 61, 3016-3033
12. Martin, A. F. (1981) Turnover of cardiac troponin subunits. Kinetic evidence for a precursor pool of troponin-I. J Biol Chem 256, 964-968
13. Odronitz, F., and Kollmar, M. (2007) Drawing the tree of eukaryotic life based on the analysis of 2,269 manually annotated myosins from 328 species. Genome Biol 8, R196

44
14. Biró, N. A. S., L. Bálint, M. (1972) Studies on the helical segment of the myosin molecule. Cold Spring Harb Symp Quant Biol, 55
15. Huxley, H. E. (1963) Electron Microscope Studies on the Structure of Natural and Synthetic Protein Filaments from Striated Muscle. J Mol Biol 7, 281-308
16. Mornet, D., Bertrand, R. U., Pantel, P., Audemard, E., and Kassab, R. (1981) Proteolytic approach to structure and function of actin recognition site in myosin heads. Biochemistry 20, 2110-2120
17. Sutoh, K. (1982) An actin-binding site on the 20K fragment of myosin subfragment 1. Biochemistry 21, 4800-4804
18. Sutoh, K. (1983) Mapping of actin-binding sites on the heavy chain of myosin subfragment 1. Biochemistry 22, 1579-1585
19. Rayment, I., Holden, H. M., Whittaker, M., Yohn, C. B., Lorenz, M., Holmes, K. C., and Milligan, R. A. (1993) Structure of the actin-myosin complex and its implications for muscle contraction. Science 261, 58-65
20. Sweeney, H. L., and Houdusse, A. (2010) Structural and functional insights into the Myosin motor mechanism. Annu Rev Biophys 39, 539-557
21. Vibert, P. J. (1988) Domain structure of the myosin head in correlation-averaged images of shadowed molecules. Journal of muscle research and cell motility 9, 147-155
22. Rayment, I., Rypniewski, W. R., Schmidt-Base, K., Smith, R., Tomchick, D. R., Benning, M. M., Winkelmann, D. A., Wesenberg, G., and Holden, H. M. (1993) Three-dimensional structure of myosin subfragment-1: a molecular motor. Science 261, 50-58
23. Coureux, P. D., Wells, A. L., Menetrey, J., Yengo, C. M., Morris, C. A., Sweeney, H. L., and Houdusse, A. (2003) A structural state of the myosin V motor without bound nucleotide. Nature 425, 419-423
24. Metzger, J. M., and Moss, R. L. (1992) Myosin light chain 2 modulates calcium-sensitive cross-bridge transitions in vertebrate skeletal muscle. Biophys J 63, 460-468
25. Greaser, M. L., Moss, R. L., and Reiser, P. J. (1988) Variations in contractile properties of rabbit single muscle fibres in relation to troponin T isoforms and myosin light chains. J Physiol 406, 85-98
26. Sweeney, H. L., Kushmerick, M. J., Mabuchi, K., Sreter, F. A., and Gergely, J. (1988) Myosin alkali light chain and heavy chain variations correlate with altered shortening velocity of isolated skeletal muscle fibers. J Biol Chem 263, 9034-9039
27. Li, X., and Larsson, L. (1996) Maximum shortening velocity and myosin isoforms in single muscle fibers from young and old rats. Am J Physiol 270, C352-360
28. Larsson, L., Li, X., and Frontera, W. R. (1997) Effects of aging on shortening velocity and myosin isoform composition in single human skeletal muscle cells. Am J Physiol 272, C638-649

45
29. Desjardins, P. R., Burkman, J. M., Shrager, J. B., Allmond, L. A., and Stedman, H. H. (2002) Evolutionary implications of three novel members of the human sarcomeric myosin heavy chain gene family. Mol Biol Evol 19, 375-393
30. Smerdu, V., Karsch-Mizrachi, I., Campione, M., Leinwand, L., and Schiaffino, S. (1994) Type IIx myosin heavy chain transcripts are expressed in type IIb fibers of human skeletal muscle. Am J Physiol 267, C1723-1728
31. Schiaffino, S., Gorza, L., Sartore, S., Saggin, L., Ausoni, S., Vianello, M., Gundersen, K., and Lomo, T. (1989) Three myosin heavy chain isoforms in type 2 skeletal muscle fibres. J Muscle Res Cell Motil 10, 197-205
32. DeNardi, C., Ausoni, S., Moretti, P., Gorza, L., Velleca, M., Buckingham, M., and Schiaffino, S. (1993) Type 2X-myosin heavy chain is coded by a muscle fiber type-specific and developmentally regulated gene. J Cell Biol 123, 823-835
33. Termin, A., Staron, R. S., and Pette, D. (1989) Myosin heavy chain isoforms in histochemically defined fiber types of rat muscle. Histochemistry 92, 453-457
34. Schiaffino, S., and Reggiani, C. (2011) Fiber types in mammalian skeletal muscles. Physiol Rev 91, 1447-1531
35. Squire, J. M. (1975) Muscle filament structure and muscle contraction. Annual review of biophysics and bioengineering 4, 137-163
36. Dantzig, J. A., Goldman, Y. E., Millar, N. C., Lacktis, J., and Homsher, E. (1992) Reversal of the cross-bridge force-generating transition by photogeneration of phosphate in rabbit psoas muscle fibres. J Physiol 451, 247-278
37. Martyn, D. A., Smith, L., Kreutziger, K. L., Xu, S., Yu, L. C., and Regnier, M. (2007) The effects of force inhibition by sodium vanadate on cross-bridge binding, force redevelopment, and Ca2+ activation in cardiac muscle. Biophys J 92, 4379-4390
38. McDonald, K. S., and Fitts, R. H. (1995) Effect of hindlimb unloading on rat soleus fiber force, stiffness, and calcium sensitivity. J Appl Physiol 79, 1796-1802
39. Toyoshima, C., Nakasako, M., Nomura, H., and Ogawa, H. (2000) Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature 405, 647-655
40. Goll, D. E., Neti, G., Mares, S. W., and Thompson, V. F. (2008) Myofibrillar protein turnover: the proteasome and the calpains. J Anim Sci 86, E19-35
41. Sacheck, J. M., Hyatt, J. P., Raffaello, A., Jagoe, R. T., Roy, R. R., Edgerton, V. R., Lecker, S. H., and Goldberg, A. L. (2007) Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J 21, 140-155

46
42. Bodine, S. C., Stitt, T. N., Gonzalez, M., Kline, W. O., Stover, G. L., Bauerlein, R., Zlotchenko, E., Scrimgeour, A., Lawrence, J. C., Glass, D. J., and Yancopoulos, G. D. (2001) Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3, 1014-1019
43. Leger, B., Cartoni, R., Praz, M., Lamon, S., Deriaz, O., Crettenand, A., Gobelet, C., Rohmer, P., Konzelmann, M., Luthi, F., and Russell, A. P. (2006) Akt signalling through GSK-3beta, mTOR and Foxo1 is involved in human skeletal muscle hypertrophy and atrophy. J Physiol 576, 923-933
44. Latres, E., Amini, A. R., Amini, A. A., Griffiths, J., Martin, F. J., Wei, Y., Lin, H. C., Yancopoulos, G. D., and Glass, D. J. (2005) Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem 280, 2737-2744
45. Sandri, M., Sandri, C., Gilbert, A., Skurk, C., Calabria, E., Picard, A., Walsh, K., Schiaffino, S., Lecker, S. H., and Goldberg, A. L. (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117, 399-412
46. Goldspink, G. (1999) Changes in muscle mass and phenotype and the expression of autocrine and systemic growth factors by muscle in response to stretch and overload. J Anat 194 ( Pt 3), 323-334
47. Spector, S. A., Gardiner, P. F., Zernicke, R. F., Roy, R. R., and Edgerton, V. R. (1980) Muscle architecture and force-velocity characteristics of cat soleus and medial gastrocnemius: implications for motor control. J Neurophysiol 44, 951-960
48. Tabary, J. C., Tabary, C., Tardieu, C., Tardieu, G., and Goldspink, G. (1972) Physiological and structural changes in the cat's soleus muscle due to immobilisation at different lengths by plaster casts. J Physiol 224, 231-244
49. Loughna, P., Goldspink, G., and Goldspink, D. F. (1986) Effect of inactivity and passive stretch on protein turnover in phasic and postural rat muscles. J Appl Physiol 61, 173-179
50. Goldspink, G., Scutt, A., Loughna, P. T., Wells, D. J., Jaenicke, T., and Gerlach, G. F. (1992) Gene expression in skeletal muscle in response to stretch and force generation. Am J Physiol 262, R356-363
51. Goldberg, A. L. (1968) Protein synthesis during work-induced growth of skeletal muscle. J Cell Biol 36, 653-658
52. Yarasheski, K. E., Zachwieja, J. J., and Bier, D. M. (1993) Acute effects of resistance exercise on muscle protein synthesis rate in young and elderly men and women. Am J Physiol 265, E210-214
53. MacDougall, J. D., Tarnopolsky, M. A., Chesley, A., and Atkinson, S. A. (1992) Changes in muscle protein synthesis following heavy resistance exercise in humans: a pilot study. Acta Physiol Scand 146, 403-404

47
54. Moore, D. R., Phillips, S. M., Babraj, J. A., Smith, K., and Rennie, M. J. (2005) Myofibrillar and collagen protein synthesis in human skeletal muscle in young men after maximal shortening and lengthening contractions. Am J Physiol Endocrinol Metab 288, E1153-1159
55. Phillips, S. M., Tipton, K. D., Aarsland, A., Wolf, S. E., and Wolfe, R. R. (1997) Mixed muscle protein synthesis and breakdown after resistance exercise in humans. Am J Physiol 273, E99-107
56. Goldspink, D. F. (1976) The effects of denervation on protein turnover of rat skeletal muscle. Biochem J 156, 71-80
57. Goldspink, D. F., Morton, A. J., Loughna, P., and Goldspink, G. (1986) The effect of hypokinesia and hypodynamia on protein turnover and the growth of four skeletal muscles of the rat. Pflugers Arch 407, 333-340
58. Haddad, F., Roy, R. R., Zhong, H., Edgerton, V. R., and Baldwin, K. M. (2003) Atrophy responses to muscle inactivity. II. Molecular markers of protein deficits. J Appl Physiol 95, 791-802
59. Kim, J. A., Roy, R. R., Kim, S. J., Zhong, H., Haddad, F., Baldwin, K. M., and Edgerton, V. R. (2010) Electromechanical modulation of catabolic and anabolic pathways in chronically inactive, but neurally intact, muscles. Muscle Nerve 42, 410-421
60. Ferrando, A. A., Lane, H. W., Stuart, C. A., Davis-Street, J., and Wolfe, R. R. (1996) Prolonged bed rest decreases skeletal muscle and whole body protein synthesis. Am J Physiol 270, E627-633
61. Booth, F. W., and Seider, M. J. (1979) Early change in skeletal muscle protein synthesis after limb immobilisation of rats. J Appl Physiol 47, 974-977
62. Edgerton, V. R., Roy, R. R., Allen, D. L., and Monti, R. J. (2002) Adaptations in skeletal muscle disuse or decreased-use atrophy. Am J Phys Med Rehabil 81, S127-147
63. Fong, A. J., Roy, R. R., Ichiyama, R. M., Lavrov, I., Courtine, G., Gerasimenko, Y., Tai, Y. C., Burdick, J., and Edgerton, V. R. (2009) Recovery of control of posture and locomotion after a spinal cord injury: solutions staring us in the face. Prog Brain Res 175, 393-418
64. Pfeilschifter, J., Eberhardt, W., and Huwiler, A. (2001) Nitric oxide and mechanisms of redox signalling: matrix and matrix-metabolizing enzymes as prime nitric oxide targets. Eur J Pharmacol 429, 279-286
65. D'Autreaux, B., and Toledano, M. B. (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8, 813-824
66. Thannickal, V. J., and Fanburg, B. L. (2000) Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol 279, L1005-1028
67. Moylan, J. S., and Reid, M. B. (2007) Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve 35, 411-429
68. Chandel, N. S., and Budinger, G. R. (2007) The cellular basis for diverse responses to oxygen. Free Radic Biol Med 42, 165-174

48
69. Stadtman, E. R., and Levine, R. L. (2000) Protein oxidation. Ann N Y Acad Sci 899, 191-208
70. Dalle-Donne, I., Giustarini, D., Colombo, R., Rossi, R., and Milzani, A. (2003) Protein carbonylation in human diseases. Trends Mol Med 9, 169-176
71. Goto, S., Nakamura, A., Radak, Z., Nakamoto, H., Takahashi, R., Yasuda, K., Sakurai, Y., and Ishii, N. (1999) Carbonylated proteins in aging and exercise: immunoblot approaches. Mech Ageing Dev 107, 245-253
72. Alamdari, D. H., Kostidou, E., Paletas, K., Sarigianni, M., Konstas, A. G., Karapiperidou, A., and Koliakos, G. (2005) High sensitivity enzyme-linked immunosorbent assay (ELISA) method for measuring protein carbonyl in samples with low amounts of protein. Free Radic Biol Med 39, 1362-1367
73. Conrad, C. C., Choi, J., Malakowsky, C. A., Talent, J. M., Dai, R., Marshall, P., and Gracy, R. W. (2001) Identification of protein carbonyls after two-dimensional electrophoresis. Proteomics 1, 829-834
74. Avner, B. S., Hinken, A. C., Yuan, C., and Solaro, R. J. (2010) H2O2 alters rat cardiac sarcomere function and protein phosphorylation through redox signaling. Am J Physiol Heart Circ Physiol 299, H723-730
75. Wright, V. P., Reiser, P. J., and Clanton, T. L. (2009) Redox modulation of global phosphatase activity and protein phosphorylation in intact skeletal muscle. J Physiol 587, 5767-5781
76. Deshmukh, A. S., Long, Y. C., de Castro Barbosa, T., Karlsson, H. K., Glund, S., Zavadoski, W. J., Gibbs, E. M., Koistinen, H. A., Wallberg-Henriksson, H., and Zierath, J. R. (2010) Nitric oxide increases cyclic GMP levels, AMP-activated protein kinase (AMPK)alpha1-specific activity and glucose transport in human skeletal muscle. Diabetologia 53, 1142-1150
77. Roberts, C. K., Barnard, R. J., Scheck, S. H., and Balon, T. W. (1997) Exercise-stimulated glucose transport in skeletal muscle is nitric oxide dependent. Am J Physiol 273, E220-225
78. Sandstrom, M. E., Zhang, S. J., Bruton, J., Silva, J. P., Reid, M. B., Westerblad, H., and Katz, A. (2006) Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. J Physiol 575, 251-262
79. Ristow, M., Zarse, K., Oberbach, A., Kloting, N., Birringer, M., Kiehntopf, M., Stumvoll, M., Kahn, C. R., and Bluher, M. (2009) Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A 106, 8665-8670
80. Moopanar, T. R., and Allen, D. G. (2006) The activity-induced reduction of myofibrillar Ca2+ sensitivity in mouse skeletal muscle is reversed by dithiothreitol. J Physiol 571, 191-200

49
81. Tidball, J. G., and Wehling-Henricks, M. (2007) The role of free radicals in the pathophysiology of muscular dystrophy. J Appl Physiol 102, 1677-1686
82. Yamada, T., Place, N., Kosterina, N., Ostberg, T., Zhang, S. J., Grundtman, C., Erlandsson-Harris, H., Lundberg, I. E., Glenmark, B., Bruton, J. D., and Westerblad, H. (2009) Impaired myofibrillar function in the soleus muscle of mice with collagen-induced arthritis. Arthritis Rheum 60, 3280-3289
83. Powers, S. K., and Jackson, M. J. (2008) Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev 88, 1243-1276
84. Murphy, M. P. (2009) How mitochondria produce reactive oxygen species. Biochem J 417, 1-13
85. Chance, B., Sies, H., and Boveris, A. (1979) Hydroperoxide metabolism in mammalian organs. Physiol Rev 59, 527-605
86. St-Pierre, J., Buckingham, J. A., Roebuck, S. J., and Brand, M. D. (2002) Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 277, 44784-44790
87. Okuda, M., Lee, H. C., Kumar, C., and Chance, B. (1992) Comparison of the effect of a mitochondrial uncoupler, 2,4-dinitrophenol and adrenaline on oxygen radical production in the isolated perfused rat liver. Acta Physiol Scand 145, 159-168
88. Kobzik, L., Stringer, B., Balligand, J. L., Reid, M. B., and Stamler, J. S. (1995) Endothelial type nitric oxide synthase in skeletal muscle fibers: mitochondrial relationships. Biochem Biophys Res Commun 211, 375-381
89. Lundberg, J. O., and Weitzberg, E. (2010) NO-synthase independent NO generation in mammals. Biochem Biophys Res Commun 396, 39-45
90. Stamler, J. S., and Meissner, G. (2001) Physiology of nitric oxide in skeletal muscle. Physiol Rev 81, 209-237
91. Balon, T. W., and Nadler, J. L. (1994) Nitric oxide release is present from incubated skeletal muscle preparations. J Appl Physiol 77, 2519-2521
92. Hirschfield, W., Moody, M. R., O'Brien, W. E., Gregg, A. R., Bryan, R. M., Jr., and Reid, M. B. (2000) Nitric oxide release and contractile properties of skeletal muscles from mice deficient in type III NOS. Am J Physiol Regul Integr Comp Physiol 278, R95-R100
93. Pye, D., Palomero, J., Kabayo, T., and Jackson, M. J. (2007) Real-time measurement of nitric oxide in single mature mouse skeletal muscle fibres during contractions. J Physiol 581, 309-318
94. Powers, S. K., Duarte, J., Kavazis, A. N., and Talbert, E. E. (2010) Reactive oxygen species are signalling molecules for skeletal muscle adaptation. Exp Physiol 95, 1-9
95. Powers, S. K., Kavazis, A. N., and DeRuisseau, K. C. (2005) Mechanisms of disuse muscle atrophy: role of oxidative stress. Am J Physiol Regul Integr Comp Physiol 288, R337-344

50
96. Ghezzi, P. (2005) Oxidoreduction of protein thiols in redox regulation. Biochem Soc Trans 33, 1378-1381
97. Janssen-Heininger, Y. M., Mossman, B. T., Heintz, N. H., Forman, H. J., Kalyanaraman, B., Finkel, T., Stamler, J. S., Rhee, S. G., and van der Vliet, A. (2008) Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med 45, 1-17
98. Jackson, M. J., Papa, S., Bolanos, J., Bruckdorfer, R., Carlsen, H., Elliott, R. M., Flier, J., Griffiths, H. R., Heales, S., Holst, B., Lorusso, M., Lund, E., Oivind Moskaug, J., Moser, U., Di Paola, M., Polidori, M. C., Signorile, A., Stahl, W., Vina-Ribes, J., and Astley, S. B. (2002) Antioxidants, reactive oxygen and nitrogen species, gene induction and mitochondrial function. Mol Aspects Med 23, 209-285
99. Ji, L. L., Gomez-Cabrera, M. C., Steinhafel, N., and Vina, J. (2004) Acute exercise activates nuclear factor (NF)-kappaB signaling pathway in rat skeletal muscle. FASEB J 18, 1499-1506
100. Davies, K. J. (1987) Protein damage and degradation by oxygen radicals. I. general aspects. J Biol Chem 262, 9895-9901
101. Shang, F., Gong, X., and Taylor, A. (1997) Activity of ubiquitin-dependent pathway in response to oxidative stress. Ubiquitin-activating enzyme is transiently up-regulated. J Biol Chem 272, 23086-23093
102. Smuder, A. J., Kavazis, A. N., Hudson, M. B., Nelson, W. B., and Powers, S. K. (2010) Oxidation enhances myofibrillar protein degradation via calpain and caspase-3. Free Radic Biol Med 49, 1152-1160
103. Grune, T., Merker, K., Sandig, G., and Davies, K. J. (2003) Selective degradation of oxidatively modified protein substrates by the proteasome. Biochem Biophys Res Commun 305, 709-718
104. Westerblad, H., and Allen, D. G. (2011) Emerging roles of ROS/RNS in muscle function and fatigue. Antioxid Redox Signal 15, 2487-2499
105. Smith, M. A., and Reid, M. B. (2006) Redox modulation of contractile function in respiratory and limb skeletal muscle. Respir Physiol Neurobiol 151, 229-241
106. Palomero, J., Pye, D., Kabayo, T., Spiller, D. G., and Jackson, M. J. (2008) In situ detection and measurement of intracellular reactive oxygen species in single isolated mature skeletal muscle fibers by real time fluorescence microscopy. Antioxid Redox Signal 10, 1463-1474
107. Lawler, J. M., Song, W., and Demaree, S. R. (2003) Hindlimb unloading increases oxidative stress and disrupts antioxidant capacity in skeletal muscle. Free Radic Biol Med 35, 9-16
108. Urso, M. L., and Clarkson, P. M. (2003) Oxidative stress, exercise, and antioxidant supplementation. Toxicology 189, 41-54
109. Kondo, H., Miura, M., Nakagaki, I., Sasaki, S., and Itokawa, Y. (1992) Trace element movement and oxidative stress in skeletal muscle atrophied by immobilisation. Am J Physiol 262, E583-590

51
110. Appell, H. J., Duarte, J. A., and Soares, J. M. (1997) Supplementation of vitamin E may attenuate skeletal muscle immobilisation atrophy. Int J Sports Med 18, 157-160
111. Arbogast, S., Smith, J., Matuszczak, Y., Hardin, B. J., Moylan, J. S., Smith, J. D., Ware, J., Kennedy, A. R., and Reid, M. B. (2007) Bowman-Birk inhibitor concentrate prevents atrophy, weakness, and oxidative stress in soleus muscle of hindlimb-unloaded mice. J Appl Physiol 102, 956-964
112. Lacomis, D., Zochodne, D. W., and Bird, S. J. (2000) Critical illness myopathy. Muscle Nerve 23, 1785-1788
113. MacFarlane, I. A., and Rosenthal, F. D. (1977) Severe myopathy after status asthmaticus. Lancet 2, 615
114. Lacomis, D., Petrella, J. T., and Giuliani, M. J. (1998) Causes of neuromuscular weakness in the intensive care unit: a study of ninety-two patients. Muscle Nerve 21, 610-617
115. Douglass, J. A., Tuxen, D. V., Horne, M., Scheinkestel, C. D., Weinmann, M., Czarny, D., and Bowes, G. (1992) Myopathy in severe asthma. Am Rev Respir Dis 146, 517-519
116. De Jonghe, B., Bastuji-Garin, S., Sharshar, T., Outin, H., and Brochard, L. (2004) Does ICU-acquired paresis lengthen weaning from mechanical ventilation? Intensive Care Med 30, 1117-1121
117. Maramattom, B., Wijdicks, E. F., Sundt, T. M., and Cassivi, S. D. (2004) Flaccid quadriplegia due to necrotizing myopathy following lung transplantation. Transplant Proc 36, 2830-2833
118. Rich, M. M., and Pinter, M. J. (2001) Sodium channel inactivation in an animal model of acute quadriplegic myopathy. Ann Neurol 50, 26-33
119. Larsson, L., Li, X., Edstrom, L., Eriksson, L. I., Zackrisson, H., Argentini, C., and Schiaffino, S. (2000) Acute quadriplegia and loss of muscle myosin in patients treated with nondepolarizing neuromuscular blocking agents and corticosteroids: mechanisms at the cellular and molecular levels. Crit Care Med 28, 34-45
120. Matsumoto, N., Nakamura, T., Yasui, Y., and Torii, J. (2000) Analysis of muscle proteins in acute quadriplegic myopathy. Muscle Nerve 23, 1270-1276
121. Ahlbeck, K., Fredriksson, K., Rooyackers, O., Maback, G., Remahl, S., Ansved, T., Eriksson, L., and Radell, P. (2009) Signs of critical illness polyneuropathy and myopathy can be seen early in the ICU course. Acta Anaesthesiol Scand 53, 717-723
122. Bolton, C. F. (2005) Neuromuscular manifestations of critical illness. Muscle Nerve 32, 140-163
123. Latronico, N., and Bolton, C. F. (2011) Critical illness polyneuropathy and myopathy: a major cause of muscle weakness and paralysis. Lancet Neurol 10, 931-941

52
124. Stevens, R. D., Marshall, S. A., Cornblath, D. R., Hoke, A., Needham, D. M., de Jonghe, B., Ali, N. A., and Sharshar, T. (2009) A framework for diagnosing and classifying intensive care unit-acquired weakness. Crit Care Med 37, S299-308
125. Stibler, H., Edstrom, L., Ahlbeck, K., Remahl, S., and Ansved, T. (2003) Electrophoretic determination of the myosin/actin ratio in the diagnosis of critical illness myopathy. Intensive Care Med 29, 1515-1527
126. Kaminsky, P. (2001) [Endocrine myopathies]. Rev Prat 51, 289-293 127. De Jonghe, B., Sharshar, T., Lefaucheur, J. P., Authier, F. J., Durand-
Zaleski, I., Boussarsar, M., Cerf, C., Renaud, E., Mesrati, F., Carlet, J., Raphael, J. C., Outin, H., and Bastuji-Garin, S. (2002) Paresis acquired in the intensive care unit: a prospective multicentre study. JAMA 288, 2859-2867
128. Hough, C. L., Steinberg, K. P., Taylor Thompson, B., Rubenfeld, G. D., and Hudson, L. D. (2009) Intensive care unit-acquired neuromyopathy and corticosteroids in survivors of persistent ARDS. Intensive Care Med 35, 63-68
129. Behbehani, N. A., Al-Mane, F., D'Yachkova, Y., Pare, P., and FitzGerald, J. M. (1999) Myopathy following mechanical ventilation for acute severe asthma: the role of muscle relaxants and corticosteroids. Chest 115, 1627-1631
130. Bloomfield, S. A. (1997) Changes in musculoskeletal structure and function with prolonged bed rest. Med Sci Sports Exerc 29, 197-206
131. Berg, H. E., Larsson, L., and Tesch, P. A. (1997) Lower limb skeletal muscle function after 6 wk of bed rest. J Appl Physiol 82, 182-188
132. LeBlanc, A. D., Schneider, V. S., Evans, H. J., Pientok, C., Rowe, R., and Spector, E. (1992) Regional changes in muscle mass following 17 weeks of bed rest. J Appl Physiol 73, 2172-2178
133. Winkelman, C. (2007) Inactivity and inflammation in the critically ill patient. Crit Care Clin 23, 21-34
134. Lacomis, D., Giuliani, M. J., Van Cott, A., and Kramer, D. J. (1996) Acute myopathy of intensive care: clinical, electromyographic, and pathological aspects. Ann Neurol 40, 645-654
135. Prielipp, R. C. (1998) Pharmacology, selection and complications associated with neuromuscular blocking drugs in ICU patients. Yale J Biol Med 71, 469-484
136. Segredo, V., Caldwell, J. E., Matthay, M. A., Sharma, M. L., Gruenke, L. D., and Miller, R. D. (1992) Persistent paralysis in critically ill patients after long-term administration of vecuronium. N Engl J Med 327, 524-528
137. de Letter, M. A., Schmitz, P. I., Visser, L. H., Verheul, F. A., Schellens, R. L., Op de Coul, D. A., and van der Meche, F. G. (2001) Risk factors for the development of polyneuropathy and myopathy in critically ill patients. Crit Care Med 29, 2281-2286

53
138. Brealey, D., Brand, M., Hargreaves, I., Heales, S., Land, J., Smolenski, R., Davies, N. A., Cooper, C. E., and Singer, M. (2002) Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 360, 219-223
139. Brealey, D., Karyampudi, S., Jacques, T. S., Novelli, M., Stidwill, R., Taylor, V., Smolenski, R. T., and Singer, M. (2004) Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am J Physiol Regul Integr Comp Physiol 286, R491-497
140. Witt, N. J., Zochodne, D. W., Bolton, C. F., Grand'Maison, F., Wells, G., Young, G. B., and Sibbald, W. J. (1991) Peripheral nerve function in sepsis and multiple organ failure. Chest 99, 176-184
141. Martin, U. J., Hincapie, L., Nimchuk, M., Gaughan, J., and Criner, G. J. (2005) Impact of whole-body rehabilitation in patients receiving chronic mechanical ventilation. Crit Care Med 33, 2259-2265
142. Brahmbhatt, N., Murugan, R., and Milbrandt, E. B. (2010) Early mobilization improves functional outcomes in critically ill patients. Crit Care 14, 321
143. Burtin, C., Clerckx, B., Robbeets, C., Ferdinande, P., Langer, D., Troosters, T., Hermans, G., Decramer, M., and Gosselink, R. (2009) Early exercise in critically ill patients enhances short-term functional recovery. Crit Care Med 37, 2499-2505
144. Schweickert, W. D., and Kress, J. P. (2011) Implementing early mobilization interventions in mechanically ventilated patients in the ICU. Chest 140, 1612-1617
145. Caruso, P., Denari, S. D., Ruiz, S. A., Bernal, K. G., Manfrin, G. M., Friedrich, C., and Deheinzelin, D. (2005) Inspiratory muscle training is ineffective in mechanically ventilated critically ill patients. Clinics (Sao Paulo) 60, 479-484
146. Griffiths, R. D., Palmer, T. E., Helliwell, T., MacLennan, P., and MacMillan, R. R. (1995) Effect of passive stretching on the wasting of muscle in the critically ill. Nutrition 11, 428-432
147. Lange, S., Xiang, F., Yakovenko, A., Vihola, A., Hackman, P., Rostkova, E., Kristensen, J., Brandmeier, B., Franzen, G., Hedberg, B., Gunnarsson, L. G., Hughes, S. M., Marchand, S., Sejersen, T., Richard, I., Edstrom, L., Ehler, E., Udd, B., and Gautel, M. (2005) The kinase domain of titin controls muscle gene expression and protein turnover. Science 308, 1599-1603
148. Rich, M. M., Pinter, M. J., Kraner, S. D., and Barchi, R. L. (1998) Loss of electrical excitability in an animal model of acute quadriplegic myopathy. Ann Neurol 43, 171-179
149. Rich, M. M., Kraner, S. D., and Barchi, R. L. (1999) Altered gene expression in steroid-treated denervated muscle. Neurobiol Dis 6, 515-522
150. Marquette, C. H., Mensier, E., Copin, M. C., Desmidt, A., Freitag, L., Witt, C., Petyt, L., and Ramon, P. (1995) Experimental models of tracheobronchial stenoses: a useful tool for evaluating airway stents. Ann Thorac Surg 60, 651-656

54
151. Marquette, C. H., Wermert, D., Wallet, F., Copin, M. C., and Tonnel, A. B. (1999) Characterization of an animal model of ventilator-acquired pneumonia. Chest 115, 200-209
152. Radell, P. J., Remahl, S., Nichols, D. G., and Eriksson, L. I. (2002) Effects of prolonged mechanical ventilation and inactivity on piglet diaphragm function. Intensive Care Med 28, 358-364
153. Banduseela, V. C., Ochala, J., Chen, Y. W., Goransson, H., Norman, H., Radell, P., Eriksson, L. I., Hoffman, E. P., and Larsson, L. (2009) Gene expression and muscle fiber function in a porcine ICU model. Physiol Genomics 39, 141-159
154. Norman, H., Kandala, K., Kolluri, R., Zackrisson, H., Nordquist, J., Walther, S., Eriksson, L. I., and Larsson, L. (2006) A porcine model of acute quadriplegic myopathy: a feasibility study. Acta Anaesthesiol Scand 50, 1058-1067
155. Dhand, U. K. (2006) Clinical approach to the weak patient in the intensive care unit. Respir Care 51, 1024-1040; discussion 1040-1021
156. Gutmann, L. (1999) Critical illness neuropathy and myopathy. Arch Neurol 56, 527-528
157. Hoke, A., Rewcastle, N. B., and Zochodne, D. W. (1999) Acute quadriplegic myopathy unrelated to steroids or paralyzing agents: quantitative EMG studies. Can J Neurol Sci 26, 325-329
158. Elliot, J. M., and Bion, J. F. (1995) The use of neuromuscular blocking drugs in intensive care practice. Acta Anaesthesiol Scand Suppl 106, 70-82
159. Leatherman, J. W., Fluegel, W. L., David, W. S., Davies, S. F., and Iber, C. (1996) Muscle weakness in mechanically ventilated patients with severe asthma. Am J Respir Crit Care Med 153, 1686-1690
160. Phillips, S. M., Glover, E. I., and Rennie, M. J. (2009) Alterations of protein turnover underlying disuse atrophy in human skeletal muscle. J Appl Physiol 107, 645-654
161. Cohen, S., Brault, J. J., Gygi, S. P., Glass, D. J., Valenzuela, D. M., Gartner, C., Latres, E., and Goldberg, A. L. (2009) During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol 185, 1083-1095
162. Lange, S., Ehler, E., and Gautel, M. (2006) From A to Z and back? Multicompartment proteins in the sarcomere. Trends Cell Biol 16, 11-18
163. Mearini, G., Gedicke, C., Schlossarek, S., Witt, C. C., Kramer, E., Cao, P., Gomes, M. D., Lecker, S. H., Labeit, S., Willis, M. S., Eschenhagen, T., and Carrier, L. (2010) Atrogin-1 and MuRF1 regulate cardiac MyBP-C levels via different mechanisms. Cardiovasc Res 85, 357-366
164. Cohen S, B. J., Gygi SP, Glass DJ, Valenzuela DM, Gartner C, Latres E, Goldberg AL (2009) During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol 185, 1083-1095

55
165. Nelson, W. B., Smuder, A. J., Hudson, M. B., Talbert, E. E., and Powers, S. K. (2012) Cross-talk between the calpain and caspase-3 proteolytic systems in the diaphragm during prolonged mechanical ventilation. Crit Care Med 40, 1857-1863
166. Williams, P. E. (1990) Use of intermittent stretch in the prevention of serial sarcomere loss in immobilised muscle. Ann Rheum Dis 49, 316-317
167. Sasa, T., Sairyo, K., Yoshida, N., Fukunaga, M., Koga, K., Ishikawa, M., and Yasui, N. (2004) Continuous muscle stretch prevents disuse muscle atrophy and deterioration of its oxidative capacity in rat tail-suspension models. Am J Phys Med Rehabil 83, 851-856
168. Schweickert, W. D., Pohlman, M. C., Pohlman, A. S., Nigos, C., Pawlik, A. J., Esbrook, C. L., Spears, L., Miller, M., Franczyk, M., Deprizio, D., Schmidt, G. A., Bowman, A., Barr, R., McCallister, K. E., Hall, J. B., and Kress, J. P. (2009) Early physical and occupational therapy in mechanically ventilated, critically ill patients: a randomised controlled trial. Lancet 373, 1874-1882
169. Morris, P. E., Goad, A., Thompson, C., Taylor, K., Harry, B., Passmore, L., Ross, A., Anderson, L., Baker, S., Sanchez, M., Penley, L., Howard, A., Dixon, L., Leach, S., Small, R., Hite, R. D., and Haponik, E. (2008) Early intensive care unit mobility therapy in the treatment of acute respiratory failure. Crit Care Med 36, 2238-2243
170. Meng, S. J., and Yu, L. J. (2010) Oxidative stress, molecular inflammation and sarcopenia. Int J Mol Sci 11, 1509-1526
171. Nystrom, T. (2005) Role of oxidative carbonylation in protein quality control and senescence. EMBO J 24, 1311-1317
172. Vitadello, M., Penzo, D., Petronilli, V., Michieli, G., Gomirato, S., Menabo, R., Di Lisa, F., and Gorza, L. (2003) Overexpression of the stress protein Grp94 reduces cardiomyocyte necrosis due to calcium overload and simulated ischemia. FASEB J 17, 923-925
173. Bando, Y., Katayama, T., Aleshin, A. N., Manabe, T., and Tohyama, M. (2004) GRP94 reduces cell death in SH-SY5Y cells perturbated calcium homeostasis. Apoptosis 9, 501-508
174. Ochala, J., Renaud, G., Llano Diez, M., Banduseela, V. C., Aare, S., Ahlbeck, K., Radell, P. J., Eriksson, L. I., and Larsson, L. (2011) Diaphragm muscle weakness in an experimental porcine intensive care unit model. PLoS One 6, e20558
175. Sano, K., Maeda, K., Oda, T., and Maeda, Y. (2000) The effect of single residue substitutions of serine-283 on the strength of head-to-tail interaction and actin binding properties of rabbit skeletal muscle alpha-tropomyosin. J Biochem 127, 1095-1102
176. Kollmar, M., Durrwang, U., Kliche, W., Manstein, D. J., and Kull, F. J. (2002) Crystal structure of the motor domain of a class-I myosin. EMBO J 21, 2517-2525
177. Deng, H. (2006) Nitrite anions induce nitrosative deamination of peptides and proteins. Rapid Commun Mass Spectrom 20, 3634-3638

56
178. Keira, N., Tatsumi, T., Matoba, S., Shiraishi, J., Yamanaka, S., Akashi, K., Kobara, M., Asayama, J., Fushiki, S., Fliss, H., and Nakagawa, M. (2002) Lethal effect of cytokine-induced nitric oxide and peroxynitrite on cultured rat cardiac myocytes. J Mol Cell Cardiol 34, 583-596
179. Suzuki, N., Mizuno, H., Warita, H., Takeda, S., Itoyama, Y., and Aoki, M. (2010) Neuronal NOS is dislocated during muscle atrophy in amyotrophic lateral sclerosis. J Neurol Sci 294, 95-101
180. Suzuki, N., Motohashi, N., Uezumi, A., Fukada, S., Yoshimura, T., Itoyama, Y., Aoki, M., Miyagoe-Suzuki, Y., and Takeda, S. (2007) NO production results in suspension-induced muscle atrophy through dislocation of neuronal NOS. J Clin Invest 117, 2468-2476
181. Grune, T., Jung, T., Merker, K., and Davies, K. J. (2004) Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and 'aggresomes' during oxidative stress, aging, and disease. Int J Biochem Cell Biol 36, 2519-2530
182. De Jonghe, B., Bastuji-Garin, S., Durand, M. C., Malissin, I., Rodrigues, P., Cerf, C., Outin, H., and Sharshar, T. (2007) Respiratory weakness is associated with limb weakness and delayed weaning in critical illness. Crit Care Med 35, 2007-2015
183. Gayan-Ramirez, G., de Paepe, K., Cadot, P., and Decramer, M. (2003) Detrimental effects of short-term mechanical ventilation on diaphragm function and IGF-I mRNA in rats. Intensive Care Med 29, 825-833
184. Powers, S. K., Shanely, R. A., Coombes, J. S., Koesterer, T. J., McKenzie, M., Van Gammeren, D., Cicale, M., and Dodd, S. L. (2002) Mechanical ventilation results in progressive contractile dysfunction in the diaphragm. J Appl Physiol 92, 1851-1858
185. Le Bourdelles, G., Viires, N., Boczkowski, J., Seta, N., Pavlovic, D., and Aubier, M. (1994) Effects of mechanical ventilation on diaphragmatic contractile properties in rats. Am J Respir Crit Care Med 149, 1539-1544
186. Yang, L., Luo, J., Bourdon, J., Lin, M. C., Gottfried, S. B., and Petrof, B. J. (2002) Controlled mechanical ventilation leads to remodeling of the rat diaphragm. Am J Respir Crit Care Med 166, 1135-1140
187. Anzueto, A., Peters, J. I., Tobin, M. J., de los Santos, R., Seidenfeld, J. J., Moore, G., Cox, W. J., and Coalson, J. J. (1997) Effects of prolonged controlled mechanical ventilation on diaphragmatic function in healthy adult baboons. Crit Care Med 25, 1187-1190
188. Levine, S., Nguyen, T., Taylor, N., Friscia, M. E., Budak, M. T., Rothenberg, P., Zhu, J., Sachdeva, R., Sonnad, S., Kaiser, L. R., Rubinstein, N. A., Powers, S. K., and Shrager, J. B. (2008) Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med 358, 1327-1335

57
189. Shanely, R. A., Zergeroglu, M. A., Lennon, S. L., Sugiura, T., Yimlamai, T., Enns, D., Belcastro, A., and Powers, S. K. (2002) Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am J Respir Crit Care Med 166, 1369-1374
190. Ochala, J., Gustafson, A. M., Diez, M. L., Renaud, G., Li, M., Aare, S., Qaisar, R., Banduseela, V. C., Hedstrom, Y., Tang, X., Dworkin, B., Ford, G. C., Nair, K. S., Perera, S., Gautel, M., and Larsson, L. (2011) Preferential skeletal muscle myosin loss in response to mechanical silencing in a novel rat intensive care unit model: underlying mechanisms. J Physiol 589, 2007-2026
191. Levine, S., Biswas, C., Dierov, J., Barsotti, R., Shrager, J. B., Nguyen, T., Sonnad, S., Kucharchzuk, J. C., Kaiser, L. R., Singhal, S., and Budak, M. T. (2011) Increased proteolysis, myosin depletion, and atrophic AKT-FOXO signaling in human diaphragm disuse. Am J Respir Crit Care Med 183, 483-490
192. Ramamurthy, B., Hook, P., Jones, A. D., and Larsson, L. (2001) Changes in myosin structure and function in response to glycation. FASEB J 15, 2415-2422
193. Prochniewicz, E., Lowe, D. A., Spakowicz, D. J., Higgins, L., O'Conor, K., Thompson, L. V., Ferrington, D. A., and Thomas, D. D. (2008) Functional, structural, and chemical changes in myosin associated with hydrogen peroxide treatment of skeletal muscle fibers. Am J Physiol Cell Physiol 294, C613-626
194. Zergeroglu, M. A., McKenzie, M. J., Shanely, R. A., Van Gammeren, D., DeRuisseau, K. C., and Powers, S. K. (2003) Mechanical ventilation-induced oxidative stress in the diaphragm. J Appl Physiol 95, 1116-1124
195. Whidden, M. A., Smuder, A. J., Wu, M., Hudson, M. B., Nelson, W. B., and Powers, S. K. (2010) Oxidative stress is required for mechanical ventilation-induced protease activation in the diaphragm. J Appl Physiol 108, 1376-1382
196. Suliman, I. A., Lindgren, J. U., Gillberg, P. G., Elhassan, A. M., Monneron, C., and Adem, A. (1999) Alteration of spinal cord IGF-I receptors and skeletal muscle IGF-I after hind-limb immobilisation in the rat. Neuroreport 10, 1195-1199
197. Blakemore, S. J., Rickhuss, P. K., Watt, P. W., Rennie, M. J., and Hundal, H. S. (1996) Effects of limb immobilisation on cytochrome c oxidase activity and GLUT4 and GLUT5 protein expression in human skeletal muscle. Clin Sci (Lond) 91, 591-599
198. Boudriau, S., Cote, C. H., Vincent, M., Houle, P., Tremblay, R. R., and Rogers, P. A. (1996) Remodeling of the cytoskeletal lattice in denervated skeletal muscle. Muscle Nerve 19, 1383-1390

ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
acta Universitatis Upsaliensis Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 862
editor: The Dean of the Faculty of Medicine
a doctoral dissertation from the Faculty of Medicine, Uppsala University, is usually a summary of a number of papers. a few copies of the complete dissertation are kept at major Swedish research libraries, while the summary alone is distributed internationally through the series Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine. (Prior to January, 2005, the series was published under the title “Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine”.)
Distribution: publications.uu.se 2013urn:nbn:se:uu:diva-192531




























