Embed Size (px)
Citation preview
SÉPTIMA PRÁCTICA: Fármacos antidepresivos y antipsicóticos
EXPERIMENTO Nº1: Efecto antidepresivo de los fármacos, mediante la prueba del nado forzado.Fármacos: Mirtazapina, solución al 0.2 % / Amitriptilina, solución al 0.3 % / Fluoxetina, solución al 0.5 %Especie: Ratón albino macho de 25 g.Software: Journal of Visualized Experiment JOVE on line www . j o v e . c o m
Experimento nº1, efecto antidepresivo de los fármacos, mediante la prueba del nado forzado, matriz de recolección de datos.
fármaco ratón pesodosismg
dosisml
tiempo deinmovilidad
observaciones
suero fisiológico blanco 34 0.34 1min23seg Tiempo alto que demuestra la rendición del roedor.mirtazapina cabeza 34 0.34 0.17 21seg
amitriptilina lomo 41 0.82 0.27 30seg
fluoxetina cola 38 1.14 0.23 0
Discusión:
mirtazapina es un medicamento antidepresivo de estructura tetracíclica clasificado como un antidepresivo noradrenérgico y serotonérgico especifico. actúa aumentando la liberación de noradrenalina y de la serotonina mediante el bloqueo de los receptores alfa 2 presinápticos. Tiene pocos efectos antimuscarínicos pero se comporta como un sedante debido a que tiene propiedades antihistamínicas (antagonista potente H1). También es utilizado como ansiolítico, hipnótico, antiemético y orexígeno. La mirtazapina tiene un efecto dual (en cuanto a la afectación de los sistemas serotoninérgico y noradrenérgico) así como específico, por su acción antagonista sobre los receptores α2-adrenérgicos, como así también sobre los receptores 5-ht2a, 5-ht2c, 5-ht3. La mirtazapina se receta principalmente para el tratamiento del trastorno depresivo mayor y otros trastornos del estado de ánimo.
La fluoxetina es un inhibidor selectivo de la recaptación de serotonina, actuando a nivel del transportador de serotonina de la membrana presináptica – SERT, inhibiéndola. Posee alta afinidad por receptores de monoaminas, pero no para receptores de histamina, acetilcolina y receptores adrenérgicos alfa. Su principal efecto se observa en el aumento de la actividad sináptica serotoninérgica. Tiene pocos efectos sobre la actividad neurotrófica (BDNF-factor neurotrófico derivado del cerebro). Su uso está indicado en la depresión mayor, trastornos de ansiedad (trastornos obsesivos compulsivos, de estrés traumático, de pánico, síndromes vasomotores de la perimenopausia y trastornos de alimentación o bulimia).
La amitriptilina es un antidepresivo tricíclico (TCA) ya que posee un núcleo iminobencílico y es una amina terciaria; al igual que otros TCA. Tiene una actividad inhibitoria muy fuerte sobre los recaptadores de serotonina (SERT), así como de noradrenalina (NET), pudiendo ocasionar efectos ortostáticos (hipotensión) en pacientes de edad avanzada. Se caracterizan por, además de inhibidores de SERT y NET, tener acción antimuscarínica ocasionando boca seca y estreñimiento. También tiende a ser un potente antagonista del receptor H1 de histamina, pudiendo ocasionar aumento de peso y sedación.
Conclusiones:
El periodo de rendición o tiempo de inmovilización es el tiempo que transcurre entre las acciones de reconocimiento y escape que el ratón manifiesta desde que es introducido al recipiente. Este periodo nos muestra la acción del fármaco sobre el roedor.
A mayor tiempo de inmovilización, mayor efecto depresivo o de rendición. Se espera que los fármacos disminuyan el tiempo de inmovilización.
En estado basal el roedor presentará (con suero fisiológico) notamos como el tiempo de inmovilización es sumamente largo 1min23seg, lo cual sirvió de base para comparar los otros resultados.
Se observa que después de la aplicación de los fármacos a los roedores, estos no van a presentar un tiempo de inmovilización largo, lo cual demuestra la acción antidepresiva de los fármacos. Ya que al no presentar depresión, no estarían “rindiéndose” por lo cual actúan tratando de escapar del recipiente y demostrando el efecto farmacológico deseado.
EXPERIMENTO Nº2: Efecto antipsicótico de los fármacos, mediante la prueba del nado forzado.

Fármacos: Clorpromazina, solución al 0.2 % / Haloperidol, solución de 5 mg/5ml / Clozapina, solución al 0.2 %Especie: Ratón albino macho de 25 g.Software: Journal of Visualized Experiment JOVE on line www . j o v e . c o m
to antipsicótico de los fármacos, mediante la prueba del nado forzado, matriz de recolección de datos.
FÀRMACO Ratón PesoDosis
mgDosis
mlTiempo de
inmovilidad observacionesSuero fisiológico Blanco 43 49seg
Cloropromacina Cabeza
Haloperidol Lomo 31 0.155 0.031 2min24seg Mayor tiempo por perdida de la movilidad
Clozapina Cola 41 0.4 0.2 6seg
Discusión:
La Cloropromacina es un antipsicótico típico (produce efectos secundarios extrapiramidales - EPS) derivado Fenotiazinico tiene potente efecto de sedación y aumento de peso (bloqueo del receptor H1) y menos potente efecto antipsicótico. Su mecanismo de acción se basa en el bloqueo de receptores D2 (dopaminérgicos) y también en el bloqueo de receptor 5HT2A serotoninérgico, de manera más leve. También bloquea receptores muscarinicos, pudiendo producir boca seca y estreñimiento
La Clozapina es un antipsicótico atípico (no produce efectos secundarios extrapiramidales - EPS) agonista inverso de los receptores 5-HT2A, bloqueando la actividad constitutiva de estos receptores (estos receptores regulan la liberación de dopamina en la corteza, la región límbica y el cuerpo estriado), también bloquea receptores D2. También bloquea receptores muscarinicos y bloqueo variable de receptores H1.
El Haloperidol es un antipsicótico típico derivado de butirofenona que bloquea receptores D2 más que a los receptores 5-HT2A. posee elevados efectos secundarios extrapiramidales y produce cierto grado de sedación por bloqueo variable de H1.
Conclusiones:
La psicosis es un conjunto de trastornos relacionados, en el caso de roedores, a funciones sensoriales alteradas y maximizadas. En estado basal el ratón no experimentará psicosis, sin embargo el ratón tendrá acciones de reconocimiento y escape del recipiente.
Las observaciones buscadas en los efectos farmacológicos inciden principalmente en los efectos secundarios extrapiramidales – EPS presentes en los fármacos usados, teniendo en cuenta que los fármacos que presentan estos efectos son llamados típicos.
Los fármacos típicos como el Haloperidol, tiene efecto sobre las manifestaciones motoras produciendo catalepsia sobre el animal de experimentación, caracterizado por perdida de la movilidad, aumentando así el tiempo de inmovilidad.
Los fármacos atípicos como la Clozapina no producen efectos motores de catalepsia por lo cual el tiempo de inmovilidad se ve reducido.
Estos resultados demostrarían que lo que se busca en un fármaco son las menores manifestaciones de efectos secundarios; y en este caso se correlaciona con el uso preferente de la Clozapina como antipsicótico de preferencia.
OCTAVA PRÁCTICA: fármacos anestésicos generales, anestésicos locales y bloqueantes neuromusculares

EXPERI MENTO Nº3: Anestesia General con Fentanilo en rata. Fármacos: Fentanilo / Midazolam / Flunixin (AINE) Especie:Rata de 250 g
Software: Software Microlabs, Dr. Henk van Wilgenburg, Dept. of Pharmacology, University of AmsterdamMetodología: Tarea y demostración en mesa de laboratorio, mediante simulación en Software.
ANESTESIA GENERAL CON FENTANILO EN RATA.
Dosis(ug/kg)
Compartimiento Central de inmediato
Ritmo Cardiaco (lat/min)
Ritmo Respiratorio (resp/min)
LocomociónReflejo al Pinchamiento (dolor)
Reflejo Postural Enderezamiento
ReflejoCorneal
Reflejo Estiramiento de la Pata
mg [ng/l] [mg/l]
Basal=0 ----- ------- 350 81 +++ +++ +++ +++ +++
1 0.00025 3.5 0.0035 345 79 ++ +++ +++ +++ +++
3 0.00075 10 0.0010 334 75 ++ +++ +++ +++ +++
6 0.0015 20 0.0020 320 70 + +++ +++ +++ +++
9 0.00225 30 0.0030 307 66 + ++ ++ ++ ++
12 0.003 40 0.0040 265 62 + 0 + + +
15 0.00375 50 0.0050 284 58 0 0 0 0 0
18 0.0045 60 0.0060 273 55 0 0 0 0 0
21 0.00525 70 0.0070 264 53 0 0 0 0 0
24 0.006 80 0.0080 255 51 0 0 0 0 0
27 0.00675 90 0.0090 0 0 0 0 0 0 0
Efectos: +++ = PRESENTE. ++ = DISMINUCION. + = MAYOR DISMINUCION. 0 = AUSENTE

Discusión:
El Fentanilo es un fármaco opioide sintético derivado de la meperidina usado para el manejo del dolor moderado a severo por su efectividad, fácil dosificación y relación riesgo/beneficio favorable. Es un agonista de alta afinidad por los receptores mu, también actúa sobre receptores delta, pero no sobre receptores kappa. Es de alta potencia farmacológica, para el tratamiento del dolor agudo.
Sus efectos dependen del receptor donde actúa; por lo cual en el receptor mu del SNC producirá analgesia, reduciendo el dolor en sus componentes sensoriales y afectivos, aumentando la tolerancia sin pérdida de la conciencia. Produce sedación dependiente de la dosis, también euforia (interpretación subjetiva del dolor como bienestar) y disforia (alteración en percepción del dolor).
Los receptores mu-2 deprimen la actividad respiratoria al deprimir el centro respiratorio bulboprotuberancial. El mecanismo consiste en la reducción de la sensibilidad al CO2 y a la hipoxia, lo cual provoca vasodilatación a nivel cerebral, aumentando la presión intracraneana. El volumen minuto respiratorio disminuye, afectándose más la frecuencia más que la amplitud. Puede llegar a la apnea. Puede desarrollarse consecuentemente acidosis respiratoria.
Tiene efecto de miosis sobre la pupila al actuar a nivel de receptores mu del nervio parasimpático que inerva la pupila (desinhibición del núcleo de Edinger y Wesphal).
Otros efectos incluyen: antitusígeno, por depresión del núcleo gatillo de la tos; nausea y vómitos, por estimulación del área postrema a nivel bulbar; y también afecta la temperatura, a dosis bajas la disminuyen y a dosis altas la aumentan.
Los efectos neuroendocrinos se dan a dosis altas por su acción sobre el hipotálamo e hipófisis; se da la inhibición de la liberación de GnRH por el hipotálamo, lo que reduce la secreción de FSH y LH. Inhibe también la liberación de CRF hipotalámico. Estimula la liberación de ACTH, somatotropina, prolactina, beta-MSH y ADH.
Por su alta afinidad a los receptores mu, produce en el TGI la disminución de secreciones gástricas, pancreáticas e intestinales. Disminuye el peristaltismo intestinal. Aumenta el tono esfinteriano (pilórico, cecal y de Oddi)
A nivel cardiovascular producen indirectamente, por liberación de histamina, y directamente una reducción de la RVP y de la PA

debido a la vasodilatación. También hipotensión ortostática por inhibición de reflejos barorreceptores y una bradicardia de origen vagal.
Por inducción de la liberación de histamina, produce calor, enrojecimiento facial y prurito en zonas de la cara. A dosis altas produce un estado de excitación general del SNC, con temblores, calambres y convulsiones previas a una depresión
respiratoria. El Fentanilo actúa sobre receptores mu y delta. El responsable de la depresión respiratoria y de la bradicardia es el receptor mu-
2, que se encuentra a nivel espinal y en el SNP, produciendo, además de lo mencionado, analgesia espinal y sedación. El receptor delta tiene un papel adicional sobre la depresión respiratoria incrementando los efectos del Fentanilo.
Conclusiones:
Se observa que hay disminución del ritmo cardiaco y respiratorio por los efectos de bradicardia y depresión respiratoria, derivados del Fentanilo, cuando actúa a nivel de receptores mu-2 y delta.
Los efectos de reducción en la locomoción y reducción en los reflejos se producen por actividad de sedación y narcolepsia inducido por el Fentanilo, principalmente sobre los receptores mu-1, que se ubica a nivel supraespinal, produciendo además analgesia supraespinal, hipotermia y liberación de prolactina.
Los efectos observados se deben a las dosis mayores, ya que a la sedación le sigue el estupor, el sueño profundo y el coma metabólico, finalmente la muerte.
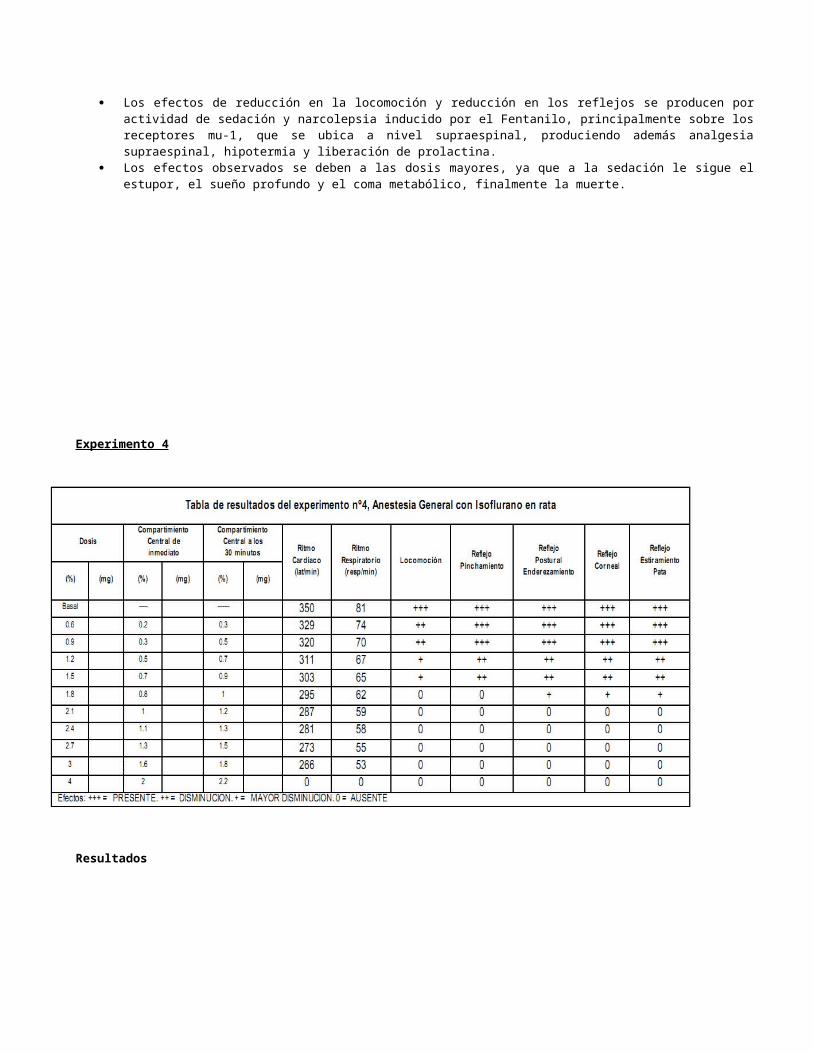
Experimento 4
Resultados
Discusión:
El isoflurano es un anestésico general, el cual necesita vaporizarse con un aparato especial para luego administrarse por vía inhalatoria. Con la primera dosis, se evidencia solo una pérdida ligera de la locomoción, esto indica que se encuentra en el inicio del periodo 1 de la
anestesia: Inducción y analgesia. Aquí es donde comienzan los trastornos de la percepción, no hay pérdida de la conciencia, disminuyen las sensaciones dolorosas.
Conforme se va aumentando la dosis de isoflurano se evidencia una disminución del ritmo cardiaco y respiratorio, además de menor locomoción y una disminución de todos los reflejos. Se puede decir que el periodo 1 sigue hasta la dosis que se administró de isoflurano al 1.8%, debido a que recién ahí perdió completamente la locomoción y por tanto la conciencia, es ahí cuando comienza el periodo 2: Excitación y delirio. Aquí se evidenció la presencia del reflejo postural de enderezamiento, reflejo corneal y reflejo de estiramiento de pata. Además hay respiración rápida e irregular, además de taquisfigmia.
No se puede evidenciar el inicio del periodo 3: Anestesia quirúrgica; plano 1. Debido a que se inicia con la estabilización del ritmo cardiaco y la pérdida del reflejo palpebral, signos que no se evidencian en la tabla de resultados. Lo que sí se puede señalar es el inicio del plano 2 del periodo 3, que coincide con la administración de isoflurano al 2.1%, se puede señalar esto porque desaparece a este nivel el reflejo corneal; en este nivel todos los reflejos están abolidos, la respiración disminuye más haciéndose más abdominal, hay ausencia de movimientos oculares y hay un aumento del pulso.
Conforme se va aumentando más la dosis se va disminuyendo más el ritmo respiratorio, esto es probablemente debido a la depresión de movimiento muscular de los músculos intercostales, dejando al diafragma todo el peso del movimiento respiratorio, originando una respiración más abdominal. Es aquí donde se puede señalar que empieza el plano 4 del periodo 3 de la anestesia.
Finalmente, con la última dosis, de isoflurano al 4%, aparte de estar abolidos todos los reflejos, desaparecieron por completo los ritmos cardiacos y respiratorios. El periodo 4 comenzó con la depresión bulbar del centro respiratorio y terminó con la muerte del animal, con el paro cardiaco.
Las razones por las que el aumento de dosis generó dichos resultados fueron: a mayor concentración del anestésico en el aire inspirado se genera mayor presión parcial del anestésico en los alveolos, luego mayor presión parcial en la sangre, para finalizar con una mayor presión parcial en el cerebro. Además, la ventilación pulmonar de alta frecuencia, favorece a la absorción del anestésico para su posterior llegada al cerebro. Cabe resaltar que la concentración alveolar mínima que se necesitaba para que ocurran estos efectos, para el isoflurano es del 1.15%.
El mecanismo de acción de los anestésicos en general es bien discutido, habiéndose generado 3 teorías hasta la actualidad: la teoría lipídica sostiene que a mayor liposolubilidad del anestésico hay mayor expansión del volumen de la membrana celular y mayor fluidifiación de esta, ambos procesos contribuyen a la disiminución de las transmisiones nerviosas. La 2da teoría es de la hidratación; la tercera teoría es la proteica, que sostiene que el anestésico se une a las proteínas de membrana de las neuronas, modificándolas y por tanto alterando los flujos iónicos de entrada y de salida finalizando con trastornos de la conducción nerviosa.
El isoflurano tiene un efecto de corta duración, pero de inicio rápido. El isoflurano disminuyó las transmisiones nerviosas del tronco encefálico, disminuyendo los reflejos; de la médula, también disminuyendo reflejos; y específicamente del bulbo raquídeo, deprimiendo el centro respiratorio, mediante los siguientes mecanismos: incrementó el umbral del potencial de acción, aumentando la permeabilidad al sodio y al potasio y modulando indirectamente la permeabilidad al calcio y al cloro. Además, dicho anestésico alteró el estado físico de la membrana celular neuronal. Las consecuencias finales son: menor respuesta simpática y parasimpática, aumento en la concentración de serotonina, y una disminución de la producción de segundos mensajeros.
Entre los efectos en general del isoflurano, mejor evidenciados en pacientes humanos, son: disminución del metabolismo cerebral (neuroprotector), aumento del flujo sanguíneo cerebral por vasodilatación. Ejerce además una acción depresora en el sistema respiratorio, con una vasodilatación pulmonar. A nivel sistémico también origina vasodilatación con caída de la presión arterial diastólica. Y a nivel muscular ocasiona relajación. Cabe hacer énfasis en que a diferencia del enflurano, el isoflurano no origina convulsiones ni arritimias.
Conclusiones:
El isoflurano es neuroprotector debido a que disminuye el metabolismo cerebral. El isoflurano aumenta el flujo sanguíneo cerebral. El isoflurano ejerce una depresión del sistema respiratorio, actuando principalmente en el centro respiratorio. El isoflurano origina vasodilatación pulmonar y vasodilatación sistémica, disminuyendo la presión arterial pulmonar y la presión arterial
diastólica sistémica. El isoflurano es un miorrelajante.
Experimento 5
Resultados
El porcentaje de elevación del umbral de antestesia local se calculó de la siguiente manera: se consideró que en estado basal, 1.00 mV era el umbral de excitación normal, es decir, llegando a 1.00 mV de estimulación eléctrica se lograba la contracción de la piel del animal; por tanto en el caso de la lidocaína, recién se evidenció contracción de la piel del animal a los 2.00 mV, es decir, al doble de voltaje se llegó al umbral, por tanto se consideró en dicho caso que la elevación del umbral originada por la lidocaína fue de 100%, porque se duplicó el voltaje necesario.
Discusión:

La lidocaína es un anestésico local, al igual que la bupicaína.Lo resaltante es el mecanismo de acción: ambos bloquean canales de sodio dependientes de voltaje, de tal manera que se bloquea la
despolarización neuronal y por tanto la transmisión nerviosa. Este bloque del canal se da en la superficie interna de la membrana celular. En los axones mielinizados (nódulos de Ranvier) y en axones no mielinizacos de las fibras C es donde se bloquea la transmisión. En el experimento se explica por tanto cómo al darle descargas eléctricas al abdomen del conejo, como se activan canales de sodio dependientes de voltaje, es posible mediante el anestésico local bloquear la transmisión y por tanto la reacción refleja de contracción o retracción de la piel.
La lidocaína tiene un pka de 7.7, con un inicio de acción rápido, duración de acción intermedia y toxicidad intermedia; todos los parámetros tomando como referencia los demás anestésicos locales. Además tiene una potencia de 4, tiempo de vida media de 2 horas aproximadamente y origina una marcada vasodilatación.
La bupivacaína tiene una potencia de 16, un periodo de latencia largo, duración de acción largo, toxicidad fuerte, tiempo de vida media de 3 horas y origina una ligera vasodilatación.
La efectividad del anestésico local recae en su farmacocinética en gran parte, debido a que es necesario que el fármaco se quede bloqueando la transmisión nerviosa sólo en la zona de administración. Por tanto es fundamental que haya la menor absorción posible, cabe mencionar entonces que a mayor flujo sanguíneo de la zona hay mayor absorción, a menor proporción de tejido adiposo hay mayor absorción y a menor vasoconstricción y mayor absorción.
Considerando que el único anestésico local que produce vasoconstricción es la cocaína, la mayoría son propensos a una absorción relativamente rápida y por tanto corta duración de la acción farmacológica. Es por dicho motivo que se asocia frecuentemente la administración con una sustancia vasoconstrictora, como la epinefrina. En el experimento se administró dicha droga y los resultados coincidieron con la teoría; en el caso de la lidocaína, la administración conjunta con la epinefrina en el control 2 se evidenció un aumento del efecto anestésico, además de que se mantuvo hasta el control 4. En el caso de la bupivacaína, lo resaltante a parte del aumento del efecto en el control 3, fue que en el control 4 no se mantuvo el efecto, sino que más bien aumentó, esto se dio debido a que se deposita cierta cantidad de bupivacaína en los adipocitos debido a la liposolubilidad, de tal manera que salen para actuar con los receptores poco a poco y se prolonga la acción.
Cabe resaltar que la potencia anestésica intrínseca depende de la liposolubilidad, que permite atravesar la membrana celular de la fibra nerviosa para unirse al canal de sodio dependiente de voltaje y bloquearlo. Es por esta razón que la bupivacaína tiene mayor potencia, mayor liposolubilidad y por tanto mayor duración e intensidad de la acción. Además, la duración está afectada por el grado de afinidad que se tiene por el receptor, por tanto se puede decir que la bupivacaína tiene mayor afinidad que la lidocaína. Por otro lado, se puede afirmar que a menor pka, menor periodo de latencia, por tanto se puede afirmar que la lidocaína tiene menor pka que la bupivacaína. No está de más señalar que la lidocaína, en caso se absorba, puede llegar a tener efectos antiarrítmicos, por la depresión de los potenciales de acción que también origina a nivel cardiaco, originando una disminución del cronotropismo y el dromotropismo.
Conclusiones:
La bupivacaína es un anestésico local más potente, con mayor periodo de latencia, la mayor duración de acción, con mayor toxicidad y con menor efecto vasodilatador que la lidocaína.
La vasoconstricción adjunta a la administración del anestésico local origina un aumento en la duración y la intensidad del efecto.La inhibición del canal de sodio dependiente de voltaje inhibe la despolarización originada por estímulos eléctricos externos al cuerpo.
Experimento N° 6: Efecto de los fármacos Bloqueantes neuromusculares en el músculo diafragma.
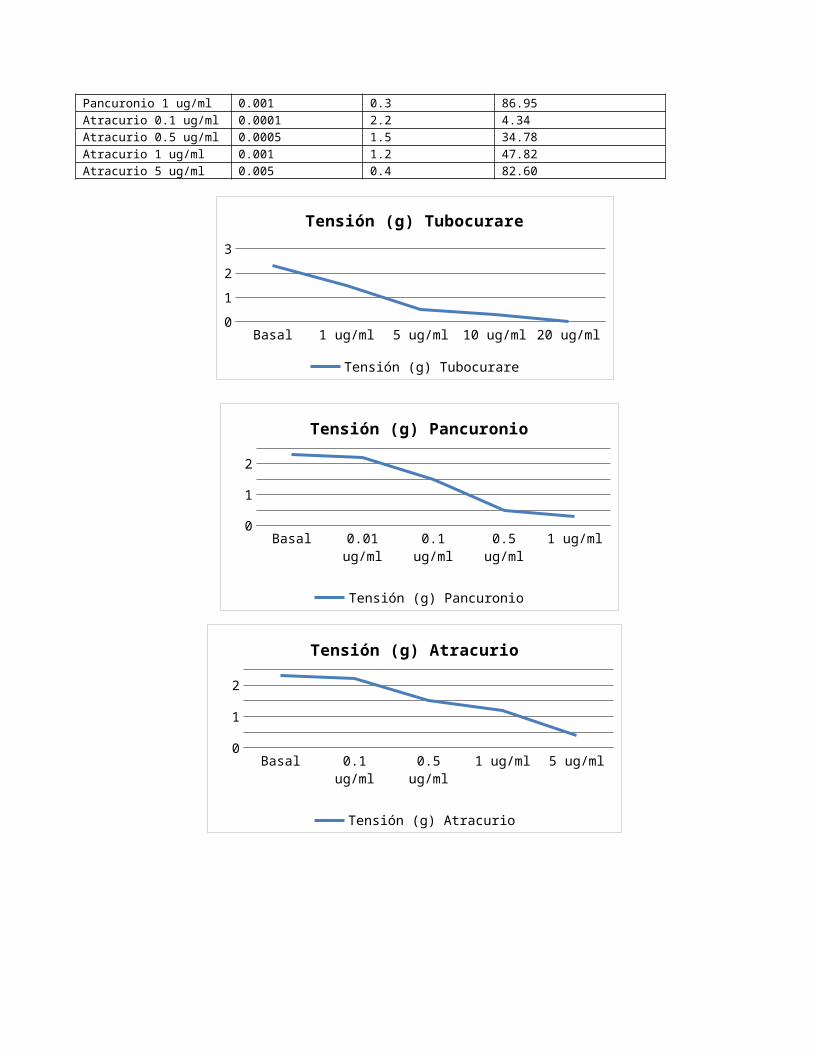
Tabla de resultados del experimento N° 6, efecto de los fármacos bloqueantes neuromusculares en el músculo diafragma.Fármacos Dosis mg/ml Tensión (g) % de inhibiciónBasal - 2.3 0Tubocurare 1 ug/ml 0.001 1.5 34.78Tubocurare 5 ug/ml 0.005 0.5 78.26Tubocurare 10 ug/ml 0.01 0.3 86.95Tubocurare 20 ug/ml 0.02 0.0 100Pancuronio 0.01 ug/ml 0.00001 2.2 4.34Pancuronio 0.1 ug/ml 0.0001 1.5 34.78Pancuronio 0.5 ug/ml 0.0005 0.5 78.26Pancuronio 1 ug/ml 0.001 0.3 86.95Atracurio 0.1 ug/ml 0.0001 2.2 4.34

Atracurio 0.5 ug/ml 0.0005 1.5 34.78Atracurio 1 ug/ml 0.001 1.2 47.82Atracurio 5 ug/ml 0.005 0.4 82.60
Basal 1 ug/ml 5 ug/ml 10 ug/ml 20 ug/ml0
0.51
1.52
2.5
Tensión (g) Tubocurare
Tensión (g) Tubocurare
Basal 0.1 ug/ml 0.5 ug/ml 1 ug/ml 5 ug/ml0
0.5
1
1.5
2
2.5
Tensión (g) Atracurio
Tensión (g) Atracurio
Basal 0.01 ug/ml 0.1 ug/ml 0.5 ug/ml 1 ug/ml0
0.5
1
1.5
2
2.5
Tensión (g) Pancuronio
Tensión (g) Pancuronio

Basal 1 ug/ml 5 ug/ml 10 ug/ml 20 ug/ml0
20
40
60
80
100
% de inhibición Tubocurare
% de inhibición Tubocurare
Basal 0.01 ug/ml 0.1 ug/ml 0.5 ug/ml 1 ug/ml0
20
40
60
80
100
% de inhibición Pancuronio
% de inhibición Pancuronio
Esquema de las interacciones de los fármacos con el receptor de ACh sobre el conducto de la placa terminal.
Arriba: la acción del agonista normal, ACh, para la abertura del conducto.
Abajo, a la izquierda: se muestra un bloqueador no despolarizante, p.ej., pancuronio, que impide la abertura del conducto cuando se une al receptor.
Abajo, a la derecha: un bloqueador despolarizante, p.ej., succinilcolina, ocupa el receptor y bloquea el conducto. Se evita el cierre normal de la entrada del conducto y el bloqueador puede moverse más rápidamente hacia el interior y exterior del poro.
Discusión:
El curare, nombre con que se designaban diversos venenos elaborados por aborígenes sudamericanos para cazar animales, fue la primera sustancia que demostró producir parálisis muscular por un mecanismo dependiente del bloqueo competitivo del receptor nicotínico muscular. Obtenido de la planta Chondodendrum tomentosum, se purificó el alcaloide tubocurare.
El tubocurare y los restantes agentes no despolarizantes se comportan como antagonistas competitivos de la ACh. Se fijan específicamente al receptor nicotínico muscular y reducen la frecuencia de apertura del canal y la amplitud del potencial postsináptico al impedir la unión de ACh. Esta amplitud debe disminuir por debajo del 70 % de su valor inicial para que se bloquee la propagación del potencial de acción muscular, lo que constituye un mecanismo de seguridad para garantizar la transmisión neuromuscular en circunstancias adversas. Puesto que el bloqueo es competitivo, el aumento del número de moléculas de ACh en la vecindad del receptor nicotínico desplaza a los bloqueantes de su unión al receptor, restaura el potencial de placa motora y, por consiguiente, se recuperan la transmisión y la contracción muscular. Estos fármacos pueden bloquear también los receptores nicotínicos facilitadores de la liberación de ACh. De esta forma pueden inhibir la liberación de ACh durante la estimulación del nervio motor.
Acciones farmacológicas: Derivan fundamentalmente del bloqueo del receptor nicotínico del músculo esquelético. Todos ellos producen, inicialmente, una sensación de debilidad muscular, seguida de parálisis flácida. En condiciones normales, los músculos intercostales y el diafragma son los últimos en ser afectados lo que lo hace letal, tal como se aprecia en las gráficas del experimento donde cada aumento de dosis ocasiona una mayor inhibición muscular, logrando cesar toda actividad a los 20 ug/ml. Mientras que el atracurio y el pancuronio generan menor bloqueo ganglionar.
Conclusiones:
El pancuronio y el atracurio alcanzan una mayor inhibición que el tubocurare a dosis de 1ug/ml. Se puede concluir que tanto el pancuronio como el atracurio poseen una mayor potencia que el tubocurare. Los fármacos tratados en este experimento son bloqueadores musculares no despolarizantes que actúan predominantemente en el receptor nicotínico por competencia con la ACh. En grandes dosis, como las administradas en el caso del tubocurare (20 ug/ml), el fármaco entra al poro del conducto iónico para producir un bloqueo motor más intenso (100 % inhibición).





























