Embed Size (px)
DESCRIPTION
CM infernal with GPU slides
Citation preview

Infernal-GPU:CUDA-Accelerated RNA Alignment
Adam Bazinet

Infernal
• Infernal: "INFERence of RNA ALignment"
• A software package for searching DNA sequence databases for RNA structure and sequence similarities
• Written and maintained by the Sean Eddy laboratory at Janelia Farm
Ribonucleic Acid (RNA)
• Much like DNA, RNA consistsof nucleotides (A, C, G, U)
• Unlike DNA, RNA is usuallysingle-stranded
• RNA molecules play a centralrole in many cellular processes
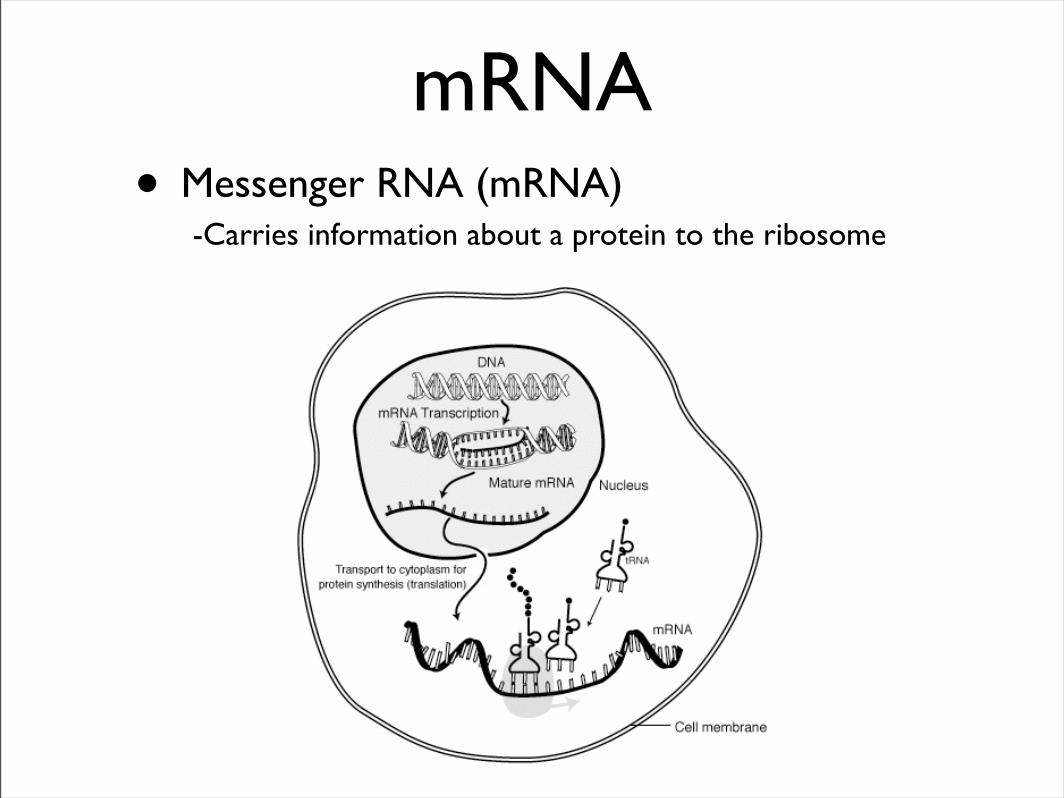
mRNA• Messenger RNA (mRNA)
-Carries information about a protein to the ribosome

ncRNA
• Non-coding RNA (ncRNA) -a functional RNA molecule that is not translated into a protein
• Many different sub-types; two of the most abundant are ribosomal RNA (rRNA) and transfer RNA (tRNA)
• Infernal is primarily concerned withthese functional, non-coding RNAs
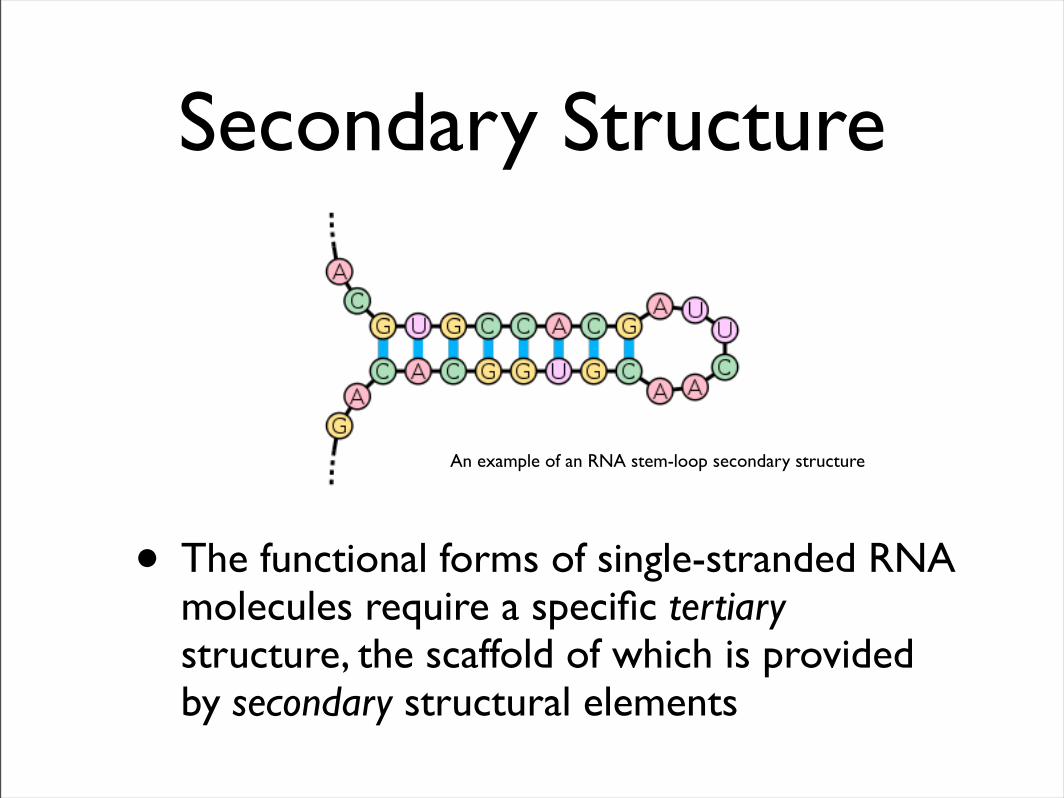
Secondary Structure
• The functional forms of single-stranded RNA molecules require a specific tertiary structure, the scaffold of which is provided by secondary structural elements
An example of an RNA stem-loop secondary structure
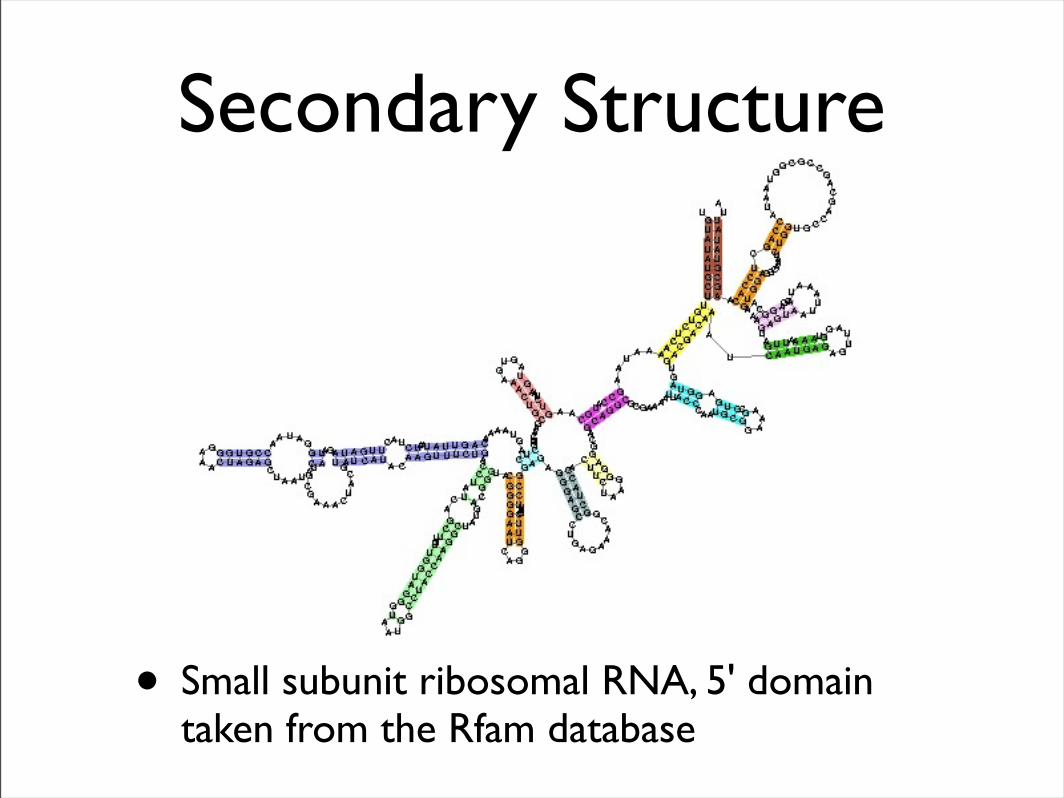
• Small subunit ribosomal RNA, 5' domain taken from the Rfam database
Secondary Structure

RNA Multiple Alignment

RNA Folding Algorithms
• Use a dynamic programming algorithm to computationally predict secondary structure according to a thermodynamic model
• Drawbacks: -only call 50-70% of base pairs correctly, on average -can be very computationally intensive (i.e., slow)
• Excellent summary article by Sean Eddy-Nature Biotechnology 22, 1457 - 1458 (2004)

Infernal
• Takes an RNA multiple alignment as input, and secondary structure must be provided! -the secondary structure is often determined in the laboratory
• Builds a covariance model (CM) from it
• Searches a target sequence database for possible matches to the input model

Covariance Model
• CMs are a type of stochastic context-free grammar (SCFG)
• Each residue in the query RNA is represented by a state, arranged in a tree-like structure that mirrors the secondary structure of the RNA, along with additional states to model insertions and deletions
• Dynamic programming calculates the probability that a substructure of the query rooted at state v aligns to a subsequence i..j in the target sequence
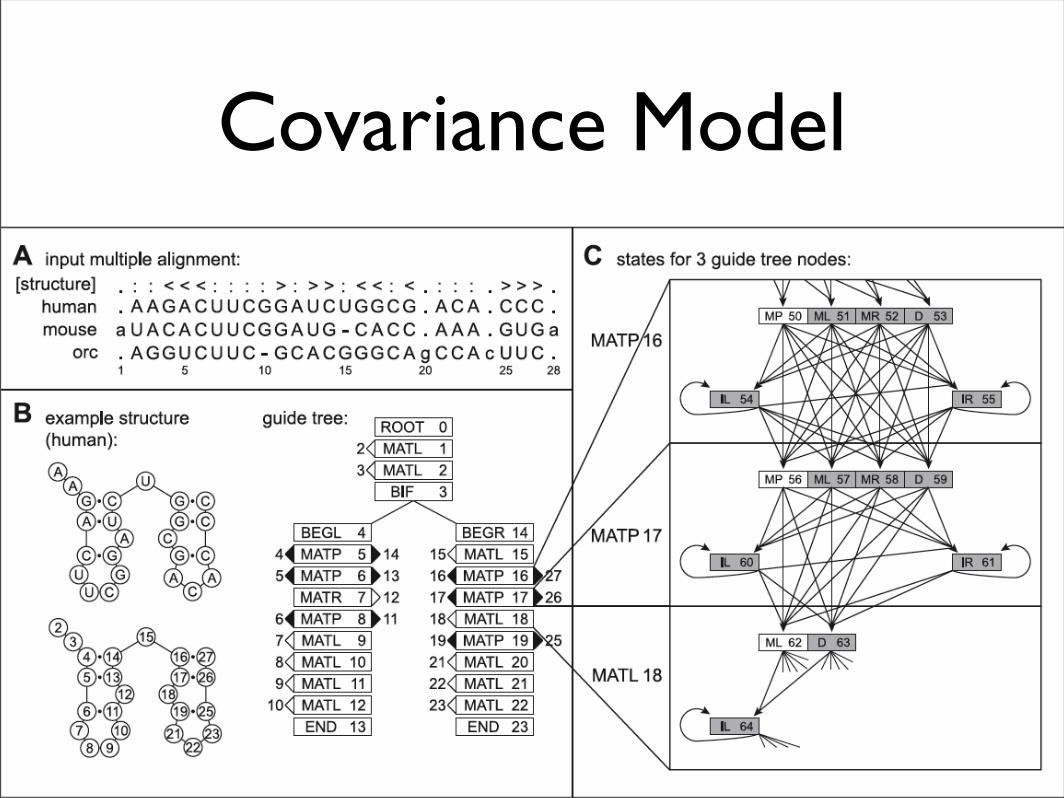
Covariance Model

Computational Complexity
• The most noteworthy limitation of SCFGs is their computational complexity
• SCFG-based RNA analysis algorithms require time and memory proportional to at least L3 (where L is the sequence length), because every possible pair of residues (L2) must be tried against up to L/2 base‐pairing states in the model (and in most RNA SCFGs, the time required more typically scales as L4)
• The latest version of Infernal incorporates some heuristics to ameliorate the situation, but the computational cost can still be considerable

Accelerating Infernal• There are two programs that would benefit
the most from speedup: -cmcalibrate (part of model building) -cmsearch (database searching)
• Both use a banded version of the ‘Inside’ algorithm, which is nearly identical to the Cocke-Younger-Kasami (CYK) database search dynamic programming algorithm for CMs
• CYK returns the optimal derivation, whereasInside returns the probability of the observation

Banded CYK Algorithm

Profiling Infernal
• Used a short test run of cmsearch as a test case
• ~25% of runtime was in FastIInsideScan, and ~22% of runtime was in ILogsum
• FastIInsideScan is optimized for the CPU, so there were 13 blocks of ILogsum calls - each of which is a potential target for parallelization

Parallelizing Infernal
• Each block of ILogsum calls was inside a loop
• Assigned each loop iteration to a separate GPU thread
• Ensured there were no redundant memory transfers
• However, the GPU version was ~9x slower than the optimized CPU version – why?
• Answer: with 22 billion kernel invocations, the overhead of invoking the kernel was greater than the work the kernel was actually doing!

Parallelizing Infernal
• Switched to working with RefIInsideScan, a simpler, non-optimized reference implementation
• Saw an opportunity for parallelization at the level of the ‘v-loop’ (loop over CM states)
• The v-loop was 229 iterations, each of which was assigned to a separate GPU thread
• v-loop was nested inside the j-loop (loop over database sequence positions) – j-loop was ~17,000 iterations, which means far fewer kernel invocations than in FastIInsideScan

Parallelizing Infernal
• Even after moving all memory transfers outside the j-loop, the program still ran ~7x slower than the reference CPU program
• Best current explanation is that the kernel is not optimized – there are large numbers of incoherent reads/writes
• Perhaps with additional work, a speedup can be attained – source code is available: http://www.cbcb.umd.edu/~pknut777/

Takeaways
• It was difficult to dive into complex scientific code and attempt to parallelize it
• Spent a LOT of time profiling the application, determining the extents of host arrays, chasing down runtime errors, etc.
• Very much enjoyed learning about this unique problem area of bioinformatics

Acknowledgments
• Eric Nawrocki, a graduate student who develops Infernal in the Eddy Lab, provided helpful information along the way
• Many thanks, Eric!

Questions?







![[height=3.0cm]img/teciplogoeng OpenMP and GPU …retis.sssup.it/~giorgio/slides/cbsd/Ruffaldi-gpu1.pdf · OpenMP and GPU Programming GPU Intro Emanuele Ru aldi ... CUDA scalar version](https://img.dokumen.tips/doc/110x75/5b6542857f8b9ab63a8b557a/height30cmimgteciplogoeng-openmp-and-gpu-retissssupitgiorgioslidescbsdruffaldi-gpu1pdf.jpg)





















