Embed Size (px)
Citation preview

Improving blood donor screening by nucleic acid technology(NAT)M. Schmidt & E. SeifriedInstitute of Transfusion Medicine and Immunohematology, German Red Cross, Johann Wolfgang Goethe University, Frankfurt, Germany
The description of the ABO blood group system by Landsteiner and coworkersmarked a sea change in making blood transfusions feasible and safe for a broadrange of indications. Nevertheless, with an increase in blood transfusions, side-effects such as transfusion-transmitted infections (TTIs) became more and moreimportant. A major challenge in transfusion medicine was (and is) to developscreening assays with maximum analytical sensitivity and analytical specificity toreduce the diagnostic window period as much as possible. Until the late 1990s,blood screening for TTIs depended entirely on serological assays. Except for HBV,where the virus can be detected using HBs-antigen assays, tests for the detection ofother TTIs relied almost exclusively on antibody detection. These tests, however, areassociated with a relatively long diagnostic window period because they detect theresponse of the immune system to an infection.
In the mid 1990s, the residual risk of transfusion-associated
HCV infection was estimated higher than 1:5000. New
upcoming molecular technologies, such as the polymerase
chain reaction (PCR), were examined to investigate how
these methods could be implemented in blood donor
screening to reduce this risk.
The German Red Cross Blood donor service, Frankfurt
am Main, developed its own ‘in-house’ method and was the
first to publish feasibility and efficiency of nucleic acid
technology (NAT) blood donor screening resulting in the
release of all blood components including packed red cells,
fresh frozen plasma and platelet concentrates being free of
HIV-1, HBV and HCV [1].
This review reports on the development of nucleic acid
amplification tests and describes the current state of tech-
nology. Functional principles of the different nucleic
amplification technologies (NAT) are depicted, and blood
donor screening by NAT for different viruses is described.
Additionally, the special situation of bacterial detection by
NAT is discussed. Blood donor screening by NAT was
started using in-house methods. Over the last decade, these
systems were significantly improved and certified by the
FDA or the EU. Currently, three fully automated, barcode-
controlled NAT systems are available for blood donor
screening (detection in individual donations or in mini-
pools up to 96 samples per pool). In most developed coun-
tries, NAT screening for HCV-RNA and HIV-1-RNA is per-
formed.
Depending on the screening strategy, blood donor testing
by NAT is able to reduce the diagnostic window period for
transfusion-transmitted infections (TTIs) such as HCV to a
minimum of 4–6 days. This has led to a residual risk of TTIs
of less than 1:1 million for HCV and HIV-1 in countries
using NAT and underlines the efficiency of these methods.
However, in some cases, NAT detection may fail owing
to mutations in the genome of the pathogen. Several cases
of TTIs have been reported in the literature in which muta-
tions in primer and probe binding regions were the major
cause for a reduced analytical sensitivity and for screening
failures. Amplification in at least two conserved genomic
regions is a promising approach to overcome this risk. Gen-
eric bacterial detection can also be carried out by NAT, but
there are a few drawbacks.
Serological combination assays may be economic alter-
natives to NAT but are associated with a longer diagnostic
window period compared to NAT systems. Pathogen-inacti-
vation methods are feasible for platelets and plasma prod-
ucts, but general inactivation methods for all three blood
products are still eagerly awaited.
Correspondence: Michael Schmidt, MD, Institute of Transfusion Medicineand Immunohematology, Johann Wolfgang Goethe University, GermanRed Cross, Sandhofstr. 1, 60528 Frankfurt am Main, GermanyE-mail: [email protected]
ISBT Science Series (2010) 5, 219–229
STATE OF THE ART 5C-S36-02 ª 2010 The Authors.Journal compilation ª 2010 International Society of Blood Transfusion
219

The description of the ABO blood group systems by Land-
steiner et al. [2] was an enormous milestone for making
transfusions of whole blood or blood components feasible
and safe for a broad range of indications. However, concern-
ing the principle of Hippocrates [3] of ‘primum nihil nocere’,
this placed physicians in a difficult situation. On the one
hand, they needed blood components for the successful
treatment of many diseases, but on the other hand, the
adverse side-effects, such as infections, were (and still are)
potentially problematic often causing severe life-threaten-
ing disease. To avoid infection, the most critical point is
the diagnostic window period [4], which is defined as the
time period between the start of an infection and the first
opportunity to recognize the infection by diagnostic testing.
Shortening the diagnostic window period has been the focus
of the last three decades of transfusion medicine. Therefore,
many general safety procedures were implemented in blood
donor screening, including critical donor selection [5], a
donor self-exclusion opportunity [6], the storage of quaran-
tined plasma and the development of new screening sys-
tems.
Mullis et al. [7,8] discovered a new molecular detection
method, named polymerase chain reaction (PCR), that is
able to produce multiple genome copies after 40–50 ampli-
fication cycles. A final concentration of approximately 1
billion genome replicates can easily be detected by agarose
gel electrophoresis followed by staining with ethidium bro-
mide. The disadvantage of first PCR version was the neces-
sity to reopen sample tubes after amplification for
detection by gel electrophoresis. This was time-consuming,
and there was an associated risk of cross-contamination
between different samples. In the beginning, PCR was only
feasible for the analysis of individual samples and not for
blood donor screening programmes. A subsequent advance
was the development of real-time-NAT [9]. By adding
oligonucleotides (approximately 20 basepairs of nucleo-
tides) labelled with two different fluorochromes able to
absorb and emit light at different wavelengths, the real-
time NAT detection can be carried out in one step without
the reopening of any sample tubes. The development of a
new generation of enzymes facilitates one-step procedures
for DNA and RNA amplification (including a reverse tran-
scription step). In principle, the two technologies can be
differentiated and will be discussed below:
(1) transcription-mediated amplification (TMA)
(2) real-time PCR technologies (with different primer and
probe designs).
Transcription-mediated amplification (TMA)
TMA [10,11] is used to amplify portions of RNA and ⁄ or
DNA. Reverse transcriptase creates a DNA copy (cDNA) of
the target nucleic acid. The RNA polymerase initiates
transcription, synthesizing RNA. Some of the newly syn-
thesised RNA amplification products re-enter the TMA pro-
cess and serve as templates for new rounds of
amplification. The amplification process is mediated by a
T7 promoter. More than 1000 amplification products are
produced in one cycle, and potentially billions of copies are
generated in < 1 h. Detection is carried out by acridinium
ester (AE)–labelled probes specifically hybridized to the
amplification products. Different AE variants are used to
label the internal control specific (IC-specific) and viral-
specific probes. The hybridization protection assay process
selectively inactivates the AE label on unhybridized probes
to minimize the background signal. Dual kinetic assay tech-
nology enables simultaneous detection of both IC-encoded
RNA, through a flash of light, and viral-encoded RNA,
through a longer lasting glow.
Real-time PCR technologies
Real-time PCR technologies can be classified into systems
using intercalation dyes (e.g. SYBR green or ethidium bro-
mide), systems with fluorescence resonance energy transfer
probes [12–14] (FRET-probes) and others [15,16].
In principle, all systems use at least one sense primer,
one antisense primer and any kind of probe, enzyme and
nucleotide.
Systems based on FRET use specific probes labelled with
one or two fluorochromes. During amplification, the DNA
polymerase works also as an exonuclease. Therefore, probes
bound at the target can be degraded, and the distance
between both dyes can be enlarged. This changes the
energy transfer between the reporter dye and the quencher
dye. Classic real-time NAT systems are available as Taq-
Man� assays with TaqMan� probes (hydrolysis probes) or
with hybridization probes. Two small hybridisation probes
are labelled, each with one dye. With this configuration, a
melting-curve analysis [17,18] (determination of the spe-
cific temperature at which probes bind to templates) can
also be performed. Another real-time NAT system uses spe-
cific probes named molecular beacons [19,20]. These are
small molecules with changing physical shapes depending
on the temperature. At lower temperatures, the molecular
beacons exist in a close state, the fluorophore and the
quencher are held in close proximity to each other by the
hairpin stem, and there is no fluorescence. However, at high
temperatures, the helical order of the stem gives way to a
random-coil configuration, separating the fluorophore
from the quencher and restoring fluorescence. The temper-
ature at which the stem melts depends upon the GC content
and the length of the stem sequence. If a target is added to
a solution containing a molecular beacon at temperatures
below the melting temperature of its stem, the molecular
beacon spontaneously binds to its target, thereby
220 M. Schmidt & E. Seifried
� 2010 The Authors.Journal compilation � 2010 International Society of Blood Transfusion, ISBT Science Series (2010) 5, 219–229
dissociating the stem and turning on the fluorescence. The
manner in which the fluorescence of the probe–target
hybrid varies with the temperature is indicated by the red
fluorescence vs. the temperature trace. At low temperatures,
the probe–target hybrid remains brightly fluorescent, but
as the temperature is raised, the probe dissociates from the
target and tends to return to its hairpin state, diminishing
the fluorescence significantly. The temperature at which
the probe–target hybrid melts apart depends upon the GC
content and the length of the probe sequence. Other real-
time NAT systems working with FRET technology (e.g.
scorpion probes [21,22]) are slightly different but work on
the same principle. The major benefits of real-time NAT
systems compared to classical PCR systems are an improved
linear range and a closed technology, as the sample cups do
not need to be reopened after amplification.
Pooling and extraction methods
Blood donor services are responsible for releasing life-
saving blood components for many kinds of different
therapeutic strategies all over the world. Therefore, some
countries centralize all blood donor screening tests in
one or two laboratories. These test centres have to
screen up to 10 000 samples per day. Analysing such a
large number of samples by NAT daily is a big chal-
lenge. Therefore, pooling procedures were developed to
reduce the total number of samples. Countries like
Japan started in 1999 with mini-pools of up to 500
samples per pool for HBV, HCV and HIV-1 [23]. Other
countries or blood services like ours very soon in
1996 ⁄ 1997 developed a mini-pool NAT (MP-NAT) sys-
tem with up to 96 samples per pool [1,24,25]. After the
pooling process, high-speed centrifugation was used to
enrich viruses at the bottom of a centrifugation tube,
followed by a manual extraction procedure using chao-
tropic salts [26]. At the beginning of blood donor
screening by NAT, no commercial systems were avail-
able. Therefore, blood banks developed their own
‘in-house’ systems to improve blood safety by this new
technology. Now, with more than 10 years of experience
with NAT, the situation has completely changed. So far
to our knowledge, three fully automated barcode-con-
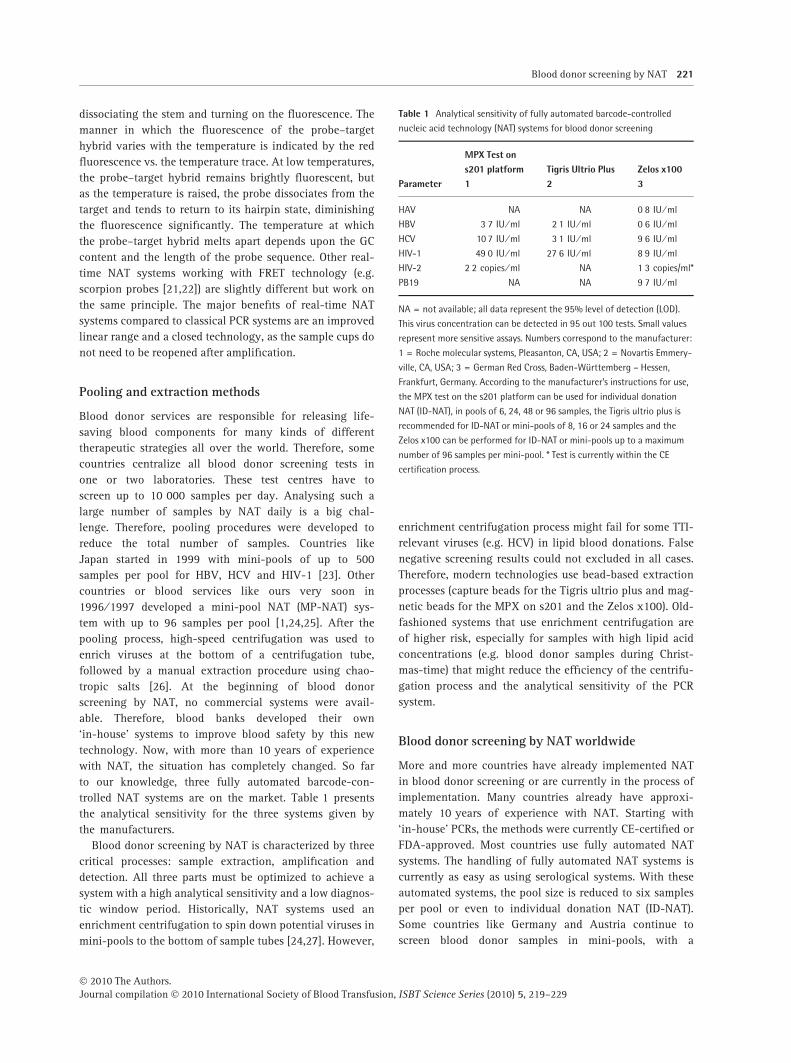
trolled NAT systems are on the market. Table 1 presents
the analytical sensitivity for the three systems given by
the manufacturers.
Blood donor screening by NAT is characterized by three
critical processes: sample extraction, amplification and
detection. All three parts must be optimized to achieve a
system with a high analytical sensitivity and a low diagnos-
tic window period. Historically, NAT systems used an
enrichment centrifugation to spin down potential viruses in
mini-pools to the bottom of sample tubes [24,27]. However,
enrichment centrifugation process might fail for some TTI-
relevant viruses (e.g. HCV) in lipid blood donations. False
negative screening results could not excluded in all cases.
Therefore, modern technologies use bead-based extraction
processes (capture beads for the Tigris ultrio plus and mag-
netic beads for the MPX on s201 and the Zelos x100). Old-
fashioned systems that use enrichment centrifugation are
of higher risk, especially for samples with high lipid acid
concentrations (e.g. blood donor samples during Christ-
mas-time) that might reduce the efficiency of the centrifu-
gation process and the analytical sensitivity of the PCR
system.
Blood donor screening by NAT worldwide
More and more countries have already implemented NAT
in blood donor screening or are currently in the process of
implementation. Many countries already have approxi-
mately 10 years of experience with NAT. Starting with
‘in-house’ PCRs, the methods were currently CE-certified or
FDA-approved. Most countries use fully automated NAT
systems. The handling of fully automated NAT systems is
currently as easy as using serological systems. With these
automated systems, the pool size is reduced to six samples
per pool or even to individual donation NAT (ID-NAT).
Some countries like Germany and Austria continue to
screen blood donor samples in mini-pools, with a
Table 1 Analytical sensitivity of fully automated barcode-controlled
nucleic acid technology (NAT) systems for blood donor screening
Parameter
MPX Test ons201 platform1
Tigris Ultrio Plus2
Zelos x1003
HAV NA NA 0Æ8 IU ⁄ ml
HBV 3Æ7 IU ⁄ ml 2Æ1 IU ⁄ ml 0Æ6 IU ⁄ ml
HCV 10Æ7 IU ⁄ ml 3Æ1 IU ⁄ ml 9Æ6 IU ⁄ ml
HIV-1 49Æ0 IU ⁄ ml 27Æ6 IU ⁄ ml 8Æ9 IU ⁄ ml
HIV-2 2Æ2 copies ⁄ ml NA 1Æ3 copies/ml*
PB19 NA NA 9Æ7 IU ⁄ ml
NA = not available; all data represent the 95% level of detection (LOD).
This virus concentration can be detected in 95 out 100 tests. Small values
represent more sensitive assays. Numbers correspond to the manufacturer:
1 = Roche molecular systems, Pleasanton, CA, USA; 2 = Novartis Emmery-
ville, CA, USA; 3 = German Red Cross, Baden-Württemberg – Hessen,
Frankfurt, Germany. According to the manufacturer’s instructions for use,
the MPX test on the s201 platform can be used for individual donation
NAT (ID-NAT), in pools of 6, 24, 48 or 96 samples, the Tigris ultrio plus is
recommended for ID-NAT or mini-pools of 8, 16 or 24 samples and the
Zelos x100 can be performed for ID-NAT or mini-pools up to a maximum
number of 96 samples per mini-pool. * Test is currently within the CE
certification process.
Blood donor screening by NAT 221
� 2010 The Authors.Journal compilation � 2010 International Society of Blood Transfusion, ISBT Science Series (2010) 5, 219–229

maximum pool size of 96. Blood donor screening by NAT
for at least HIV-1 and HCV has been implemented in differ-
ent countries (e. g. USA, Canada, parts of Brazil, Spain,
France, the UK, Denmark, Germany, the Netherlands, Bel-
gium, Greece, Slovenia, the Czech Republic, South Africa,
Ghana, Luxembourg, Switzerland, Italy, Japan, parts of
China, Australia, Poland, Norway, Finnland and New Zeal-
and). One exception in Europe is Sweden. Based on the very
low incidence of HIV-1 and HCV in their donor population,
they decided to stop blood donor screening by NAT in
2008.
Blood donor screening for pathogens by NAT can be
divided into four groups:
(1) transfusion-relevant pathogens that are generally tes-
ted for in many countries (HBV, HCV and HIV-1),
(2) transfusion-relevant pathogens that are tested for only
in some countries with special circumstances (WNV,
HAV, B19V, Chikungunya virus and HIV-2),
(3) hepatitis A virus (HAV) pathogens that are probably
transfusion-relevant but that are currently not indicated
as special risks for blood transfusions and not tested for
in blood donor screening programmes (SARS CoV and
Influenza viruses),
(4) bacterial screening by NAT.
Transfusion-relevant pathogens that aregenerally tested
Human immunodeficiency virus 1 (HIV-1)
The first cases of immune deficiency after blood transfusion
were reported in 1982. In 1983, HIV was described as the
cause of acquired immune deficiency syndrome (AIDS). A
detection assay was available in the mid 1980s, but at this
time, many haemophilia A and haemophilia B patients were
already infected from the transfusion of contaminated
blood components. HIV is an RNA virus and can be divided
into three groups (M group, O group and N group). The M
group can be characterized into subtypes (A, B, C, D, F, G,
H, J and K) and circulating recombinant forms. The virus
doubling time is approximately 17 h. Therefore, the diag-
nostic window period is approximately 8–9 days by screen-
ing in mini-pools of up to 96 samples per pool and can be
reduced to 5–6 days for ID-NAT. Blood donor screening by
MP-NAT for HIV-1 was mandated in Germany in 2004.
After this time-point, only one case of a transfusion-trans-
mitted HIV-1 infection was reported (see below risk analy-
sis of NAT systems) [28].
Hepatitis C virus infections
Hepatitis C viruses belong to the flavivirus family. The virus
was first described in 1989, but it was known since the
1970s that a virus other than HAV and HBV existed; it was
originally named ‘non-A-non-B’ hepatitis. The first anti-
body screening assays were available in 1990 (EIA first
generation). The diagnostic window period was approxi-
mately 80 days for these assays. The virus doubling time is
very short (approximately 10–11 h). Therefore, blood donor
screening by NAT was able to reduce the diagnostic win-
dow period to 6–7 days for screening in mini-pools (with a
maximum pool size of 96 samples per pool) or to 4–5 days
for ID-NAT. Before the introduction of blood donor screen-
ing by NAT, the residual transfusion-transmitted infectious
risk was estimated to be 1:200 [29], and it is currently cal-
culated at 1:10Æ88 million [30]. After blood donor screening
by NAT was mandated in Germany, only one single case
was reported as a TTI [31]. The donation was performed in
the very early infection period with a virus load of only
10 IU ⁄ ml, which was below the analytical sensitivity of the
MP-NAT.
Hepatitis B virus infections
Blood donor screening for HBV is performed on a voluntary
basis, although most of the fully automated NAT systems
enable the detection of this pathogen. HBV belongs to the
hepadnavirus family and is a DNA retrovirus. Compared to
HIV-1 or HCV, the virus doubling time is very low, at
approximately 2Æ56 days [32]. The virus can be integrated
into the genome of hepatocytes, which are the primary tar-
get cells. Approximately 90% of infected patients suffer
from acute infection. The immune system is able to elimi-
nate the virus from plasma in 90% of cases. In approxi-
mately 10% of cases, HBsAg and ⁄ or HBV DNA can be
detected for more than 6 months. These cases are defined
as chronic HBV infections or occult HBV infections (OBI).
The virus load can be very low (< 10 IU ⁄ ml), which repre-
sents special challenges for diagnostic assays. Patients with
OBIs are at a higher risk of developing liver cirrhosis or
liver cancer after 10–15 years. OBIs can be detected by
anti-HBc, which is also tested for in blood donor screening
in low epidemic areas such as the USA or Germany. Screen-
ing for anti-HBc is not feasible in high epidemic areas such
as Asia because the percentage of anti-HBc reactive donors
might cause an unacceptable loss of necessary life-saving
blood components. General HBV vaccination programmes
for infants were implemented in the mid 1990s. These
might reduce the risk of HBV TTIs in the near future. Unfor-
tunately, most vaccines induce HBV-neutralizing antibod-
ies against genotype A. In this context, it is important that
the majority of HBV infections in Asia are of genotype B or
C. These HBV infections might not be sufficiently prevented
by the current vaccination programmes. Therefore, blood
donor screening by NAT with a high analytical sensitivity
is recommended.
222 M. Schmidt & E. Seifried
� 2010 The Authors.Journal compilation � 2010 International Society of Blood Transfusion, ISBT Science Series (2010) 5, 219–229

Transfusion-relevant pathogens that aretested for only in some countries with specialcircumstances (WNV, HAV, HEV, B19V,Chikungunya virus and HIV-2)
West Nile virus infections
West Nile virus infections (WNV) occurred in birds and
humans in the USA in 1999, spreading from the east coast
to the west coast within 3 years [33]. In 2003, the FDA
mandated blood donor screening by NAT [34] for all blood
donations. The incidence of WNV infections increases in
the summer season. Based on the low virus load, especially
in the preseroconversion period, blood donor screening was
implemented in mini-pools of eight in the winter season
and changed to ID-NAT blood donor screening with the
increasing WNV incidence in local districts. WNV infec-
tions were also reported in Europe in some regions, but
general screening is currently not recommended [35–37].
Hepatitis A virus infections
Hepatitis A viruses are small, non-enveloped RNA viruses
belonging to the picornaviridae family. Pathogen reduction
by solvent and detergent methods are less efficient for HAV.
The major infection route for HAV is the faecal–oral path-
way, but HAV can also be detected in blood components
[38–40]. Chronic infections are not described, but in rare
cases, an acute HAV virus infection can cause a fulminant
liver dysfunction. Although the incidence of HAV is low,
blood safety can be improved by real-time NAT systems [41].
Hepatitis E virus infection
Hepatitis E virus is a small RNA virus that belongs to the
caliciviridae family. Infections are frequent in Asia, the
Near East, Africa and Middle America. The virus originates
in drinking water contaminated with faeces or in infected
animals (pigs). TTIs were reported in Japan and the UK
[42–44]. Blood donor screening by NAT might possibly pre-
vent these infections, but they are rare events. Cost–benefit
analyses are still needed to calculate the value of this
parameter. General screening of blood donations by NAT
for HEV is not recommended.
Parvovirus B19 virus infections
Parvovirus B19 is a non-enveloped DNA virus that was
detected in 1975. The virus grows to very high virus con-
centrations (up to 1014 IU ⁄ ml) with only mild symptoms,
such as tiredness, in most cases. The virus binds to the P
antigen at erythrocyte precursor cells and induces apopto-
sis. The B19 virus can cause haemolysis, which might be
clinically relevant for infants and newborns. Transfusion
transmissions by blood components are described in case
reports [45]. The infections depend on the immune response
of the recipients. Approximately 60% of adults 30 years of
age will have relevant levels of neutralizing antibodies
owing to a past infection. A recently published retrospec-
tive study by Kleinman et al. [46] could not confirm a TTI
in recipients transfused with blood products with a low
virus load.
Chikungunya virus infections
In recent years, large Chikungunya virus (CHIKV) outbreaks
originating in Kenya have spread to islands of the Indian
Ocean and parts of India, Southeast Asia and Europe [47].
The concern of transfusion transmission has been height-
ened for this mosquito-borne arbovirus because of high
population infection incidence during outbreaks and the
high-titre viraemia lasting approximately 6 days. CHIKV
produces a fever–arthralgia syndrome, resulting in consid-
erable morbidity and some mortality, particularly among
older age groups and ⁄ or those with pre-existing conditions.
Possible measures to prevent possible CHIKV transfusion
transmission include the deferral of symptomatic donors,
discontinuing blood collections in affected areas and
CHIKV nucleic acid screening of donations. Even a rela-
tively small outbreak in Italy [48] resulted in a considerable
adverse impact on blood collections and economic conse-
quences. Assays suitable for testing donations for CHIKV
RNA are available as ‘in-house’ systems. Although there
were many cases of potentially transfusion-transmitted
CHIKV infections between 2005 and 2007 during the mas-
sive epidemic on Reunion Island, no cases are known to
have been confirmed by phylogenetic analysis.
Human immunodeficiency virus 2 infections
The global distribution of the two causes of acquired immu-
nodeficiency syndrome (AIDS), human immunodeficiency
virus type 1 (HIV-1) and HIV-2, are remarkably different. In
the Americas, Europe and Asia, there has been an epidemic
spread of HIV-1 in certain risk groups, mostly through
homosexual sex and injection drug use. In contrast, HIV-2
has been found predominantly in heterosexual populations
in West Africa but has spread very little to other areas
[49,50]. Based on reports from 2008 from the WHO, 33Æ4million people (range 31Æ1–35Æ8 million) were living with
AIDS, 2Æ7 million were newly infected in 2008 (range 2Æ4–
3Æ0 million) and 2Æ0 million people died in 2008 as a conse-
quence of AIDS (range 1Æ7–2Æ4 million). The residual risk
from blood donation for HIV-1 is very low, especially in
countries where blood donor screening by NAT is imple-
mented. Although the infectious risk for HIV-2 outside the
Blood donor screening by NAT 223
� 2010 The Authors.Journal compilation � 2010 International Society of Blood Transfusion, ISBT Science Series (2010) 5, 219–229

middle of Africa is very low, two fully automated NAT sys-
tems (Roche MPX test on the s201 platform and DRK HIV
1 ⁄ 2 PCR Kit on the Zelos ·100 platform) have already
added HIV-2 to a multiplex screening procedure.
Pathogens that are probably transfusionrelevant but that are currently not indicatedas special risks for blood transfusions and arenot implemented into blood donor screeningprogrammes (SARS CoV, Influenza viruses andH1N1)
Every year, a ‘new’ well-known pathogen gains attention in
the daily news. In 2003, an epidemic of corona viruses was
reported in Asia. Because of the general travel behaviour of
the human population, infections spread all over the world
within some days. The major transmission pathway of
SARS CoV was airborne infection, and at the end the epi-
demic, it could be curtailed efficiently by compliance to
strict quarantine procedures. However, in the beginning, no
information existed as to whether asymptomatic patients in
the early infection period were viraemic (in this case, blood
products could be infectious). SARS CoV was a good exam-
ple of the powerful opportunities provided by NAT systems.
After sequencing a genome, a specific real-time NAT sys-
tem can be developed within a few weeks [51,52].
A new antigen combination of influenza A viruses
(H1N1) was found in 2009 in a young child in Mexico. The
new virus supposedly originated in pigs. In history, an
influenza epidemic with the same antigens occurred in
1918 (i.e. the ‘Spanish flu’) that caused the death of approx-
imately 50 million people. Therefore, people all over the
world were alert and developed risk strategies to prevent
the global spreading of the new infection. In this special sit-
uation, the first effort was aimed at developing vaccines
against this new influenza infection, but later on, the sec-
ond or third activities were aimed at developing diagnostic
NAT systems for blood and sputum [53,54].
Bacterial screening by NAT
Improvements in blood donor screening systems, e.g. the
introduction of third and fourth generation antibody assays
and the introduction of nucleic acid testing (NAT) [55],
have reduced the risks of the transmission of clinically
relevant viral infections to far below the risk of the trans-
mission of bacterial infections. Therefore, bacterial contam-
ination of blood products represents an ongoing challenge
in transfusion medicine. Blood donor screening for bacte-
rial contamination is difficult because of the very low bac-
terial concentration after the production process. Donors
with relevant bacteraemia should have clinical symptoms
and are eliminated from blood donation. Transient or
resident skin bacteria in deep areas could be a source of the
bacterial contamination of blood components. Leucocytes
from the buffy coat and complement factors will also
reduce residual bacteria. Based on reverse calculations,
the amount of bacteria in contaminated platelet product is
estimated at 10 CFU ⁄ bag.
Many countries have implemented culture methods such
as BacT ⁄ ALERT to detect bacterial contamination of plate-
lets. As a result of the long incubation time, platelets were
released as ‘negative-to-date’. Based on the very low bacte-
rial concentration in the platelet products after production,
sample failures were reported in different countries with
negative screening results and severe TTIs owing to bacte-
rial contamination [56–58]. In Germany, the Paul-Ehrlich-
Institute reduced the shelf-life of platelet products from 5
to 4 days in 2009 as a result of a statistical analysis where
transfusion-transmitted fatalities were associated in four
out of five cases with a transfusion of platelets on day 5
after production. Additionally, rapid bacterial detection
systems were developed within the last 10 years and
include NAT and FACS systems.
Bacterial detection by NAT
Target genes for the development of generic bacterial
NAT systems are ribosomal structures such as 16s RNA
or 23s RNA. Unfortunately, the PCR enzymes were
extracted from thermoresistant bacteria such as Thermus
aquaticus, and these enzymes might be contaminated
with bacterial ribosomal genes, causing false-reactive
results. Feng et al. described one of the first assays for
the detection of Yersinia enterocolitica in blood with a
sensitivity of 5000 CFU ⁄ ml [59]. This sensitivity is not
acceptable for a blood screening test because a donor
with 2Æ5 million bacteria in 500 ml of blood
(5000 CFU · 500 ml) would have clinical symptoms that
would exclude the donor from blood donation. Newly
developed oligonucleotides with fluorescent molecules at
their 5¢ and 3¢ ends enable detection in a closed system
with improved sensitivity compared to PCR detection via
agarose gel electrophoresis. This real-time PCR system
for bacterial detection was recently described by Nadkar-
ni et al. [60] and has an analytical sensitivity between
30 and 100 CFU ⁄ ml; in principle, however, it did not
overcome the problem of non-specific signals. Moham-
madi et al. [61] solved this challenge by pretreating the
PCR mixture with the restriction enzyme Sau3AI prior
to the addition of template DNA. These authors were
able to improve the detection limit to 1 CFU equivalent ⁄PCR. Another solution might be an additional filtration
of all NAT reagents with plasmid binding columns [62].
Both methods can be combined to optimize the results.
Other investigators have attempted to decontaminate
224 M. Schmidt & E. Seifried
� 2010 The Authors.Journal compilation � 2010 International Society of Blood Transfusion, ISBT Science Series (2010) 5, 219–229

PCR materials and reagents by UV irradiation, 8-meth-
oxypsoralen treatment, DNase treatment or combinations
of these methods [62–66]. However, most of these meth-
ods also reduce the analytical sensitivity. Therefore,
some investigators recommend reductions in the number
of PCR cycles as the most effective and reproducible
way of avoiding false-positive results [60,64]. Real-time
NAT is a powerful tool in the clinical diagnosis of bac-
terial contamination in blood products. The extraction
method can be completely automated [67,68] and bar-
code-controlled to enable screening of a huge number
of donations. DNA ⁄ RNA extraction can be performed
with material from platelet concentrates and whole
blood to include all blood products (erythrocytes, plate-
let concentrates and plasma) in the bacterial screening
process. The analytical sensitivity is currently between
10 and 50 CFU ⁄ ml and thus is slightly behind the sensi-
tivity of culture methods. The total screening time for
NAT systems (extraction and amplification) takes
approximately 4 h. Therefore, these methods offer oppor-
tunities for a late sample collection to overcome sample
errors. In this context, rapid bacterial detection systems
can be used for bacterial detection on day 4 platelets. In
the case of negative results, the shelf-life can be
extended to 5 days (see Fig. 1).
Special NAT risks and NAT failures
Blood safety was significantly improved by the introduc-
tion of NAT systems through the reduction of the diagnos-
tic window period, especially for transfusion-transmitted
virus infections. Nevertheless, the new technologies are not
risk free, which will be discussed below.
Diagnostic window period of donation
The introduction of NAT systems into blood donor screen-
ing was able to reduce the diagnostic window period to
only a few days for HCV and HIV-1. Currently, at least three
fully automated NAT systems are available on the market
that enable blood banks to reduce pool sizes even to
ID-NAT. This topic is currently a point of discussion in the
Transfusion Medicine Society. On the one hand, the highest
analytical sensitivity will achieve the maximum blood
safety. However, the benefit for ID-NAT compared to MP-
NAT is limited for HCV and HIV-1. For HBV (doubling time
of 2Æ56 days), the situation is different. NAT systems with a
very high analytical sensitivity are still needed to detect
infected blood donors in the preseroconversion time period
as well as in the second diagnostic window period in a rea-
sonable time. On the other hand, ID-NAT increases the total
costs for public health systems. In this context, each coun-
try must come to its own decision. On a related note, it is of
interest that Sweden stopped blood donor screening by
NAT for HCV and HIV-1 in 2008. Currently, all countries
that have implemented NAT systems into their blood donor
screening programmes are using mini-pool sizes between
individual donations and pools with up to 24 samples per
pool. Only Germany and Austria, two countries with low
incidences for HCV, HIV-1 and HBV, are screening blood
donors by NAT in mini-pools up to a maximum pool size of
96 samples per pool. Independent of the maximum pool
size, it should be kept in mind that a very small number of
blood donors might be infected with virus concentrations
below the analytical sensitivity of the test of record, as
described for HCV by Kretzschmar et al. [31]. Therefore,
NAT cannot guarantee 100% safety.
Virus mutation in primer ⁄probe regions
All transfusion-transmission-relevant viruses can be subdi-
vided into different genotypes and subtypes. RNA viruses
are of higher risk of genetic diversity because the RNA must
be reverse-transcribed into DNA during the amplification
process. The enzyme responsible for this lacks a proofread-
ing function. Therefore, any mistakes during the process
will not be corrected. These mutations represent a risk for
NAT systems if they occur in the primer and probe binding
regions. In 2007, the first transmission of HIV-1 in Ger-
many after the introduction of mandatory MP-NAT was
reported by Schmidt et al. [28]. The infective donation was
probably missed by MP-NAT owing to mutations in the
probe binding region and the antisense primer binding
Days0 1 2 3 4 5
Release of plateletsproducts form day 1
to day 3 withoutany bacterial tests
Blo
od d
onat
ion
(don
or s
elec
tion,
sta
bdar
d sk
in d
esin
fect
ion,
pre-
dona
tion
sam
oplin
g)
Pro
duct
ion
of p
late
lets
(sel
f-st
erili
zing
effe
cts)
Rapid bacterial test on day 4
Self life forplatelets withneg. testresults for5 days
Shelf life afterpathogen inactivation for 5 days
Pat
ho
gen
in
acti
vati
on (a)
(b)
Fig. 1 Blood donor screening for bacterial contamination of platelets.
Blood safety for platelets can be improved by two methods. Scheme a:
Pathogen-inactivation method directly after the production of platelet
products. Scheme b: Release of platelet products on day 1 to day 3 without
any additional screening. Retesting of all platelets on day 4 with rapid bac-
terial screening assays (FACS or nucleic acid technology) and release of all
platelets with negative screening results on day 4 to day 5. This procedure
is in accordance with the guidelines from the Paul-Ehrlich-Institute.
Blood donor screening by NAT 225
� 2010 The Authors.Journal compilation � 2010 International Society of Blood Transfusion, ISBT Science Series (2010) 5, 219–229

region. The manufacturers are aware of the methodological
risk by real-time NAT and have developed screening sys-
tems with the parallel amplification of at least two con-
served regions.
NAT alternatives
Within the last 5 years, new combination assays for the
parallel detection of antigens and antibodies were devel-
oped for HCV and HIV, respectively. Barbara et al. com-
pared the analytical sensitivities of different assays. The
optimized antigen tests for HCV requires an additional
3 days for the diagnostic window period compared to
NAT. The best HCV combo test was reactive 5 days after
NAT. Such data will be comparable for HIV. These data
clearly demonstrate that blood donor screening by NAT
will reduce the diagnostic window to a minimum; on
the other hand, if NAT technology cannot be imple-
mented, a combo test could be a fairly good alternative
to improve blood safety and should be in these cases of
state-of-the-art methods.
Other alternatives to screening methods could include
pathogen inactivation or reduction methods. Three differ-
ent methods have been developed for platelet products
and for plasma products with different pathomecha-
nisms. Pathogen-inactivation methods can be divided into
photochemical systems [69,70] (e.g. S59 ⁄ Amotosalen,
Intercept�, Cerus), photodynamic systems [71–73] (e.g.
Riboflavin, Mirasol�, Gambro BCT) and systems using
only UV-C light [74]. Independent of the method, patho-
gen-inactivation technologies can inactivate viruses or
bacteria up to 6 log phases. For most of the pathogens,
the capacity will be sufficient, especially in the early
infection period. Only some viruses such as Parvovirus
B19 could occur in asymptomatic donors in concentra-
tions up to 1014 IU ⁄ ml. Pathogens such as Bacillus cereus
could be other exceptions, as they can occur in both veg-
etative and spore states. Spores are extremely resistant
against environmental conditions. Unfortunately, patho-
gen-inactivation reagents penetrate into spores less effi-
ciently. These are two reasons to keep in mind that
inactivation could be incomplete for pathogen reduction
methods. The different types of blood products are
another challenge for the inactivation methods; different
systems are recommended for plasma, platelets and ery-
throcytes. However, there is already some experimental
data that one system, such as the Mirasol system, can be
used for both platelets and red cells. Based on these
points, a combination of pathogen inactivation together
with MP-NAT could be the blood screening procedure of
the future. New unknown pathogens will be inactivated,
and high concentrations of viruses can be detected by
MP-NAT. In combination with pathogen-inactivation
methods, the maximum number of samples pooled
together for NAT will be the subject of future discussions.
Summary
Blood donor screening by NAT reduces the diagnostic win-
dow period to only a few days. Therefore, the residual
transfusion transmission risk is very low. The implementa-
tion of the NAT system is as easy as the implementation of
serological systems, as three fully automated NAT systems
are already available on the market. NAT systems are also
available for bacterial detection in platelets. This special
situation requires a late sample collection. NAT systems
can be used for new pathogens, as real-time NAT systems
will be available immediately after the sequencing of new
pathogens. The maximum pool size used for blood donor
screening is still a point of discussion. On the one hand,
blood donor screening by ID-NAT reduces the diagnostic
window period to a minimum, but on the other hand, MP-
NAT in combination with pathogen-inactivation methods
may represent a new standard for blood donor screening
and will probably be feasible for all three blood compo-
nents in the near future.
Disclosure
No potential conflicts declared.
References
1 Roth WK, Weber M, Seifried E: Feasibility and efficacy of rou-
tine PCR screening of blood donations for hepatitis C virus,
hepatitis B virus, and HIV-1 in a blood-bank setting. Lancet
1999; 353:359–363
2 Landsteiner K: Individual differences in human blood. Science
1931; 73:403–409
3 Rosahl SK, Samii M: ‘‘Nihil nocere’’ and beyond: the issue of
leadership in healthcare. Healthc Pap 2003; 4:78–82; discus-
sion 8–90
4 Dodd R, Kurt Roth W, Ashford P, Dax EM, Vyas G: Transfusion
medicine and safety. Biologicals 2009; 37:62–70
5 Epstein JS: Alternative strategies in assuring blood safety: an
overview. Biologicals 2010; 38(1):31–35
6 Brennan MT, Hewitt PE, Moore C, Hall G, Barbara JA: Confiden-
tial unit exclusion: the North London Blood Transfusion Cen-
tre’s experience. Transfus Med 1995; 5:51–56
7 Mullis PE, Brickell PM: The use of the polymerase chain reac-
tion in prenatal diagnosis of growth hormone gene deletions.
Clin Endocrinol (Oxf) 1992; 37:89–95
8 Mullis KB: Target amplification for DNA analysis by the
polymerase chain reaction. Ann Biol Clin (Paris) 1990;
48:579–582
9 Fronhoffs S, Bruning T, Ortiz-Pallardo E, Brode P, Koch B,
Harth V, Sachinidis A, Bolt HM, Herberhold C, Vetter H, Ko Y:
Real-time PCR analysis of the N-acetyltransferase NAT1 allele
226 M. Schmidt & E. Seifried
� 2010 The Authors.Journal compilation � 2010 International Society of Blood Transfusion, ISBT Science Series (2010) 5, 219–229

*3, *4, *10, *11, *14 and *17 polymorphism in squamous cell
cancer of head and neck. Carcinogenesis 2001; 22:1405–1412
10 Benjamin RJ: Nucleic acid testing: update and applications.
Semin Hematol 2001; 38:11–16
11 Allain JP: Genomic screening for blood-borne viruses in trans-
fusion settings. Clin Lab Haematol 2000; 22:1–10
12 Kashida H, Takatsu T, Sekiguchi K, Asanuma H: An efficient
fluorescence resonance energy transfer (FRET) between pyrene
and perylene assembled in a DNA duplex and its potential
for discriminating single-base changes. Chemistry 2010; 16(8):
2479–2486
13 Lassauniere R, Kresfelder T, Venter M: A novel multiplex real-
time RT-PCR assay with FRET hybridization probes for the
detection and quantitation of 13 respiratory viruses. J Virol
Methods 2010; 165(2):254–260
14 Olsen EV, Gibbins CS, Grayson JK: Real-time FRET PCR assay
for Salmonella enterica serotype detection in food. Mil Med
2009; 174:983–990
15 Buh Gasparic M, Tengs T, La Paz JL, Holst-Jensen A, Pla M,
Esteve T, Zel J, Gruden K: Comparison of nine different
real-time PCR chemistries for qualitative and quantitative
applications in GMO detection. Anal Bioanal Chem 2010;
396(6):2023–2029
16 Kusser W: Use of self-quenched, fluorogenic LUX primers
for gene expression profiling. Methods Mol Biol 2006;
335:115–33
17 Erali M, Wittwer CT: High resolution melting analysis for gene
scanning. Methods 2010; 50(4):250–261
18 Crews N, Wittwer CT, Montgomery J, Pryor R, Gale B: Spatial
DNA melting analysis for genotyping and variant scanning.
Anal Chem 2009; 81:2053–2058
19 Sandhya S, Chen W, Mulchandani A: Molecular beacons: a
real-time polymerase chain reaction assay for detecting Escher-
ichia coli from fresh produce and water. Anal Chim Acta 2008;
614:208–212
20 Gubala AJ, Proll DF: Molecular-beacon multiplex real-time
PCR assay for detection of Vibrio cholerae. Appl Environ
Microbiol 2006; 72:6424–6428
21 Stroup SE, Roy S, McHele J, Maro V, Ntabaguzi S, Siddique A,
Kang G, Guerrant RL, Kirkpatrick BD, Fayer R, Herbein J, Ward
H, Haque R, Houpt ER: Real-time PCR detection and speciation
of Cryptosporidium infection using Scorpion probes. J Med
Microbiol 2006; 55:1217–1222
22 Arya M, Shergill IS, Williamson M, Gommersall L, Arya N, Patel
HR: Basic principles of real-time quantitative PCR. Expert Rev
Mol Diagn 2005; 5:209–219
23 Mine H, Emura H, Miyamoto M, Tomono T, Minegishi K,
Murokawa H, Yamanaka R, Yoshikawa A, Nishioka K: High
throughput screening of 16 million serologically negative blood
donors for hepatitis B virus, hepatitis C virus and human immu-
nodeficiency virus type-1 by nucleic acid amplification testing
with specific and sensitive multiplex reagent in Japan. J Virol
Methods 2003; 112:145–151
24 Roth WK, Weber M, Buhr S, Drosten C, Weichert W, Sireis W,
Hedges D, Seifried E: Yield of HCV and HIV-1 NAT after screen-
ing of 3.6 million blood donations in central Europe. Transfu-
sion 2002; 42:862–868
25 Roth WK, Seifried E: The German experience with NAT. Trans-
fus Med 2002; 12:255–258
26 Roth WK, Buhr S, Drosten C, Seifried E: NAT and viral safety in
blood transfusion. Vox Sang 2000; 78(Suppl. 2):257–259
27 Roth WK, Weber M, Petersen D, Drosten C, Buhr S, Sireis W,
Weichert W, Hedges D, Seifried E: NAT for HBV and anti-HBc
testing increase blood safety. Transfusion 2002; 42:869–875
28 Schmidt M, Korn K, Nubling CM, Chudy M, Kress J, Horst HA,
Geusendam G, Hennig H, Sireis W, Rabenau HF, Doerr HW, Ber-
ger A, Hourfar MK, Gubbe K, Karl A, Fickenscher H, Tischer BK,
Babiel R, Seifried E, Gurtler L: First transmission of human
immunodeficiency virus Type 1 by a cellular blood product
after mandatory nucleic acid screening in Germany. Transfu-
sion 2009; 49:1836–1844
29 Kubanek B, Cardoso M, Gluck D, Koerner K: [Risk of infection
transmission by blood components]. Infusionsther Transfu-
sionsmed 1993; 20:54–59
30 Hourfar MK, Jork C, Schottstedt V, Weber-Schehl M, Brixner V,
Busch MP, Geusendam G, Gubbe K, Mahnhardt C, Mayr-Wohlf-
art U, Pichl L, Roth WK, Schmidt M, Seifried E, Wright DJ:
Experience of German Red Cross blood donor services with
nucleic acid testing: results of screening more than 30 million
blood donations for human immunodeficiency virus-1, hepati-
tis C virus, and hepatitis B virus. Transfusion 2008; 48:1558–
1566
31 Kretzschmar E, Chudy M, Nubling CM, Ross RS, Kruse F, Tro-
bisch H: First case of hepatitis C virus transmission by a red
blood cell concentrate after introduction of nucleic acid ampli-
fication technique screening in Germany: a comparative study
with various assays. Vox Sang 2007; 92:297–301
32 Biswas R, Tabor E, Hsia CC, Wright DJ, Laycock ME, Fiebig EW,
Peddada L, Smith R, Schreiber GB, Epstein JS, Nemo GJ, Busch
MP: Comparative sensitivity of HBV NATs and HBsAg assays
for detection of acute HBV infection. Transfusion 2003;
43:788–798
33 Busch MP, Wright DJ, Custer B, Tobler LH, Stramer SL, Klein-
man SH, Prince HE, Bianco C, Foster G, Petersen LR, Nemo G,
Glynn SA: West Nile virus infections projected from blood
donor screening data, United States, 2003. Emerg Infect Dis
2006; 12:395–402
34 Busch MP, Caglioti S, Robertson EF, McAuley JD, Tobler LH,
Kamel H, Linnen JM, Shyamala V, Tomasulo P, Kleinman SH:
Screening the blood supply for West Nile virus RNA by nucleic
acid amplification testing. N Engl J Med 2005; 353:460–467
35 Kantzanou MN, Moschidis ZM, Kremastinou G, Levidiotou S,
Karafoulidou A, Politis C, Marantidou O, Kavallierou L, Kape-
roni A, Veneti C, Hatzakis A: Searching for West Nile virus
(WNV) in Greece. Transfus Med 2010; 20(2):113–117
36 Pfleiderer C, Blumel J, Schmidt M, Roth WK, Houfar MK, Eckert
J, Chudy M, Menichetti E, Lechner S, Nubling CM: West Nile
virus and blood product safety in Germany. J Med Virol 2008;
80:557–563
37 Costa AN, Grossi P, Porta E, Venettoni S, Fehily D: Measures
taken to reduce the risk of West Nile virus transmission by
transplantation in Italy. Euro Surveill 2008; 13(42):19009
38 Nainan OV, Armstrong GL, Han XH, Williams I, Bell BP, Margo-
lis HS: Hepatitis a molecular epidemiology in the United States,
Blood donor screening by NAT 227
� 2010 The Authors.Journal compilation � 2010 International Society of Blood Transfusion, ISBT Science Series (2010) 5, 219–229

1996–1997: sources of infection and implications of vaccina-
tion policy. J Infect Dis 2005; 191:957–963
39 Henriques I, Monteiro F, Meireles E, Cruz A, Tavares G, Ferreira
M, Araujo F: Prevalence of Parvovirus B19 and Hepatitis A
virus in Portuguese blood donors. Transfus Apher Sci 2005;
33:305–309
40 Heitmann A, Laue T, Schottstedt V, Dotzauer A, Pichl L: Occur-
rence of hepatitis A virus genotype III in Germany requires the
adaptation of commercially available diagnostic test systems.
Transfusion 2005; 45:1097–1105
41 Lee DH, Prince AM: Automation of nucleic acid extraction for
NAT screening of individual blood units. Transfusion 2001;
41:483–487
42 Tamura A, Shimizu YK, Tanaka T, Kuroda K, Arakawa Y, Takah-
ashi K, Mishiro S, Shimizu K, Moriyama M: Persistent infection
of hepatitis E virus transmitted by blood transfusion in a patient
with T-cell lymphoma. Hepatol Res. 2007; 37:113–120
43 Boxall E, Herborn A, Kochethu G, Pratt G, Adams D, Ijaz S, Teo
CG: Transfusion-transmitted hepatitis E in a ‘nonhyperendemic’
country. Transfus Med 2006; 16:79–83
44 Matsubayashi K, Nagaoka Y, Sakata H, Sato S, Fukai K, Kato T,
Takahashi K, Mishiro S, Imai M, Takeda N, Ikeda H: Transfu-
sion-transmitted hepatitis E caused by apparently indigenous
hepatitis E virus strain in Hokkaido, Japan. Transfusion 2004;
44:934–940
45 Yee TT, Cohen BJ, Pasi KJ, Lee CA: Transmission of symptom-
atic parvovirus B19 infection by clotting factor concentrate. Br
J Haematol 1996; 93:457–459
46 Kleinman SH, Glynn SA, Lee TH, Tobler LH, Schlumpf KS, Todd
DS, Qiao H, Yu MY, Busch MP: A linked donor–recipient study
to evaluate parvovirus B19 transmission by blood component
transfusion. Blood 2009; 114:3677–3683
47 Brouard C, Bernillon P, Quatresous I, Pillonel J, Assal A, De
Valk H, Desenclos JC: Estimated risk of Chikungunya viremic
blood donation during an epidemic on Reunion Island in the
Indian Ocean, 2005 to 2007. Transfusion 2008; 48:1333–1341
48 Liumbruno GM, Calteri D, Petropulacos K, Mattivi A, Po C,
Macini P, Tomasini I, Zucchelli P, Silvestri AR, Sambri V,
Pupella S, Catalano L, Piccinini V, Calizzani G, Grazzini G: The
Chikungunya epidemic in Italy and its repercussion on the
blood system. Blood Transfus 2008; 6:199–210
49 De Cock KM, Brun-Vezinet F: Epidemiology of HIV-2 infection.
AIDS 1989; 3(Suppl. 1):S89–S95
50 De Cock KM, Brun-Vezinet F, Soro B: HIV-1 and HIV-2
infections and AIDS in West Africa. AIDS 1991; 5(Suppl.
1):S21–S28
51 Schmidt M, Brixner V, Ruster B, Hourfar MK, Drosten C, Preiser
W, Seifried E, Roth WK: NAT screening of blood donors for
severe acute respiratory syndrome coronavirus can potentially
prevent transfusion associated transmissions. Transfusion
2004; 44:470–475
52 Hourfar MK, Roth WK, Seifried E, Schmidt M: Comparison of
two real-time quantitative assays for detection of severe acute
respiratory syndrome coronavirus. J Clin Microbiol 2004;
42:2094–2100
53 Hindiyeh M, Ram D, Mandelboim M, Meningher T, Hirsh S,
Robinov J, Levy V, Orzitzer S, Azar R, Grossman Z, Mendelson
E: Rapid Detection of Influenza A Pandemic (H1N1) 2009 Virus
Neuraminidase Resistance Mutation H275Y by Real-Time
RT-PCR. J Clin Microbiol 2010; 48(5):1884–1887
54 Gimeno C, Navarro D: Real-time reverse-transcription PCR in
the diagnosis of influenza A (H1N1)v in intensive care unit
adult patients. Crit Care 2009; 13:428; author reply
55 Busch MP, Glynn SA, Stramer SL, Strong DM, Caglioti S,
Wright DJ, Pappalardo B, Kleinman SH: A new strategy for esti-
mating risks of transfusion-transmitted viral infections based
on rates of detection of recently infected donors. Transfusion
2005; 45:254–264
56 Eder AF, Kennedy JM, Dy BA, Notari EP, Weiss JW, Fang CT,
Wagner S, Dodd RY, Benjamin RJ: Bacterial screening of apher-
esis platelets and the residual risk of septic transfusion reac-
tions: the American Red Cross experience (2004–2006).
Transfusion 2007; 47:1134–1142
57 Munksgaard L, Albjerg L, Lillevang ST, Gahrn-Hansen B,
Georgsen J: Detection of bacterial contamination of platelet
components: six years’ experience with the BacT ⁄ ALERT sys-
tem. Transfusion 2004; 44:1166–1173
58 te Boekhorst PA, Beckers EA, Vos MC, Vermeij H, van Rhenen
DJ: Clinical significance of bacteriologic screening in platelet
concentrates. Transfusion 2005; 45:514–519
59 Feng P, Keasler SP, Hill WE: Direct identification of Yersinia
enterocolitica in blood by polymerase chain reaction amplifica-
tion. Transfusion 1992; 32:850–854
60 Nadkarni MA, Martin FE, Jacques NA, Hunter N: Determination
of bacterial load by real-time PCR using a broad-range (univer-
sal) probe and primers set. Microbiology 2002; 148:257–266
61 Mohammadi T, Reesink HW, Vandenbroucke-Grauls CM, Save-
lkoul PH: Optimization of real-time PCR assay for rapid and
sensitive detection of eubacterial 16S ribosomal DNA in platelet
concentrates. J Clin Microbiol 2003; 41:4796–4798
62 Mohammadi T, Reesink HW, Vandenbroucke-Grauls CM, Save-
lkoul PH: Removal of contaminating DNA from commercial
nucleic acid extraction kit reagents. J Microbiol Methods 2005;
61:285–288
63 Dreier J, Stormer M, Kleesiek K: Two novel real-time reverse
transcriptase PCR assays for rapid detection of bacterial con-
tamination in platelet concentrates. J Clin Microbiol 2004;
42:4759–4764
64 Harris KA, Hartley JC: Development of broad-range 16S rDNA
PCR for use in the routine diagnostic clinical microbiology ser-
vice. J Med Microbiol 2003; 52:685–691
65 Schmidt M, Hourfar MK, Nicol SB, Wahl A, Heck J, Weis C,
Tonn T, Spengler HP, Montag T, Seifried E, Roth WK: A
comparison of three rapid bacterial detection methods under
simulated real-life conditions. Transfusion 2006; 46:1367–
1373
66 Corless CE, Guiver M, Borrow R, Edwards-Jones V, Kaczmarski
EB, Fox AJ: Contamination and sensitivity issues with a real-
time universal 16S rRNA PCR. J Clin Microbiol 2000; 38:1747–
1752
67 Hourfar MK, Schmidt M, Seifried E, Roth WK: Evaluation of an
automated high-volume extraction method for viral nucleic
acids in comparison to a manual procedure with preceding
enrichment. Vox Sang 2005; 89:71–76
228 M. Schmidt & E. Seifried
� 2010 The Authors.Journal compilation � 2010 International Society of Blood Transfusion, ISBT Science Series (2010) 5, 219–229

68 Stormer M, Kleesiek K, Dreier J: High-volume extraction of
nucleic acids by magnetic bead technology for ultrasensitive
detection of bacteria in blood components. Clin Chem 2007;
53:104–110
69 Osselaer JC, Messe N, Hervig T, Bueno J, Castro E, Espinosa
A, Accorsi P, Junge K, Jacquet M, Flament J, Corash L: A
prospective observational cohort safety study of 5106 plate-
let transfusions with components prepared with photochemi-
cal pathogen inactivation treatment. Transfusion 2008;
48:1061–1071
70 Janetzko K, Cazenave JP, Kluter H, Kientz D, Michel M,
Beris P, Lioure B, Hastka J, Marblie S, Mayaudon V, Lin L,
Lin JS, Conlan MG, Flament J: Therapeutic efficacy and
safety of photochemically treated apheresis platelets pro-
cessed with an optimized integrated set. Transfusion 2005;
45:1443–1452
71 Janetzko K, Hinz K, Marschner S, Goodrich R, Kluter H: Patho-
gen reduction technology (Mirasol) treated single-donor plate-
lets resuspended in a mixture of autologous plasma and PAS.
Vox Sang 2009; 97:234–239
72 Goodrich RP, Edrich RA, Li J, Seghatchian J: The Mirasol PRT
system for pathogen reduction of platelets and plasma: an over-
view of current status and future trends. Transfus Apher Sci
2006; 35:5–17
73 AuBuchon JP, Herschel L, Roger J, Taylor H, Whitley P, Li J,
Edrich R, Goodrich RP: Efficacy of apheresis platelets treated
with riboflavin and ultraviolet light for pathogen reduction.
Transfusion 2005; 45:1335–1341
74 Mohr H, Steil L, Gravemann U, Thiele T, Hammer E, Greinacher
A, Muller TH, Volker U: A novel approach to pathogen reduc-
tion in platelet concentrates using short-wave ultraviolet light.
Transfusion 2009; 49:2612–2624
Blood donor screening by NAT 229
� 2010 The Authors.Journal compilation � 2010 International Society of Blood Transfusion, ISBT Science Series (2010) 5, 219–229





























