Embed Size (px)
Citation preview
CHM 1302 page 54
IV- LES ALCOOLS ET LES ÉTHERS
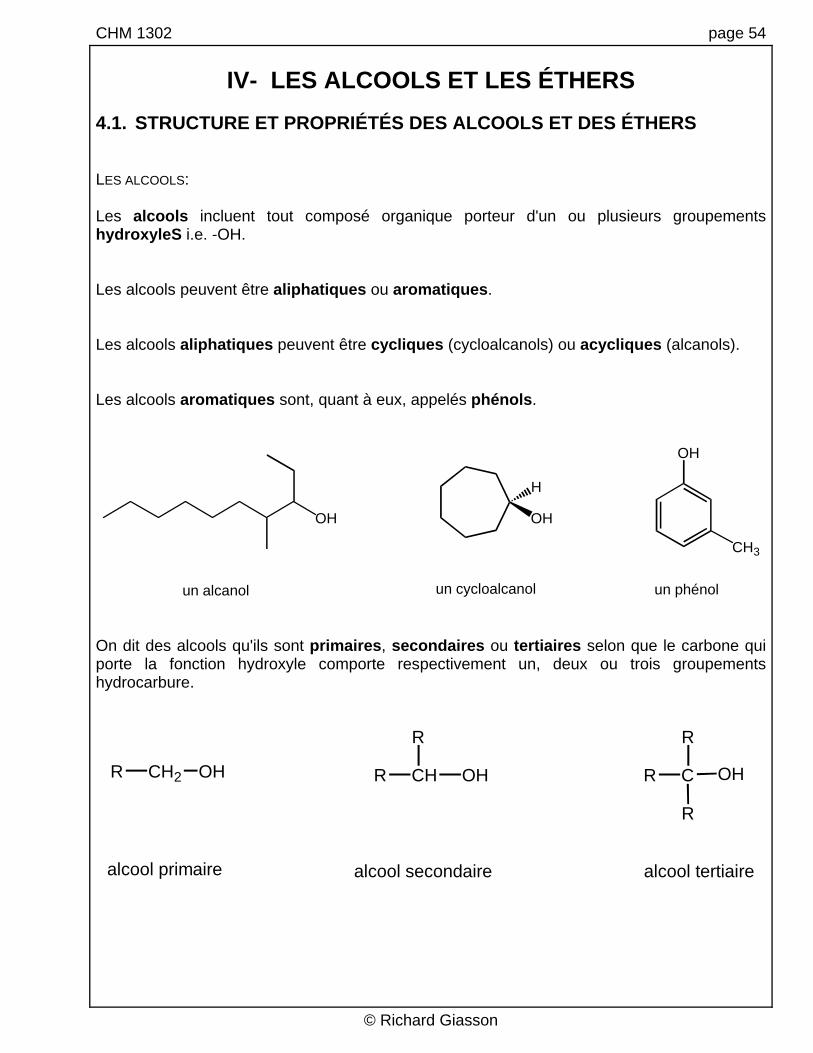
4.1. STRUCTURE ET PROPRIÉTÉS DES ALCOOLS ET DES ÉTHERS LES ALCOOLS: Les alcools incluent tout composé organique porteur d'un ou plusieurs groupements hydroxyleS i.e. -OH. Les alcools peuvent être aliphatiques ou aromatiques. Les alcools aliphatiques peuvent être cycliques (cycloalcanols) ou acycliques (alcanols). Les alcools aromatiques sont, quant à eux, appelés phénols.
OH OH
H
OH
CH3
un alcanol un cycloalcanol un phénol On dit des alcools qu'ils sont primaires, secondaires ou tertiaires selon que le carbone qui porte la fonction hydroxyle comporte respectivement un, deux ou trois groupements hydrocarbure.
R CH2 OH R CH OH R C OH
R R
R
alcool primaire alcool secondaire alcool tertiaire
© Richard Giasson
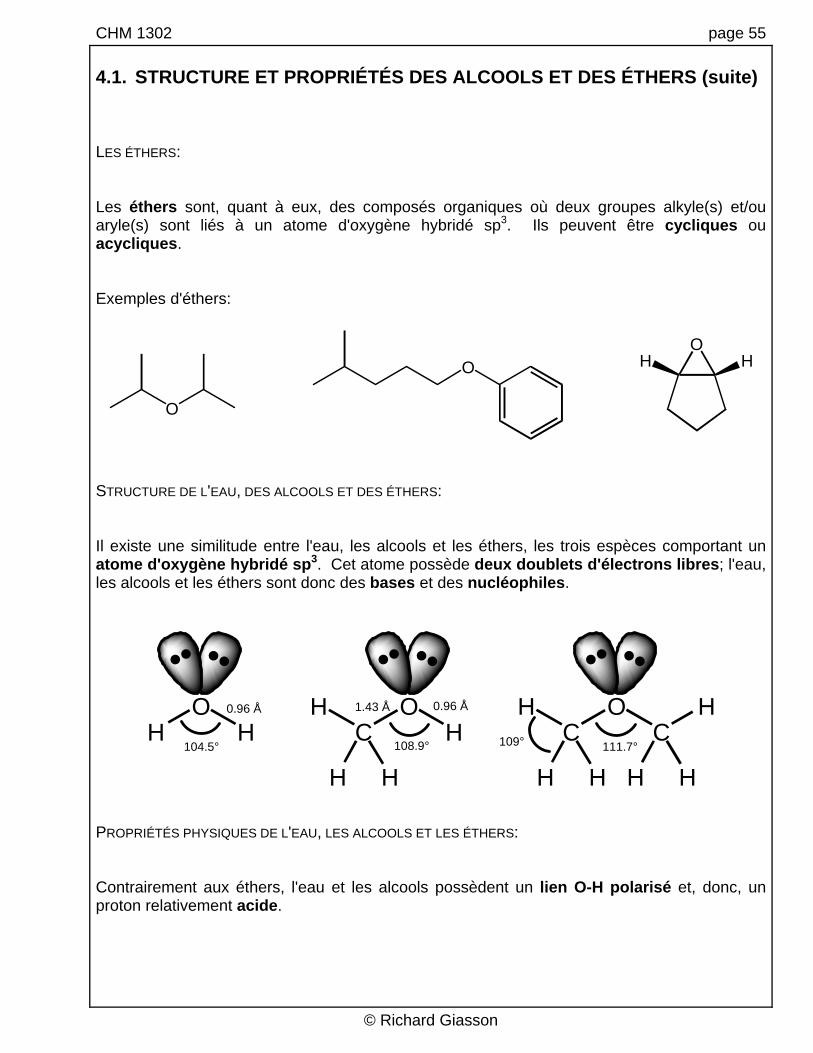
CHM 1302 page 55 4.1. STRUCTURE ET PROPRIÉTÉS DES ALCOOLS ET DES ÉTHERS (suite) LES ÉTHERS: Les éthers sont, quant à eux, des composés organiques où deux groupes alkyle(s) et/ou aryle(s) sont liés à un atome d'oxygène hybridé sp3. Ils peuvent être cycliques ou acycliques. Exemples d'éthers:
O
OO
HH
STRUCTURE DE L'EAU, DES ALCOOLS ET DES ÉTHERS: Il existe une similitude entre l'eau, les alcools et les éthers, les trois espèces comportant un atome d'oxygène hybridé sp3. Cet atome possède deux doublets d'électrons libres; l'eau, les alcools et les éthers sont donc des bases et des nucléophiles.
H HO
C HO
C COH H
H H H H H H104.5° 108.9° 111.7°109°
0.96 Å 0.96 Å1.43 Å H
PROPRIÉTÉS PHYSIQUES DE L'EAU, LES ALCOOLS ET LES ÉTHERS: Contrairement aux éthers, l'eau et les alcools possèdent un lien O-H polarisé et, donc, un proton relativement acide.
© Richard Giasson

CHM 1302 page 56 4.1. STRUCTURE ET PROPRIÉTÉS DES ALCOOLS ET DES ÉTHERS (suite) PROPRIÉTÉS PHYSIQUES DE L'EAU, LES ALCOOLS ET LES ÉTHERS (suite): Acidité de l'eau et des alcools:
H HO
H3C HO
Ph HO
H
H
H
+
+
+
HO
CH3O
O O
etc...
pKa = 15.7
pKa = 15.5
pKa = 10
Notez que les phénols sont beaucoup plus acides que les alcools aliphatiques car la charge négative de leurs bases conjuguées, les anions phénolates, est stabilisée par délocalisation à travers le cycle aromatique. Finalement, l'eau et les alcools peuvent agir comme donneurs et accepteurs de ponts hydrogène.
O
R H
OH
OH
R
R Cette caractéristique explique pourquoi les alcools ont des points d'ébullition et des solubilités dans l'eau très supérieurs à ceux des éthers de même formule moléculaire.
CH3CH2CH2CH2OH CH3CH2OCH2CH3
n-butanol (C4H10O) éther diéthylique (C4H10O) p. éb. 117°C p. éb. 35°C
soluble dans l'eau insoluble dans l'eau
© Richard Giasson
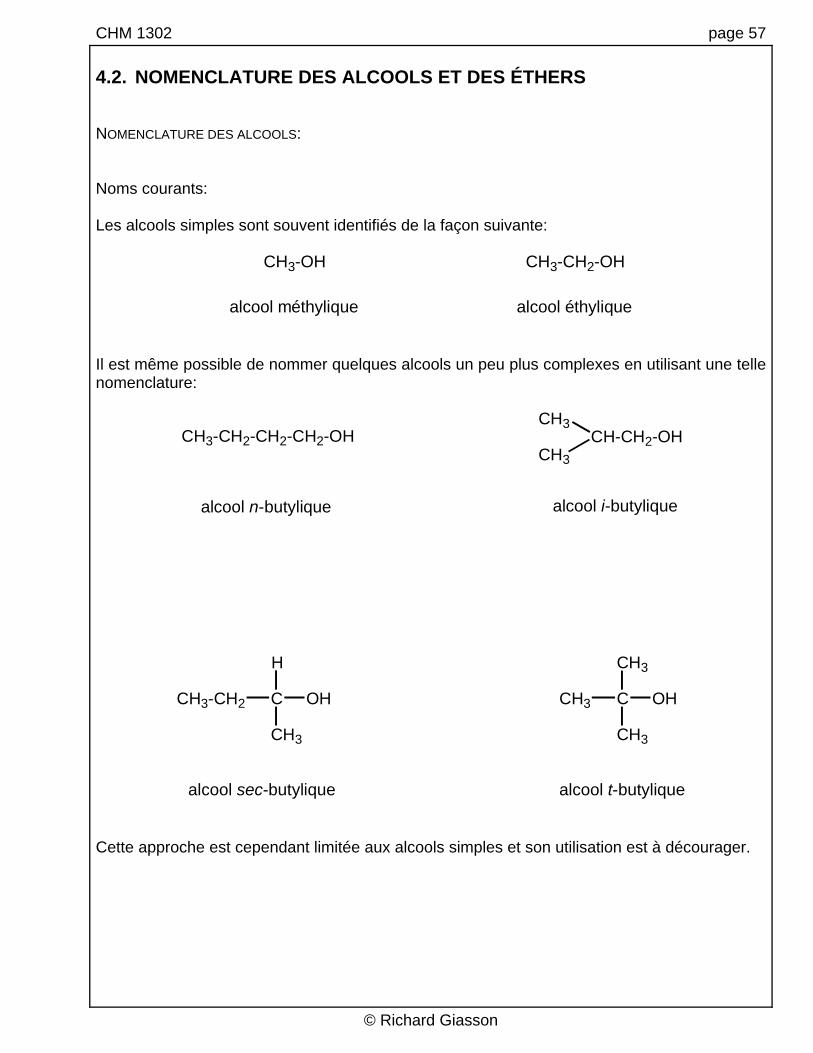
CHM 1302 page 57 4.2. NOMENCLATURE DES ALCOOLS ET DES ÉTHERS NOMENCLATURE DES ALCOOLS: Noms courants: Les alcools simples sont souvent identifiés de la façon suivante:
CH3-OH CH3-CH2-OH
alcool méthylique alcool éthylique Il est même possible de nommer quelques alcools un peu plus complexes en utilisant une telle nomenclature:
CH3-CH2-CH2-CH2-OH
alcool n-butylique
alcool sec-butylique
C
alcool t-butylique
CH3
CH3
CH3
OHC
CH3
CH3-CH2
H
OH
CH-CH2-OHCH3
CH3
alcool i-butylique
Cette approche est cependant limitée aux alcools simples et son utilisation est à décourager.
© Richard Giasson

CHM 1302 page 58 4.2. NOMENCLATURE DES ALCOOLS ET DES ÉTHERS (suite) NOMENCLATURE DES ALCOOLS: Noms systématiques: Les noms des alcools sont obtenus en remplaçant la terminaison "e" des alcanes correspondants par le suffixe "ol". Pour identifier la position du groupe hydroxyle, les atomes de carbone de la chaîne la plus longue sont numérotés en commençant par l'extrémité la plus proche du groupe OH.
CH3 CH2 CH2 OH CH3 CH CH2 CH2
OH
CH3
CH3 CH CH2 CH2
CH3
OH CH3 CH CH2 CH
CH3 OH
C CH3
CH3
CH3
1 2 3 4 5123
1234561234
propan-1-ol pentan-2-ol
3-méthylbutan-1-ol 2,2,5-triméthylhexan-3-ol Les alcools cycliques sont, quant à eux, numérotés de manière à ce que le carbone porteur du groupe hydroxyle reçoive d'office le numéro un.
12
34
5
12
3 4
cyclohexanol cis-3-chlorocyclobutanol1-éthylcyclopentanol
OH CH2CH3HO OH
Cl
© Richard Giasson

CHM 1302 page 59 4.2. NOMENCLATURE DES ALCOOLS ET DES ÉTHERS (suite) NOMENCLATURE DES ÉTHERS: Les noms tels "éther diéthylique" ou "diéthyl éther" sont erronés et ne devraient pas être utilisés. Noms de classe fonctionnelle: Les noms de classe fonctionnelle peuvent être utilisés pour identifier les éthers simples. Notez que l'on utilise le terme "oxyde de" et non pas "éther".
CH3-O-CH3 CH3-O-CH2-CH3
oxyde de diméthyle oxyde d'éthyle et de méthyle Noms systématiques: Dans le système IUPAC de nomenclature, les éthers sont considérés comme des hydrocarbures porteurs de groupements alkoxyles. La chaîne principale est généralement celle qui est la plus longue.
CH3-O-CH2-CH3
méthoxyéthane
méthoxybenzène 1,2-diméthoxyéthane
2-éthoxy-2-méthylpropane
CH3 CH2 O C CH3
CH3
CH31
2
3
OCH3
CH3 O CH2 CH2 O CH3
© Richard Giasson
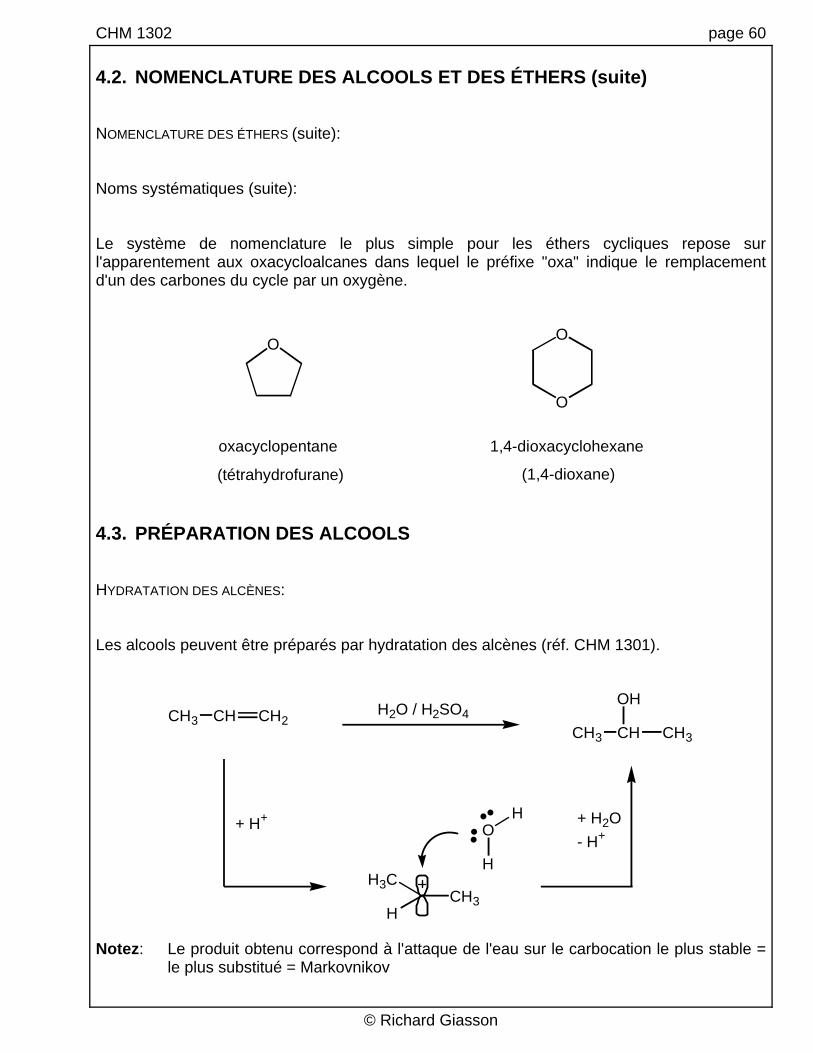
CHM 1302 page 60 4.2. NOMENCLATURE DES ALCOOLS ET DES ÉTHERS (suite) NOMENCLATURE DES ÉTHERS (suite): Noms systématiques (suite): Le système de nomenclature le plus simple pour les éthers cycliques repose sur l'apparentement aux oxacycloalcanes dans lequel le préfixe "oxa" indique le remplacement d'un des carbones du cycle par un oxygène.
O
O
O
oxacyclopentane 1,4-dioxacyclohexane
(tétrahydrofurane) (1,4-dioxane) 4.3. PRÉPARATION DES ALCOOLS HYDRATATION DES ALCÈNES: Les alcools peuvent être préparés par hydratation des alcènes (réf. CHM 1301).
CH3 CH CH2H2O / H2SO4
CH3 CH CH3
OH
CH3H
H3C
O
H
H+ H+ + H2O
- H+
Notez: Le produit obtenu correspond à l'attaque de l'eau sur le carbocation le plus stable =
le plus substitué = Markovnikov
© Richard Giasson
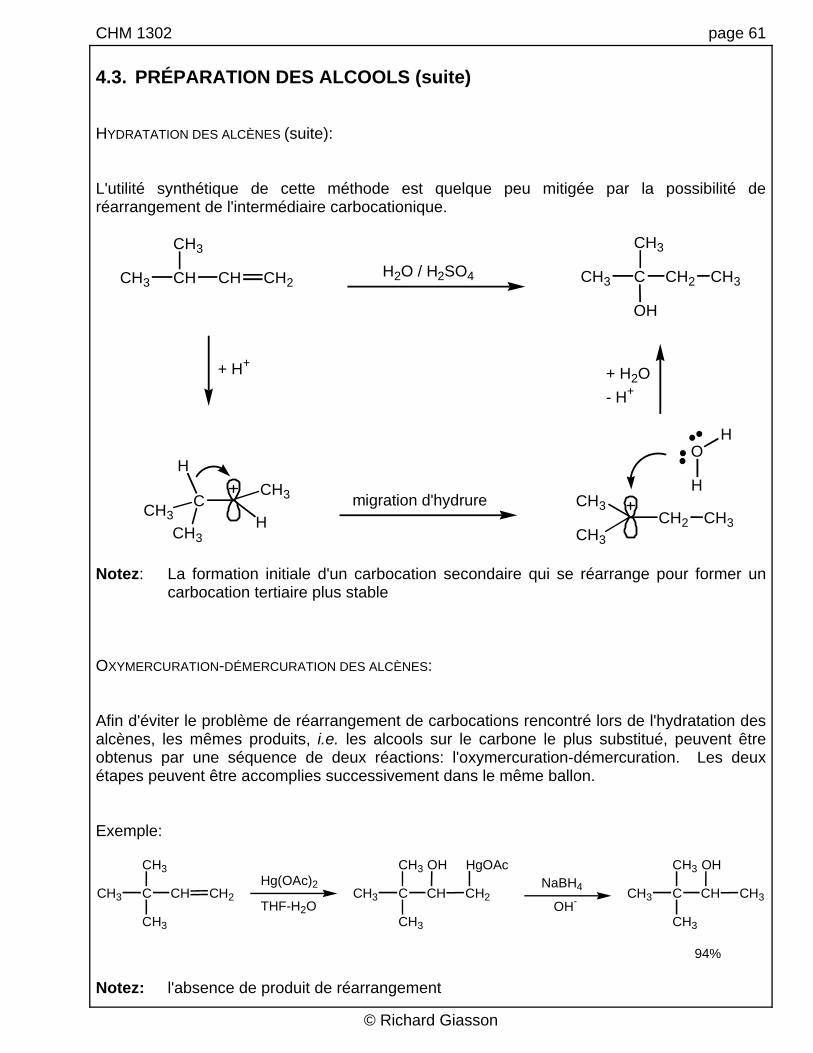
CHM 1302 page 61 4.3. PRÉPARATION DES ALCOOLS (suite) HYDRATATION DES ALCÈNES (suite): L'utilité synthétique de cette méthode est quelque peu mitigée par la possibilité de réarrangement de l'intermédiaire carbocationique.
CH CH CH2H2O / H2SO4
CH2CH3
CH3
O
H
H
+ H++ H2O- H+
CH3
CH3
C CH2 CH3CH3
CH3
OH
CH3
CH3
HC
H
CH3CH3
migration d'hydrure
Notez: La formation initiale d'un carbocation secondaire qui se réarrange pour former un
carbocation tertiaire plus stable OXYMERCURATION-DÉMERCURATION DES ALCÈNES: Afin d'éviter le problème de réarrangement de carbocations rencontré lors de l'hydratation des alcènes, les mêmes produits, i.e. les alcools sur le carbone le plus substitué, peuvent être obtenus par une séquence de deux réactions: l'oxymercuration-démercuration. Les deux étapes peuvent être accomplies successivement dans le même ballon. Exemple:
CCH3
CH3
CH3
CH CH2Hg(OAc)2
THF-H2OCCH3
CH3
CH3
CH CH2
HgOAcOHNaBH4
OH- CCH3
CH3
CH3
CH CH3
OH
94% Notez: l'absence de produit de réarrangement
© Richard Giasson
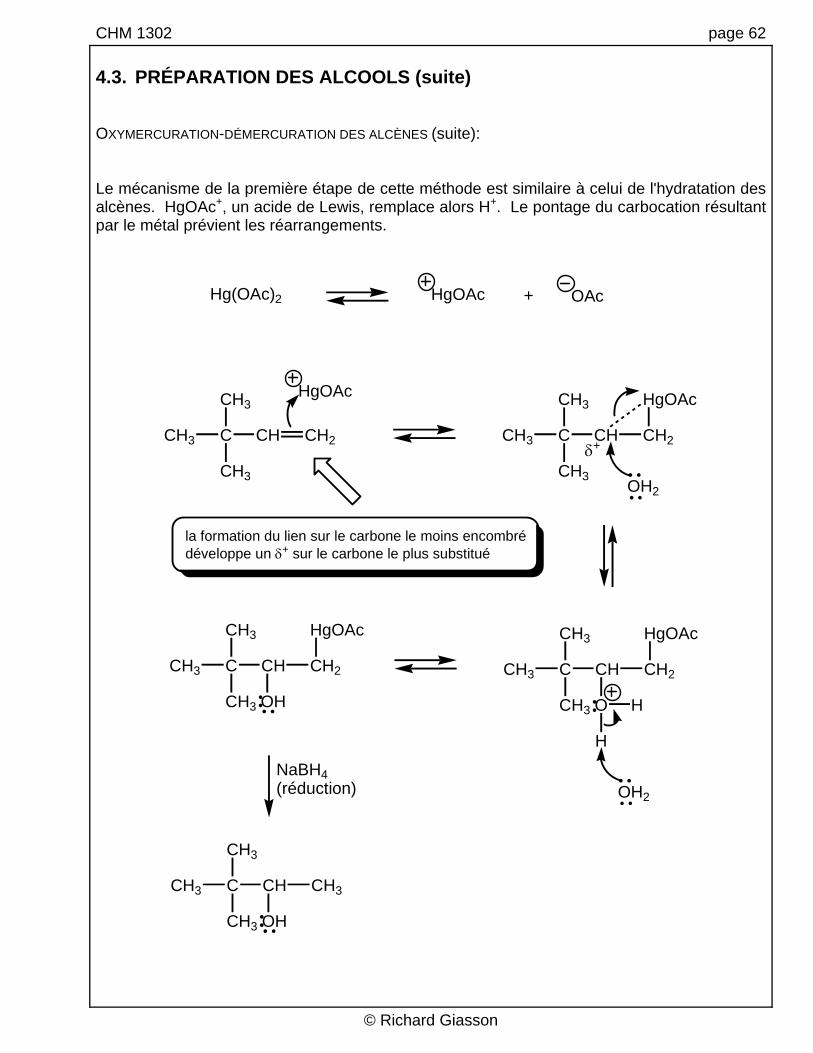
CHM 1302 page 62 4.3. PRÉPARATION DES ALCOOLS (suite) OXYMERCURATION-DÉMERCURATION DES ALCÈNES (suite): Le mécanisme de la première étape de cette méthode est similaire à celui de l'hydratation des alcènes. HgOAc+, un acide de Lewis, remplace alors H+. Le pontage du carbocation résultant par le métal prévient les réarrangements.
Hg(OAc)2
CCH3
CH3
CH3
CH CH2
HgOAc
OH
NaBH4(réduction)
CCH3
CH3
CH3
CH CH2 CCH3
CH3
CH3
CH CH2
HgOAc
CCH3
CH3
CH3
CH CH3
OH
CCH3
CH3
CH3
CH CH2
HgOAc
O
H
H
la formation du lien sur le carbone le moins encombrédéveloppe un δ+ sur le carbone le plus substitué
HgOAc
OH2
OH2
δ+
HgOAc OAc+
© Richard Giasson

CHM 1302 page 63 4.3. PRÉPARATION DES ALCOOLS (suite) HYDROBORATION DES ALCÈNES: Il est aussi possible de préparer les alcools à partir des alcènes par la méthode de l'hydroboration (réf. CHM 1301). Il s'agit ici aussi d'une approche en deux étapes. Les alcools obtenus par cette méthode sont cependant ceux où le groupe OH se retrouvent sur le carbone le moins substitué i.e. anti-Markovnikov. Exemple:
BH3•THFCH3-CH CH2
3 H2O2
BH3•THF B O
H
HH
3 NaOH
B O
H
HH
3 B(CH2-CH2-CH3)3 Na3BO3 3 H2O+ +3 CH3-CH2-CH2-OH
Les phénomènes à l'origine de la régiosélectivité observée se font sentir à la première des deux étapes et sont doubles. L'approche du bore vers le carbone le moins substitué est favorisée par le plus faible encombrement stérique, ainsi que par la charge positive partielle qui se développe sur le carbone le plus substitué à l'état de transition. Regardons donc le mécanisme de cette première étape.
© Richard Giasson
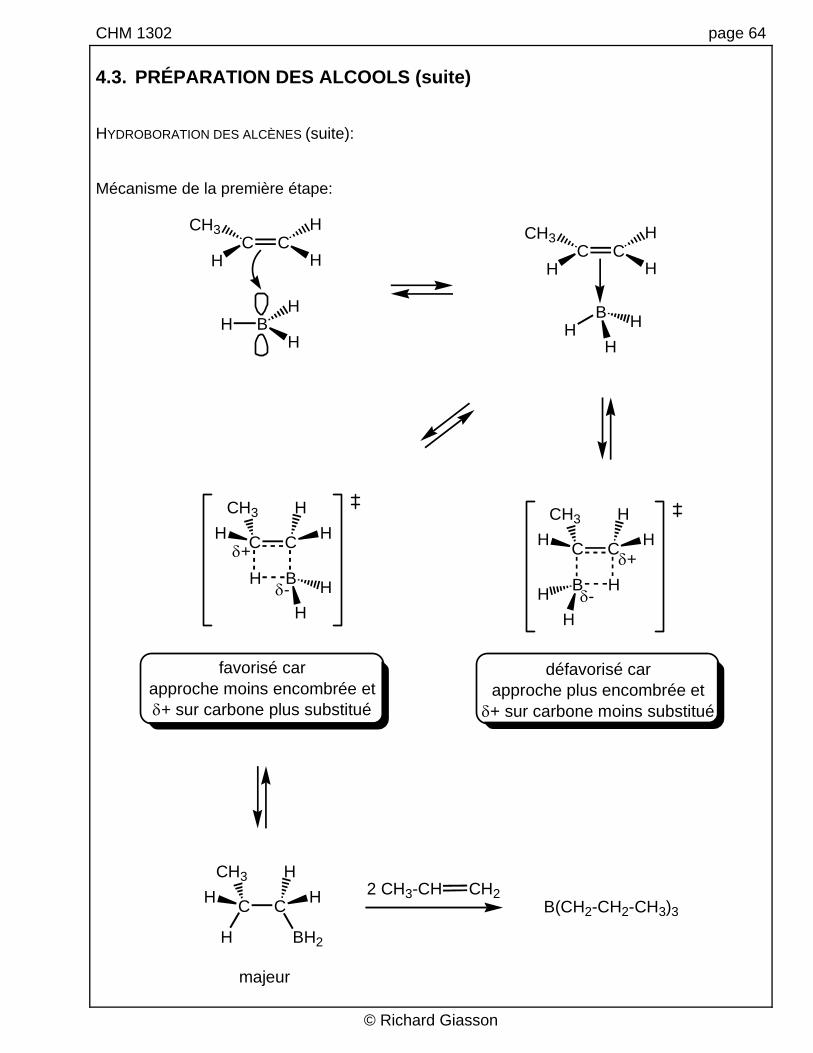
CHM 1302 page 64 4.3. PRÉPARATION DES ALCOOLS (suite) HYDROBORATION DES ALCÈNES (suite): Mécanisme de la première étape:
C CH
HCH3
H C CH
HCH3
H
C
H B
C HHCH3
H
HH
C
B H
C HHCH3
H
HH
défavorisé carapproche plus encombrée et
δ+ sur carbone moins substitué
favorisé carapproche moins encombrée etδ+ sur carbone plus substitué
BHH
HH
B
HH
δ+
δ-
δ+
δ-
C
H BH2
C HHCH3
H 2 CH3-CH CH2B(CH2-CH2-CH3)3
majeur
© Richard Giasson
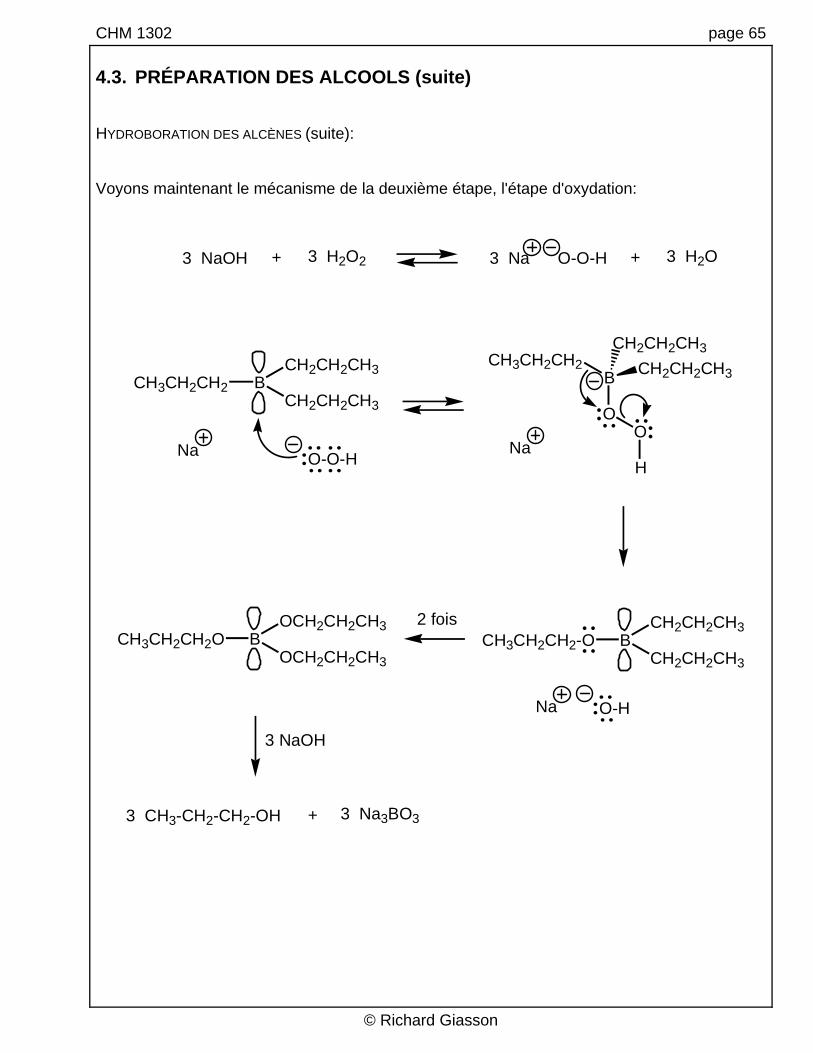
CHM 1302 page 65 4.3. PRÉPARATION DES ALCOOLS (suite) HYDROBORATION DES ALCÈNES (suite): Voyons maintenant le mécanisme de la deuxième étape, l'étape d'oxydation:
3 NaOH
BCH3CH2CH2CH2CH2CH3
CH2CH2CH3
O
BCH3CH2CH2 CH2CH2CH3
CH2CH2CH3
O
H
O-H
BCH3CH2CH2OOCH2CH2CH3
OCH2CH2CH3
3 CH3-CH2-CH2-OH
3 H2O2
O-O-H
3 Na3BO3
3 Na O-O-H
BCH3CH2CH2-OCH2CH2CH3
CH2CH2CH3
3 H2O+ +
+
Na
3 NaOH
Na
Na
2 fois
© Richard Giasson

CHM 1302 page 66 4.3. PRÉPARATION DES ALCOOLS (suite) HYDROBORATION DES ALCÈNES (suite): Pour des raisons pratiques, le réactif 9-borabicyclo[3.3.1]nonane est souvent utilisé dans les hydroborations à la place du complexe BH3•THF.
1) 9-BBN / THF / 65°C
2) H2O2 / NaOH aq.
H3C HH3C
H OHH
H3CH B
H
B
H
H2O2NaOH aq.
+ énantiomère
+ énantiomère Notez les aspects suivant de cette réaction qui découlent du mécanisme que l'on vient de voir:
• L'addition des atomes de bore et d'hydrogène est syn.
• Le bore se lie au carbone le moins substitué.
• L'oxydation de l'intermédiaire borane se fait avec rétention de configuration au niveau de l'atome de carbone.
• Globalement, on obtient l'alcool correspondant à une addition syn de l'eau sur le
carbone le moins substitué.
© Richard Giasson
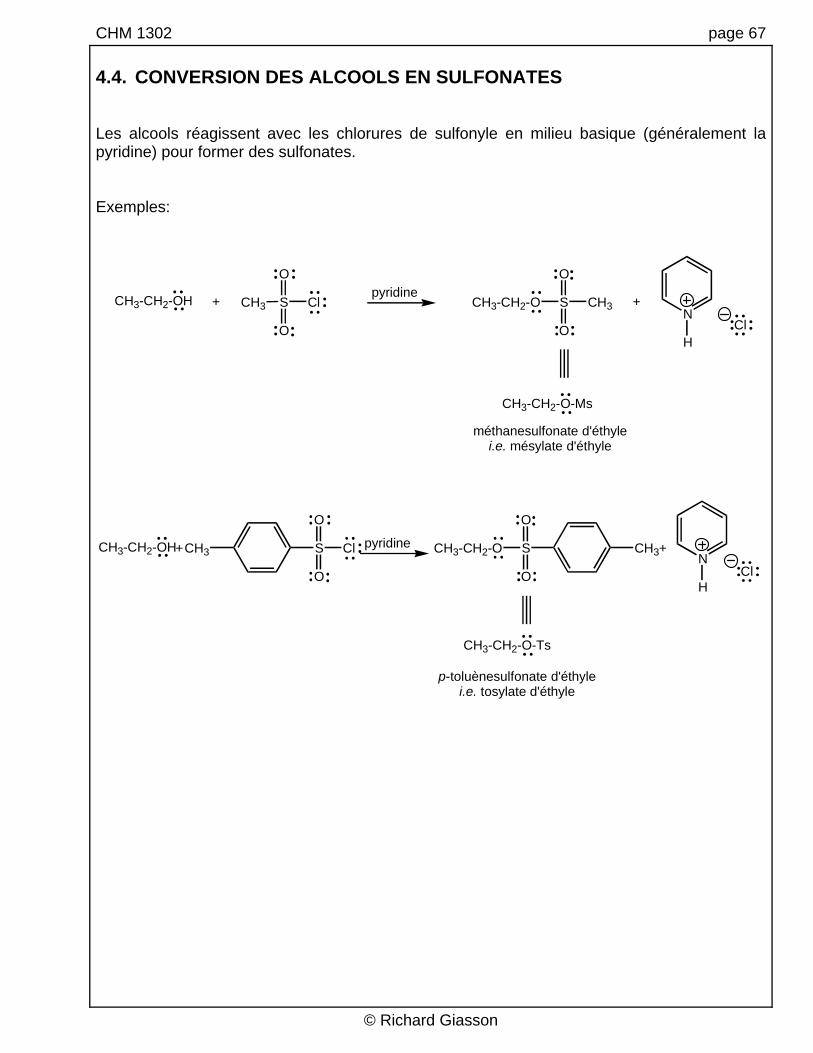
CHM 1302 page 67 4.4. CONVERSION DES ALCOOLS EN SULFONATES Les alcools réagissent avec les chlorures de sulfonyle en milieu basique (généralement la pyridine) pour former des sulfonates. Exemples:
pyridine
CH3-CH2-OH
CH3-CH2-OH Cl
O
O
CH3 S
S Cl
O
O
CH3
S CH3
O
O
CH3-CH2-O
S
O
O
CH3-CH2-O CH3
N
H
N
H
Cl
Cl
+ +
+ +pyridine
CH3-CH2-O-Ts
CH3-CH2-O-Ms
méthanesulfonate d'éthylei.e. mésylate d'éthyle
p-toluènesulfonate d'éthylei.e. tosylate d'éthyle
© Richard Giasson
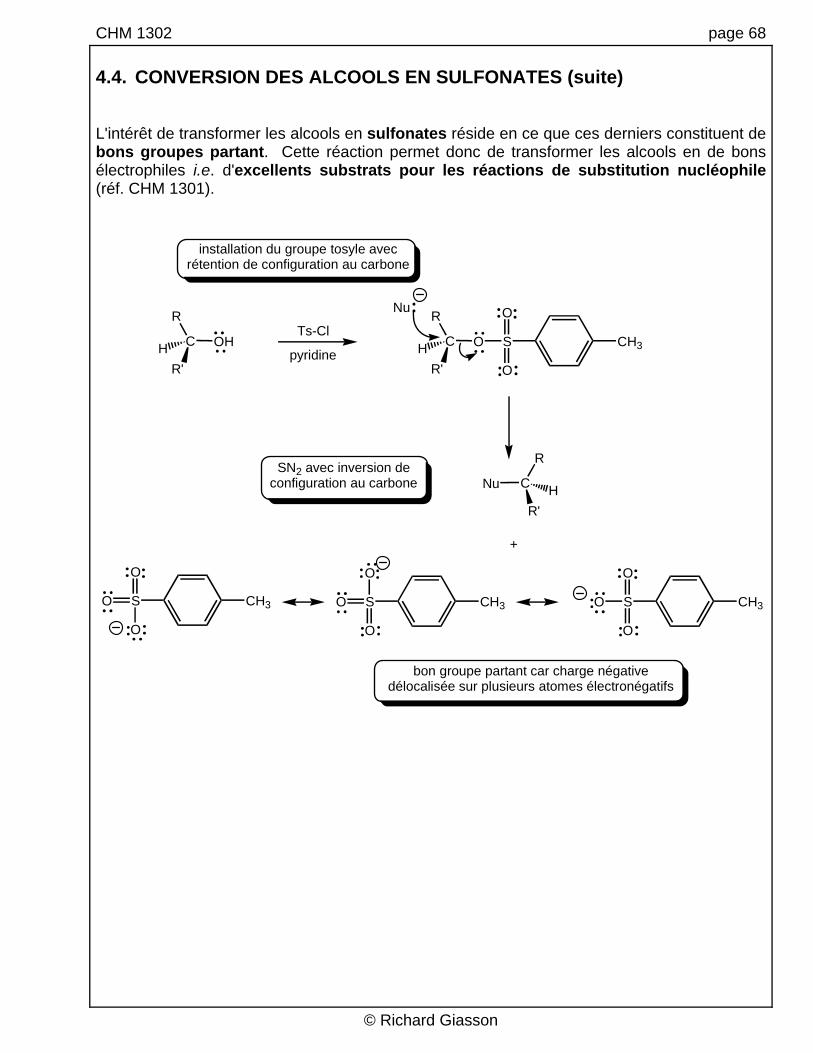
CHM 1302 page 68 4.4. CONVERSION DES ALCOOLS EN SULFONATES (suite) L'intérêt de transformer les alcools en sulfonates réside en ce que ces derniers constituent de bons groupes partant. Cette réaction permet donc de transformer les alcools en de bons électrophiles i.e. d'excellents substrats pour les réactions de substitution nucléophile (réf. CHM 1301).
installation du groupe tosyle avecrétention de configuration au carbone
SN2 avec inversion deconfiguration au carbone
S
O
O
CH3OS
O
O
CH3OS
O
O
CH3O
R
C OHHR'
Ts-Cl
R
CNu HR'
S
O
O
CH3
R
C OHR'
Nu
pyridine
+
bon groupe partant car charge négativedélocalisée sur plusieurs atomes électronégatifs
© Richard Giasson
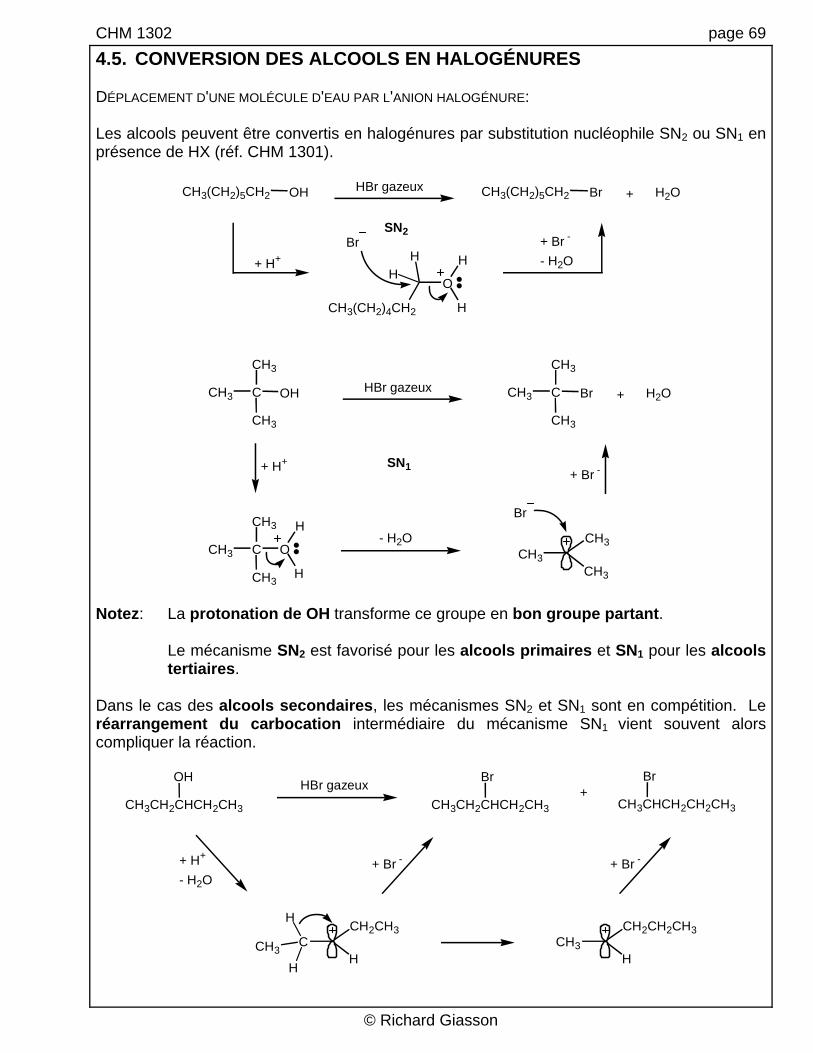
CHM 1302 page 69
4.5. CONVERSION DES ALCOOLS EN HALOGÉNURES DÉPLACEMENT D'UNE MOLÉCULE D'EAU PAR L'ANION HALOGÉNURE: Les alcools peuvent être convertis en halogénures par substitution nucléophile SN2 ou SN1 en présence de HX (réf. CHM 1301).
CH3(CH2)5CH2 OH CH3(CH2)5CH2 Br + H2OHBr gazeux
O
CH3(CH2)4CH2
HH
H
H
Br+ H+
+ Br -
- H2O
SN2
C OH C Br + H2OHBr gazeux
C O
H
H
Br
+ H++ Br -
SN1
CH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3
CH3
- H2OCH3
CH3
CH3
Notez: La protonation de OH transforme ce groupe en bon groupe partant. Le mécanisme SN2 est favorisé pour les alcools primaires et SN1 pour les alcools
tertiaires. Dans le cas des alcools secondaires, les mécanismes SN2 et SN1 sont en compétition. Le réarrangement du carbocation intermédiaire du mécanisme SN1 vient souvent alors compliquer la réaction.
+HBr gazeux
+ H+
- H2O+ Br -
CH
CH2CH3
CH3CH2CHCH2CH3
OH
CH3CH2CHCH2CH3
Br
CH3H
CH2CH2CH3H
CH3
H
CH3CHCH2CH2CH3
Br
+ Br -
© Richard Giasson

CHM 1302 page 70
4.5. CONVERSION DES ALCOOLS EN HALOGÉNURES (suite) DÉPLACEMENT D'UN TOSYLATE PAR L'ANION HALOGÉNURE: Une autre façon d'effectuer cette transformation sur les alcools primaires et secondaires consiste à convertir le groupe hydroxyle en tosylate (réf. section 4.4) pour ensuite déplacer ce dernier avec l'anion halogénure par un mécanisme SN2 (réf. CHM 1301).
CH3CH2CHCH2CH3
OH
CH3CH2CHCH2CH3
OTsTsCl
pyridine CH3CH2CHCH2CH3
BrNaBr
DMSO
85%
SO
OClCH3 = TsCl
Notez: On évite ici la formation d'un intermédiaire carbocationique qui pourrait se
réarranger. Un seul régioisomère est donc obtenu. DÉPLACEMENT D'UN GROUPE PARTANT FORMÉ IN SITU PAR L'ANION HALOGÉNURE: Les chlorures peuvent être formés à partir des alcools par traitement avec le chlorure de thionyle, SOCl2.
CH3CH2CHCH2CH3
OH
CH3CH2CHCH2CH3
ClSOCl2pyridine
Les bromures, quant à eux, peuvent êtres obtenus en utilisant PBr3 qui agit selon un mécanisme semblable i.e. activation in situ suivie d'une SN2 avec Br-.
CH3CHCH2OH
CH3
CH3CHCH2Br
CH3PBr3 (O.33 eq.)
0°C+ H3PO3 (0.33 eq.)
© Richard Giasson
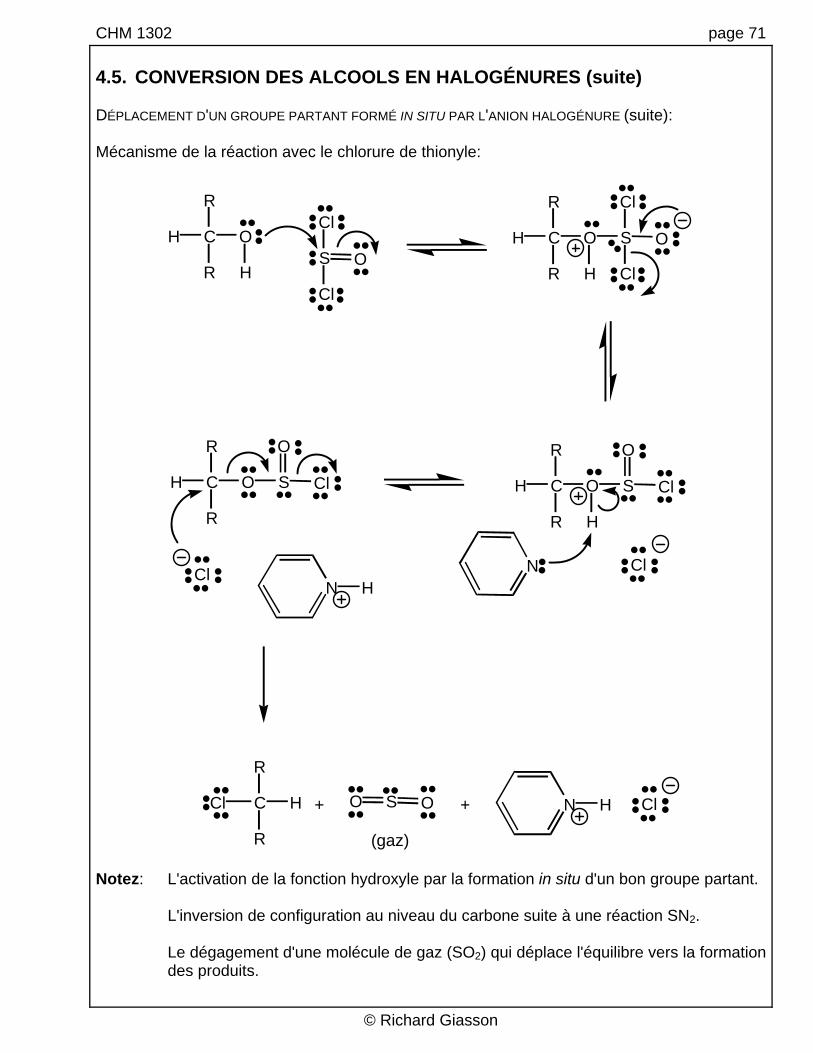
CHM 1302 page 71 4.5. CONVERSION DES ALCOOLS EN HALOGÉNURES (suite) DÉPLACEMENT D'UN GROUPE PARTANT FORMÉ IN SITU PAR L'ANION HALOGÉNURE (suite): Mécanisme de la réaction avec le chlorure de thionyle:
H C
R
R
O
HS
Cl
Cl
OH C
R
R
O
H
Cl
Cl
OS
H C
R
R
O
H
O
Cl
ClSH C
R
R
O
O
Cl
ClS
NN H
HC
R
R
Cl ClOS N HO
(gaz)
+ +
Notez: L'activation de la fonction hydroxyle par la formation in situ d'un bon groupe partant. L'inversion de configuration au niveau du carbone suite à une réaction SN2. Le dégagement d'une molécule de gaz (SO2) qui déplace l'équilibre vers la formation
des produits.
© Richard Giasson

CHM 1302 page 72 4.6. PRÉPARATION DES ÉTHERS PRÉPARATION DE L'ANION ALKOXYDE: Les alkoxydes sont obtenus en traitant les alcools avec des bases, telles NaOH et NaH, ou des métaux réducteurs, tel Na.
CH3OH
CH3OH
CH3OH
+
+
+ +
+
+NaOH
NaH
Na
CH3ONa
CH3ONa
CH3ONa
H2O
H2 (gaz)
1/2 H2 (gaz)
pKa ≈ 15.5
pKa = 35
pKa ≈ 15.7
RÉACTION DE L'ANION ALKOXYDE AVEC LES ÉLECTROPHILES; SYNTHÈSE D'ÉTHERS DE WILLIAMSON: Les alkoxydes sont des nucléophiles puissants qui peuvent réagir par SN2 avec les électrophiles, tels les halogénures (réf. CHM 1301). Cette méthode de synthèse des éthers se nomme "la synthèse des éthers de Williamson".
CH3CHCH2O Na
CH3
+ CH2Br CH3CHCH2OCH2Ph
CH3
Notez: Les alkoxydes sont aussi des bases fortes. Conséquemment, cette réaction est
limitée aux halogénures primaires afin de minimiser la réaction d'élimination E2 compétitive (réf. CHM 1301).
© Richard Giasson
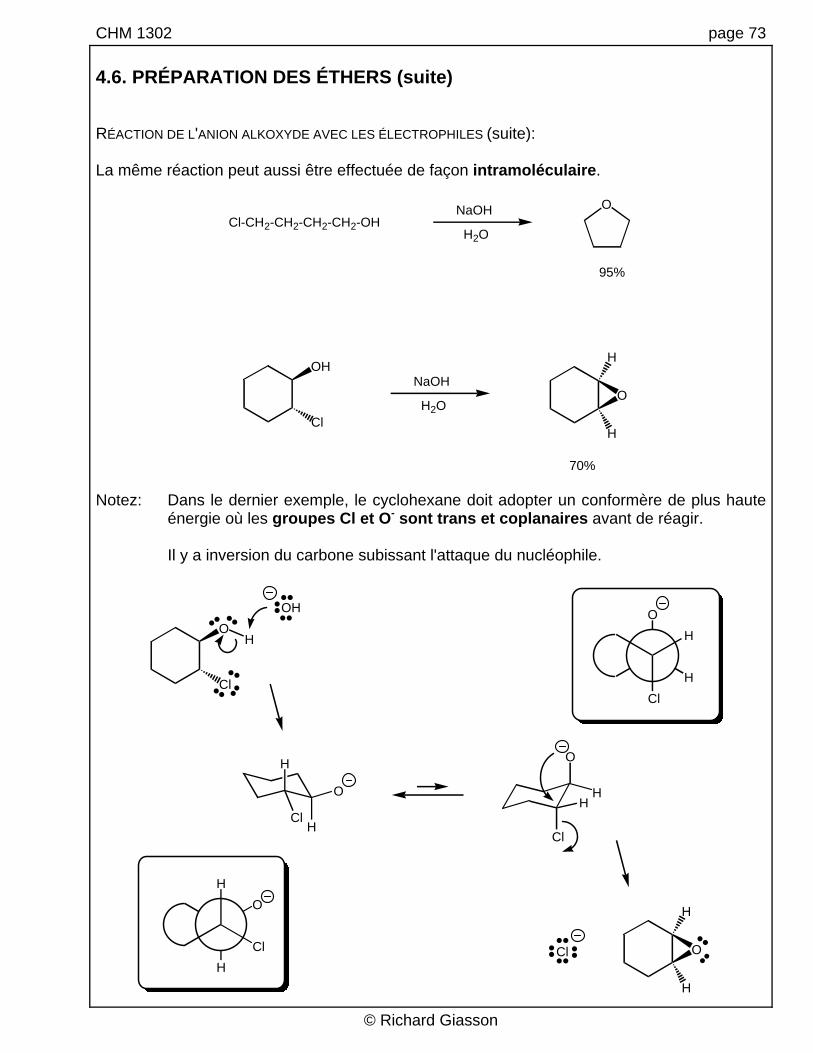
CHM 1302 page 73 4.6. PRÉPARATION DES ÉTHERS (suite) RÉACTION DE L'ANION ALKOXYDE AVEC LES ÉLECTROPHILES (suite): La même réaction peut aussi être effectuée de façon intramoléculaire.
Cl-CH2-CH2-CH2-CH2-OHNaOH
H2O
O
Cl
OHNaOH
H2OO
H
H
95%
70% Notez: Dans le dernier exemple, le cyclohexane doit adopter un conformère de plus haute
énergie où les groupes Cl et O- sont trans et coplanaires avant de réagir. Il y a inversion du carbone subissant l'attaque du nucléophile.
Cl
O
O
H
H
H
OH
H
O
H
Cl
O
H
Cl
H
O
H
H
Cl
H
OH
Cl Cl
© Richard Giasson
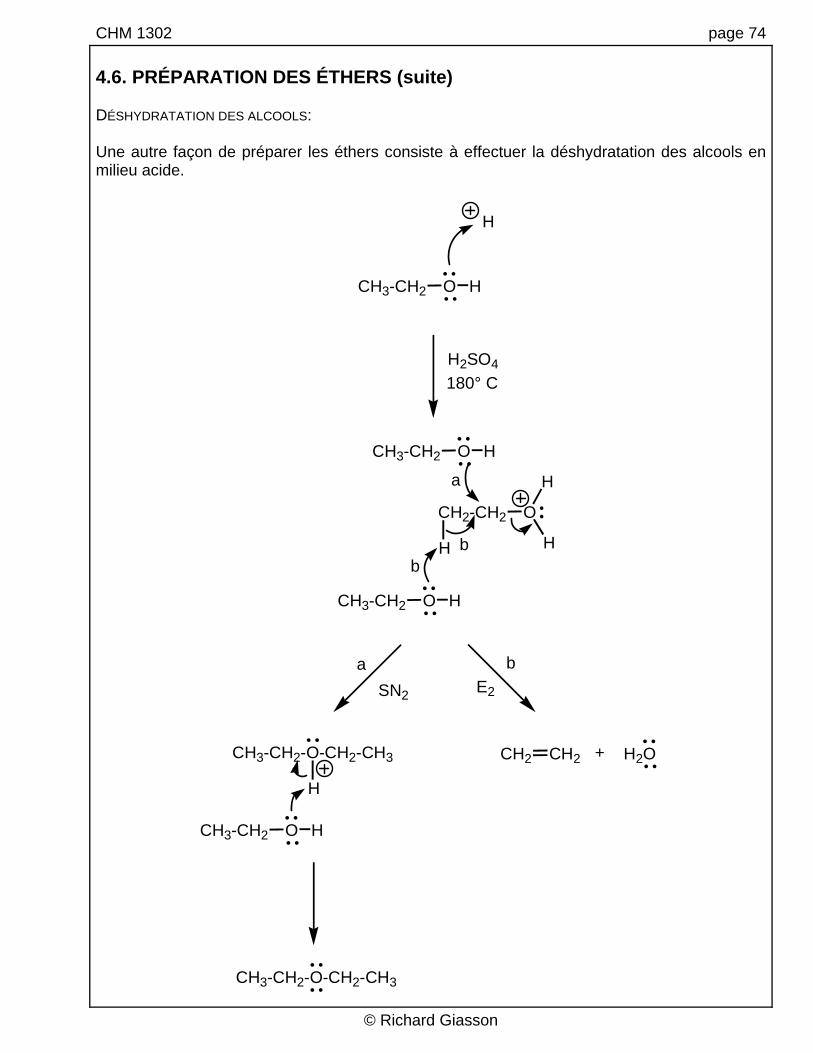
CHM 1302 page 74 4.6. PRÉPARATION DES ÉTHERS (suite) DÉSHYDRATATION DES ALCOOLS: Une autre façon de préparer les éthers consiste à effectuer la déshydratation des alcools en milieu acide.
H2SO4180° C
H
CH3-CH2 O H
CH3-CH2 O H
CH3-CH2 O H
CH2-CH2
H
O
H
H
CH3-CH2-O-CH2-CH3
H
SN2
CH3-CH2 O H
CH3-CH2-O-CH2-CH3
CH2 CH2 H2O
a
bb
a bE2
+
© Richard Giasson

CHM 1302 page 75 4.6. PRÉPARATION DES ÉTHERS (suite) DÉSHYDRATATION DES ALCOOLS (suite): Bien que d'intérêt industriel, cette méthode est d'utilité limitée à cause des complications potentielles suivantes:
• Formation compétitive de produits d'élimination.
• Préparation d'éthers dissymétriques par couplages mixtes impossible.
• Possibilité, dans certains cas, de réarrangements de carbocations. Conséquemment, la synthèse d'éther de Williamson, qui est une méthode plus générale, est souvent préférée à la simple déshydratation des alcools. 4.7. RÉACTIONS DES ÉTHERS La fonction éther est une fonction relativement inerte. Par exemple, les éthers résistent bien aux nucléophiles en milieu non-acide car RO- constitue un mauvais groupe partant. Voilà pourquoi l'éther diéthylique et le THF sont souvent utilisés comme solvant en chimie organique.
CH3 ORNu Nu CH3 OR+
mauvais groupe partant La protonation d'un éther, cependant, accroît la réactivité des éthers vis-à-vis les nucléophiles. Les éthers seront donc clivés par HX (X = Br, I, etc.).
CH3-CH2 O CH2-CH3
Br
CH3-CH2 OHCH3-CH2 Br
H
CH3-CH2 O CH2-CH3
H
+
© Richard Giasson
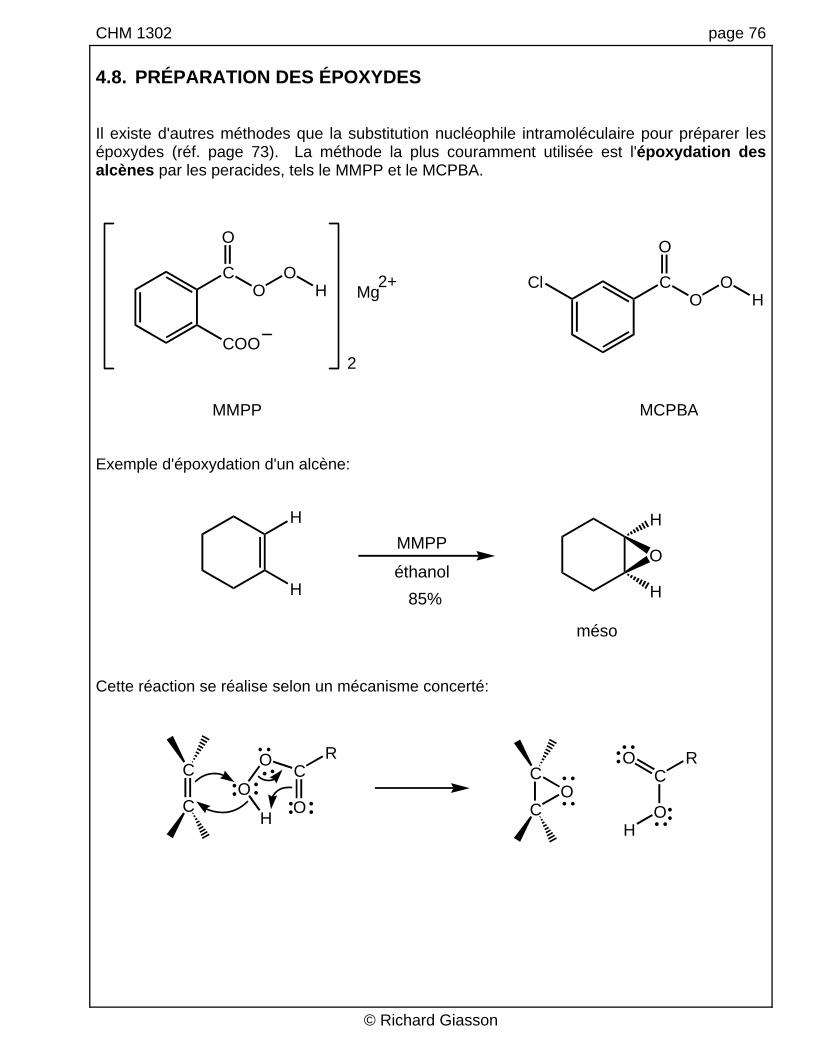
CHM 1302 page 76 4.8. PRÉPARATION DES ÉPOXYDES Il existe d'autres méthodes que la substitution nucléophile intramoléculaire pour préparer les époxydes (réf. page 73). La méthode la plus couramment utilisée est l'époxydation des alcènes par les peracides, tels le MMPP et le MCPBA.
CO
O
O
H
COO
Mg
2
2+ CO
O
O
HCl
MMPP MCPBA Exemple d'époxydation d'un alcène:
MMPP
éthanol
H
H
H
H
O
méso
85%
Cette réaction se réalise selon un mécanisme concerté:
C
C C
OH
O
O RC
CO
C
O
RO
H
© Richard Giasson

CHM 1302 page 77 4.9. RÉACTION D'OUVERTURE DES ÉPOXYDES RÉACTION D'OUVERTURE DES ÉPOXYDES EN MILIEU ACIDE: Les époxydes comportent un cycle tendu (tension de cycle ~25 kcal mol-1). Ces composés sont donc très susceptibles aux réactions menant à l'ouverture du cycle et au relâchement de cette tension. Une telle réaction se produit aisément sous l'action de HX.
O
CH3
HH
H3C
HBr aq. 48%
0°CBr
OHHH3C
CH3
HBr
HO HCH3
H3CH+
(2S,3S)-3-bromobutan-2-ol (2R,3R)-3-bromobutan-2-ol
composé méso mélange racémique
85% Notez: Le 3-bromobutan-2-ol est obtenu sous forme de mélange racémique dans
l'exemple ci-dessus. Ceci est dû à l'équiprobabilité de l'attaque de Br- sur un ou l'autre des atomes de carbone de cet époxyde symétrique. (voir le mécanisme à la page suivante)
© Richard Giasson
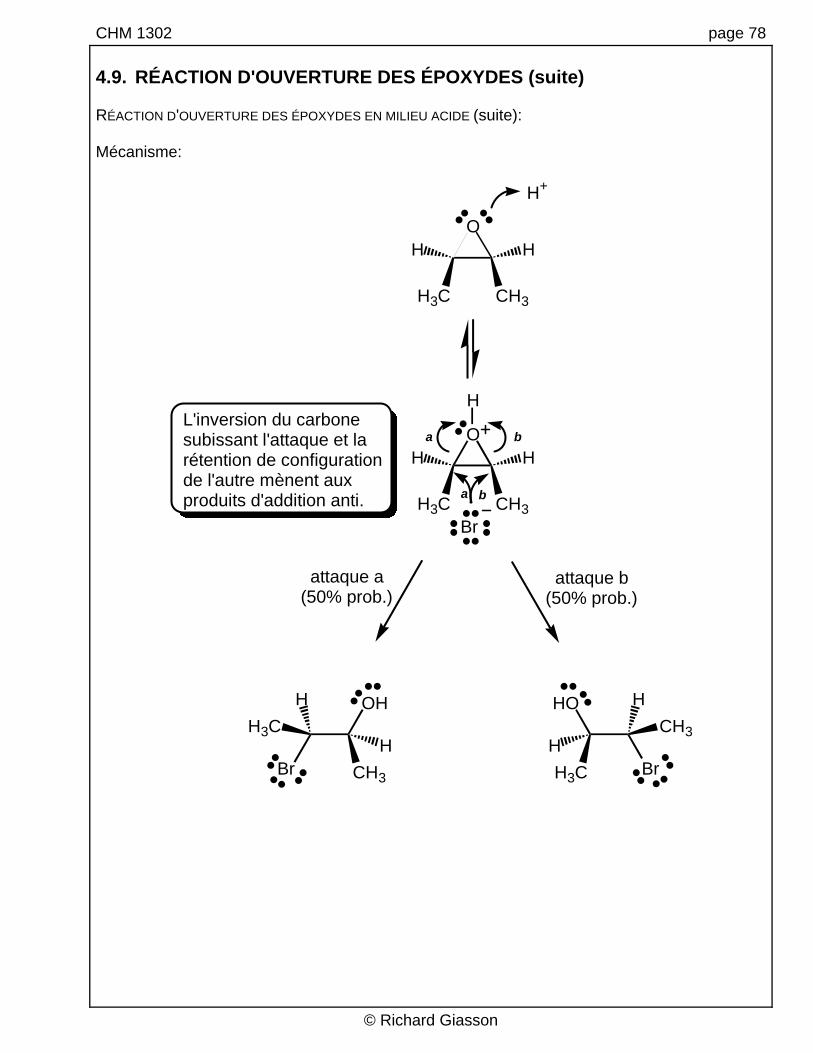
CHM 1302 page 78 4.9. RÉACTION D'OUVERTURE DES ÉPOXYDES (suite) RÉACTION D'OUVERTURE DES ÉPOXYDES EN MILIEU ACIDE (suite): Mécanisme:
O
CH3
HH
H3C
Br
OHHH3C
CH3
HBr
HO HCH3
H3CH
O
CH3
HH
H3C
H
H+
a b
a b
attaque a(50% prob.)
attaque b(50% prob.)
L'inversion du carbone subissant l'attaque et la rétention de configuration de l'autre mènent aux produits d'addition anti.
Br
© Richard Giasson
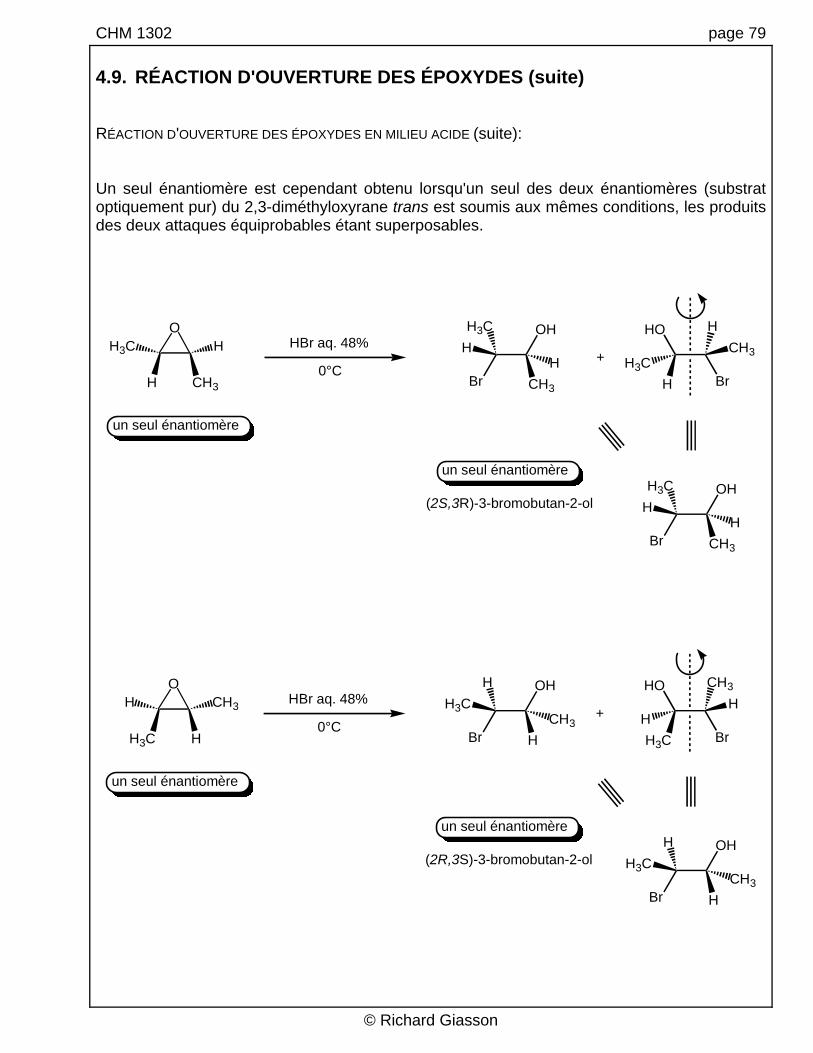
CHM 1302 page 79 4.9. RÉACTION D'OUVERTURE DES ÉPOXYDES (suite) RÉACTION D'OUVERTURE DES ÉPOXYDES EN MILIEU ACIDE (suite): Un seul énantiomère est cependant obtenu lorsqu'un seul des deux énantiomères (substrat optiquement pur) du 2,3-diméthyloxyrane trans est soumis aux mêmes conditions, les produits des deux attaques équiprobables étant superposables.
O
CH3
HH3C
H
HBr aq. 48%
0°CBr
OHH3CH
CH3
HBr
HO HCH3
HH3C+
(2S,3R)-3-bromobutan-2-ol
un seul énantiomère
un seul énantiomère
Br
OHH3CH
CH3
H
O
H
CH3H
H3C
HBr aq. 48%
0°CBr
OHHH3C
HCH3
Br
HO CH3
H
H3CH+
(2R,3S)-3-bromobutan-2-ol
un seul énantiomère
un seul énantiomère
Br
OHHH3C
HCH3
© Richard Giasson

CHM 1302 page 80 4.9. RÉACTION D'OUVERTURE DES ÉPOXYDES (suite) RÉACTION D'OUVERTURE DES ÉPOXYDES EN MILIEU ACIDE (suite): Les attaques d'un nucléophile, tel le méthanol, sur un ou l'autre des atomes de carbone d'un époxyde protoné ne sont cependant pas équiprobables pour les époxydes asymétriques et le produit de l'attaque sur le carbone le plus substitué est alors majoritaire.
O
H
CH3H3C
H3CH2SO4 CH3O
OHH3CH3C
HCH3
CH3OH
L'explication de ce phénomène est que la protonation de l'oxygène d'un époxyde asymétrique n'affaiblit pas les deux liens C-O dans la même mesure. L'élongation d'un lien C-O, suite à la protonation de l'oxygène, diminue la densité électronique sur l'atome de carbone. Un atome de carbone plus substitué stabilisera davantage la charge positive partielle qui se développe. Conséquemment, le lien C-O le plus substitué est celui qui est le plus affaibli par la protonation de l'oxygène et donc le plus réactif.
O
H
CH3H3C
H3C
CH3O
OH3CH3C
HCH3
H
O
H
CH3H3C
H3C
H
O
OH3CH3C
HCH3
H
H+
δ+
H
O CH3
HCH3
HO
CH3
Notez le lien C-O le plus substituédavantage affaibli à cause de lastabilité accrue de la charge δ+
© Richard Giasson
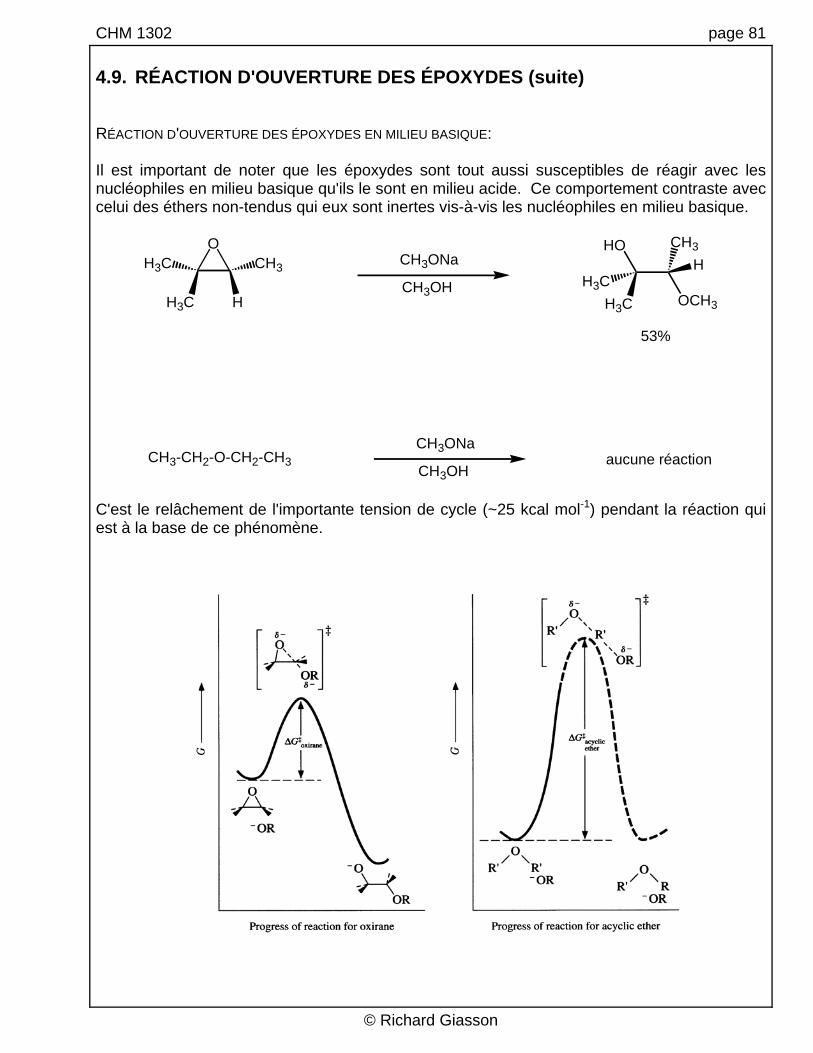
CHM 1302 page 81 4.9. RÉACTION D'OUVERTURE DES ÉPOXYDES (suite) RÉACTION D'OUVERTURE DES ÉPOXYDES EN MILIEU BASIQUE: Il est important de noter que les époxydes sont tout aussi susceptibles de réagir avec les nucléophiles en milieu basique qu'ils le sont en milieu acide. Ce comportement contraste avec celui des éthers non-tendus qui eux sont inertes vis-à-vis les nucléophiles en milieu basique.
O
H
CH3H3C
H3C OCH3
HO CH3
H
H3CH3C
CH3ONa
CH3OH
CH3ONa
CH3OHCH3-CH2-O-CH2-CH3 aucune réaction
53%
C'est le relâchement de l'importante tension de cycle (~25 kcal mol-1) pendant la réaction qui est à la base de ce phénomène.
© Richard Giasson
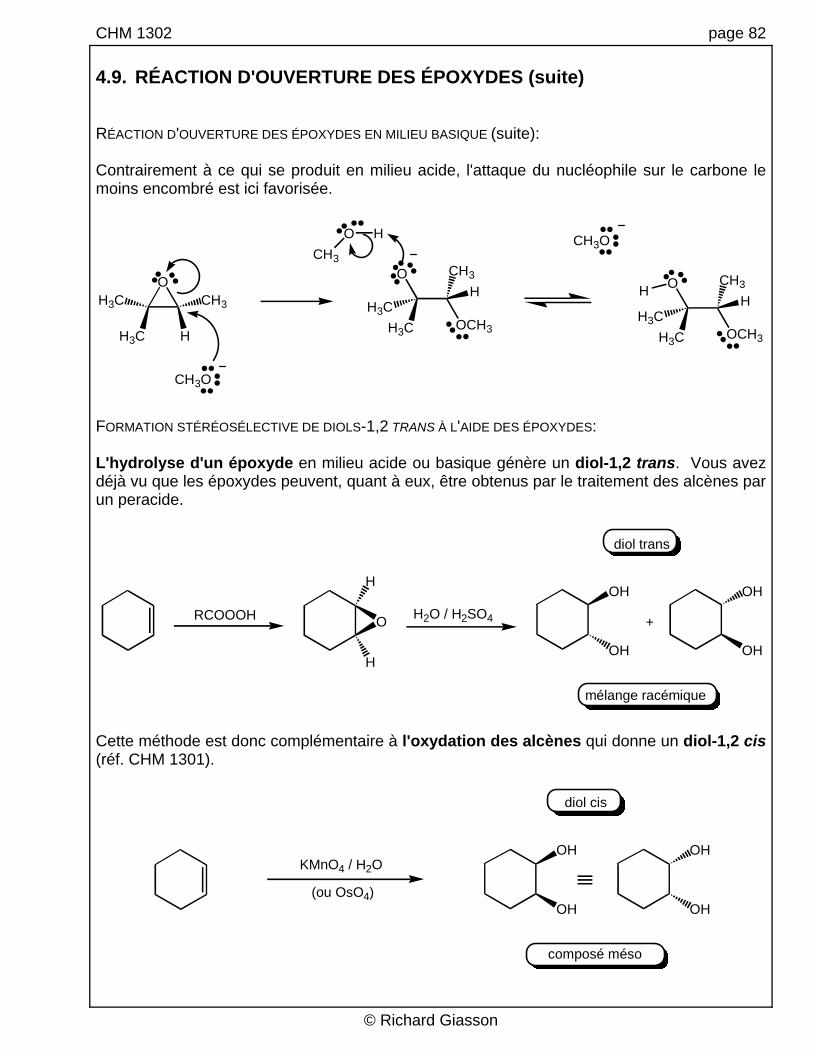
CHM 1302 page 82 4.9. RÉACTION D'OUVERTURE DES ÉPOXYDES (suite) RÉACTION D'OUVERTURE DES ÉPOXYDES EN MILIEU BASIQUE (suite): Contrairement à ce qui se produit en milieu acide, l'attaque du nucléophile sur le carbone le moins encombré est ici favorisée.
O
H
CH3H3C
H3COCH3
O CH3
H
H3CH3C
CH3O
OCH3
O CH3
H
H3CH3C
CH3
O H CH3O
H
FORMATION STÉRÉOSÉLECTIVE DE DIOLS-1,2 TRANS À L'AIDE DES ÉPOXYDES: L'hydrolyse d'un époxyde en milieu acide ou basique génère un diol-1,2 trans. Vous avez déjà vu que les époxydes peuvent, quant à eux, être obtenus par le traitement des alcènes par un peracide.
RCOOOH O
H
H
H2O / H2SO4
OH
OH
OH
OH
+
mélange racémique
diol trans
Cette méthode est donc complémentaire à l'oxydation des alcènes qui donne un diol-1,2 cis (réf. CHM 1301).
OH
OH
OH
OH
composé méso
diol cis
KMnO4 / H2O
(ou OsO4)
© Richard Giasson
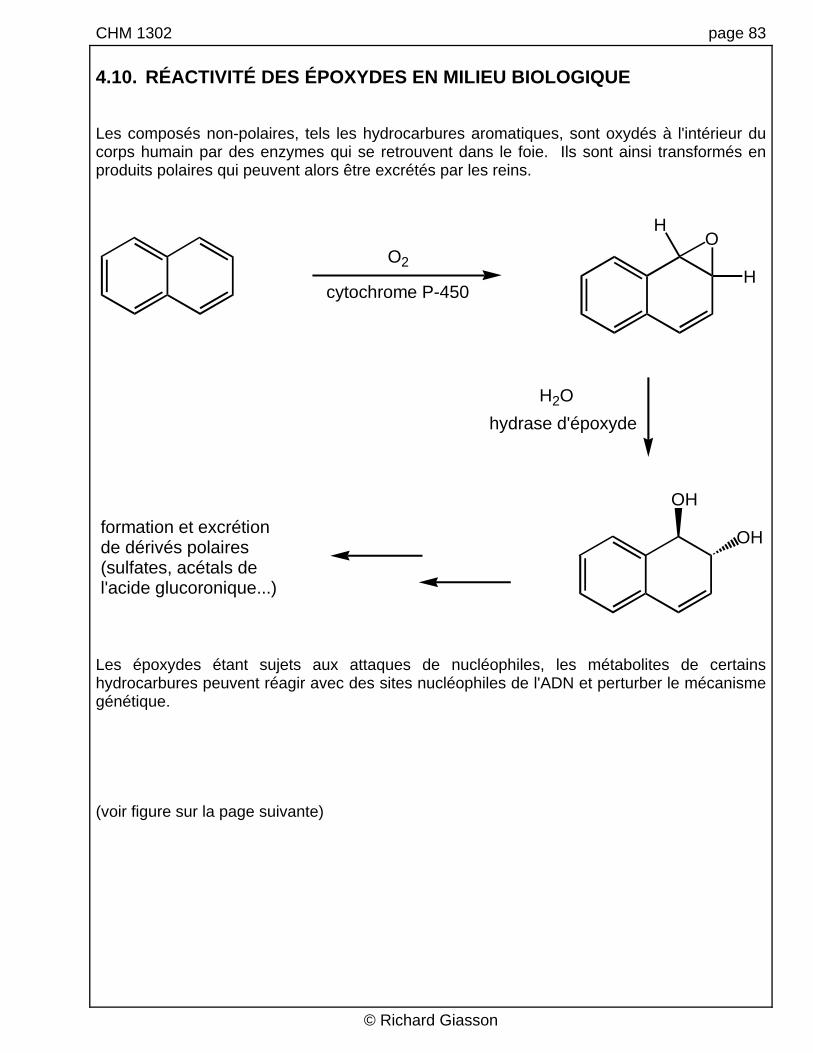
CHM 1302 page 83 4.10. RÉACTIVITÉ DES ÉPOXYDES EN MILIEU BIOLOGIQUE Les composés non-polaires, tels les hydrocarbures aromatiques, sont oxydés à l'intérieur du corps humain par des enzymes qui se retrouvent dans le foie. Ils sont ainsi transformés en produits polaires qui peuvent alors être excrétés par les reins.
O2
cytochrome P-450
O
H
H
OH
OH
H2Ohydrase d'époxyde
formation et excrétionde dérivés polaires(sulfates, acétals del'acide glucoronique...)
Les époxydes étant sujets aux attaques de nucléophiles, les métabolites de certains hydrocarbures peuvent réagir avec des sites nucléophiles de l'ADN et perturber le mécanisme génétique. (voir figure sur la page suivante)
© Richard Giasson
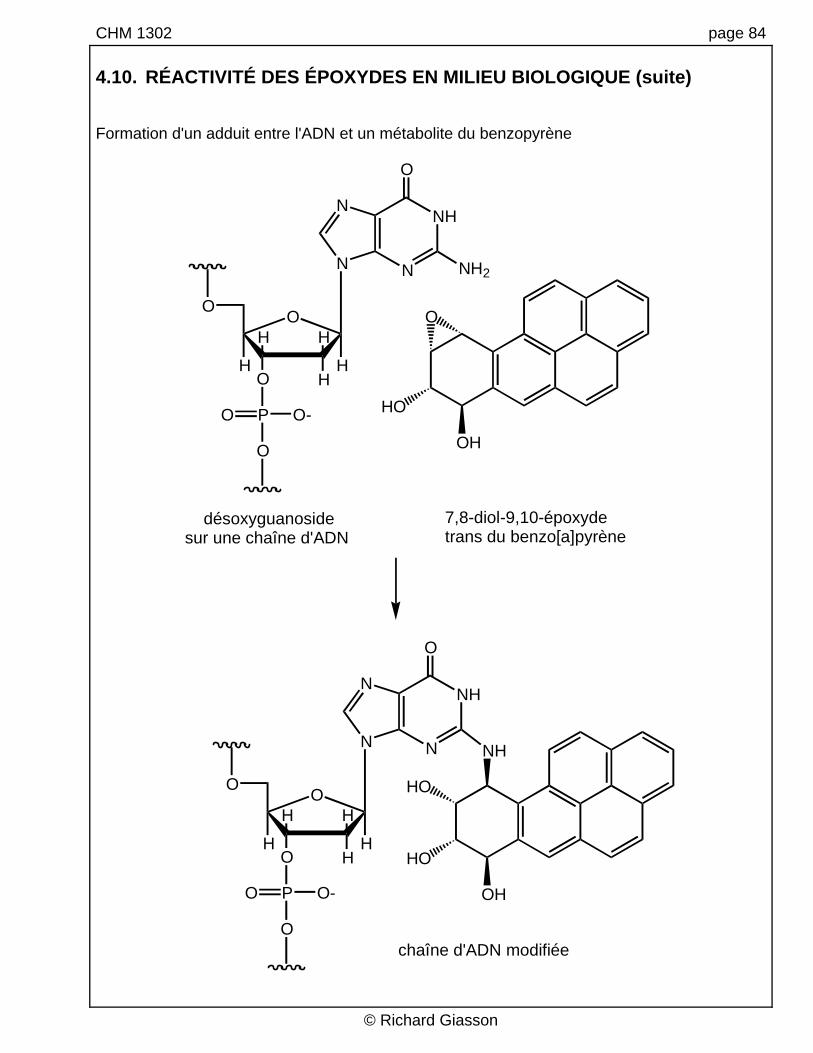
CHM 1302 page 84 4.10. RÉACTIVITÉ DES ÉPOXYDES EN MILIEU BIOLOGIQUE (suite) Formation d'un adduit entre l'ADN et un métabolite du benzopyrène
O
NH
N
N
O
NH2N
O
H
HH
HHO
PO
O
O-
O
HO
OH
O
NH
N
N
O
N
O
H
HH
HHO
PO
O
O-
HO
OH
NH
HO
désoxyguanosidesur une chaîne d'ADN
7,8-diol-9,10-époxyde trans du benzo[a]pyrène
chaîne d'ADN modifiée
© Richard Giasson
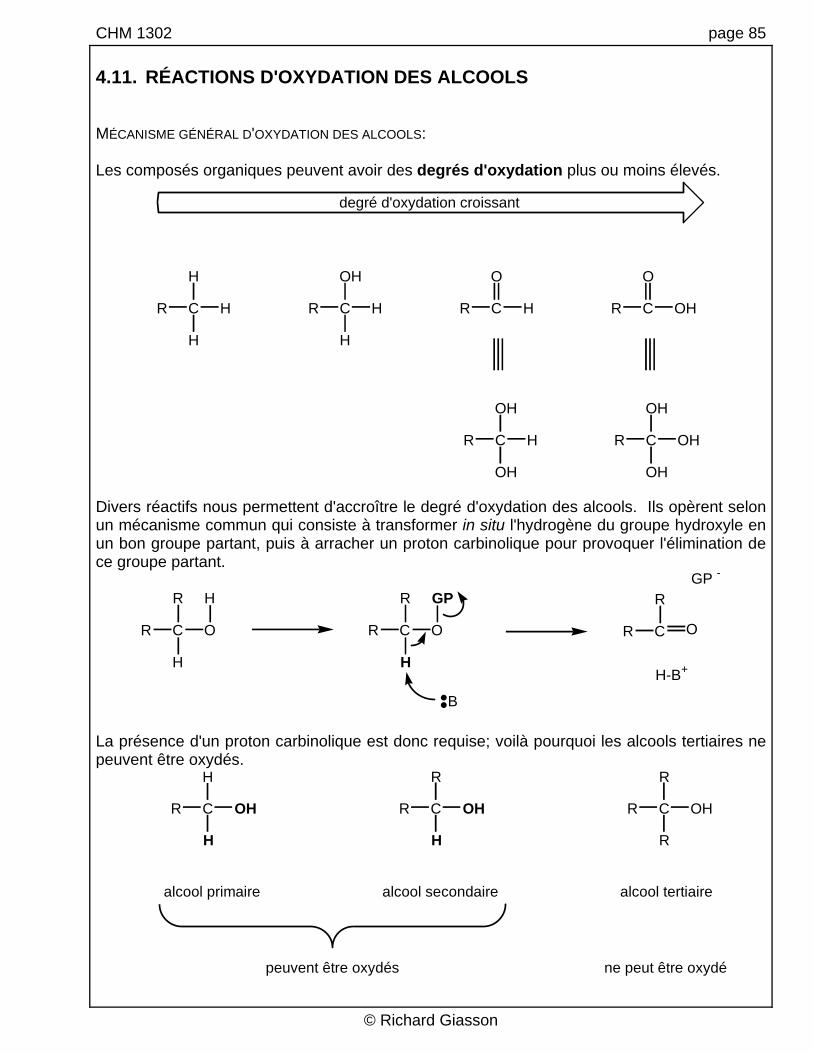
CHM 1302 page 85 4.11. RÉACTIONS D'OXYDATION DES ALCOOLS MÉCANISME GÉNÉRAL D'OXYDATION DES ALCOOLS: Les composés organiques peuvent avoir des degrés d'oxydation plus ou moins élevés.
R C
H
H
H
R C
OH
H
H
R C
O
H R C
O
OH
R C
OH
OH
OH
R C
OH
H
OH
degré d'oxydation croissant
Divers réactifs nous permettent d'accroître le degré d'oxydation des alcools. Ils opèrent selon un mécanisme commun qui consiste à transformer in situ l'hydrogène du groupe hydroxyle en un bon groupe partant, puis à arracher un proton carbinolique pour provoquer l'élimination de ce groupe partant.
R C
R
O
H
H
R C
R
O
H
GP
R C
R
O
H-B+
GP -
B La présence d'un proton carbinolique est donc requise; voilà pourquoi les alcools tertiaires ne peuvent être oxydés.
R C
H
OH
H
R C
R
OH
H
R C
R
OH
R
alcool primaire alcool secondaire alcool tertiaire
peuvent être oxydés ne peut être oxydé
© Richard Giasson
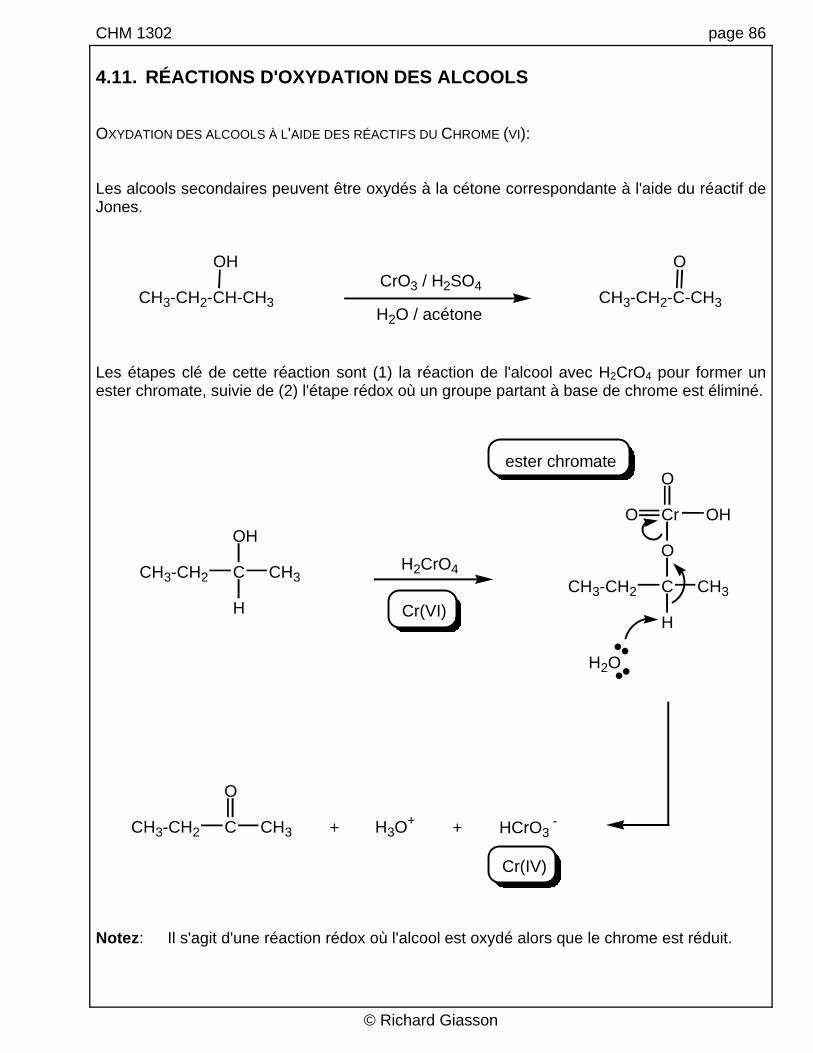
CHM 1302 page 86 4.11. RÉACTIONS D'OXYDATION DES ALCOOLS OXYDATION DES ALCOOLS À L'AIDE DES RÉACTIFS DU CHROME (VI): Les alcools secondaires peuvent être oxydés à la cétone correspondante à l'aide du réactif de Jones.
CH3-CH2-CH-CH3 CH3-CH2-C-CH3
OH OCrO3 / H2SO4
H2O / acétone Les étapes clé de cette réaction sont (1) la réaction de l'alcool avec H2CrO4 pour former un ester chromate, suivie de (2) l'étape rédox où un groupe partant à base de chrome est éliminé.
CCH3-CH2
H
OH
CH3 CCH3-CH2
H
O
CH3
CrO
O
OH
H2O
H2CrO4
CCH3-CH2
O
CH3 + H3O+ + HCrO3 -
Cr(VI)
Cr(IV)
ester chromate
Notez: Il s'agit d'une réaction rédox où l'alcool est oxydé alors que le chrome est réduit.
© Richard Giasson

CHM 1302 page 87 4.11. RÉACTIONS D'OXYDATION DES ALCOOLS (suite) OXYDATION DES ALCOOLS À L'AIDE DES RÉACTIFS DU CHROME (VI) (suite): Les alcools primaires, quant à eux, sont oxydés à l'acide carboxylique correspondant dans ces mêmes conditions, l'aldéhyde initialement formé étant lui aussi réactif en présence de H2CrO4.
CH3-CH2-CH2-CH2-OH CH3-CH2-CH2-CHO CH3-CH2-CH2-COOHCrO3 / H2SO4
H2O / acétone Pour arrêter l'oxydation d'un alcool primaire au niveau de l'aldéhyde, il est nécessaire d'utiliser un réactif du chrome (VI) sous une autre forme que H2CrO4 i.e. en absence d'eau. Un réactif de choix est alors le chlorochromate de pyridinium utilisé en solvant organique anhydre, tel le dichlorométhane.
CH3(CH2)8CH2OH CH3(CH2)8CHO
N
H
CrO3Cl
PCC
CH2Cl292%
Notez: Ce réactif peut aussi être utilisé pour oxyder les alcools secondaires. OXYDATION DE SWERN: L'utilisation de réactifs toxiques du Cr(VI) peut cependant être évitée. L'oxydation de Swern est une méthode d'oxydation des alcools primaires et secondaires attrayante.
CH3(CH2)8CH2OH CH3(CH2)8CHO
95%
CH3-S-CH3 / Cl-C-C-Cl
O O O
Et3N / CH2Cl2 / -60°C
Notez: L'oxydation d'un alcool primaire dans ces conditions arrête au niveau de l'aldéhyde.
© Richard Giasson
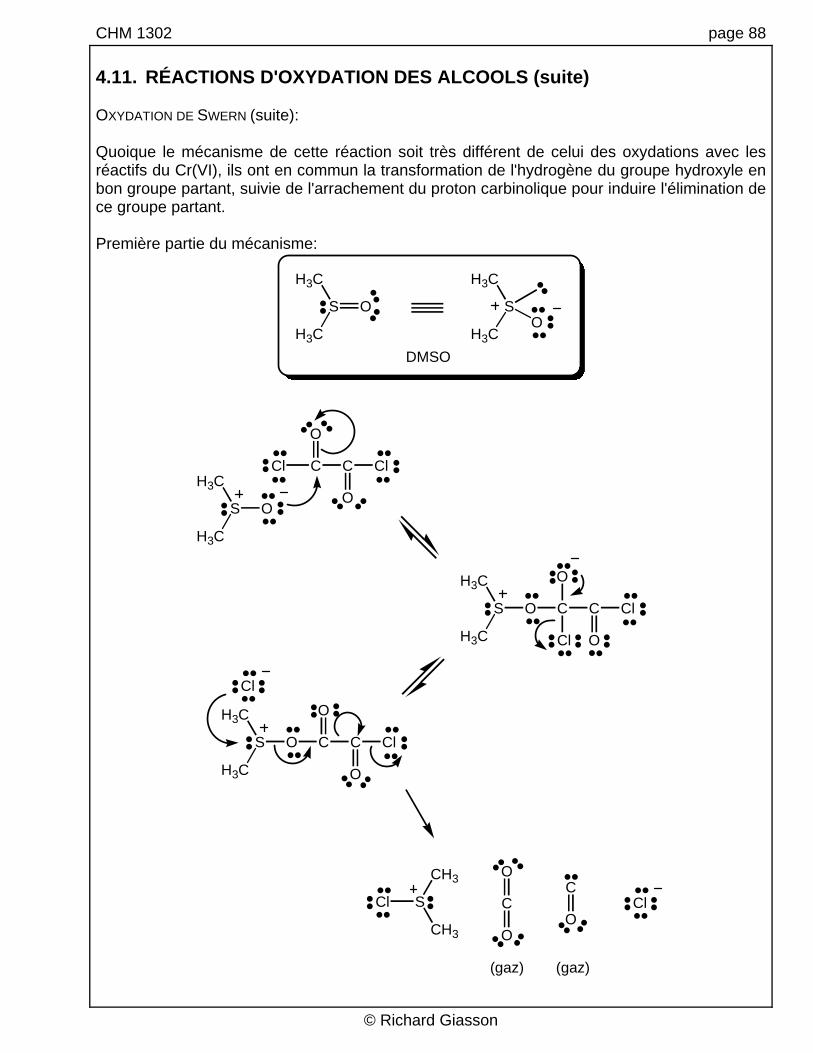
CHM 1302 page 88 4.11. RÉACTIONS D'OXYDATION DES ALCOOLS (suite) OXYDATION DE SWERN (suite): Quoique le mécanisme de cette réaction soit très différent de celui des oxydations avec les réactifs du Cr(VI), ils ont en commun la transformation de l'hydrogène du groupe hydroxyle en bon groupe partant, suivie de l'arrachement du proton carbinolique pour induire l'élimination de ce groupe partant. Première partie du mécanisme:
S
H3C
O
H3C
S
H3C
H3C
O
DMSO
S
H3C
O
H3CCl C
O
C
O
Cl
S
H3C
H3C
O C
O
C
O
Cl
Cl
S
H3C
H3C
O C
O
C
O
Cl
ClCl
CH3
S
CH3
C
O
OO
C
Cl
(gaz) (gaz)
© Richard Giasson
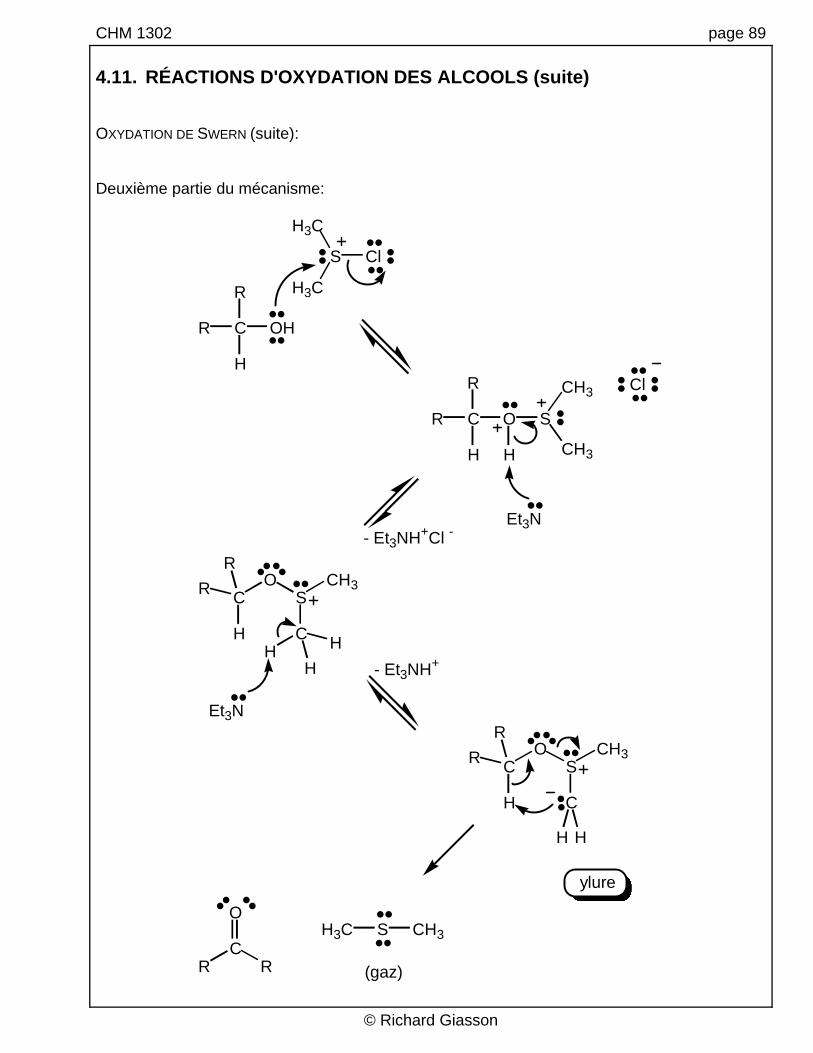
CHM 1302 page 89 4.11. RÉACTIONS D'OXYDATION DES ALCOOLS (suite) OXYDATION DE SWERN (suite): Deuxième partie du mécanisme:
Cl
H3C
S
H3C
R C
H
R
OH
Cl
CH3
CH3
R C
H
R
O S
H
C
HH
C
SO
RR CH3
HH
C
H C
SO
RR CH3
H H
O
CRR
H3C S CH3
(gaz)
- Et3NH+Cl -
- Et3NH+
Et3N
Et3N
ylure
© Richard Giasson

CHM 1302 page 90 4.12. SPECTROSCOPIE INFRAROUGE DES ALCOOLS ET ÉTHERS EFFET DES PONTS HYDROGÈNE: Les bandes caractéristiques de l'élongation des liens O-H sont influencées par la formation de ponts hydrogène entre molécules d'alcools. En phase gazeuse ou en solution très diluée, il y a absence de ponts hydrogène; la bande d'élongation du lien O-H située entre 3650 et 3584 cm-1 est alors fine. De nombreux ponts hydrogène existent en solution concentrée; l'affaiblissement des liens O-H qui en résulte fait apparaître des bandes à des fréquences inférieures. Le résultat est une bande large et intense entre 3550 et 3200 cm-1.
R O
H
RO
H
MODES VIBRATIONNELS:
Élongation du lien O-H
O H
• Groupe OH libre: Bande fine d'intensité variable vers 3600 cm-1.
• Groupe OH lié: Bande large et intense entre 3550 et 3200 cm-1.
• Bande absente chez les éthers.
Élongations du lien C-O
C O
• Alcools: Bande intense entre 1260 et 1000 cm-1. La position de la bande varie selon que l'alcool soit primaire (~1050 cm-
1), secondaire (~1150 cm-1), tertiaire (~1200 cm-1) ou aromatique (~1200 cm-1).
• Éthers aliphatiques: Bande intense entre 1150 et 1085 cm-1.
• Éthers aromatiques (Ar-O-R): Deux bandes intenses vers 1275-1200 cm-1 (élongation asymétrique) et 1075-1020 cm-1 (élongation symétrique).
Déformation angulaire de C-O-H
C O H
• Alcools: Bande(s) peu caractéristique(s) entre 1420 et 1330 cm-1. Les alcools primaires et secondaires ont deux bandes, alors que les tertiaires en ont qu'une.
• Éthers: Bande absente chez les éthers
© Richard Giasson
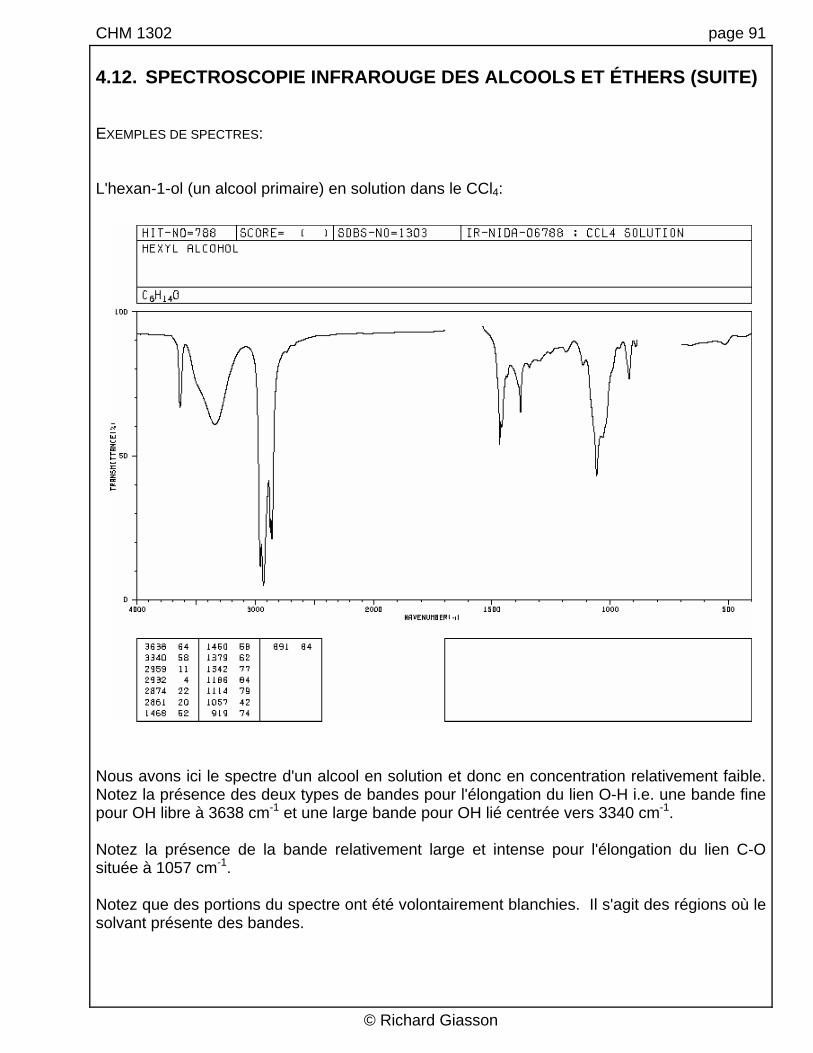
CHM 1302 page 91 4.12. SPECTROSCOPIE INFRAROUGE DES ALCOOLS ET ÉTHERS (SUITE) EXEMPLES DE SPECTRES: L'hexan-1-ol (un alcool primaire) en solution dans le CCl4:
Nous avons ici le spectre d'un alcool en solution et donc en concentration relativement faible. Notez la présence des deux types de bandes pour l'élongation du lien O-H i.e. une bande fine pour OH libre à 3638 cm-1 et une large bande pour OH lié centrée vers 3340 cm-1. Notez la présence de la bande relativement large et intense pour l'élongation du lien C-O située à 1057 cm-1. Notez que des portions du spectre ont été volontairement blanchies. Il s'agit des régions où le solvant présente des bandes.
© Richard Giasson
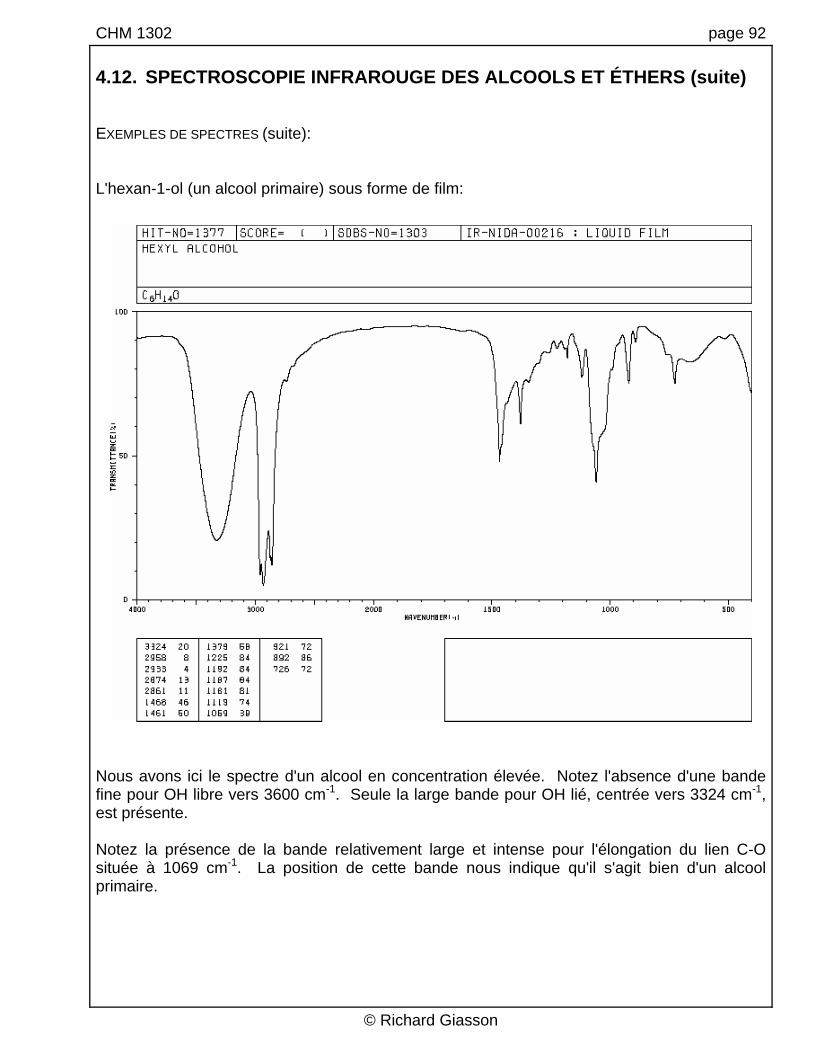
CHM 1302 page 92 4.12. SPECTROSCOPIE INFRAROUGE DES ALCOOLS ET ÉTHERS (suite) EXEMPLES DE SPECTRES (suite): L'hexan-1-ol (un alcool primaire) sous forme de film:
Nous avons ici le spectre d'un alcool en concentration élevée. Notez l'absence d'une bande fine pour OH libre vers 3600 cm-1. Seule la large bande pour OH lié, centrée vers 3324 cm-1, est présente. Notez la présence de la bande relativement large et intense pour l'élongation du lien C-O située à 1069 cm-1. La position de cette bande nous indique qu'il s'agit bien d'un alcool primaire.
© Richard Giasson
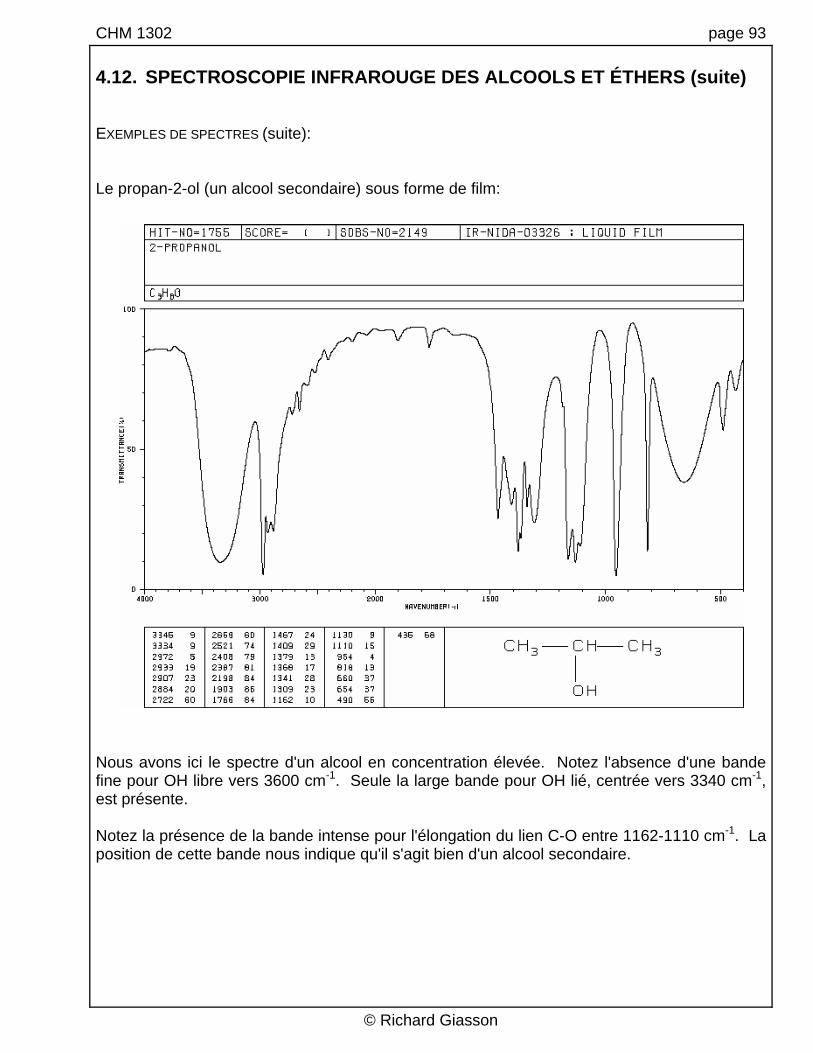
CHM 1302 page 93 4.12. SPECTROSCOPIE INFRAROUGE DES ALCOOLS ET ÉTHERS (suite) EXEMPLES DE SPECTRES (suite): Le propan-2-ol (un alcool secondaire) sous forme de film:
Nous avons ici le spectre d'un alcool en concentration élevée. Notez l'absence d'une bande fine pour OH libre vers 3600 cm-1. Seule la large bande pour OH lié, centrée vers 3340 cm-1, est présente. Notez la présence de la bande intense pour l'élongation du lien C-O entre 1162-1110 cm-1. La position de cette bande nous indique qu'il s'agit bien d'un alcool secondaire.
© Richard Giasson
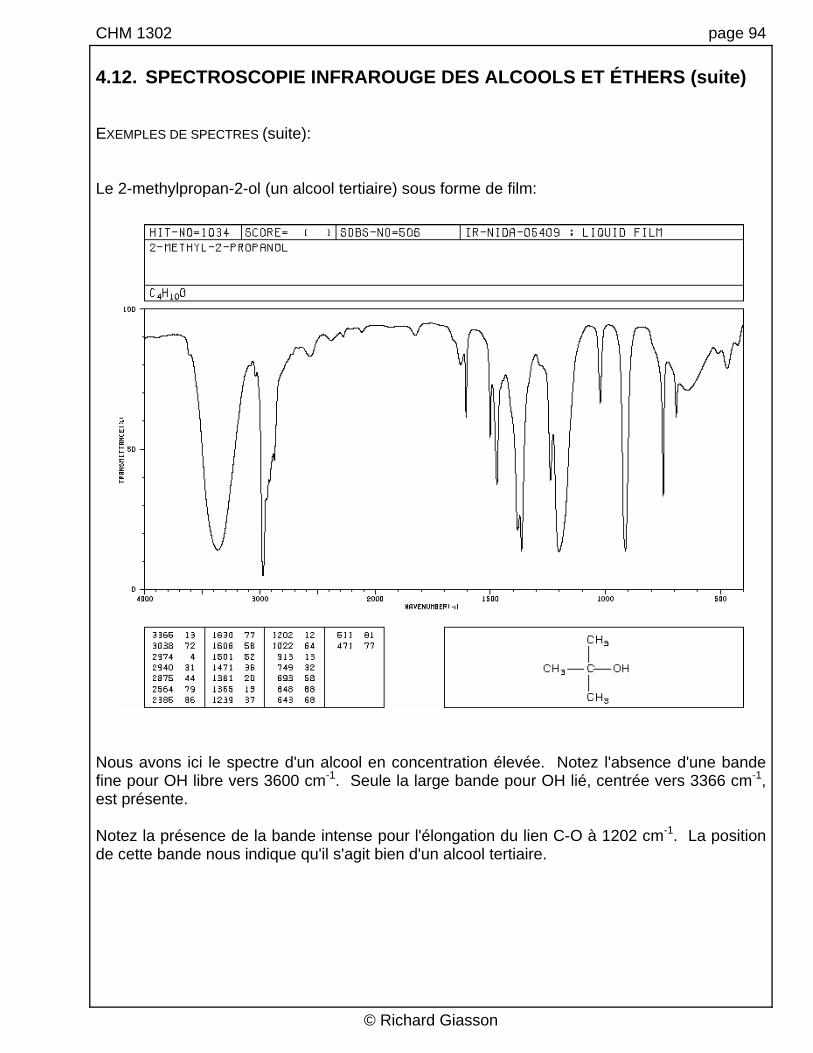
CHM 1302 page 94 4.12. SPECTROSCOPIE INFRAROUGE DES ALCOOLS ET ÉTHERS (suite) EXEMPLES DE SPECTRES (suite): Le 2-methylpropan-2-ol (un alcool tertiaire) sous forme de film:
Nous avons ici le spectre d'un alcool en concentration élevée. Notez l'absence d'une bande fine pour OH libre vers 3600 cm-1. Seule la large bande pour OH lié, centrée vers 3366 cm-1, est présente. Notez la présence de la bande intense pour l'élongation du lien C-O à 1202 cm-1. La position de cette bande nous indique qu'il s'agit bien d'un alcool tertiaire.
© Richard Giasson
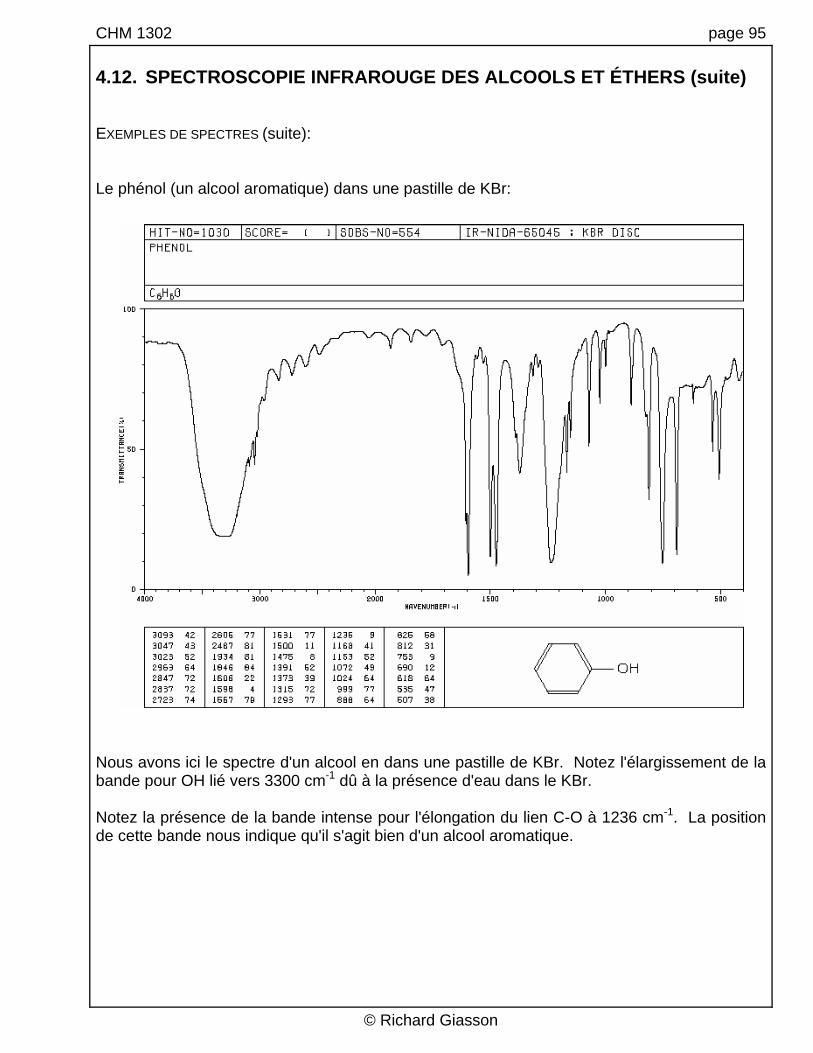
CHM 1302 page 95 4.12. SPECTROSCOPIE INFRAROUGE DES ALCOOLS ET ÉTHERS (suite) EXEMPLES DE SPECTRES (suite): Le phénol (un alcool aromatique) dans une pastille de KBr:
Nous avons ici le spectre d'un alcool en dans une pastille de KBr. Notez l'élargissement de la bande pour OH lié vers 3300 cm-1 dû à la présence d'eau dans le KBr. Notez la présence de la bande intense pour l'élongation du lien C-O à 1236 cm-1. La position de cette bande nous indique qu'il s'agit bien d'un alcool aromatique.
© Richard Giasson
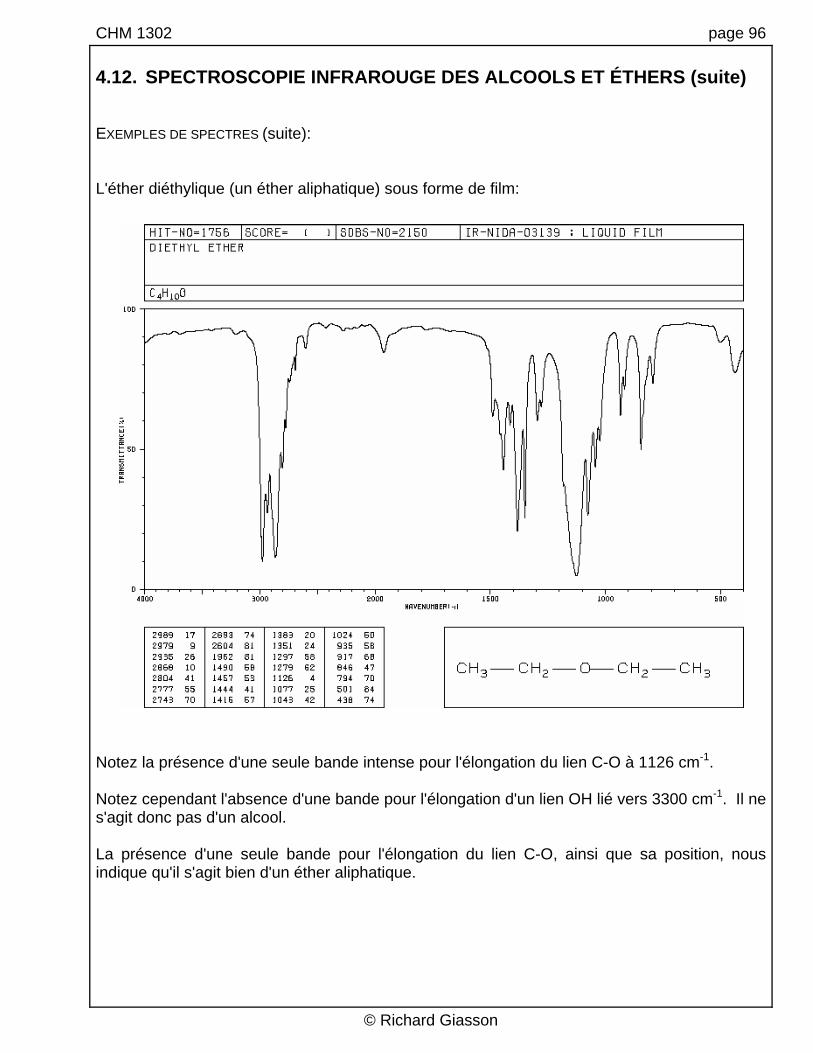
CHM 1302 page 96 4.12. SPECTROSCOPIE INFRAROUGE DES ALCOOLS ET ÉTHERS (suite) EXEMPLES DE SPECTRES (suite): L'éther diéthylique (un éther aliphatique) sous forme de film:
Notez la présence d'une seule bande intense pour l'élongation du lien C-O à 1126 cm-1. Notez cependant l'absence d'une bande pour l'élongation d'un lien OH lié vers 3300 cm-1. Il ne s'agit donc pas d'un alcool. La présence d'une seule bande pour l'élongation du lien C-O, ainsi que sa position, nous indique qu'il s'agit bien d'un éther aliphatique.
© Richard Giasson
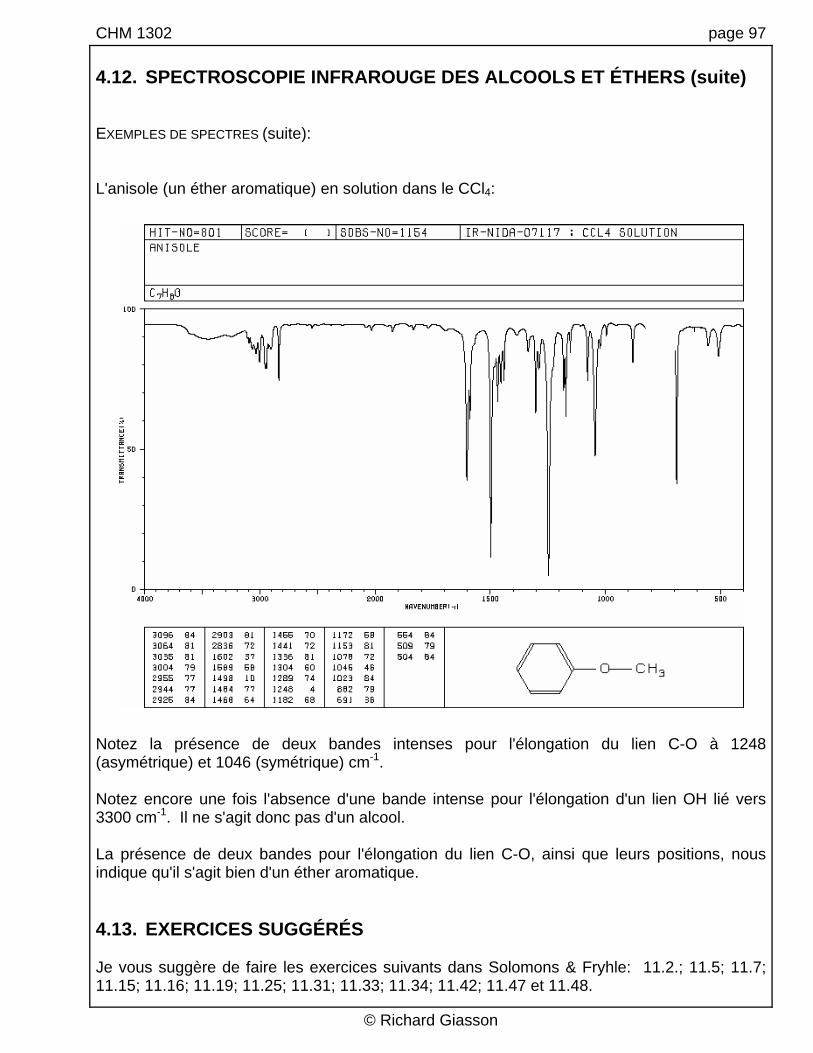
CHM 1302 page 97 4.12. SPECTROSCOPIE INFRAROUGE DES ALCOOLS ET ÉTHERS (suite) EXEMPLES DE SPECTRES (suite): L'anisole (un éther aromatique) en solution dans le CCl4:
Notez la présence de deux bandes intenses pour l'élongation du lien C-O à 1248 (asymétrique) et 1046 (symétrique) cm-1. Notez encore une fois l'absence d'une bande intense pour l'élongation d'un lien OH lié vers 3300 cm-1. Il ne s'agit donc pas d'un alcool. La présence de deux bandes pour l'élongation du lien C-O, ainsi que leurs positions, nous indique qu'il s'agit bien d'un éther aromatique. 4.13. EXERCICES SUGGÉRÉS Je vous suggère de faire les exercices suivants dans Solomons & Fryhle: 11.2.; 11.5; 11.7; 11.15; 11.16; 11.19; 11.25; 11.31; 11.33; 11.34; 11.42; 11.47 et 11.48.
© Richard Giasson





























