Embed Size (px)
Citation preview

Tumor and Stem Cell Biology
IFN-g Inhibits Gastric Carcinogenesis by Inducing EpithelialCell Autophagy and T-Cell Apoptosis
Shui Ping Tu1,3, Michael Quante1, Govind Bhagat2, Shigeo Takaishi1, Guanglin Cui1, Xiang Dong Yang1,Sureshkumar Muthuplani4, Wataru Shibata1, James G. Fox4, D. Mark Pritchard5, and Timothy C. Wang1
AbstractIFN-g mediates responses to bacterial infection and autoimmune disease, but it is also an important tumor
suppressor. It is upregulated in the gastric mucosa by chronic Helicobacter infection; however, whether it plays apositive or negative role in inflammation-associated gastric carcinogenesis is unexplored. To study this question,we generated an Hþ/Kþ-ATPase-IFN-g transgenic mouse that overexpresses murine IFN-g in the stomachmucosa. In contrast to the expected proinflammatory role during infection, we found that IFN-g overexpressionfailed to induce gastritis and instead inhibited gastric carcinogenesis induced by interleukin-1beta (IL-1b) and/or Helicobacter infection. Helper T cell (Th) 1 and Th17 immune responses were inhibited by IFN-g through Fasinduction and apoptosis in CD4 T cells. IFN-g also induced autophagy in gastric epithelial cells throughincreased expression of Beclin-1. Finally, in the gastric epithelium, IFN-g also inhibited IL-1b- and Helicobacter-induced epithelial apoptosis, proliferation, and Dckl1þ cell expansion. Taken together, our results suggest thatIFN-g coordinately inhibits bacterial infection and carcinogenesis in the gastric mucosa by suppressing putativegastric progenitor cell expansion and reducing epithelial cell apoptosis via induction of an autophagic program.Cancer Res; 71(12); 4247–59. �2011 AACR.
Introduction
IFN-g is a cytokine produced primarily by activated CD4þ orCD8þ T cells and natural killer cells and is recognized as animportant mediator of innate and adaptive immunity. Itinduces a variety of immunomodulatory molecules (1) andhas been identified as a critical effector in numerous models ofinflammatory and autoimmune diseases. Proposed roles ofIFN-g include orchestrating defense responses against intra-and extracellular bacteria. IFN-g primes mononuclear phago-cytes for production of monokines, and in concert with TNF-acan augment the bacteriostatic activity of phagocytes. Inter-leukin (IL)-12 produced by monocytes can further polarizetoward a helper T-cell (Th) 1 response resulting in additionalIFN-g production.
Helicobacter pylori represents one of the world's mostcommon chronic bacterial infection, and IFN-g has beenshown to be upregulated in the stomach of humans and miceinfected with Helicobacter sp. (2, 3). Helicobacter pylori bac-teria survive for decades in the human stomach, occasionallyshowing intracellular invasion of gastric epithelial cells (4).Chronic infection with Helicobacter represents an importantrisk factor for gastric cancer (5), and growing evidence sug-gests that it is the host immune response that is predictive ofsusceptibility to gastric cancer. Proinflammatory IL-1b, TNF-a, and IL-10 genotypes are associated with an increased riskfor developing gastric cancer in the setting of H. pylori infec-tion (6, 7), and overexpression of IL-1b alone can inducegastric cancer in mice (8).
Interestingly, there has been no reported human associa-tion between IFN-g genotypes and gastric cancer risk, and theprecise role of IFN-g in gastric carcinogenesis remains unclear.Although several studies have suggested that neutralization ordeletion of IFN-g protects against the development of gastricatrophy, others could not confirm the importance of IFN-g inpreneoplasia (9, 10). Furthermore, studies of IFN-g�/� micesuggest a role in protection from H. pylori infection (3, 11),supported by findings that IFN-g can inhibit the developmentof IL-17–producing Th17 cells. IL-17 is overexpressed in H.pylori–infected stomachs in mice and human (12), and likelyplays a role in the clearance of extracellular pathogens (13).
One way that IFN-g defends against intracellular pathogensis by activation of immunity-related GTPases (IRG), some ofwhich (Irgm1/LRG-47) have been shown to eliminate myco-bacteria from macrophages through autophagy (14). Autop-hagy is a highly conserved, bulk degradation process in which
Authors' Affiliations: Departments of 1Medicine and 2Pathology and CellBiology, College of Physicians & Surgeons, Columbia University, NewYork; 3Department of Chemical Biology, Center for Cancer Prevention,Susam L. Cullman Laboratory of Cancer Research, Rutgers University,Piscataway, New Jersey; 4Division of Comparative Medicine, Massachu-setts Institute of Technology, Cambridge, Massachusetts; and 5Division ofGastroenterology, School of Clinical Sciences, University of Liverpool,Liverpool, United Kingdom
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
S-P. Tu and M. Quante contributed equally to this work.
Corresponding Author: Timothy C. Wang, Department of Medicine,Columbia University Medical Center, New York, NY 10032. Phone: 212-851-4581; Fax: 212-851-4590; E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-10-4009
�2011 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 4247
Research. on May 6, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst April 21, 2011; DOI: 10.1158/0008-5472.CAN-10-4009

portions of cytoplasm are sequestered into autophagosomes.Several studies have linked autophagy to host defense againstintracellular bacterial pathogens (15); however, the precisemechanisms remain to be elucidated. In addition, autophagyis thought to protect against cancer, in part, through removalof cellular debris that promotes chronic inflammation (16).
IFN-g has also been linked to antitumor immunity and, insome models, the immune system plays an active role insuppressing the development of incipient tumors (17). Workfrom a number of groups has shown that IFN-g preventstumor induction (18, 19), although whether IFN-g suppressestumorigenesis by stimulation of "cancer immunosurveillance"is debatable. Although IFN-gR–deficient animals are moresusceptible to tumorigenesis, immunodeficient Rag-2–defi-cient mice do not consistently show increased tumor suscept-ibility (19, 20), and spontaneous tumor development in micelacking IFN-g and granulocyte macrophage colony-stimulat-ing factor can be inhibited by treatment with antibiotics (21),suggesting that carcinogenesis in immunodeficient mice maybe mediated in part by changes in bacterial flora.
To investigate the potential role of IFN-g in gastric inflam-mation and carcinogenesis, we generated a transgenic mousewith stomach-specific overexpression of IFN-g by using theHþ/Kþ-ATPase promoter. Surprisingly, IFN-g inhibited thedevelopment of IL-1b- and H. felis–induced gastritis and neo-plasia by induction of apoptosis, Beclin-1–mediated autop-hagy, and suppression of potential progenitor cell expansion.
Materials and Methods
Generation of Hþ/Kþ-ATPase and IFN-g transgenicmice
The 1,060-bp fragments of themouse Hþ/Kþ-ATPase b-sub-unit promoter (8) and the 450-bp fragments of maturesecreted form mouse IFN-g cDNA (Howard Young, NCI) weresubcloned together with human growth hormone polyadeny-lation sequence into pBluescript vector. The transgenic con-struct was used for pronuclear injection of C57BL/6 � SJL F2hybrid zygotes. A total of 5 positive founders were obtainedand backcrossed to C57BL/6J mice. A high IFN-g–expressingline (line 54) and a low FN-g–expressing line (line 32) wereselected for further study. IFN-g�/� (B6.129S7-Ifngtm1Ts/J)mice were purchased from Jackson Laboratories.
Helicobacter felis infection and detectionHelicobacter felis infection was carried out as previously
described (8). Pieces of gastric mucosa were streaked onto aBrucella agar plate, which was incubated at 35�C in a micro-aerophilic atmosphere until pinhead-sized colonies with ayellow appearance were noted. These gram-negative bacteriahad spiral rodmorphology and expressed urease, catalase, andoxidase. Warthin–Starry and hematoxyline and eosin (H&E)-stained sections of gastric tissue were examined for thepresence of H. felis- and bacteria-associated pathology.
IFN-g infusionRecombinant mouse IFN-g (10,714 units/d; ref. 22; R&D)
was diluted in 2% bovine serum albumin (BSA) and PBS and
loaded into Alzet 15-day micro-osmotic pumps according tothe manufacturer's instructions. Control pumps containedequivalent volumes of 2% BSA in PBS. Pumps were insertedinto male C57BL/6 IFN-g�/� and IL-1b mice (n ¼ 4 per timepoint).
Measurement of cytokine levelsThe levels of IL-1b, TNF-a, IL-6, and IFN-g in serum or
gastric tissues of transgenic mice were determined using amouse ELISA kit (BD Biosciences). Tissues were sonicated inmedium containing a protease inhibitor cocktail (RocheApplied Science). Absorbance was measured at 450 nm bya Multiscan MC reader, and the samples were analyzed byDeltaSoft software (BioMetallics, Inc.).
Cell linesThe human gastric cancer cell line MKN28, which was
established from amoderately differentiated adenocarcinoma,was recently obtained from the Riken Cell Bank, and cells werestored in multiple aliquots in liquid nitrogen and passaged forfewer than 3 months after resuscitation (MKN28 cells wereregularly tested by short tandem repeat polymorphism ana-lysis test by Riken). These cells were maintained in Dulbecco'smodified Eagle's medium supplemented with 10% fetal calfserum (Sigma), 100 units/mL penicillin, and 100 mg/mLstreptomycin (Life Technologies, Inc.).
Quantitative and semiquantitative PCRsReverse transcription was carried out by using the Super-
Script III System. Quantitative reverse transcriptase PCR(qRT-PCR) was carried out with a 3-step method by usingthe Bio-Rad iCycler qRT-PCR detection system (Bio-RadLaboratories) and QuantiTect SYBR Green PCR (Qiagen).The PCR conditions were as follows: 95�C for 3 minutes,followed by 40 cycles at 95�C for 30 seconds, 60�C for 30seconds, and 72�C for 30 seconds.
Western blotThe gastric cancer MNK-28 cell line was transfected with a
green fluorescent protein (GFP)-LC3 construct (Cell Biolabs,Inc.) and then treated with 20 ng IFN-g for 48 hours. Cells wereharvested and protein was extracted. GFP-LC3 was detectedby Western blot by using an anti-LC3 antibody (Abcam).
Histopathologic analysisSections of stomach and other tissues from transgenic and
control mice were fixed in 10% formalin and embedded inparaffin. Histopathologic indices were scored as previouslydescribed (8).
Analysis of apoptosisDeparaffinized, colonic tissue slides were stained with an In
Situ Apoptosis Detection kit (Chemicon) according to themanufacturer's instructions. The apoptotic index was calcu-lated by assessing the mean of the total number of TUNEL(terminal deoxynucleotidyl transferase–mediated dUTP nickend labeling)-positive cells per field at 10 different locations ofthe stomach section under a light microscope (400�).
Tu et al.
Cancer Res; 71(12) June 15, 2011 Cancer Research4248
Research. on May 6, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst April 21, 2011; DOI: 10.1158/0008-5472.CAN-10-4009

Analysis of autophagyThe gastric cancer cell line MNK-28 was transfected with
GFP-LC3 plasmids in the presence or absence of 20 ng IFN-gfor 48 hours. Autophagy was detected by visualizing punctuatedots in cells under a fluorescence microscope. MKN-28-GFP-LC3 stable transfectants were transfected with Beclin-1 siRNAand control siRNA (Santa Cruz Biotechnology) for 6 hours. Thetransfectants were cultured in the presence or absence of20 ng IFN-g for 48 hours. Autophagy was assessed by detectionof GFP-LC3-II punctuate dots in cells by fluorescence micro-scopy. Cells were harvested for Western blotting to detect theexpression of LC3-II and Beclin-1.
Reporter construct and luciferase assayA 950-bp fragment of the Beclin-1 promoter was cloned into
the pGL3-basic vector through PCR amplification of humangenomic DNA (Promega) by using synthetic primers (23).MKN-28 cells (1.0 � 105 cells/12-well) were transfected with1.5 mg Beclin-1 luciferase or 1.5 mg empty reporter vector DNAby using the FuGene HD transfection reagent (Promega). Tocontrol for background luciferase activity, 0.05 mg/well of aRenilla luciferase reporter vector DNA, driven by a minimallyactive thymidine kinase promoter (pRLTK; Promega), wascotransfected. After 6 hours, the transfected cells were cul-tured with or without 20 ng/mL IFN-g for 24 hours. Luciferaseactivity was determined as previously described (23). Allassays were conducted in triplicate and repeated 3 times.
Immunohistochemical stainingParaffin sections fixed in 10% formalin were incubated with
the following primary antibodies: Ki-67 (Dako), Dclk-1 orDCAMKL-1 (Abgent), F4/80 (Santa Cruz), LC3 (Abcam), andcontrol rat IgG2a. Biotinylated secondary antibodies (JacksonImmunoresearch Laboratories Inc.) and ABC avidin–biotin–DAB (3,30-diaminobenzidine) detection kit (Vector Labs) wereused for detection and visualization according to suppliedprotocol.
Single-cell preparation and fluorescence-activated cell-sorting analysisFor single-cell suspension preparation from stomach tissue,
the mucosa was gently scraped free from the serosa, mincedand digested for 1 hour in 1 mmol/L dithiothreitol, 1 mmol/LEDTA, 5% FBS in PBS at 37�C, filtered through a 40-mm nylonmesh strainer, and then resuspended in Dulbecco's PBS (D-PBS; ref. 24). For splenic single-cell suspensions, spleens weredisaggregated in cold Hanks’ balanced salt solution. Erythro-cytes were removed by hypotonic lysis. Splenocytes wereresuspended in D-PBS. Single-cell suspensions were stainedwith fluorescent labeled anti-CD45, CD3, CD19, CD8, CD4,CD11b, Ly-6G, c-kit, FLK-1, and Scal-1 antibodies (BD Phar-mingen) and detected using a LSAII flow cytometer (BDBioscience). Data were analyzed by FlowJo7 software (TreeStar, Inc.).
StatisticsData are presented as the mean � SD. The significance of
the difference between groups was evaluated with the
Student's t test or ANOVA test; P < 0.05 was consideredsignificant.
Results
Hþ/Kþ�-ATPase-IFN-gmice fail to develop spontaneousgastritis or metaplasia
To investigate the role of IFN-g in gastric inflammation andcarcinogenesis, we generated Hþ/Kþ-ATPase-IFN-g trans-genic mice, in which the mouse IFN-g cDNA (kind gift ofHoward A Young, NCI) was placed downstream of the mouseparietal cell–specific Hþ/Kþ-ATPase promoter (Fig. 1A). Weidentified 2 lines of IFN-g transgenic mice (line 54, high; line32, low; Fig. 1B) that showed elevated levels of mouse IFN-g(line 32, 2-fold; line 54, 3.5-fold), specifically in the stomach byqRT-PCR and ELISA assays (Supplementary Fig. S1). IFN-gbioactivity was confirmed by elevated levels of IFN-g–regulated genes such as IFN-g–inducible protein 10 (IP-10) and monokine induced by IFN-g (Mig; Fig. 1C). IFN-gexpression in the Hþ/Kþ-ATPase-IFN-g mice (line 54, 3.5-fold)was comparable with that found in agedH. felis–infected mice,providing an opportunity to study the role of moderateelevation of IFN-g in the absence of prior inflammation orinfection.
Overexpression of IFN-g in the gastric mucosa did notinduce spontaneous atrophic gastritis or dysplasia (Fig. 1D),and these mice had near normal histopathology scores (Sup-plementary Table S1). Consistent with these observations,mRNA expression levels of the proinflammatory cytokines(IL-6, TNF-a, and IL-1b) were not significantly differentbetween Hþ/Kþ-ATPase-IFN-g and wild-type (WT) mice (datanot shown).
IFN-g inhibits the development of H. felis- or IL-1b–induced gastric dysplasia
Because IFN-g overexpression induced minimal gastritis inthe mouse stomach, we next examined its role in the setting ofH. felis infection, as H. felis–infected WT mice develop gastricinflammation, atrophy metaplasia, and dysplasia (8). Surpris-ingly, we found that H. felis–infected Hþ/Kþ-ATPase-IFN-gmice developed only mild gastritis, with minimal degrees ofatrophy and metaplasia after 12 or 24 months (Fig. 2A–C).Histopathology scores were significantly lower in H. felis–infected Hþ/Kþ-ATPase-IFN-g mice compared with H. felis–infectedWTmice (Supplementary Table S1, Fig. 2C and D). NoIFN-g transgenic mice developed dysplasia (Fig. 2D), suggest-ing that elevated levels of IFN-g inhibit carcinogenesis. Despiteprevious observations that IFN-g may reduce H. pylori colo-nization (3), in our model, there were no differences incolonization when we quantified H. felis infection levels byH&E staining (Fig. 2E) and qRT-PCR (Fig. 2F).
We next examined the effects of IFN-g overexpression in asecond model of gastric cancer, the Hþ/Kþ-ATPase-IL-1btransgenic mouse model, in which we had shown inflamma-tion-induced gastric carcinogenesis (8). IL-1b transgenic micewere crossed with Hþ/Kþ-ATPase-IFNg mice and followed for12 to 14 months with or without H. felis infection. IL-1b;IFN-gmice developed less severe hyperplasia, chronic inflammation,
IFN-g Inhibits Gastric Inflammation and Carcinoma
www.aacrjournals.org Cancer Res; 71(12) June 15, 2011 4249
Research. on May 6, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst April 21, 2011; DOI: 10.1158/0008-5472.CAN-10-4009

atrophy, metaplasia, and dysplasia compared with their IL-1blittermates (Supplementary Table S1). No IL-1b;IFN-g micedeveloped dysplasia (Fig. 3C; Supplementary Table S1),whereas H. felis–infected IL-1b mice progressed rapidly tometaplasia and high-grade dysplasia. These findings againindicate that overexpression of IFN-g inhibits inflamma-tion-dependent carcinogenesis.
IFN-g inhibits gastric epithelial cell proliferation andDclk-1þ cell expansion
The development of Helicobacter-dependent gastric canceris preceded by increased epithelial proliferation. Ki-67þ cells inthe stomach were significantly reduced in IFN-g mice com-pared with WT mice (Fig. 3A). Consistently, the number of Ki-67þ cells in H. felis–infected Hþ/Kþ-ATPase-IFN-g mice weremuch lower than those present in infected WT mice (Fig. 3B,top), andproliferation rateswere also lower in IL-1b;IFN-gmicethan in IL-1b mice without (Fig. 3A) and with H. felis (Fig. 3B)infection, showing that both H. felis- and IL-1b–induced cellproliferation could be inhibited by overexpression of IFN-g .
Double cortin and CaM kinase-like-1 (Dclk-1) has beenassociated with the proliferative zone of the stomach andhas been proposed as a putative gastrointestinal progenitorcell marker (24). In the absence of H. felis infection, Dclk-1þ
cells were not significantly different between WT and Hþ/Kþ-ATPase-IFN-g mice (Fig. 3A). Dclk-1þ cells were markedlyelevated in H. felis–infected WT mice (Fig. 3B), an effect thatwas abrogated by IFN-g overexpression. In addition, Dclk-1þ
cells were significantly increased in stomachs of uninfected IL-1b mice compared with WT mice (Fig. 3A), and even further
increased in H. felis–infected IL-1b mice compared with H.felis–infected WT mice (Fig. 3B). The increase in Dclk-1þ cellswas inhibited by overexpression of IFN-g (Fig. 3B). IFN-g alsoinhibited IL-1b–induced inflammation, cell proliferation, andDckl1þ cell expansion in very young IL-1b;IFN-g mice (<3months old) in the absence or presence of H. felis infection(Supplementary Table S1; Supplementary Fig. S2A and B).Overall, these results suggest that inflammatory signals suchas IL-1b and H. felis infection can stimulate cell proliferationand Dclk-1þ cell expansion and that IFN-g inhibits the expan-sion of these cells.
IFN-g overexpression accelerates apoptosis of gastric Tlymphocytes and inhibits the production ofproinflammatory Th1 and Th17 cytokines
IFN-g has been shown to induce CD4þ T-cell apoptosis(25), and CD4þ T cells are required for H. pylori–inducedatrophic gastritis (8). In uninfected WT and IFN-g mice,TUNEL staining showed low levels of gastric epithelial andinflammatory cell apoptosis (Fig. 4A; SupplementaryFig. S3A). However, in the setting of H. felis infection orIL-1b overexpression, the mean number of apoptotic epithe-lial cells per hpf and cleaved caspase-3–stained cells weremarkedly decreased by IFN-g overexpression (Fig. 4B; Sup-plementary Fig. S3B). The IFN-g-dependent decrease inepithelial apoptosis showed an inverse correlation withleukocyte apoptosis. In both WT and Hþ/Kþ-ATPase-IFN-g mice, H. felis infection resulted in an increase in leukocyteapoptosis, but there were significantly more apoptotic cellsin the H. felis–infected Hþ/Kþ-ATPase-IFN-g mice than in
ScaI
6,000 8 WT Line 32
Rel
ativ
e m
RN
Aex
pres
sion
(fo
ld)
Line 547
6
54
32
10
IP-10 Mig
IFN
-γ (
pg/m
g pr
otei
n)
5,000
4,000
3,000
2,000
1,000
D
A
B C
WTWT
200× 200×
IFN-γ (Line 54)IFN-γ (line 54)
0
WT Line 32Stomach
Line 54
XabI
mHK-ATP βpromoter
Mouse IFN-γcDNA
hGH polyAsequences
BamH I BamH I EcoR I ScaI
Figure 1. Hþ/Kþ-ATPase-IFN-gmice fail to develop spontaneousgastric metaplasia. A, the IFN-gconstruct contains the mouse Hþ/Kþ-ATPase b-subunit gene andmurine IFN-g cDNA. B, ELISAdetermined murine IFN-g instomachs of 4-month-old Hþ/Kþ-ATPase-IFN-g mice and controlmice. Data are mean � SD of 10mice. C, gene expression levels ofmouse IP-10 andMigmRNA in thestomach. D, representativemacroscopic and histologicappearance of IFN-g transgenicmouse stomach at 24 months.
Tu et al.
Cancer Res; 71(12) June 15, 2011 Cancer Research4250
Research. on May 6, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst April 21, 2011; DOI: 10.1158/0008-5472.CAN-10-4009

the H. felis–infected WT mice. Fluorescence-activated cell-sorting (FACS) analysis confirmed this finding and revealedthat apoptosis was primarily occurring in gastric CD4þ andCD8þ T cells. An increase in Annexin V staining was noted inboth CD4þ and CD8þ T cells in Hþ/Kþ-ATPase-IFN-g micecompared with WT mice, with the biggest increase observedin the setting of H. felis infection (Fig. 4C) and in IL-1b micecompared with double transgenic IL-1b;IFN-g mice (Supple-mentary Fig. S4A). T-cell apoptosis correlated with an abro-gated Th1 immune response, because the expression ofproinflammatory cytokines (IL-1b, TNF-a, and IL-6) wassignificantly decreased in H. felis–infected Hþ/Kþ-ATPase-IFNg compared with H. felis–infected WT mice (Fig. 4D).Although expression of Th2 cytokines (IL-4 and IL-10) wasnot significantly different between IFN-g and WT micewithout H. felis infection (Supplementary Fig. S4B), IL-10was significantly upregulated in IFN-g mice after H. felisinfection. Furthermore, Th17-related cytokines (IL-17A andIL-17F) were significantly upregulated in IL-1b mice and H.
felis–infected WT mice compared with uninfected WT micebut downregulated by IFN-g overexpression (Fig. 4E). IL-12and IL-23 stimulate either Th1 or Th17 immune responsesthrough T-cell activation, and we observed a downregulationof IL12, and even more so of IL-23, in association with IFN-goverexpression (Fig. 4E), indicating that IFNg overexpres-sion leads to a reduction of the Th17 response. Moreover,macrophages were diminished in IFN-g mice compared withWT mice, in H. felis–infected IFN-g mice compared with H.felis–infected WT mice, and in IL-1b/IFNg mice comparedwith IL-1bmice (Supplementary Fig. S4C), indicating that inour model IFN-g reduces not only the adaptive immuneresponse but possibly also the innate immune response.
FAS, TRAIL, and XAF1 can all mediate IFN-g–inducedapoptosis (26), in particular, FAS mediates IFN-g–dependentapoptosis of T cells (27). Indeed, in both H. felis–infected anduninfected Hþ/Kþ-ATPase-IFN-g mice, qRT-PCR showedupregulated FAS expression in the gastric mucosa (Supple-mentary Fig. S5C). The gene expression of IFN-g downstream
Figure 2. Overexpression of IFN-galleviates development of H. felis–induced gastric inflammation andneoplasia. Representativephotomicrographs of H. felis–infected Hþ/Kþ-ATPase-IFN-gmouse stomach at 12 months (A)and 24 months (B). C,representative macroscopicphotographs of H. felis–infectedHþ/Kþ-ATPase-IFN-g and WTmice at 12 months. D,histopathologic scores forstomachs of indicated mice; datarepresent the mean � SD of 20mice [*, P < 0.05, WT vs. IFN-gmice (both lines)]. E,overexpression of IFN-g does notaffect the colonization of H. felis inthe stomach. Stomach sectionsfrom Hþ/Kþ-ATPase-IFN-g andWT mice infected for 2 monthswere stained with H&E. Arrow andinsert indicate H. felis localizationin the lumen of gastric glands. F,quantification ofH. felisDNA in thestomach from indicated mice byPCR. Data represent the mean �SD of 5 mice.
A
B
D
EF
2.5 WT + H. felis
* * ** *
*
Sco
re o
f inf
lam
mat
ion
and
path
olog
y
Line 32 + H. felis Line 54 + H. felis
2
1.5
1
0.5
Acuteinflamation
Chronicinflamation
Hyperplasia Atrophy Metaplasia Dysplasia0
C
WT + H. felis 12m
WT + H. felis
WT + H. felis
WT + H. felis
200×
200×
600× 600×36
Rel
ativ
e D
NA
co
nten
t of H
. fel
is
3024181260
WT
WT +
H. fe
lis
Line
54
Line
54 +
H. fe
lis
200×
200×
IFN-γ + H. felis
IFN-γ + H. felis 12m
IFN-γ + H. felis
IFN-γ + H. felis
IFN-g Inhibits Gastric Inflammation and Carcinoma
www.aacrjournals.org Cancer Res; 71(12) June 15, 2011 4251
Research. on May 6, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst April 21, 2011; DOI: 10.1158/0008-5472.CAN-10-4009

target genes TRAIL and XAF1 was also slightly increased (1.5-to 3-fold) in Hþ/Kþ-ATPase-IFN-g compared with WT mice(Supplementary Fig. S5A and B).
IFN-g induces autophagy in gastric epithelial cellsthrough upregulation of Beclin-1
Current thinking suggests that in early stages of carcino-genesis, autophagy protects from carcinogenesis (28) and IFN-g can induce autophagy in epithelial cells and immune cells(29). We found that the number of cells expressing LC3-II, themammalian homologue of yeast Atg8, were significantlyincreased in the stomachs of Hþ/Kþ-ATPase-IFN-g micecompared with WT mice (Fig. 5A). With H. felis infection,
inducing similar tissue levels of IFN-g , the number of LC3-IIþ
cells was also increased in WT mice (Fig. 5A), confirming thatH. felis infection and/or the immune response to Helicobacterinfection may trigger autophagy in gastric epithelial cells (30,31). Western blot analysis showed that LC3-II levels wereincreased in Hþ/Kþ-ATPase-IFN-g mice compared with WTmice, with or without H. felis infection (Fig. 5B), and werehigher in IL-1b;IFN-g than in the IL-1b mice (Fig. 5C, Supple-mentary Fig. S6A). Importantly, to confirm that the accumula-tion of LC3-II was due to increased autophagosome formation,rather than decreased autophagosome degradation, we trea-ted Hþ/Kþ-ATPase-IFN-g mice with chloroquine for 10 daysto block lysosomal turnover (32), observing a further increase
40
A
B
C
20
DC
AM
KL
-1+
cells
(%
)D
CA
MK
L-1
+ ce
lls (
%)
15
10
5
0
*353025 *
*
* **
**
*
* **
2015
Ki-
67+
cells
(%
)
1050
30
25
20
15
10
5
0
8035
70605040
Ki-
67+
cells
(%
)
302010
0
WT
IFN-γ
IL-1
β;IFN-γ
IL-1
β
WT
IFN-γ
IL-1
β;IFN-γ
IL-1
βW
TIF
N-γ
IL-1
β;IFN-γ
IL-1β/IFN-γ 12mIL-1β 12m
IL-1
β
WT
IFN-γ
IL-1
β;IFN-γ
IL-1
β
Figure 3. Overexpression of IFN-ginhibits IL-1b–induced gastricinflammation, cell proliferation,and progenitor cell expansion.Twelve-month-old uninfectedmice (A) and H. felis–infected mice(B). Proliferation index wascalculated using the number ofpositive epithelial cells per totalepithelial cell number per gland in10 different hpfs (400�). Thepresented data represent themean � SD of 5 mice (*, P < 0.05).C, representative macroscopicphotographs of IL-1bmice and IL-1b/Hþ/Kþ-ATPase-IFN-gstomachs at 12 months.
Tu et al.
Cancer Res; 71(12) June 15, 2011 Cancer Research4252
Research. on May 6, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst April 21, 2011; DOI: 10.1158/0008-5472.CAN-10-4009

of the LC3 signal in Hþ/Kþ-ATPase-IFN-g mice (Fig. 5D) incontrast to IL-1b mice in which we did not detect autophagy.To confirm that IFN-g can directly induce autophagy in
gastric epithelial cells, we transfected gastric cancer cells
(MNK-28) with a GFP-LC3 construct. Transfected cells weretreated with IFN-g (20 ng/mL) for 24 or 48 hours, whichresulted in characteristic morphologic changes of autophagy.The percentage of GFP-LC3-II dots (Fig. 5E and G) and protein
2.59 28.23
A
C
D E
B
53.34
46.67 0.00 7.22
4.8187.97
9.03
1.2943.01
4.15
13.820.21
WT
WT
CD
4C
D8
25 14
1210
8
Cas
pas
e-3
and
in H
&E
6
42
0
Total apoptotic cells Mean apopotic epithelial cells
20
15
10
Ap
op
toti
c in
dex
(%
)
5
0IFN-γ
IFN-γγ
WT+H. felis
WT + H. felis
IFN-γ+ H. felis
IFN-γγ + H. felis
II-1β
IL-1ββ
IL-124,500 600
876543210
500
400
300
200
100
0
4,0003,5003,0002,5002,0001,5001,000
Cop
ies
/ 1,0
00 G
AP
DH
5000
IL-23 IL-17β IL17α
IL-6IFN-γγ TNF-αα
II-1β:IFN-γ WT+H. felis IFN-γ + H. felis
105 10
5 3.51 44.97
27.5122.01
2.84 19.34
17.4358.53
105
105
105
0.32 6.56
6.19
6.2
1.7
Annexin V
20 WTWT+H. felisLine 54Line 54+H. felis
10
15
5
Rel
ativ
e m
RN
A e
xpre
ssio
n(f
old
)
0
1.3
4.22.4
PI
4.8
27.5
17.4
105
104 10
4
104
104
104
104
103 10
3
103
103
103
103
102
102
101
105
104
103
102
101
0
105
104
103
0
105
104
103
0
105
104
103
00
105
104
103
0
0105
104
103
0105
104
103
0
10510
410
300
101
86.93
105
104
103
102
101
Com
p-P
E-T
R-A
::PI
Com
p-P
E-T
R-A
::PI
Com
p-P
E-T
R-A
::PI
Com
p-P
E-T
R-A
::PI
Com
p-P
E-T
R-A
::PI
Com
p-P
E-T
R-A
::PI
Com
p-P
E-T
R-A
::PI
Com
p-P
E-T
R-A
::PI
Comp-FITC-A: AnnexComp-FITC-A: AnnexComp-FITC-A: Annex
Comp-FITC-A: Annex Comp-FITC-A: Annex10
510
410
30
Comp-FITC-A: Annex Comp-FITC-A: Annex
Comp-FITC-A: Annex
94.83
0.08 2.50
1.7295.69
2.38
**
**
*
**
*
*
*
#
**
##
**
WT
WT H
. felis
IFN H
. felis
IL1-
β
IFN/IL
1-β
WT
WT H
. felis
IFN H
. felis
IL1-
β
IFN/IL
1-β
WT
WT H
. felis
IFN H
. felis
IL1-
β
IFN/IL
1-β
Figure 4. Overexpression of IFN-g induces apoptosis of T cells and inhibits the production of proinflammatory cytokines. A, TUNEL staining ofstomach sections from 12-month-old mice showing percentage of TUNELþ cells in different locations from each of the groups (n ¼ 5). All data represent themean � SD. *, P < 0.05. B, apoptotic epithelial cells were assessed in H&E-stained sections, and caspase-3–stained cells were counted in 10 hpfsand numbers are presented as mean � SD. *, P < 0.05. C, immune cells isolated from the indicated mice were stained with PerCP-CD4, APC-CD8,TITC-Annexin V, and PI, and analyzed by FACS (n¼ 5) to analyze apoptosis. D and E, mRNA expression levels of cytokines in 12-month-oldmice, as indicated.GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
IFN-g Inhibits Gastric Inflammation and Carcinoma
www.aacrjournals.org Cancer Res; 71(12) June 15, 2011 4253
Research. on May 6, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst April 21, 2011; DOI: 10.1158/0008-5472.CAN-10-4009

expression (Fig. 5F) was significantly increased in IFN-g–treated cells, compared with untreated cells, confirmingthat IFN-g can directly induce autophagy in gastric epithelialcells.
Beclin-1 is known to play an important role in the regula-tion of autophagy (33, 34). Overexpression of IFN-g upregu-lated Beclin-1 mRNA expression (Fig. 6B) and protein (Fig. 6A)levels in the stomachs of mice with or without H. felis infec-tion. Immunohistochemical staining localized Beclin-1 pro-tein to epithelial cells (Fig. 6A; Supplementary Fig. S6A), andoverexpression of IL-1b correlated with inhibited expressionof Beclin-1 in the stomach (Supplementary Fig. S6A and B).Beclin-1 upregulation correlated with the induction of autop-hagy in our mouse model. Indeed, knockdown of Beclin-1expression by siRNA in MKN-28 cells reduced IFN-g–inducedautophagy (Fig. 6C–E), confirming a direct mediation of
Beclin-1 in IFN-g–induced autophagy. Moreover, Beclin-1seemed to be a direct target of IFN-g because IFN-g activatedBeclin-1 promoter activity (Fig. 6F). By using the transcrip-tional regulatory element database (35), we located IFN-gactivation site (GAS) elements, which are short stretches ofDNA required for the rapid transcriptional induction of genesin response to IFN-g . Three GAS elements were located in thefirst 1,000 bp of the Beclin-1 promoter 50 of the ATG and 13GAS elements within the different introns of the Beclin-1 gene(data not shown). These results suggest that IFN-g inducesautophagy in part through upregulation of Beclin-1.
IFN-g infusion inhibits progenitor cell expansion andinduces autophagy
Although constitutive overexpression of IFN-g inhibits pro-liferation and carcinogenesis and induces autophagy, our
Figure 5. Overexpression of IFN-ginduces autophagy in gastricepithelial cells in vivo. A,immunohistochemical staining forLC3 in stomach sections from 12-month-old mice, as indicated.Representative results from 8different groups are shown(original magnification � 200). Band C, Western blots showingexpression of LC3 in the stomachtissue of indicated mice. D,accumulation of LC3-II due toincreased autophagosomeformation after chloroquinetreatment (30 mg/kg for 10 daysintraperitoneal). E, gastric cancerMKN-28 cell line was transfectedwith GFP-LC3 and then treatedwith 40 mg IFN-g for 24 and 48hours (original magnification �400). F, IFN-g increased LC3-IIexpression in MKN-28 cells aftertreatment with 40 mg IFN-g for 48hours. GFP-LC3 was detected byWestern blotting. G, GFP-LC3–positive punctate dots per cell.Data are from 100 to 200 cellsfrom 3 independent experiments.Data are mean � SEM, n ¼ 100 to200 cells from 3 independentexperiments; *, P < 0.01.
Tu et al.
Cancer Res; 71(12) June 15, 2011 Cancer Research4254
Research. on May 6, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst April 21, 2011; DOI: 10.1158/0008-5472.CAN-10-4009

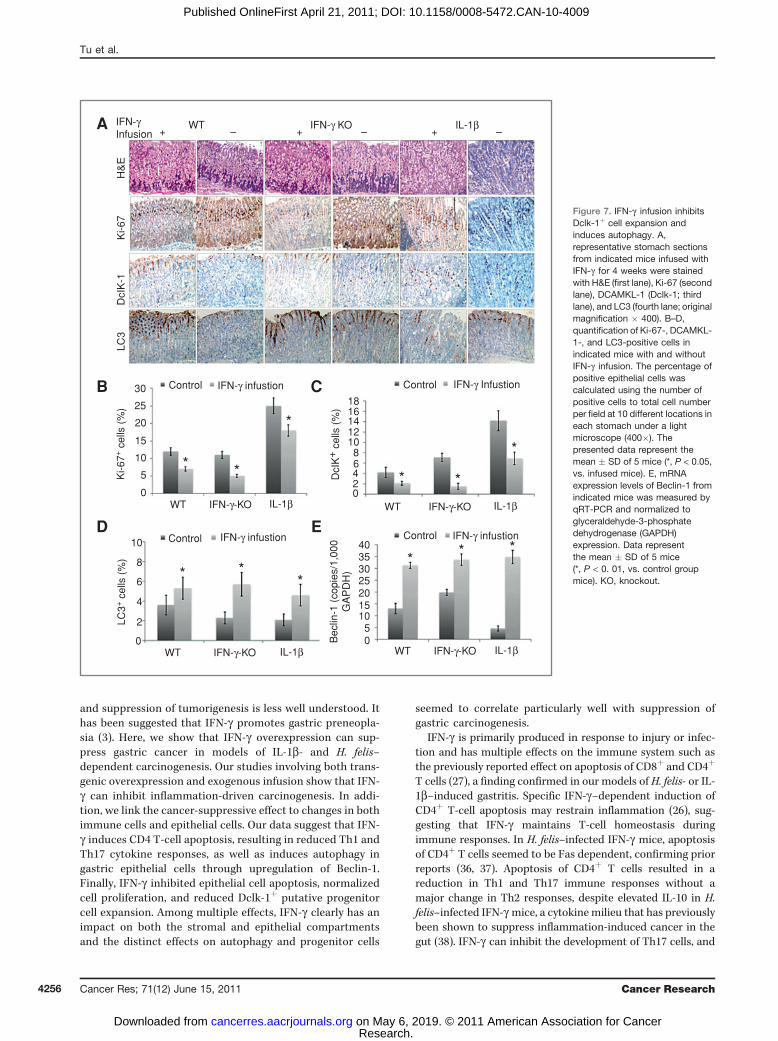
transgenic model does have the possible limitation of lifelongchronic expression that could potentially result in compen-satory responses. Thus, we infused IFN-g into 2-month-oldWT B6, IFN-g-knockout, and IL-1b mice for 4 weeks andconfirmed significantly increased levels of IFN-g in the sto-mach (Supplementary Fig. S7A) and serum (data not shown)in all infusedmice. Two weeks postcessation of IFN-g infusion,we found that the treatment induced only mild gastritis inboth the WT mice and IFN-g knockout mice (inflammationscore <0.5; data not shown). No mice developed gastricatrophy or metaplasia, a finding confirmed by Alcian bluestaining (Supplementary Fig. S7B). The short-term IFN-ginfusion did result in reduced cell proliferation (Ki-67 staining;Fig. 7A and C) in all infused mice, consistent with previousreports (8). Moreover, IFN-g infusion abrogated the develop-
ment of gastric metaplasia in the IL-1b transgenic mice(Supplementary Fig. S7B), which was confirmed by histo-pathologic analysis (data not shown), a result that was con-sistent with previous findings from the double IL-1b;IFN-gtransgenic mice. Interestingly, IFN-g infusion also reduced thenumber of Dclk-1þ cells (Fig. 7A and B), induced autophagy ingastric epithelial cells (Fig. 7A and D), and upregulated theexpression of Beclin-1 (Fig. 7E). Finally, IFN-g infusion inhib-ited the production of IL-1b, TNF-a, and IL-6 in all treatedmice (Supplementary Fig. S7C).
Discussion
Although IFN-g is a signature Th1 cytokine mediatingprotection from pathogens, its role in cancer surveillance
Figure 6. Overexpression of IFN-ginduces autophagy via Beclin-1 ingastric cancer cells in vitro. A,immunohistochemical staining forBeclin-1 in stomach sections from12-month-old mice.Representative photomicrographsare shown (original magnification� 200). B, mRNA expression ofBeclin-1 in the stomach fromindicated mice, normalized toglyceraldehyde-3-phosphatedehydrogenase (GAPDH)expression. Data representthe mean � SD of 5 mice(*, P < 0.001, vs. indicated group ofmice). C, MKN-28 cells weretransfected with control andBeclin-1 siRNA for 6 hours andcultured with or without 20 ng/mLIFN-g for another 48 hours.D, Western blot analysis showsIFN-dependent upregulation ofBeclin and LC3-II as well asBeclin-dependent expression ofLC3-II. E, downregulation ofBeclin-1 expression by siRNAdecreased IFN-g–inducedautophagy in MKN-28 cells.*, P < 0.05. F, IFN-g activated thepromoter activity of Beclin-1 inMKN-28 cotransfected with theindicated Beclin-1 promoterplasmids or empty pGL3 vectorand Renilla luciferase vector for6 hours before IFN-g treatment.Data are shown as means � SD of3 independent experiments.*, P < 0.05.
F8
Untreated
Rel
ativ
e B
eclin
-1pr
omot
er a
ctiv
ity (
fold
)
IRN-γ
pGL-3 vector pGL-3-Beclin-1
76543210
WTA
B C
D E
IFN-γ
IFN-γ
IFN-γ + H. felisWT + H. felis
400×
180Untreated
ControlsiRNA
Control siRNA
IFN-γUntreated IFN-γUntreated
Beclin-1siRNA
Beclin 1 siRNA
Bec
lin-1
Beclin-1
Cop
ies
/1,0
00 G
AP
DH
Beclin-15 Control siRNA
Beclin-1 siRNA
UntreatedEG
FP
-LC
3-II
punc
tate
dots
/cel
l
IFN-γ
432
10
160140120100
80604020
0
400× 400× 400×
WT IFN-γ WTH. felis
IFNH. felis
*
*
**
*
LC3-ILC3-II
β-Actin
IFN-g Inhibits Gastric Inflammation and Carcinoma
www.aacrjournals.org Cancer Res; 71(12) June 15, 2011 4255
Research. on May 6, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst April 21, 2011; DOI: 10.1158/0008-5472.CAN-10-4009
and suppression of tumorigenesis is less well understood. Ithas been suggested that IFN-g promotes gastric preneopla-sia (3). Here, we show that IFN-g overexpression can sup-press gastric cancer in models of IL-1b- and H. felis–dependent carcinogenesis. Our studies involving both trans-genic overexpression and exogenous infusion show that IFN-g can inhibit inflammation-driven carcinogenesis. In addi-tion, we link the cancer-suppressive effect to changes in bothimmune cells and epithelial cells. Our data suggest that IFN-g induces CD4 T-cell apoptosis, resulting in reduced Th1 andTh17 cytokine responses, as well as induces autophagy ingastric epithelial cells through upregulation of Beclin-1.Finally, IFN-g inhibited epithelial cell apoptosis, normalizedcell proliferation, and reduced Dclk-1þ putative progenitorcell expansion. Among multiple effects, IFN-g clearly has animpact on both the stromal and epithelial compartmentsand the distinct effects on autophagy and progenitor cells
seemed to correlate particularly well with suppression ofgastric carcinogenesis.
IFN-g is primarily produced in response to injury or infec-tion and has multiple effects on the immune system such asthe previously reported effect on apoptosis of CD8þ and CD4þ
T cells (27), a finding confirmed in our models of H. felis- or IL-1b–induced gastritis. Specific IFN-g–dependent induction ofCD4þ T-cell apoptosis may restrain inflammation (26), sug-gesting that IFN-g maintains T-cell homeostasis duringimmune responses. In H. felis–infected IFN-g mice, apoptosisof CD4þ T cells seemed to be Fas dependent, confirming priorreports (36, 37). Apoptosis of CD4þ T cells resulted in areduction in Th1 and Th17 immune responses without amajor change in Th2 responses, despite elevated IL-10 in H.felis–infected IFN-g mice, a cytokinemilieu that has previouslybeen shown to suppress inflammation-induced cancer in thegut (38). IFN-g can inhibit the development of Th17 cells, and
3018
4035302520151050
1614121086420
A
B C
D E
25
Control
WTH
&E
Ki-6
7D
cIK
-1LC
3+
IFN-γ KOIFN-γ Infusion
IL-1β– + – + –
Control
IFN-γ infustion Control IFN-γ Infustion
Control
Bec
lin-1
(co
pies
/1,0
00G
AP
DH
)
IFN-γ infustionIFN-γ infustion
20
15
Ki-6
7+ c
ells
(%
)
DcI
K+
cel
ls (
%)
LC3+
cel
ls (
%)
10
5
2
4
6
8
10
0
0
IL-1β
IL-1β
IFN-γ-KO
IFN-γ-KO
WT
IL-1βIFN-γ-KOWT
IL-1βIFN-γ-KOWT
WT
* * * *
*
****
**
*
Figure 7. IFN-g infusion inhibitsDclk-1þ cell expansion andinduces autophagy. A,representative stomach sectionsfrom indicated mice infused withIFN-g for 4 weeks were stainedwith H&E (first lane), Ki-67 (secondlane), DCAMKL-1 (Dclk-1; thirdlane), and LC3 (fourth lane; originalmagnification � 400). B–D,quantification of Ki-67-, DCAMKL-1-, and LC3-positive cells inindicated mice with and withoutIFN-g infusion. The percentage ofpositive epithelial cells wascalculated using the number ofpositive cells to total cell numberper field at 10 different locations ineach stomach under a lightmicroscope (400�). Thepresented data represent themean � SD of 5 mice (*, P < 0.05,vs. infused mice). E, mRNAexpression levels of Beclin-1 fromindicated mice was measured byqRT-PCR and normalized toglyceraldehyde-3-phosphatedehydrogenase (GAPDH)expression. Data representthe mean � SD of 5 mice(*, P < 0. 01, vs. control groupmice). KO, knockout.
Tu et al.
Cancer Res; 71(12) June 15, 2011 Cancer Research4256
Research. on May 6, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst April 21, 2011; DOI: 10.1158/0008-5472.CAN-10-4009

recent studies have pointed that Th17 cells play a key role notonly in the clearance of extracellular pathogens such as H. felis(13) but also in cancer. IL-17 is overexpressed in H. pylori–infected gastric mucosa in both mice and humans (12, 39),whereas inhibition of the Th17 response prevents gastricdysplasia (40). Interestingly, macrophages, representing theinnate immune response, also seemed to be reduced innumber, suggesting that IFN-g inhibits recruitment of macro-phages, a potential source of Th1- and Th17-inducing cyto-kines such as IL-12 and IL-23, which are downregulated inIFN-g transgenic mice. Because we have shown that IFN-g canaffect adaptive and innate immune responses in H. felis–induced and in IL-1b–induced gastric carcinogenesis, it istempting to speculate that the changes in the cytokine profileare due to either directly expression of IFN-g or to missingrecruitment of tumor-promoting immune cells, such as imma-ture myeloid cells, which contribute to the promotion ofcarcinogenesis in the IL-1b model of gastric cancer (8).Although IFN-g is clearly able to promote T-cell apoptosis
and potentially affect innate immunity in the stomach, it seemsto have a different effect on the epithelium, inhibiting gastricepithelial apoptosis while promoting autophagy during earlystages of inflammation-associated gastric carcinogenesis. Theinduction of autophagy seemshighly relevant to the role of IFN-g in protection from pathogens, because Helicobacter canoccasionally be found intracellularly (41), and publishedreports have indicated that autophagy represents an importantmechanism for elimination of intracellular pathogens (29)independent of apoptosis (42). This new mechanism shouldbe considered in Helicobacter infection, especially becauserecent studies have shown that IFN-g can enhance the degra-dation of Mycobacterium tuberculosis and Ricksettia conorii byinduction of autophagy in infected cells (29, 33). AlthoughNod1and Nod2 have been shown to comprise potential intracellularsignals for triggering autophagy and bacterial elimination (43),the induction of IFN-g may represent a second pathway.Autophagymayhave arisenduring evolutionas a response to
infectionorstarvationstress,butithasalsobeenassociatedwithprotection fromneoplasia at early stages of carcinogenesis (44).Thus, while overexpression of IL-1b correlated with reducedautophagy and increased apoptosis of epithelial cells, over-expression of IFN-g , in contrast, increased autophagy, reducedepithelial cell apoptosis, and inhibited progression to cancer.Interestingly,H. felis infection itself could inducesomedegreeofautophagy in the stomach as suggested by recent reports (30,31), as well as by elevated levels of IFN-g , thus explaining thesomewhat slower progression to cancer inH. felis–infectedWTmice compared with uninfected IL-1b mice. Moreover, theswitch to autophagy was associated with decreased epithelialcell apoptosis, a critical factor in gastric carcinogenesis (45).Our data suggest that IFN-g–induced autophagy involves
upregulation of Beclin-1, a haploinsufficient tumor suppressorand a key regulator of autophagosome formation (34, 46).Beclin-1 expression was upregulated by IFN-g overexpressionor infusion and was directly associated with the induction ofautophagy in gastric epithelial cells. Computer-based searchescould identify interferon-responsive GAS elements in theBeclin-1 promoter, suggesting bindingmotifs for direct regula-
tion through IFN-g–activated STAT1, as shown previously (47).Because downregulation of Beclin-1 expression reduced IFN-g–induced autophagy, consistent with previous reports on theeffect of IFN-g on phagosome–lysosome fusion in infectedmurine macrophages (48), our findings suggest that IFN-g–induced phagosome maturation may be responsible forthe autophagic response.
The expansion of cells expressing Dclk-1, a putative pro-genitor cell marker also known as DCAMKL-1, was stronglyassociated with inflammation-related carcinogenesis and pre-ceded the development of gastric cancer. Although formallineage tracing has not been reported, Dclk-1 is expressed inthe progenitor zone of the stomach and intestine and has beenshown to be upregulated in response to carcinogen treatmentin the colon. Notably, overexpression or infusion of IFN-greduces both cell proliferation and the number of Dclk-1þ cellsand therefore raises the possibility of a direct effect of IFN-g ongastric progenitor cells. These results are consistent withprevious report that IFN-g can inhibit hematopoietic stemor progenitor cell expansion (49).
In summary, overexpression of IFN-g inhibits H. felis- andIL-1b–dependent gastric inflammation and carcinoma.Although previous studies have shown that tissue-specificIFN-g transgenic mice developed severe chronic inflamma-tion, including chronic active hepatitis (50), no IFN-g trans-genic mouse model has yet shown development of carcinoma(50). Our data suggest that rather than contributing to therejection or destruction of incipient tumors, IFN-g mediates aswitch from epithelial apoptosis to Beclin-1–mediated epithe-lial cell autophagy early in the process of inflammation-induced carcinogenesis. Decreased epithelial cell apoptosisreduces the need for cell and tissue replacement, leads todecreased progenitor or stem cell proliferation, and reducesinflammation. In addition, IFN-g induces CD4þ T-cell apop-tosis, resulting in decreased Th1 and Th17 immune responsesand less epithelial stress that might decrease genetic altera-tions. The overall tumor suppressor function of the immunesystem seems indeed critically dependent on the actions ofIFN-g , and greater attention should be given in the future tothe potential role of IFN-g in cancer prevention.
Disclosure of Potential Conflicts of Interests
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Shengwen Wang, Bethany Diprete, Ashley Whelan, and JustinDeGrazia for technical assistance and genotyping mice.
Grant Support
This project was supported by NIH grants 1U54CA126513, RO1CA093405, andR01CA120979 (T.C. Wang), NIH grant R21CA149865 (S-P. Tu). M. Quante wassupported by a grant from the Mildred-Scheel-Stiftung, Deutsche Krebshilfe.
The costs of publication of this article were defrayed in part by the paymentof page charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received November 4, 2010; revised April 14, 2011; accepted April 18, 2011;published OnlineFirst April 21, 2011.
IFN-g Inhibits Gastric Inflammation and Carcinoma
www.aacrjournals.org Cancer Res; 71(12) June 15, 2011 4257
Research. on May 6, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst April 21, 2011; DOI: 10.1158/0008-5472.CAN-10-4009

References1. Billiau A, Heremans H, Vermeire K, Matthys P. Immunomodulatory
properties of interferon-gamma. An update. Ann N Y Acad Sci1998;856:22–32.
2. Fan XJ, Chua A, O’Connell MA, Kelleher D, Keeling PW. Interferon-gamma and tumour necrosis factor production in patients with Heli-cobacter pylori infection. Ir J Med Sci 1993;162:408–11.
3. Sawai N, KitaM, Kodama T, Tanahashi T, Yamaoka Y, TagawaY, et al.Role of gamma interferon in Helicobacter pylori-induced gastricinflammatory responses in a mouse model. Infect Immun 1999;67:279–85.
4. Necchi V, Candusso ME, Tava F, Luinetti O, Ventura U, Fiocca R, et al.Intracellular, intercellular, and stromal invasion of gastric mucosa,preneoplastic lesions, and cancer by Helicobacter pylori. Gastroen-terology 2007;132:1009–23.
5. Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J ClinInvest 2007;117:60–9.
6. Figueiredo C, Machado JC, Pharoah P, Seruca R, Sousa S, CarvalhoR, et al. Helicobacter pylori and interleukin 1 genotyping: an oppor-tunity to identify high-risk individuals for gastric carcinoma. J NatlCancer Inst 2002;94:1680–7.
7. Mohammadi M, Nedrud J, Redline R, Lycke N, Czinn SJ. Murine CD4T-cell response to Helicobacter infection: TH1 cells enhance gastritisand TH2 cells reduce bacterial load. Gastroenterology 1997;113:1848–57.
8. Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, et al.Overexpression of interleukin-1beta induces gastric inflammation andcancer and mobilizes myeloid-derived suppressor cells in mice.Cancer Cell 2008;14:408–19.
9. Eaton KA, Benson LH, Haeger J, Gray BM. Role of transcription factorT-bet expression by CD4þ cells in gastritis due to Helicobacter pyloriin mice. Infect Immun 2006;74:4673–84.
10. Sutton P, Kolesnikow T, Danon S, Wilson J, Lee A. Dominant non-responsiveness to Helicobacter pylori infection is associated withproduction of interleukin 10 but not gamma interferon. Infect Immun2000;68:4802–4.
11. Mohammadi M, Czinn S, Redline R, Nedrud J. Helicobacter-specificcell-mediated immune responses display a predominant Th1 pheno-type and promote a delayed-type hypersensitivity response in thestomachs of mice. J Immunol 1996;156:4729–38.
12. Caruso R, Pallone F, Monteleone G. Emerging role of IL-23/IL-17 axisin H. pylori-associated pathology.World J Gastroenterol 2007;13:5547–51.
13. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al.Reciprocal developmental pathways for the generation of pathogeniceffector TH17 and regulatory T cells. Nature 2006;441:235–8.
14. Taylor GA, Feng CG, Sher A. Control of IFN-gamma-mediated hostresistance to intracellular pathogens by immunity-related GTPases(p47 GTPases). Microbes Infect 2007;9:1644–51.
15. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fightsdisease through cellular self-digestion. Nature 2008;451:1069–75.
16. White E, DiPaola RS. The double-edged sword of autophagy mod-ulation in cancer. Clin Cancer Res 2009;15:5308–16.
17. Willimsky G, Cz�eh M, Loddenkemper C, Gellermann J, Schmidt K,Wust P, et al. Immunogenicity of premalignant lesions is the primarycause of general cytotoxic T lymphocyte unresponsiveness. J ExpMed 2008;205:1687–700.
18. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ,et al. IFNgamma and lymphocytes prevent primary tumour devel-opment and shape tumour immunogenicity. Nature 2001;410:1107–11.
19. Qin Z, Blankenstein T. A cancer immunosurveillance controversy. NatImmunol 2004;5:3–4; author reply 5.
20. Erdman SE, Poutahidis T, Tomczak M, Rogers AB, Cormier K, PlankB, et al. CD4þ CD25þ regulatory T lymphocytes inhibit microbiallyinduced colon cancer in Rag2-deficient mice. Am J Pathol 2003;162:691–702.
21. Enzler T, Gillessen S, Manis JP, Ferguson D, Fleming J, Alt FW, et al.Deficiencies of GM-CSF and interferon gamma link inflammation andcancer. J Exp Med 2003;197:1213–9.
22. Kang W, Rathinavelu S, Samuelson LC, Merchant JL. Interferongamma induction of gastric mucous neck cell hypertrophy. Lab Invest2005;85:702–15.
23. Tu SP, Chi AL, Ai W, Takaishi S, Dubeykovskaya Z, Quante M, et al.p53 inhibition of AP1-dependent TFF2 expression induces apoptosisand inhibits cell migration in gastric cancer cells. Am J PhysiolGastrointest Liver Physiol 2009;297:G385–96.
24. May R, Riehl TE, Hunt C, Sureban SM, Anant S, Houchen CW.Identification of a novel putative gastrointestinal stem cell and ade-noma stem cell marker, doublecortin and CaM kinase-like-1, followingradiation injury and in adenomatous polyposis coli/multiple intestinalneoplasia mice.Stem Cells 2008;26:630–7.
25. Muhl H, Pfeilschifter J. Anti-inflammatory properties of pro-inflamma-tory interferon-gamma. Int Immunopharmacol 2003;3:1247–55.
26. Chawla-Sarkar M, Lindner DJ, Liu YF,WilliamsBR, SenGC, SilvermanRH, et al. Apoptosis and interferons: role of interferon-stimulatedgenes as mediators of apoptosis. Apoptosis 2003;8:237–49.
27. Liesenfeld O, Kosek JC, Suzuki Y. Gamma interferon induces Fas-dependent apoptosis of Peyer's patch T cells in mice followingperoral infection with Toxoplasma gondii. Infect Immun 1997;65:4682–9.
28. Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G,et al. Autophagy promotes tumor cell survival and restricts necrosis,inflammation, and tumorigenesis. Cancer Cell 2006;10:51–64.
29. Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM inducesautophagy to eliminate intracellular mycobacteria. Science2006;313:1438–41.
30. Wang YH, Wu JJ, Lei HY. When Helicobacter pylori invades andreplicates in the cells. Autophagy 2009;5:540–2.
31. Terebiznik MR, Raju D, V�azquez CL, Torbricki K, Kulkarni R, BlankeSR, et al. Effect of Helicobacter pylori's vacuolating cytotoxin on theautophagy pathway in gastric epithelial cells. AutophagyR 20095:370–9.
32. Mizushima N, Yoshimori T, Levine B. Methods in mammalian autop-hagy research. Cell 140:313–26.
33. Feng CG, Zheng L, Lenardo MJ, Sher A. Interferon-inducible immu-nity-related GTPase Irgm1 regulates IFN gamma-dependent hostdefense, lymphocyte survival and autophagy. Autophagy 2009;5:232–4.
34. Liang XH, Jackson S, SeamanM, Brown K, Kempkes B, Hibshoosh H,et al. Induction of autophagy and inhibition of tumorigenesis by beclin1. Nature 1999;402:672–6.
35. Michael Zhang Lab, Cold Spring Harbor Laboratory. TranscriptionalRegulatory Element Database [cited 2010 Apr]. Available from: http://rulai.cshl.edu/cgi-bin/TRED/tred.cgi?process¼home.
36. Okamoto T, Yamakawa T, Yamamura K, Hino O. Induction of Fasligand and Fas antigen mRNA expressions in interferon-gammatransgenic mouse liver. Jpn J Pharmacol 1998;78:233–5.
37. Ishihara S, Fukuda R, Kawashima K, Moriyama N, Suetsugu H,Ishimura N, et al. T cell-mediated cytotoxicity via Fas/Fas ligandsignaling in Helicobacter pylori-infected gastric corpus.Helicobacter2001;6:283–93.
38. Erdman SE, Rao VP, Poutahidis T, Ihrig MM, Ge Z, Feng Y, et al. CD4(þ)CD25(þ) regulatory lymphocytes require interleukin 10 to interruptcolon carcinogenesis in mice. Cancer Res 2003;63:6042–50.
39. Mizuno T, Ando T, Nobata K, Tsuzuki T, Maeda O, Watanabe O, et al.Interleukin-17 levels in Helicobacter pylori-infected gastric mucosaand pathologic sequelae of colonization. World J Gastroenterol2005;11:6305–11.
40. Wang SW, Asfaha S, Okumura T, et al. Bone marrow derived Lin-Cd44hisca1-CkitþCD34- can give rise tomesenchymal stem cells anddelay progression to dysplasia in a murine Helicobacter model ofgastric cancer. Gastroenterology 2009;136Suppl 1:58.
41. Dubois A, Boren T. Helicobacter pylori is invasive and it may be afacultative intracellular organism. Cell Microbiol 2007;9:1108–16.
42. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, DereticV. Autophagy is a defense mechanism inhibiting BCG and Mycobac-terium tuberculosis survival in infected macrophages. Cell 2004;119:753–66.
Tu et al.
Cancer Res; 71(12) June 15, 2011 Cancer Research4258
Research. on May 6, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst April 21, 2011; DOI: 10.1158/0008-5472.CAN-10-4009

43. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG,Magalh~aes JG, et al. Nod1 and Nod2 direct autophagy by recruitingATG16L1 to the plasma membrane at the site of bacterial entry. NatImmunol 2009;11:55–62.
44. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al.Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to theplasma membrane at the site of bacterial entry. Nat Immunol2010;11:55–62.
45. Correa P, Miller MJ. Carcinogenesis, apoptosis and cell proliferation.Br Med Bull 1998;54:151–62.
46. Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y,et al. Bif-1 interacts with Beclin 1 through UVRAG and regulatesautophagy and tumorigenesis. Nat Cell Biol 2007;9:1142–51.
47. Contursi C, Wang IM, Gabriele L, Gadina M, O'Shea J, Morse HC,et al. IFN consensus sequence binding protein potentiates STAT1-dependent activation of IFNgamma-responsive promoters in macro-phages. Proc Natl Acad Sci U S A 2000;97:91–6.
48. Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, ,et al. T helper 2 cytokines inhibit autophagic control of intracellularMycobacterium tuberculosis. Immunity 2007;27:505–17.
49. Yang L, Dybedal I, Bryder D, Nilsson L, Sitnicka E, Sasaki Y, et al. IFN-gamma negatively modulates self-renewal of repopulating humanhemopoietic stem cells. J Immunol 2005;174:752–7.
50. Toyonaga T, Hino O, Sugai S, Wakasugi S, Abe K, Shichiri M, et al.Chronic active hepatitis in transgenic mice expressing interferon-gamma in the liver. Proc Natl Acad Sci U S A 1994;91:614–8.
IFN-g Inhibits Gastric Inflammation and Carcinoma
www.aacrjournals.org Cancer Res; 71(12) June 15, 2011 4259
Research. on May 6, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst April 21, 2011; DOI: 10.1158/0008-5472.CAN-10-4009

2011;71:4247-4259. Published OnlineFirst April 21, 2011.Cancer Res Shui Ping Tu, Michael Quante, Govind Bhagat, et al. Cell Autophagy and T-Cell Apoptosis
Inhibits Gastric Carcinogenesis by Inducing EpithelialγIFN-
Updated version
10.1158/0008-5472.CAN-10-4009doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2011/04/21/0008-5472.CAN-10-4009.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/71/12/4247.full#ref-list-1
This article cites 48 articles, 13 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/71/12/4247.full#related-urls
This article has been cited by 11 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/71/12/4247To request permission to re-use all or part of this article, use this link
Research. on May 6, 2019. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst April 21, 2011; DOI: 10.1158/0008-5472.CAN-10-4009