Embed Size (px)
Citation preview

Epilepsiu, 41(10):1269-1275, 2000 Lippincott Williams & Wilkins, Inc., Baltimore 0 International League Against Epilepsy
Clinical Research
How Well A
Can Epilepsy Reassessment
Syndromes Be Identified at Diagnosis? 2 Years After Initial Diagnosis
*Anne T. Berg, TShlomo Shinnar, $§Susan R. Levy, $§Francine M. Testa, $Susan Smith-Rapaport, and $Barbara Beckerman
*Depai%nent of Biological Sciences, Northern Illinois University, DeKalb, Illinois, U.S.A.; fDepaments of Neurology and Pediatrics and the Comprehensive Epilepsy Management Center, Montefore Medical Center, Albert Einstein College of
Medicine, Bronx, New York, U.S.A.; and Deparhents of $Pediatrics and §Neurology, Yale University, New Haven, Connecticut, U.S.A.
~
Summary: Purpose: Epilepsy syndromes can be identified very early in the course of a seizure disorder. It is unclear how accurate and resilient such early classifications are. We compared the classification of epilepsy syndromes made pre- viously on the basis of information available at diagnosis with those made 2 years later in a cohort of children with newly diagnosed epilepsy.
Methods: Children (n = 613) were prospectively identified at the t ime of initial diagnosis by participating physicians in Connecticut between 1993 and 1997. Classification of epilepsy syndrome according to International League Against Epilepsy guidelines was made previously based on all relevant informa- tion available at diagnosis. All cases were reclassified again after 2 years of additional evidence had accumulated. The dis- tributions of syndromes at diagnosis and at 2 years are com- pared and reasons for changes examined.
Results: After 2 years, syndromes remained the same in 86.3% of the cohort and changes occurred in 13.7% (n = 84). Evolution of the syndrome occurred in 24 children (3.9%), and rectification to the initial diagnosis occurred in 60 children
(9.8%). The most common scenario for evolution of a syn- drome was from West syndrome (n = 5), undetermined (n = 4), or symptomatic localization-related epilepsy (n = 3) to the Lennox-Gastaut syndrome. The most common rectification of initial classifications involved incompletely classified syn- dromes (cryptogenic localization-related and undetermined syndromes; n = 36). In a few instances, a fully specified syn- drome was reclassified to another apparently unrelated syn- drome. In these cases, initial information at diagnosis had been difficult to interpret.
Conclusions: Epilepsy syndromes can, for the most part, be identified at the time of initial diagnosis. Two years la- ter, rectifications were made in only 9.8% of cases, and most of these involved syndromes that represented incomplete classifications in the f i s t place. Significant changes were rare. The International League Against Epilepsy classifi- cation of the epilepsies can be meaningfully applied in epide- miological studies of newly diagnosed pediatric epilepsy. Key Words: Childhood-onset epilepsy-Epilepsy syndromes- Classification-Epidemiology
The syndromiC approach to classlfymg epilepsy rec- ognizes epilepsy as a set of diverse disorders (1). Ideally, knowledge of an individual’s syndrome should provide information about the underlying etiology and the prog- nosis with respect to seizures and epilepsy as well as help determine initial treatment and management strategies. To some extent, accurate classification may require ad- ditional evidence (including about the outcomes) accu- mulated over time to determine the syndrome. Three other studies, one from the Netherlands of children with
newly diagnosed epilepsy (2), one from France of chil- dren and adults with a single seizure or newly diagnosed epilepsy (3), and one from the United States of children identified at the time of a first unprovoked seizure (4), have examined this issue and found that, 2 to 5 years after diagnosis, approximately 10 to 12% of patients are reassigned to a different syndrome than was originally assigned at diagnosis. In this follow-up to our previous report (3, we examined the classification of pediatric epilepsy syndromes 2 years after initial diagnosis and the reasons for changes since diagnosis.
Acceoted June 12.2000. METHODS A prospective cohort of 613 children was recruited at
the time of the initial diagnosis with epilepsy (two or
Addrkss correspondence and reprint requests to Dr. Anne T. Berg at Department of Biological Sciences, Northern Illinois University, DeKalb, IL 601 15, U.S.A. E-mail: [email protected]
1269

1270 A. T. BERG ETAL.
more unprovoked seizures on separate days). Full details of the study methods have been provided previously (5). The following provides a synopsis. Children were re- mited in Connecticut from 1993 through 1997. Initial diagnostic assessments were those done by the treating physician, usually a child neurologist. AU but five chil- dren had electroencephalograms (EEGs), and 79.6% of the cohort had some form of neuroimaging, including 63% who underwent magnetic resonance imaging (MIU). Syndromes, seizure types, and causes .were clas- sified according to International League Against Epi- lepsy (ILAE) published criteria (C8). For more detailed criteria for individual syndromes, we relied on Roger et al. (9). Etiology was separately categorized as idiopathic, cryptogenic, and remote symptomatic. Specific disorders were recorded for remote symptomatic causes. Previ- ously, syndromes and seizures had been independently classified by each of three child neurologists (S.S., S.R.L., F.M.T.) on the basis of all relevant clinical data obtained at the time of initial diagnosis ("initial classifi- cation"), and discrepancies and concerns were resolved in conference (5,lO). All pertinent information from the medical record was used.
Study personnel conducted follow-up calls with par- ents every 3 months, and medical records were reexam- ined at 6-month intervals for subsequently obtained clinical information and test results. Discrepancies be- tween medical records and the parents' reports were re- solved through discussion with the parents and sometime the neurologist.
After 2 years of follow-up, each child's etiology and syndrome were reassessed based on baseline information as well as all subsequent information accumulated during the 2 years of follow-up. Etiology could be reclassified because of a change in the syndrome to or from an id- iopathic syndrome or because of new evidence regarding an underlying etiology. Developmental regression or failure to develop after initial diagnosis in a child con- sidered normal at the onset of epilepsy was not grounds for reclassifying the initial etiology.
For syndrome, two types of potential changes were considered: First, the initial classification of the syn- drome was reassessed based on evidence accumulated during 2 years from additional imaging studies, EEGs, and the seizure history. For example, if a child with cryptogenic etiology was initially classified with a form of epilepsy that was undetermined to be either focal or generalized (essentially unclassifable) and a subsequent EEG revealed a clear seizure focus not related to an idiopathlc partial epilepsy, the syndrome would be re- classified as symptomatic localization-related epilepsy by virtue of localization. In such a case, there is no change in the underlying epilepsy, and this represents essentially a rectification of the initial classification. In addition, there were instances in which the initial clas-
sification was not rectified but during the course of 2 years the epilepsy evolved to a different syndrome. A well-known example of this is West syndrome, which often evolves into the Lennox-Gastaut syndrome (9). Reasons for changes in the syndrome were recorded. When changes were made, we determined as accurately as possible when the changes first became apparent.
After 2 years, any information that came to light that might alter the classification of the underlying syndrome or etiology was reviewed independently by each of the three study neurologists. Any discrepancies in their clas- sifications were discussed in conference, and a consensus classification was reached. When necessary and possible, original EEG and imaging studies were obtained for clarification of findings described in the reports.
For children who were lost to follow-up before 2 years, the syndromes were reassessed based on what was available. If critical information that resulted in a change in their syndromes came to light, their syndromes were reclassified. Otherwise, they were left unchanged but flagged as having less than 2 years of follow-up.
RESULTS
The median age at onset was 5.3 years, and the ethnic distribution was comparable to that in the state. These and other characteristics of the cohort have been de- scribed previously (5). Of the initial 613 children re- cruited into the study, 20 (3.3%) were not followed for a full 2 years (2 deaths, 3 refusals, and 15 currently lost to follow-up). A re-review of all information after 2 years was necessary in 219 children (35.7%).
Etiology There were 28 changes made to etiology (Table 1).
Fourteen were attributable simply to a change in the classification of the syndrome from cryptogenic to idio-
TABLE 1 . Etiology at initial diagnosis and as reassessed afier 2 years of follow-up
Etiology as assessed after 2 years
Remote At initial diagnosis Total Idiopathic Cryptogenic symptomatic
Idiopathic 185 183 1 1" Cryptogenic 317 14 292 1 1" Remote symptomatic 11 1 1" 0 110" Total 613 198 293 122
"One child initially classified as having idiopathic etiology was recognized in retrospect to have autism predating the onset of his epilepsy. Another child initially classified as having remote symptom- atic etiology secondary to a brain cyst was reclassified as having benign rolandic epilepsy with an incidental choroid fissure cyst. Eleven chil- dren initially classified with cryptogenic etiology had their etiology reclassified as intrauterine insult (n = 3). brain malformation (n = 2). tumor (n = 2), and neurodegenerative disorder (n = 4). Five children initially assessed with remote symptomatic etiology had their etiology reclassified from atypical trisomy 15 to Angelman's syndrome (n = l), from intrauterine insult, perinatal hypoxia, or trauma to neurodegen- erative disease (n = 3), and from vascular malformation to tumor (n = 1).
Epilepsia, Vol. 41, No. 10, 2000
HOW WELL CAN EPILEPSY SYNDROMES BE IDENTIFIED AT DIAGNOSIS? 1271
pathic, and one from idiopathic to cryptogenic. One child was reclassified from remote symptomatic based on the initial interpretation of an MRI scan as showing a cyst in the temporal lobe to idiopathic when, later, after initial classifications were complete, the MRI scan became available for re-review. Ln 12 children, evidence of a remote symptomatic etiology became apparent after the initial diagnosis. In another five children initially con- sidered to have a remote symptomatic etiology, a differ- ent etiology became evident. Of note, by 2 years, seven children were reassessed as having neurodegen- erative conditions.
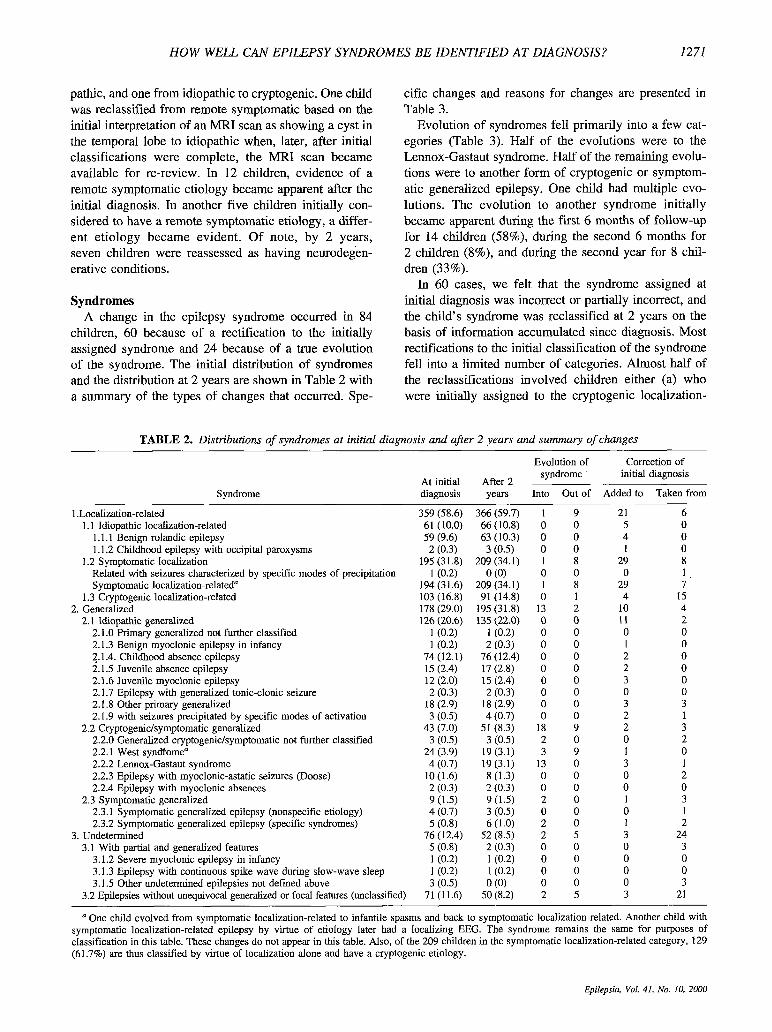
Syndromes A change in the epilepsy syndrome occurred in 84
children, 60 because of a rectification to the initially assigned syndrome and 24 because of a true evolution of the syndrome. The initial distribution of syndromes and the distribution at 2 years are shown in Table 2 with a summary of the types of changes that occurred. Spe-
cific changes and reasons for changes are presented in Table 3.
Evolution of syndromes fell primarily into a few cat- egories (Table 3) . Half of the evolutions were to the Lennox-Gastaut syndrome. Half of the remaining evolu- tions were to another form of cryptogenic or symptom- atic generalized epilepsy. One child had multiple evo- lutions. The evolution to another syndrome initially became apparent during the f i s t 6 months of follow-up for 14 children (58%), during the second 6 months for 2 children (8%), and during the second year for 8 chil- dren (33%).
In 60 cases, we felt that the syndrome assigned at initial diagnosis was incorrect or partially incorrect, and the child's syndrome was reclassified at 2 years on the basis of information accumulated since diagnosis. Most rectifications to the initial classification of the syndrome fell into a limited number of categories. Almost half of the reclassifications involved children either (a) who were initially assigned to the cryptogenic localization-
TABLE 2. Distributions of syndromes at initial diagnosis and after 2 years and summary of changes
Syndrome ~
1 .Localization-related 1.1 Idiopathic localization-related
I. 1.1 Benign rolandic epilepsy 1.1.2 Childhood epilepsy with occipital paroxysms
Related with seizures characterized by specific modes of precipitation Symptomatic localization-related"
1.3 Cryptogenic localization-related
2.1 Idiopathic generalized
1.2 Symptomatic localization
2. Generalized
2.1 .O Primary generalized not further classified 2.1.3 Benign myoclonic epilepsy in infancy 2.1.4. Childhood absence epilepsy 2.1.5 Juvenile absence epilepsy 2.1.6 Juvenile myoclonic epilepsy 2.1.7 Epilepsy with generalized tonic-clonic seizure 2.1.8 Other primary generalized 2.1.9 with seizures precipitated by specific modes of activation
2.2.0 Generalized cryptogenic/symptomatic not further classified 2.2.1 West syndfome" 2.2.2 LeMOx-GaStaUt syndrome 2.2.3 Epilepsy with myoclonic-astatic seizures (Doose) 2.2.4 Epilepsy with myoclonic absences
2.3.1 Symptomatic generalized epilepsy (nonspecific etiology) 2.3.2 Symptomatic generalized epilepsy (specific syndromes)
2.2 Cryptogenic/symptomatic generalized
2.3 Symptomatic generalized
3. Undetermined 3.1 With partial and generalized features
3.1.2 Severe myoclonic epilepsy in infancy 3.1.3 Epilepsy with continuous spike wave during slow-wave sleep 3.1.5 Other undetermined epilepsies not def ied above
3.2 Epilepsies without unequivocal generalized or focal features (unclassified)
At initial diagnosis
After 2 years
Evolution of syndrome
Into Out of
Correction of initial diagnosis
~ ~
Added to Taken from
359 (58.6) 61 (10.0) 59 (9.6) 2 (0.3)
195 (3 1.8) 1 (0.2)
194 (31.6) 103 (16.8) 178 (29.0) 126 (20.6)
l(0.2) 1 (0.2)
74 (12.1) 15 (2.4) 12 (2.0) 2 (0.3)
18 (2.9) 3 (0.5)
43 (7.0) 3 (0.5)
24 (3.9) 4 (0.7)
10 (1.6) 2 (0.3) 9 (1.5) 4 (0.7) 5 (0.8)
76 ( I 2.4) 5 (0.8) 1 (0.2) 1 (0.2) 3 (0.5)
71 (11.6)
366 (59.7) 66 (10.8) 63 (10.3) 3 (0.5)
209 (34.1) 0 (0)
209 (34.1) 91 (14.8)
195 (31.8) 135 (22.0)
1 (0.2) 2 (0.3)
76 (12.4) 17 (2.8) 15 (2.4) 2 (0.3)
18 (2.9) 4 (0.7)
51 (8.3) 3 (0.5)
19 (3.1) 19 (3.1) 8 (1.3) 2 (0.3) 9(1.5) 3 (0.5) 6 (1.0)
52 (8.5) 2 (0.3) 1 (0.2) l(0.2) 0 (0)
50 (8.2)
1 9 0 0 0 0 0 0 1 8 0 0 1 8 0 1
13 2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
18 9 2 0 3 9
13 0 0 0 0 0 2 0 0 0 2 0 2 5 0 0 0 0 0 0 0 0 2 5
21 6 5 0 4 0 1 0
29 8 0 1
29 7 4 15
10 4 11 2 0 0 1 0 2 0 2 0 3 0 0 0 3 3 2 1 2 3 0 2 1 0 3 1 0 2 0 0 1 3 0 1 1 2 3 24 0 3 0 0 0 0 0 3 3 21
a One child evolved from symptomatic localization-related to infantile spasms and back to symptomatic localization-related. Another child with symptomatic localization-related epilepsy by virtue of etiology later had a localizing EEG. The syndrome remains the same for purposes of classification in this table. These changes do not appear in this table. Also, of the 209 children in the symptomatic localization-related category, 129 (61.7%) are thus classified by virtue of localization alone and have a cryptogenic etiology.
Epilepsia, Vul. 41, Nu. 10, 2000

1272 A. T. BERG ET AL.
TABLE 3. Summary of reasons for changes to syndromes after 2 years
Reason for and type of rectification or change to initial classification
Evolution of syndrome SLRE to West syndrome (1.2.3 to 2.2.1) SLRE to Lennox-Gastaut syndrome (1.2.3 to 2.2.2) SLRE to West syndrome to SLRE (1.2.3 to 2.2.1 to 1.2.3) SLRE to SGE (1.2.3 to 2.3.2) SLRE to S/CGE not further specified (1.2.3 to 2.2.0) CLRE to Lennox-Gastaut syndrome (1.3 to 2.2.2) West syndrome to S/CGE not further specified (2.2.1 to 2.2.0) West syndrome to Lennox-Gastaut syndrome (2.2.1 to 2.2.2) West syndrome to SGE (2.2.1 to 2.3.2) West syndrome to UE2 (2.2.1 to 3.2) UE2 to SLRE (3.2 to 1.2.3) UE2 to Lennox-Gastaut syndrome (3.2 to 2.2.2)
More definitive EEG information Total rectifications
SLRE with specific mode of precipitation to IGE with specific
SLRE to benign rolandic epilepsy (1.2.3 to 1.1.1) SLRE by virtue of etiology to SLRE by virtue of localization and
SLRE to childhood absence epilepsy (1.2.3 to 2.1.4) SLRE to ICE with specific mode of precipitation (1.2.3 to 2.1.9) CLRE to childhood epilepsy with occipital paroxysms (1.3 to 1.1.2) CLRE to SLRE (1.3 to 1.2.3) Other ICE to CLRE (2.1.8 to 1.3) Epilepsy with myoclonic-astatic seizures to other ICE (2.2.3 to 2.1.8) UE2 to benign rolandic epilepsy (3.2 to 1.1.1) uE2 to SLRE (3.2 to 1.2.3) UE2 to other ICE (3.2 to 2.1.8)
subsequent seizures SLRE to juvenile myoclonic epilepsy (1.2.3 to 2.1.6) SLRE to LeMox-Gastaut syndrome (1.2.3 to 2.2.2) Other ICE to juvenile absence epilepsy (2.1.8 to 2.1.5) Other ICE to juvenile myoclonic epilepsy (2.1.8 to 2.1.6) WCGE not further classified to Lennox-Gastaut syndrome (2.2.0 to 2.2.2) Epilepsy with myoclonic-astatic seizures to Lennox-Gastaut
SCE to childhood absence epilepsy (2.3.2 to 2.1.4) UE2 to juvenile absence epilepsy (3.2 to 2.1.5) UE2 to juvenile myoclonic epilepsy (3.2 to 2.1.6)
uE2 to SLRE 3.2 to 1.2.3) UE2 to CLRE (3.2 to 1.3)
and additional results S L F to UE2 (1.2.3 to 3.2) ICE with specific mode of precipitation to UE2 (2.1.9 to 3.2) SKGE not further classified to UE2 (2.2.0 to 3.2) Lennox-Gastaut syndrome to SGE (2.2.2 to 2.3.2) SGE to West syndrome (2.3.1 to 2.2.1) SGE to SLRE (2.3.2 to 1.2.3) UEl to one each of SLRE, CLRE, benign myoclonic epilepsy in
mode of precipitation (1.2.2 to 2.1.9)
etiology (1.2.3 to 1.2.3)
More definitive EEG information and clearer information about
syndrome (2.2.3 to 2.2.2)
Seizures clearly focal or had clear auras
Reassessment of initial clinical information in light of subsequent course
infancy (3.1.5 to 1.2.3, 130, 213) Other
SLRE to benign rolandic epilepsy (1.2.3 to 1.1.1) CLRE to SLRE (1.3 to 1.2.3)
Total (n = 84)
24 3 3 1 1 1 1 1 5 1 2" 1 4
60 36
1 1
1 1 1 1
13 1 1 2
11 2
9 1 1 1 1 1
1 1 1 1 4 2 2
9 1 1 1 1 1 1
3 2 1 1
Change apparent during
First 6 months Second 6 months (n = 31) (n = 15)
14 2 3 0 1 0 1 0 0 0 1 0 0 1 1 0 2 1 1 0 1 0 0 0 3 0
17 13 9 10
0 0 0 1
1 0 0 0 0 0 0 0 3 5 0 0 0 0 0 1 3 3 2 0
3 1 0 0 0 1 1 0 0 0 1 0
0 0 0 0 1 0 0 0 3 0 2 0 1 0
1 1 0 0 0 0 0 0 0 0 1 0 0 1
0 0 1 1 1 0 0 1
Second year (n = 38)
8 0 2 0 1 0 0 0 2 0 1 1 1
30 17
1 0
0 1 1 1 5 1 1 1 5 0
5 1 0 0 1 0
1 1 0 1 1 0 1
7 1 1 1 1 0 0
3 0 0 0
SLRE, symptomatic localization-related epilepsy; SGE, symptomatic generalized epilepsy; SICGE, symptomatic/cryptogenic generalized epilepsy; CLRE, cryptogenic localization-related epilepsy; UE2, undetermined epilepsy with neither clear focal nor generalized features; ICE, idiopathic generalized epilepsy; UEl, undetermined epilepsy with both focal and generalized features.
a These children's syndromes had clearly evolved; however, it was impossible to determine to which syndrome because no EEGs had been done and the seizure types were not sufficiently revealing.
Epilepsia, Vol. 41, No. 10, 2000

HOW WELL CAN EPILEPSY SYNDROMES BE IDENTIFIED AT DIAGNOSIS? 1273
related category who were later found to have a localiz- able EEG focus or an underlying etiology and therefore were reclassified as having a symptomatic localization- related syndrome (n = 14) or (b) who were assigned to the undetermined category and were subsequently reclassified as having cryptogenic or symptomatic localization-related epilepsy (n = 15). Six other children initially classified with undetermined epilepsy were later found to have idiopathic syndromes (four idiopathic gen- eralized and two benign rolandic). These and the remain- ing cases are summarized in Table 3.
The information necessary to reclassify the initial syndromes was apparent within the first 6 months for 17 children (28%), during the second 6 months for 13 children (22%), and during the second year for 30 chil- dren (50%). In most cases (n = 45), a subsequent EEG was the basis or partial basis for the change. In 12 instances, the EEG was done only because the child was seizure-free and the physician was considering stop- ping medication.
There were 11 instances in which the initial syndrome was based on well-specified, unambiguous criteria and in which a significant change (rectification) was made. In four instances, the classification of symptomatic localization-related epilepsy was changed to a form of idiopathic generalized epilepsy. In one of these in- stances, both the initial and rectified classifications rep- resented photosensitive forms of epilepsy. Two cases of idiopathic generalized epilepsy were changed to crypto- genic localization-related epilepsy (n = 1) and unclas- sified epilepsy with autism (n = 1). Two other cases initially classified as cryptogeniclsymptomatic general- ized epilepsy were reclassified as idiopathic generalized epilepsy after 2 years. One of these children had a neu- rocutaneous syndrome that, upon re-review, appeared to be incidental to his epilepsy. Nonetheless, his etiology remained classified as remote symptomatic (5). Finally, three children with symptomatic localization-related syn- dromes were reclassified as having benign rolandic epi- lepsy (n = 2) or undetermined epilepsy (n = I). In most of these instances, there had been problems with the initial information, and, as a result, these cases had been discussed in conference at the time of initial classifica- tion of the syndrome. Of three children classified as hav- ing nonspecific forms of epilepsy with both clearly focal and generalized features, all were reclassified with either localization-related epilepsy or a form of idiopathic gen- eralized epilepsy.
DISCUSSION
The classification of the epilepsies is increasingly be- ing used in studies of the epidemiology and prognosis of epilepsy (2-4,ll-16). Its utility, especially for prognosis, will depend on how well syndromes can be classified
early in the course of the disorder. We have already demonstrated good to excellent interrater agreement for classification at diagnosis (lo), although we noted that many children were assigned only partially specified or unspecified syndromes at that time. Our current report highlights several points.
1. The figure of 13.7% of children who had their syn- drome reclassified after 2 years is comparable to what has been reported in three other studies that used similar approaches to this issue (2-4).
2. In our study, 24 of the changes (29%) were attrib- utable to a true evolution of the underlying syndrome. These changes do not constitute errors in the classifica- tion of the syndrome at diagnosis but reflect the natural history of some of these disorders, which are know to evolve over time (9). There were only four cases of Lennox-Gastaut syndrome at the h e of initial diagno- sis. This is consistent with findings of other studies that have examined patients at the onset of epilepsy (4,17). After 2 years, this number increased to 19, largely be- cause of evolution into the syndrome. Thus, the Lennox- Gastaut syndrome does eventually account for a larger fraction of pediatric epilepsy than can be appreciated at the time of initial diagnosis, as is seen with time in other prospective follow-up studies (4,16) as well as in preva- lence studies (1 8).
3. Of the remaining changes, more than half were attributable to reclassification from an only partially specified syndrome (e.g., typically cryptogenic localization-related or undetermined) to a more specific syndrome. By definition, classification into cryptogenk localization-related and undetermined categories is an acknowledgment of inadequate information to make an accurate classification. In contrast, syndromes that were classified on the basis of well-defined objective criteria were rarely reclassified 2 years later.
The other studies that have examined this issue found that the cryptogenic localization-related and undeter- mined categories are particularly subject to rectification after 2 or more years of follow-up (24) . Clearly, one of the problems in using the classification system is that some of the distinctions are based simply on the type, extent, and quality of information available. For ex- ample, the difference between cryptogenic localization- related epilepsy and symptomatic localization-related epilepsy (by virtue of localization) may be merely how clearly the EEG abnormalities can be localized to a spe- cific region of the cortex. This can be determined simply by the decision to pursue (or not) an initial normal EEG with, for example, a 24-hour ambulatory EEG. In addi- tion, although every effort was made to obtain original EEG tracings when the EEG report was inadequate for classification purposes, this was not always possible. Consequently, the quality of the information available in the reports also influenced the classification of the syn-
Epilepsia, Vol. 41, No. 10, 2000

1274 A. T. BERG ET AL.
drome. Admittedly, it may have been possible to obtain more precise classifications of seizures and syndromes both at diagnosis and 2 years later had all children un- dergone a standardized and rigorous evaluation at an epilepsy center. This was neither feasible in the context of this observational study nor consistent with the evalu- ation of epilepsy in everyday community practice in the United States or elsewhere.
4. Some notable areas in which errors in the initial classification occurred further help defme the limitations of the classification system, especially as applied in a community-based setting. Although classifications for this study were assigned by three neurologists with spe- cial expertise in pediatric epilepsy and EEG, the clinical information on which these classifications were based was collected and interpreted by physicians from across the state. In fact, some tests were ordered and interpreted elsewhere (e.g.. when a child had the first seizure while visiting out of state). Consequently, and understandably, there was some variability in the quality and extent of information available to the study. Only in a handful of cases did this appear to result in marked and unexpected errors in the initial classification, as determined by re- view 2 years later. In 9 of these 11 instances, there were acknowledged concerns about the quality of the initial information and disagreement over its interpretation, and a conference was required to discuss the discrepancies. In comparison, only 26% of all initial classifications re- quired a conference. Although in theory one might be able to improve on this, in practice it is unlikely.
A further ramification is that, for the ILAE classifica- tion of the epilepsies to be used effectively, patients must receive adequate diagnostic assessments. These include routine use of EEG and, whenever indicated, neuroim- aging. In health care settings in which EEG is not rou- tinely available or used and neuroimaging is not part of the standard evaluation of seizures, the ILAE classifica- tion of the epilepsies cannot be as meaningfully applied. A corollary is that comparisons of syndromes across populations will need to take into account the quality and extent of the diagnostic evaluations.
The iyndromic classification of newly diagnosed pe- diatric epilepsy, as applied in a community-based setting, appears to have excellent reliability over the course of 2 years. Most changes that occurred involved true evolu- tion of syndromes or more specific classification of in- completely classified cases. Major unexpected changes tended to occur only when there were difficulties with the initial or subsequent information. With relatively few and largely explainable exceptions, specific syndromes can be reliably identified at the time of initial diagnosis. We do caution, however, that the ILAE classification system allows room for essentially unclassified and only partially classified syndromes. With longer follow-up, it is possible that more changes may occur in the assess-
ment of the underlying syndrome. In fact, this issue will be reexamined after 5 years of follow-up. Identification of the underlying syndrome after 5 years, however, is not especially helpful if the syndrome is supposed to be used for prognosis and treatment decisions early in the course of the disorder. On the other hand, it may be useful for investigations into etiology and for other purposes re- lated to treatment. Limitations on the quality, consis- tency, and extent of information could, in theory, be eliminated. In practice, this is unlikely, and our study provides a fair assessment of the current ILAE classifi- cation of the epilepsies as applied in a community-based setting in the United States.
Acknowledgment: We thank Dr. JtrBme Loiseau for his helpful and detailed comments on the manuscript. We also extend our thanks to the physicians in Connecticut who have referred their patients to this study: Drs. Robert Cerciello, Fran- cis Dimario, Barry Russman, Michelle Kleirnan, Carol Leicher, Edwin Zalneraitis, Philip Brunquell, Laura Ment, Edward No- votny, Bennet Shaywitz, S. Nallainathan, Alok Bhargava, Mar- tin Kreminitzer, Barbara Coughlin, Harriet Fellows, Jack Finkelstein, Daniel Moalli, Louise Resor, Owen Erlich, Ber- nard Giserman, John Monroe, Lawrence Rifkin, and Murray Engel. We also thank Wuthikrai Uayingsak for his exceptional programming expertise. Dr. Eugene Shapiro has kindly facili- tated many administrative issues for us. This study would not be possible without the generous participation of the many parents and their children. This study was funded by grant ROI-NS31146 from the National Institutes of Health, National Institute of Neurological Disorders and Stroke.
REFERENCES
1 . Benbadis SR, Luders HO. Epileptic syndromes: an underutilized concept. Epilepsia 1996;37: 1029-34.
2. Arts WF, Geerts A, Brouwer 0, Peters A, Stroink H, van Donse- laar C. Classification schemes in childhood epilepsy: reliability and causes of discrepancy. Epilepsia 1997;38(SuppI 3): 120.
3. Loiseau J, Picot M-C, J d o n P, Dartigues J-F, Loiseau P. Classi- fication and incidence of epileptic syndromes in a prospective study: reliability and causes of change. Epilepsia 1998;39(Suppl 6): 18 1.
4. Shinnar S , O’Dell C, Berg AT. Distribution of epilepsy syndromes in a cohort of children prospectively monitored from the time of their first unprovoked seizure. Epilepsia 1999;40: 1378-83.
5. Berg AT, Shinoar S , Levy SR, Testa Fh4. Newly-diagnosed epi- lepsy in children: presentation at diagnosis. Epilepsia 1999;40: 45-52,
6. Commission on Classification and Terminology of the Interna- tional League Against Epilepsy. Proposal for revised clinical and electrographic classification of epileptic seizures. Epilepsia 198 1 ; 22:489-501.
7. Commission on Classification and Terminology of the Interna- tional League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389-99.
8. Commission on Epidemiology and Prognosis, International League Against Epilepsy. Guidelines for epidemiologic studies on epi- lepsy. Epilepsia 1993;34:592-6.
9. Roger J, Bureau M, Dravet C, Dreifuss FE, Perret A, Wolf P. Epileptic syndromes in infancy, childhood, and adolescence. 2nd ed. London: John Libbey, 1992.
10. Berg AT, Levy SR, Testa FM, S h i ~ a r S . Classification of child- hood epilepsy syndromes in newly diagnosed epilepsy: interrater agreement and reasons for disagreement. Epilepsia 1999;40:439- 44.
Epilepsia, Vol. 41, No. 10, 2000

HOW WELL CAN EPILEPSY SYNDROMES BE IDENTIFIED AT DLAGNOSIS?
1 1. A r t s WF, Geerts AT, Brouwer OF, Boudewyn Peters AC, Stroink H, van Donselaar CA. The early prognosis of epilepsy in child- hood: the prediction of a poor outcome. The Dutch Study of Epi- lepsy in Childhood. Epilepsia 1999;40:726-34.
12. Loiseau J, Loiseau P, Guyot M, Duche B, Dartigues JF, Aublet B. Survey of seizure disorders in the French southwest. I. hcidence of epileptic syndromes. Epilepsia 1990;31:3914.
13. Sillanpaa M, Jalava M, Kaleva 0, Shinnar S. Long-term prognosis of seizures with onset in childhood. N Engl J Med 1998;338:1715- 22.
14. Shinnar S, Berg AT, Moshe SL, et al. Discontinuing antiepileptic drugs in children with epilepsy: a prospective study. Ann Neurol 1994;35:534-45.
1275
15. Jallon P. CAROLE, a French prospective cohort of newly diag- nosed epileptic seizures. Epilepsia 1997;38(Suppl 3):24 1.
16. Sillanpaa M, Jalava M, Shinnar S. Epilepsy syndromes in patients with childhood-onset seizures in Finland. Pediafr Neurol 1999;21: 533-7.
17. Groupe CAROLE. Traitement des crises tpileptiques nouvelle- ment diagnostiqutes. Une exptrience franpise. Rev Neurol (in press).
18. Viani F, Beghi E, Atza MG, Gulotta M p . Classifications of epi- leptic syndromes: advantages and limitations for evaluation of childhood epileptic syndromes in clinical practice. Epilepsia 1988; 29:4465.
Epilepsia, Vol. 41, No. 10, 2000





























