Embed Size (px)
Citation preview

Genome Sequence Informatics&
Comparative GenomeSequence Analysis
Genome Sequence Informatics&
Comparative GenomeSequence Analysis
Niclas JareborgAstraZeneca R&D Södertälje

Genome sequencing projects
• Aim: Better understanding of biology• Bioinformatics
• Manage data• Cut corners• Generate and test new hypotheses
• Make the most of the data• comparative analysis

gttaaaattcagcaggcagaatgaaaataaatgtcaataattttttattttaaaatattcatgttttactattttgatataatttttaaagaaaaaggcagaaaccactgcttattagaaggcagattttattgattttatacccctagacttgttgcatatcaaacctatgtaaaaacatctataaatcaaatcattaattgcacctagtataataattctatatatggaggtaatgtttgattcttcaggagctttaataacttgaagcccgtttgattgctttaaaatgatttctcattgtatttgtttatattgtatcattaagcaaaagtacagagtaagcaattagtgtgattaattcctcttccataatacagtaaagcactgcctccatagaccaattctctgggatccctggaaaacatctggcatccagcaagtcttgacccctctttagaaagccatggagaaactggaggcaattctgttaattatttgccctctagaggcaattgggttaattaccctcccttccctatccatgacacaatttctccagttacatgtagaatgctgttatgtgtctcctgaccagaccccttatttcatagatgtggaaactgaggccatgaaggatgaggtgactgttcacaatccacatggctagttagtgtccagagcctggcctggacttctctcttgttctggggccttgagttctctccctcttctttagtacatatggccacaggtaacgtaatctgcgtaccacatttgcatttggagtgcatctgttttgcattcatttaatcttgttgagatggtttgcttgttgacctactcagtcagttatcttttcacctttgtgagttgagagctttgtgtattaaatctgtaaaactttgcatcgtggaaagtgacataatctgtagcagacccatgctgtttttagatgcatcttcattgtggtagtgacagtgattgagaaactttacat
Where are the functional elements?Where are the functional elements?

Features in genome sequences• Genes
• Exons, introns, promoters• RNA genes• CpG islands• Enhancers• Other functional elements
• e.g. Replication origins, Nuclear matrix association
• Repeats

How to find genomic features
• Repeats, CpG islands, RNA gene• Bioinformatics programs
• Genes• Homology to known sequences• Bioinformatics prediction programs
• Transcription regulatory regions• Bioinformatics prediction programs

Finding genes by homology
• Database searches – BLAST, BLAT, SSAHA• EST and cDNA sequences• Protein sequences
High accuracy, misses unknown sequences• caveat: junk EST sequences
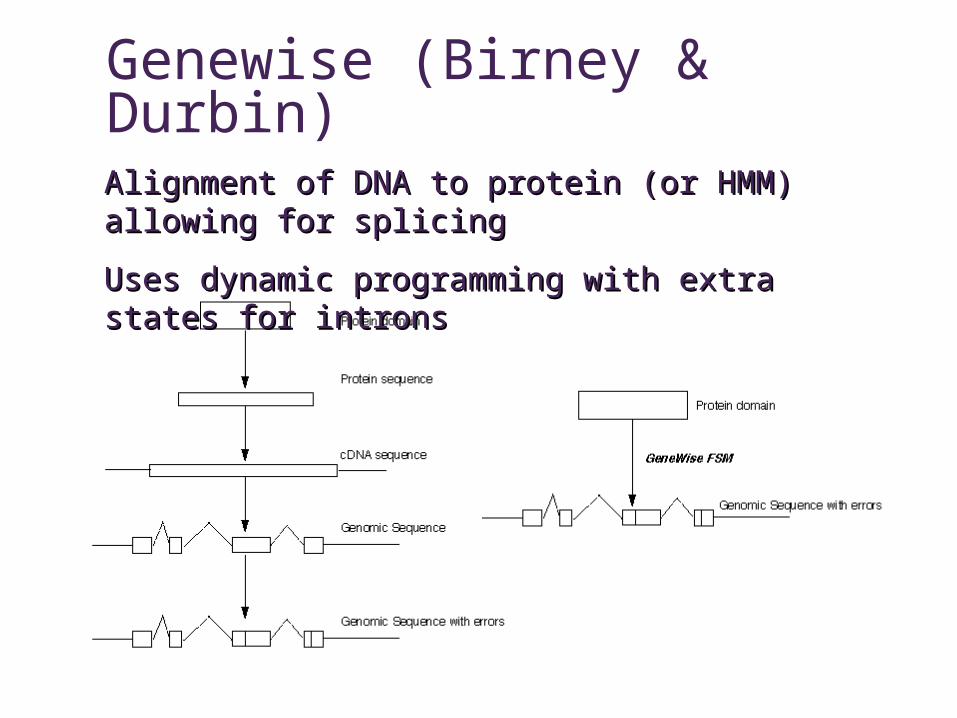
Genewise (Birney & Durbin)
Alignment of DNA to protein (or HMM) allowing for Alignment of DNA to protein (or HMM) allowing for splicingsplicing
Uses dynamic programming with extra states for intronsUses dynamic programming with extra states for introns
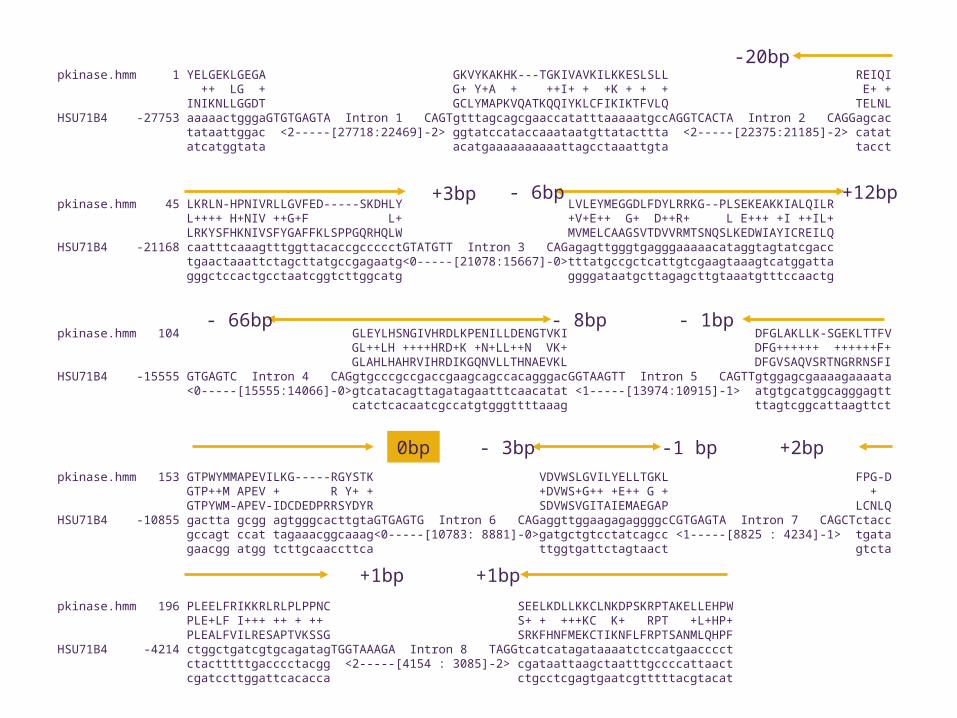
pkinase.hmm 1 YELGEKLGEGA GKVYKAKHK---TGKIVAVKILKKESLSLL REIQI ++ LG + G+ Y+A + ++I+ + +K + + + E+ + INIKNLLGGDT GCLYMAPKVQATKQQIYKLCFIKIKTFVLQ TELNLHSU71B4 -27753 aaaaactgggaGTGTGAGTA Intron 1 CAGTgtttagcagcgaaccatatttaaaaatgccAGGTCACTA Intron 2 CAGGagcac tataattggac <2-----[27718:22469]-2> ggtatccataccaaataatgttatacttta <2-----[22375:21185]-2> catat atcatggtata acatgaaaaaaaaaattagcctaaattgta tacct
pkinase.hmm 45 LKRLN-HPNIVRLLGVFED-----SKDHLY LVLEYMEGGDLFDYLRRKG--PLSEKEAKKIALQILR L++++ H+NIV ++G+F L+ +V+E++ G+ D++R+ L E+++ +I ++IL+ LRKYSFHKNIVSFYGAFFKLSPPGQRHQLW MVMELCAAGSVTDVVRMTSNQSLKEDWIAYICREILQHSU71B4 -21168 caatttcaaagtttggttacaccgccccctGTATGTT Intron 3 CAGagagttgggtgagggaaaaacataggtagtatcgacc tgaactaaattctagcttatgccgagaatg<0-----[21078:15667]-0>tttatgccgctcattgtcgaagtaaagtcatggatta gggctccactgcctaatcggtcttggcatg ggggataatgcttagagcttgtaaatgtttccaactg
pkinase.hmm 104 GLEYLHSNGIVHRDLKPENILLDENGTVKI DFGLAKLLK-SGEKLTTFV GL++LH ++++HRD+K +N+LL++N VK+ DFG++++++ ++++++F+ GLAHLHAHRVIHRDIKGQNVLLTHNAEVKL DFGVSAQVSRTNGRRNSFIHSU71B4 -15555 GTGAGTC Intron 4 CAGgtgcccgccgaccgaagcagccacagggacGGTAAGTT Intron 5 CAGTTgtggagcgaaaagaaaata <0-----[15555:14066]-0>gtcatacagttagatagaatttcaacatat <1-----[13974:10915]-1> atgtgcatggcagggagtt catctcacaatcgccatgtgggttttaaag ttagtcggcattaagttct
pkinase.hmm 153 GTPWYMMAPEVILKG-----RGYSTK VDVWSLGVILYELLTGKL FPG-D GTP++M APEV + R Y+ + +DVWS+G++ +E++ G + + GTPYWM-APEV-IDCDEDPRRSYDYR SDVWSVGITAIEMAEGAP LCNLQHSU71B4 -10855 gactta gcgg agtgggcacttgtaGTGAGTG Intron 6 CAGaggttggaagagaggggcCGTGAGTA Intron 7 CAGCTctacc gccagt ccat tagaaacggcaaag<0-----[10783: 8881]-0>gatgctgtcctatcagcc <1-----[8825 : 4234]-1> tgata gaacgg atgg tcttgcaaccttca ttggtgattctagtaact gtcta
pkinase.hmm 196 PLEELFRIKKRLRLPLPPNC SEELKDLLKKCLNKDPSKRPTAKELLEHPW PLE+LF I+++ ++ + ++ S+ + +++KC K+ RPT +L+HP+ PLEALFVILRESAPTVKSSG SRKFHNFMEKCTIKNFLFRPTSANMLQHPFHSU71B4 -4214 ctggctgatcgtgcagatagTGGTAAAGA Intron 8 TAGGtcatcatagataaaatctccatgaacccct ctactttttgacccctacgg <2-----[4154 : 3085]-2> cgataattaagctaatttgccccattaact cgatccttggattcacacca ctgcctcgagtgaatcgtttttacgtacat
+3bp - 6bp +12bp
-20bp
- 8bp- 66bp - 1bp
0bp - 3bp -1 bp
+1bp
+2bp
+1bp

Gene prediction methods
• ATGs• Stop codons• ORFs• Coding preference• Splice sites
• profiles, statistical methods, neural networks etc.
High coverage, low accuracy
easy
hard

Accuracy of gene-finding programs for 1.4 MB genomic
region BRCA2 on human
chromosome 13q
Region includes 159 true exons exact match overlap exons 5'- splice site 3'- splice site NE N acc cov N acc cov N acc cov N acc covfgenesh.masked 169 110 0.65 0.69 125 0.74 0.79 118 0.70 0.74 116 0.69 0.73fgenesh 190 109 0.57 0.69 126 0.66 0.79 117 0.62 0.74 117 0.62 0.74fgenes.masked 238 103 0.43 0.65 132 0.55 0.83 114 0.48 0.72 118 0.50 0.74fgenes 281 104 0.37 0.65 136 0.48 0.86 116 0.41 0.73 120 0.43 0.75genscan 292 105 0.36 0.66 129 0.44 0.81 116 0.40 0.73 115 0.39 0.72fgeneh 381 68 0.18 0.43 101 0.27 0.64 79 0.21 0.50 87 0.23 0.55mzef 623 95 0.15 0.60 122 0.20 0.77 106 0.17 0.67 107 0.17 0.67
fgeneshm+genescan 118 97 0.82 0.61 106 0.90 0.67 101 0.86 0.64 101 0.86 0.64fgeneshm+fgenes 89 83 0.93 0.52 86 0.97 0.54 86 0.97 0.54 83 0.93 0.52
acc - specificity (true predicted/all predicted) cov - sensitivity (true predicted/true)NE - number of predicted exons
data provided by Tim Hubbard and Richard Bruskiewich (Sanger Centre)

Repetitive elements• 1/3 of the human genome • Transposable elements
• LINEs (Long Interspersed Nuclear Elements), 6-8 kb • SINEs (Short Interspersed Nuclear Elements, e.g. Alu), 100-
400 bp • Retrovirus-like elements, 1.5-10 kb (LTRs 300-1000 bp) • DNA transposons, 80 bp-3 kb
• Tandem repeats
• Simple repeats/Microsatellites (1-5bp)n, e.g. caacaacaa
• Minisatellites (6-1000s bp)n
• Low complexity regions

Repeat masking• Repeats disturb analysis
• Homology searching • Gene prediction
• Masking exchange repeat region with N's. Will be ignored by analysis programs
• RepeatMasker (Smit & Green)
• LINEs, SINEs, LTR transposons, DNA transposons, Simple repeats, Low complexity regions
• trf (Benson)• Tandem repeats

Predicting regulatory regions
• Transcription Factor Binding Sites (TFBSs) have very low information content
• Given a long enough sequence a binding site will be predicted
• Combination of TFBSs• Even the best algorithms will overpredict

CpG islands
• Associated with transcribed genes • House keeping genes + ~50% of other genes• Often in 5' ends of genes
• >200 bp• GC content >50% • obs/exp CpG >0.6

Gene Ontology
• “Controlled vocabulary that can be applied to all organisms even as knowledge of gene and protein roles in cells is accumulating and changing.”
“Biologists would rather share a toothbrush than a gene name”- Michael Ashburner

Gene Ontology
• Organizing principles • Molecular function• Biological process• Cellular component
• Hierarchical structure

Genome resources• Genome sequence centered
• Ensembl• http://www.ensembl.org
• NCBI• http://www.ncbi.nlm.nih.gov
• UCSC Human genome browser • http://genome.ucsc.edu
• All based on NCBI assembly• Gene centered
• SOURCE• http://source.stanford.edu
• GeneLynx • http://www.genelynx.org
• GeneCards• http://bioinformatics.weizmann.ac.il/cards/

Ensembl
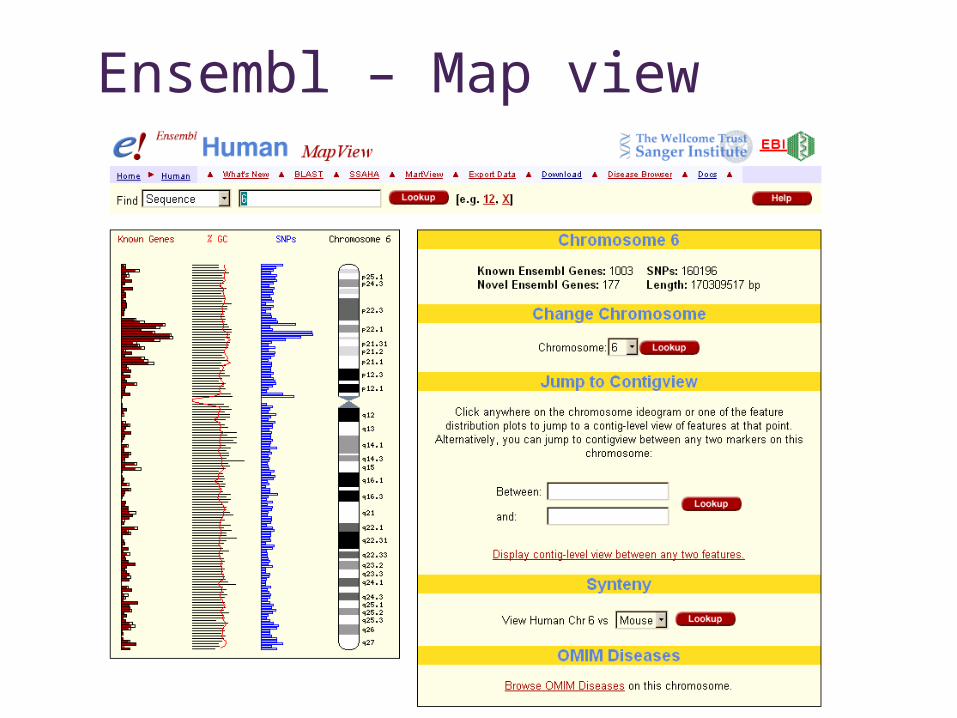
Ensembl – Map view
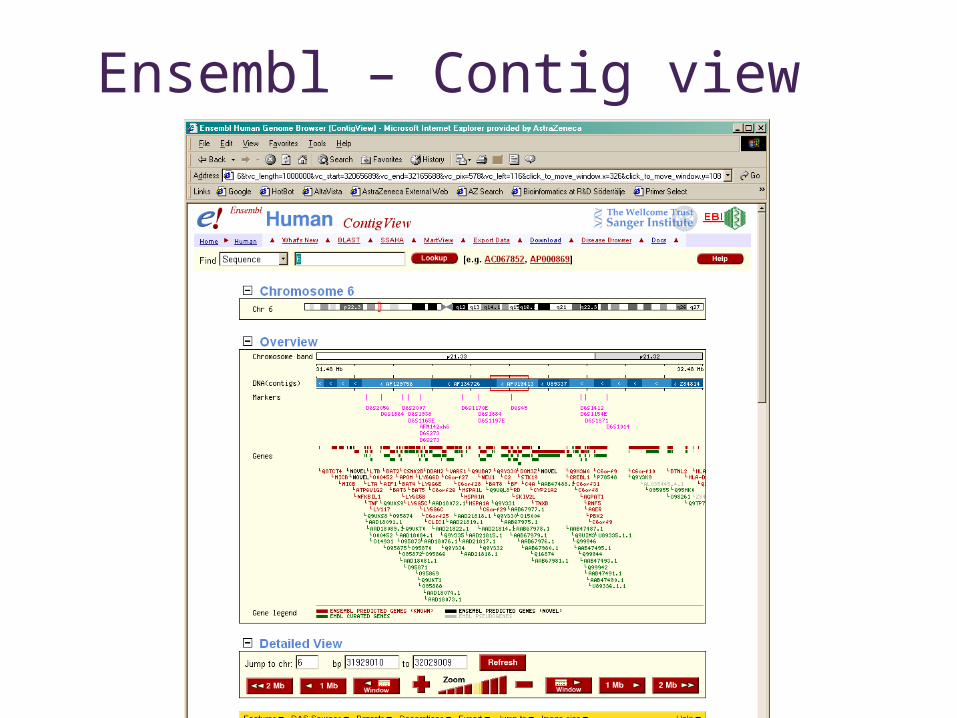
Ensembl – Contig view
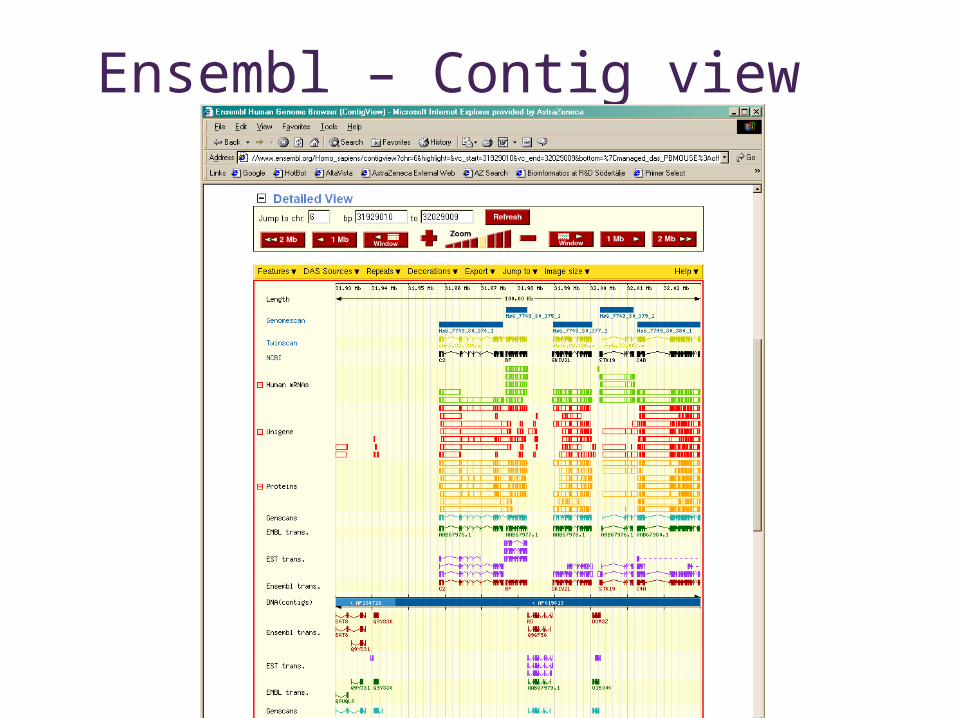
Ensembl – Contig view
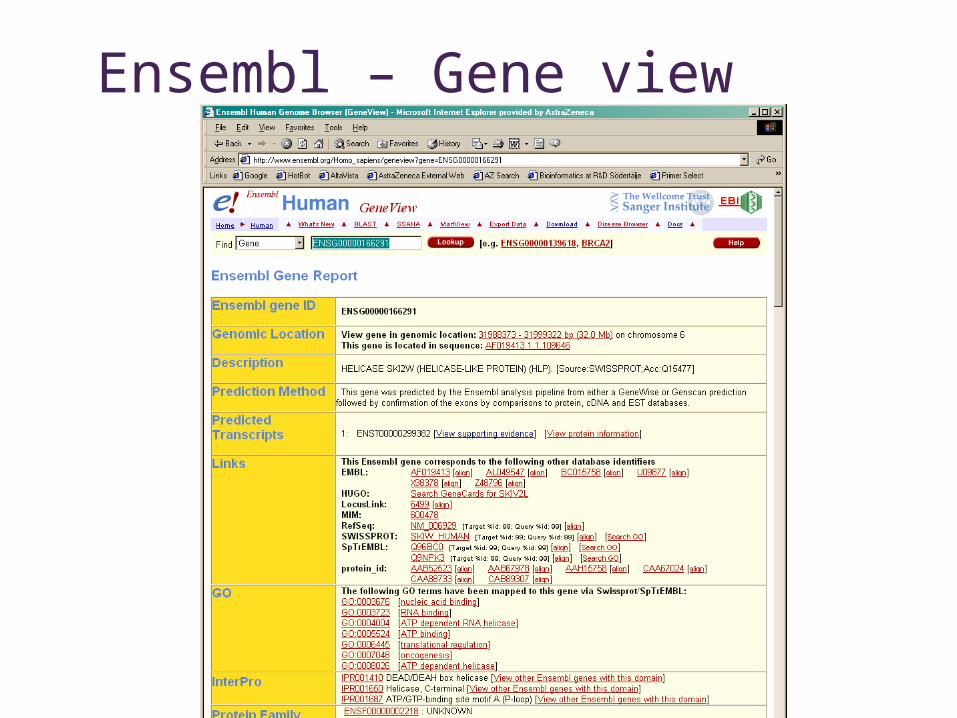
Ensembl – Gene view
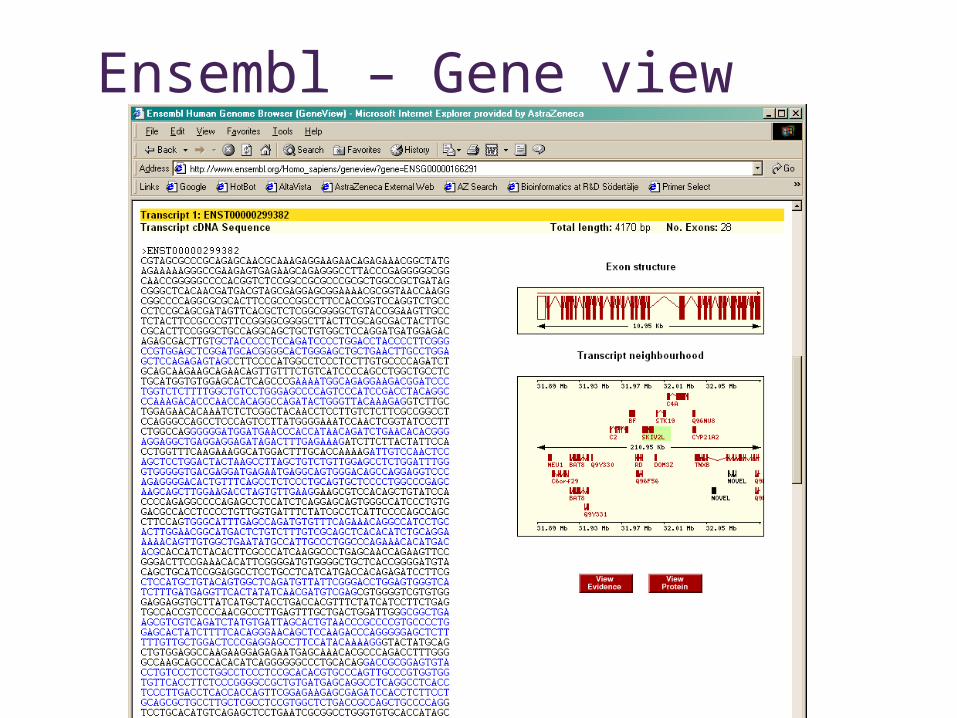
Ensembl – Gene view
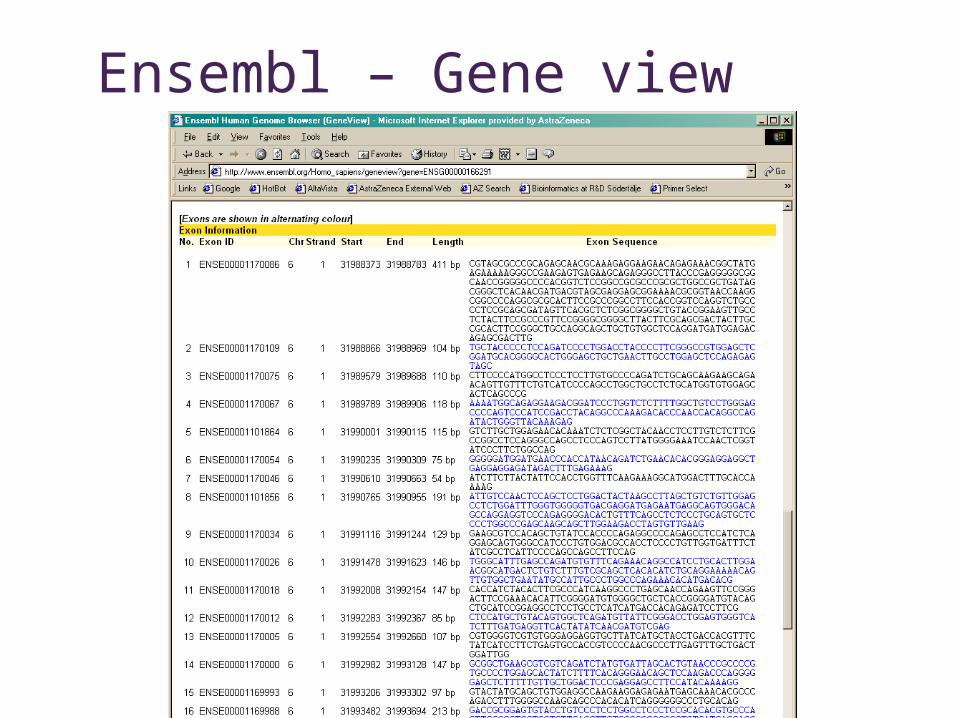
Ensembl – Gene view

NCBI Genome resources
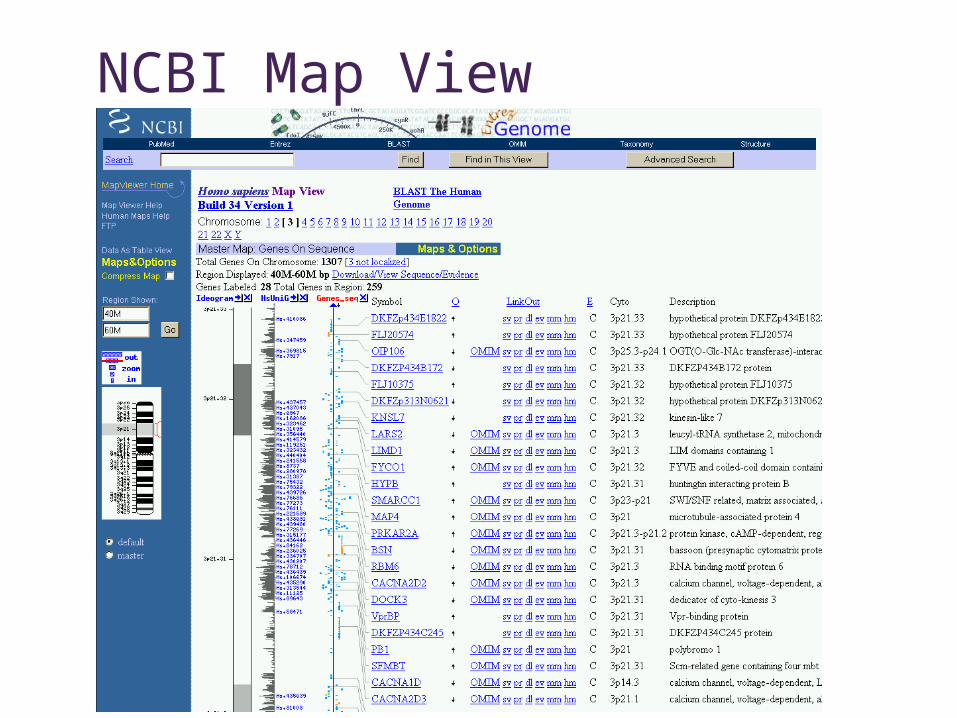
NCBI Map View
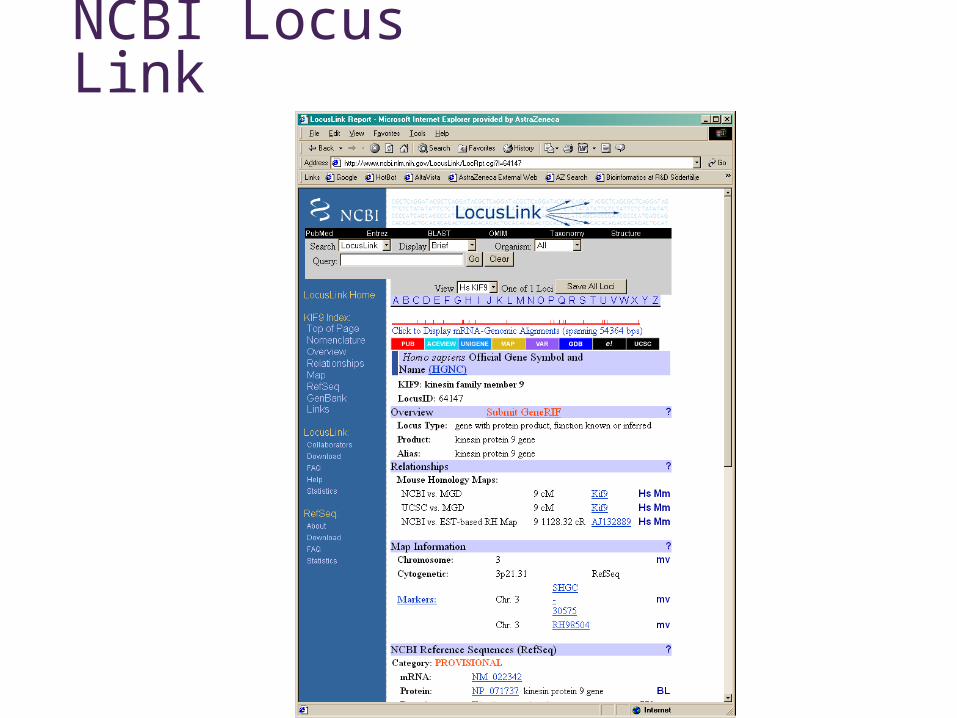
NCBI Locus Link
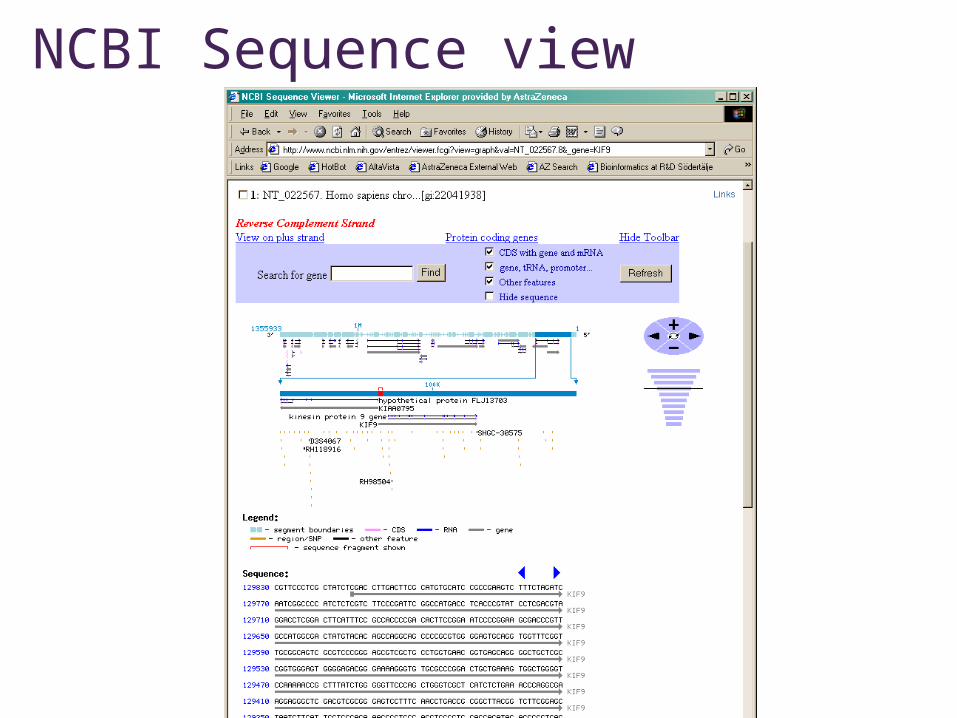
NCBI Sequence view
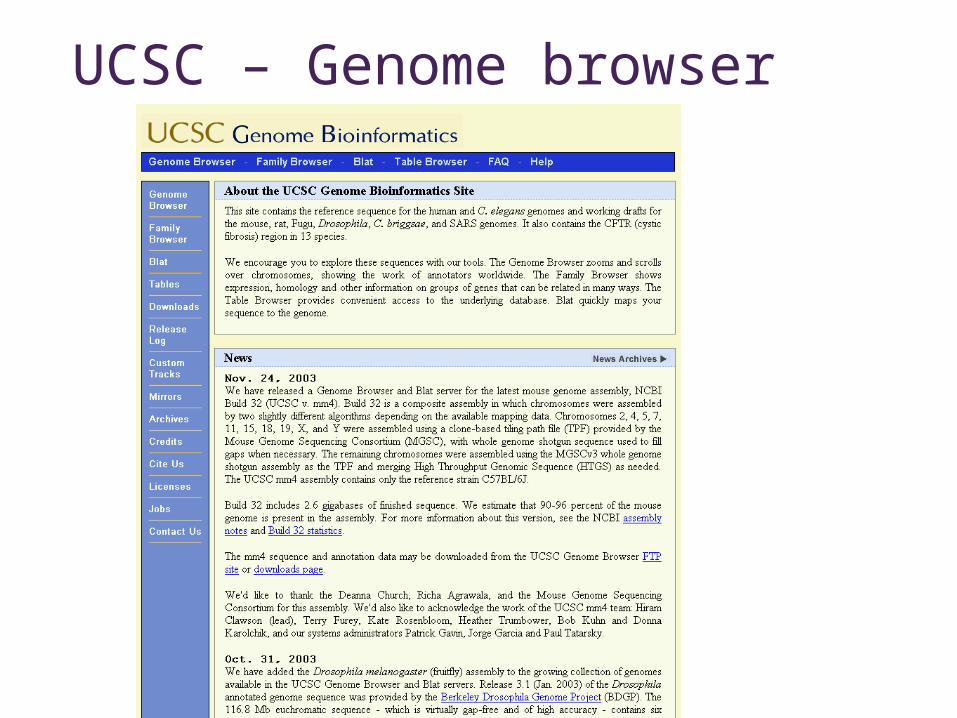
UCSC – Genome browser
UCSC – Genome browser

UCSC – Genome browser
Gene-centered resources
• Genomic resources• Transcripts• Protein sequences• Protein structure and
domains• Protein function and
disease links• Homologs• Functional/GO
classifications• Physical clones• etc

Comparative Genomic Sequence Analysis
• Aid in finding functional regions• Coding regions• Regulatory regions

Comparative Genomic Sequence Analysis
• Compare corresponding genomic sequences from different species
• Potential protein coding and/or regulatory regions can be identified by their conservation
• “Phylogenetic footprinting”
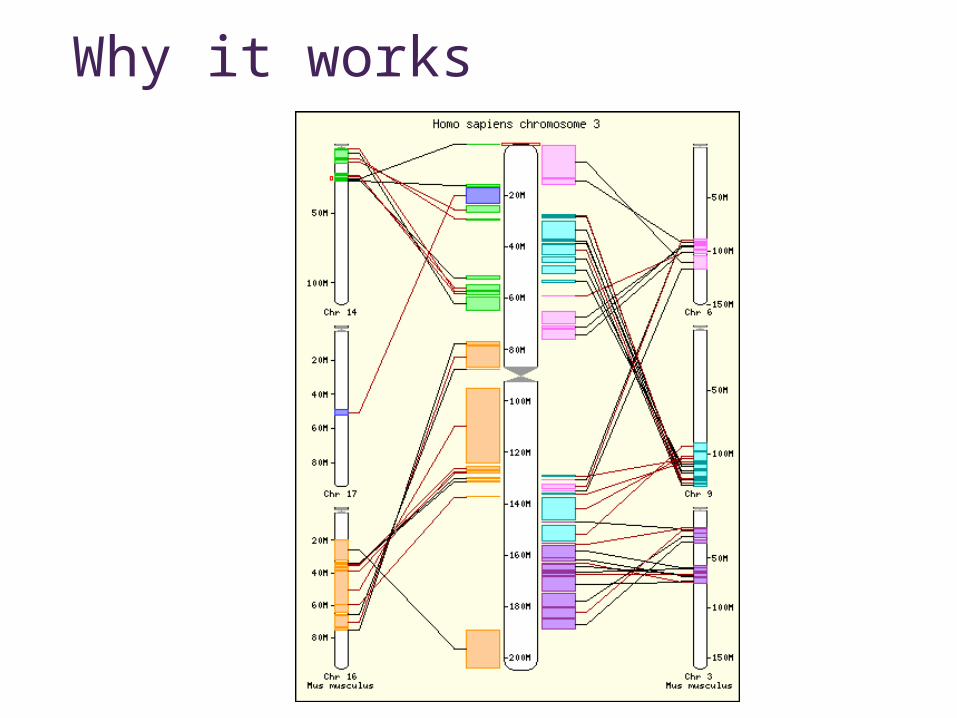
Why it works

Synteny maps• Maps corresponding regions in different genomes• Large-scale relationships• Based on
• genetics• sequence
• Available for • Human vs.
• Mouse• Rat• Dog• Chimp• etc…
• Mouse vs Rat
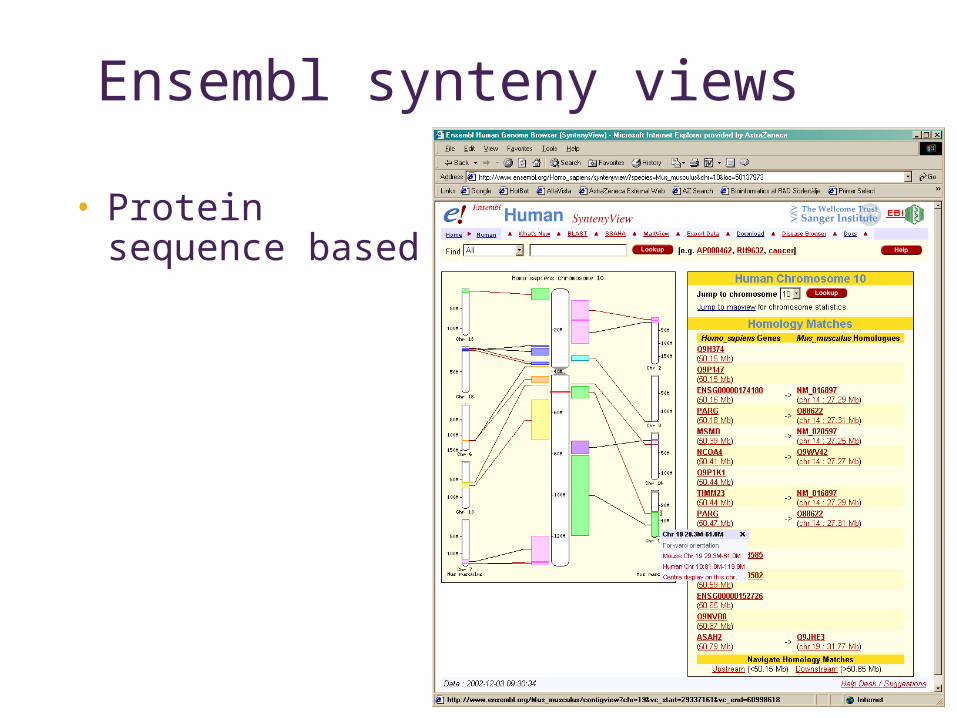
Ensembl synteny views
• Protein sequence based

NCBI comparative maps
• Based on genetics
• Several genetic maps

Human/vertebrate sequence comparisons (80-450 Myrs)
• Coding sequences generally well conserved• Non-coding regions show highly variable
levels of conservation• Conservation of non-coding regions imply a
functional role• promoters• other transcriptional regulators• replication origins• chromatin condensation• matrix association

Model organisms for vertebrate comparative analysis
• Not too evolutionary close• Impossible to identify functional regions through
conservation• Mouse 3000 Mb 80 Myrs
Genetics Sequence ”finished”
• Chicken 1200 Mb 300 Myrs Micro-chromosomes (~75% of genes) Prioritized for sequencing
• Fugu (Puffer fish) 400 Mb 450 Myrs Small genome, shorter introns and intergenic regions More or less the same gene content as higher vertebrates Sequence finished

What are we comparing?
• Homologue • common ancestor, may have similar function
• Orthologue• the “same” sequence, generated by a
speciation event, probably same function
• Paralogue• similar sequence within species, generated by
a gene duplication event, may have similar function
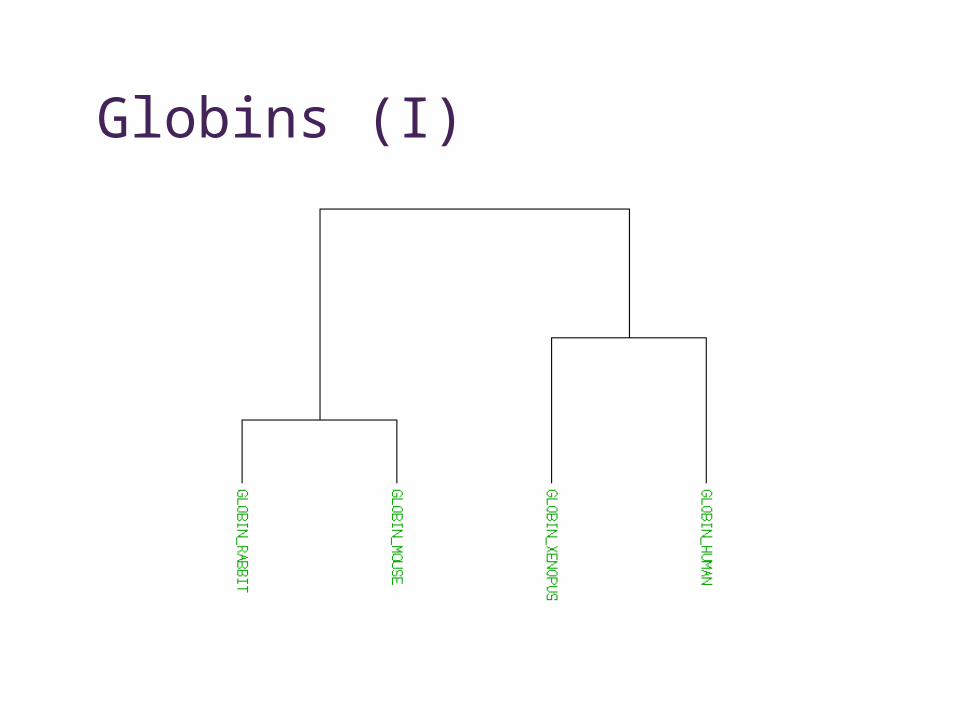
Globins (I)
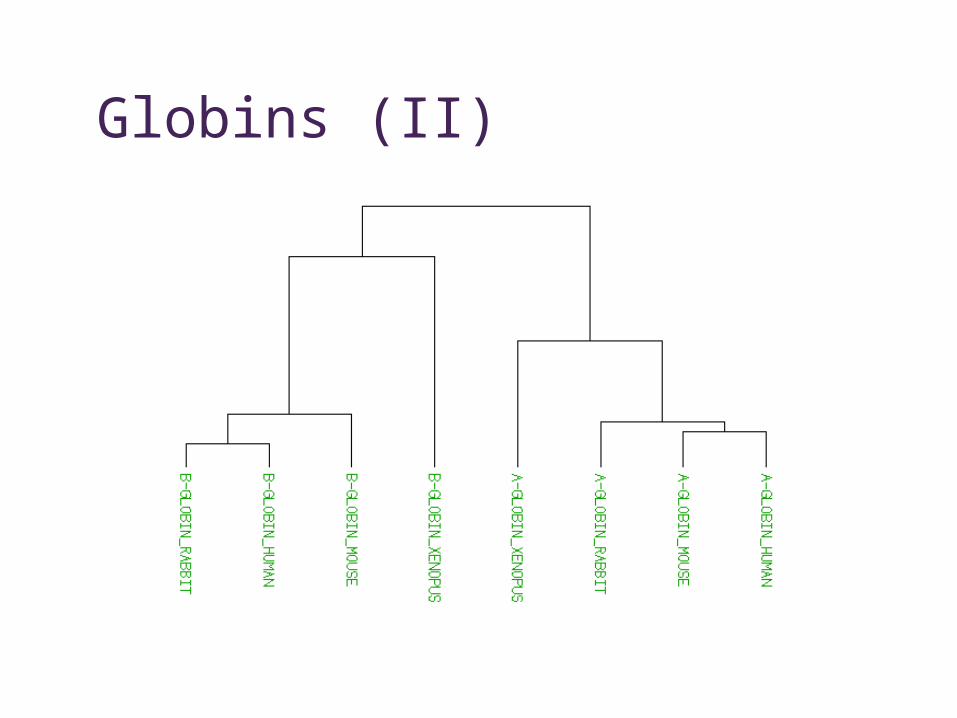
Globins (II)

Finding conserved regions• Dot plot
• Dotter• Similarity search programs
• Blast• Alignment programs
• DBA (Jareborg et al)• blastz (Schwartz et al.)• Dialign (Morgenstern et al.)• WABA (Kent & Zahler)• Avid (Bray et al.)• others

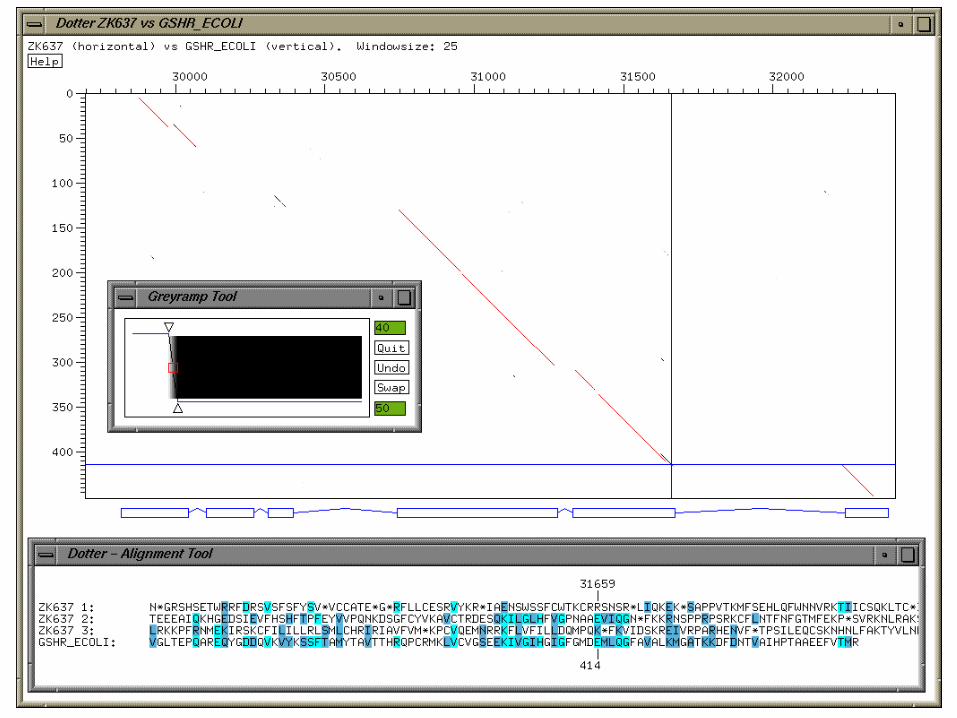
Dotter (Sonnhammer & Durbin)
• Graphical dot plot program for detailed comparison of two sequences
• Features • dynamic greyscale ramp for stringency cut-off• alignment viewer• zooming.
• Unix & Windows• http://www.cgb.ki.se/cgb/groups/sonnhammer/Dotter.html

DBA (Jareborg, Birney & Durbin)
• DNA Block Aligner• Finds co-linear blocks with high similarity• Does not try to align the sequences
between these blocks• Divides blocks into four different
categories• approx. 60-70%, 70-80%, 80-90%, 90-
100%

Comparison-based functional prediction • Gene prediction• Regulatory region predictions
”Comparative” gene prediction programs
• Twinscan• Doublescan• SGP-1
http://genes.cs.wustl.edu/
http://www.sanger.ac.uk/Software/analysis/doublescan/
http://195.37.47.237/sgp-1

Regulatory region prediction• Consite
• Detection of TFBS conserved in corresponding genomic sequences from different species
www.phylofoot.org/consite
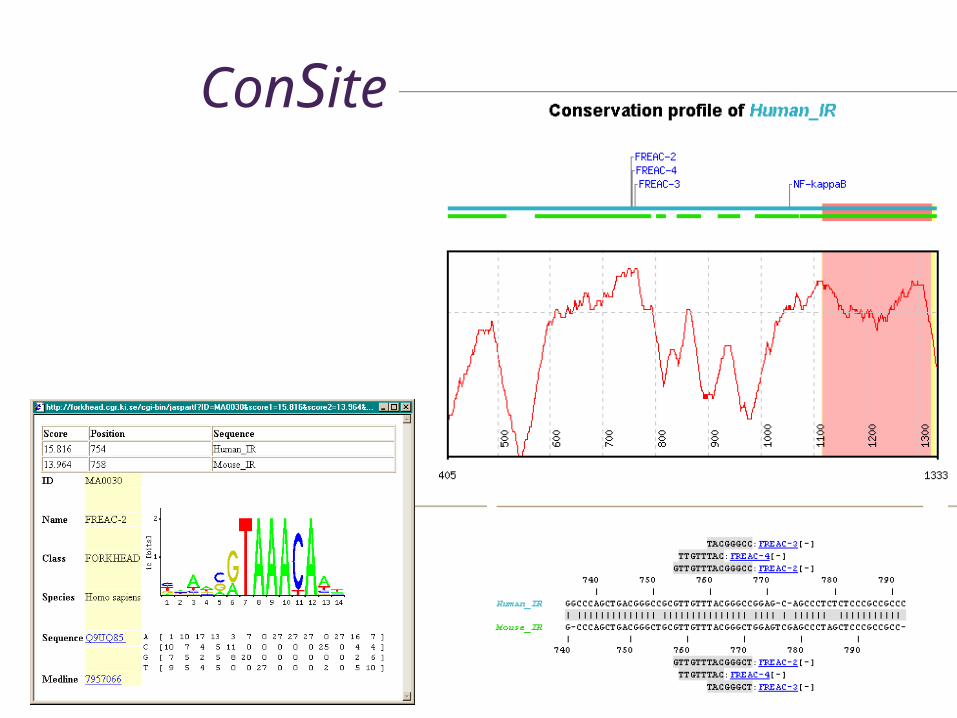
ConSite

Visualisation• Easier to grasp large data volumes• Programs
• Dot plot (e.g. Dotter)• PIP• Alfresco• VISTA
• Genome comparative resources• VISTA genome browser• UCSC• Ensembl
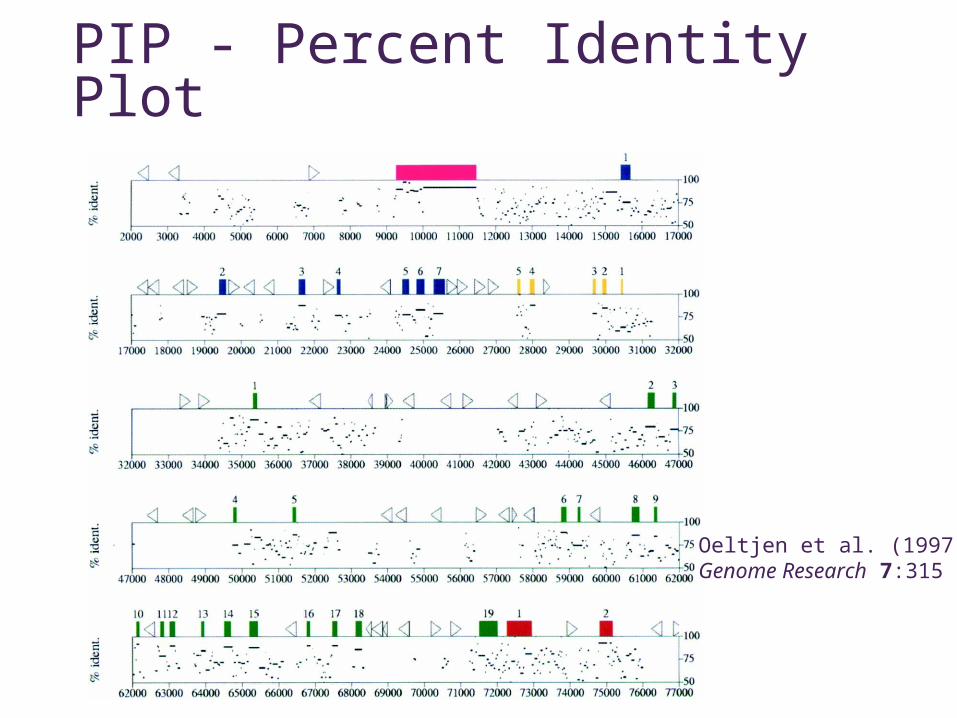
PIP - Percent Identity Plot
Oeltjen et al. (1997)Genome Research 7:315

Alfresco
• Over-all control of comparative analysis• Display and summarize results from external
analysis programs
Tool for comparative genome sequence analysisTool for comparative genome sequence analysis
Jareborg & Durbin Genome Research 10:1148–1157

Alfresco FeaturesAlfresco Features• Interactive graphical interfaceInteractive graphical interface• Uses external programs for analysisUses external programs for analysis
• Dotter - interactive dotplot programDotter - interactive dotplot program• Blastn alignments - finds conserved blocksBlastn alignments - finds conserved blocks• DBA - detects and aligns conserved blocksDBA - detects and aligns conserved blocks• Cpg - detects CpG islandsCpg - detects CpG islands• RepeatMasker - identifies repeatsRepeatMasker - identifies repeats• Genscan - gene predictionGenscan - gene prediction• GeneWise - gene prediction using homologous protein GeneWise - gene prediction using homologous protein
sequence sequence • est_genome - gene prediction using homologous RNA est_genome - gene prediction using homologous RNA
sequencesequence
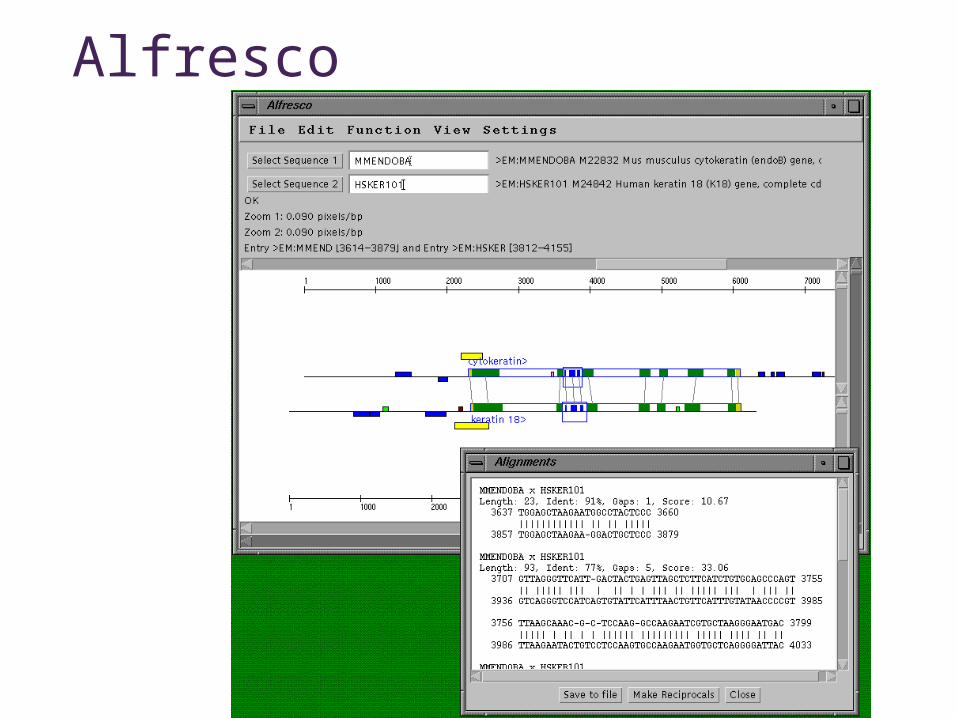
Alfresco
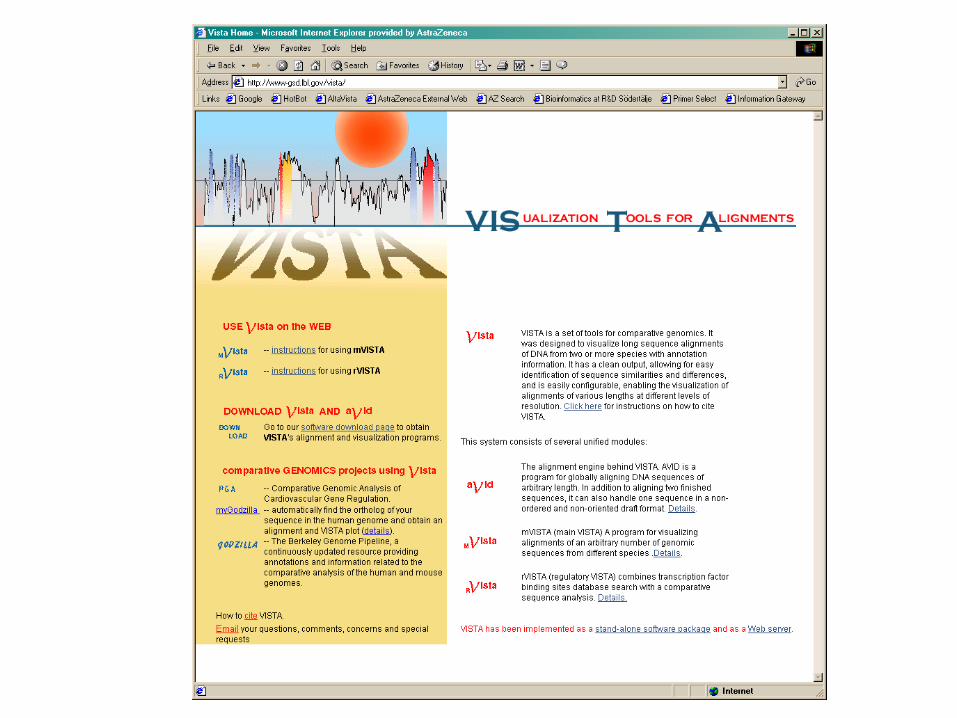

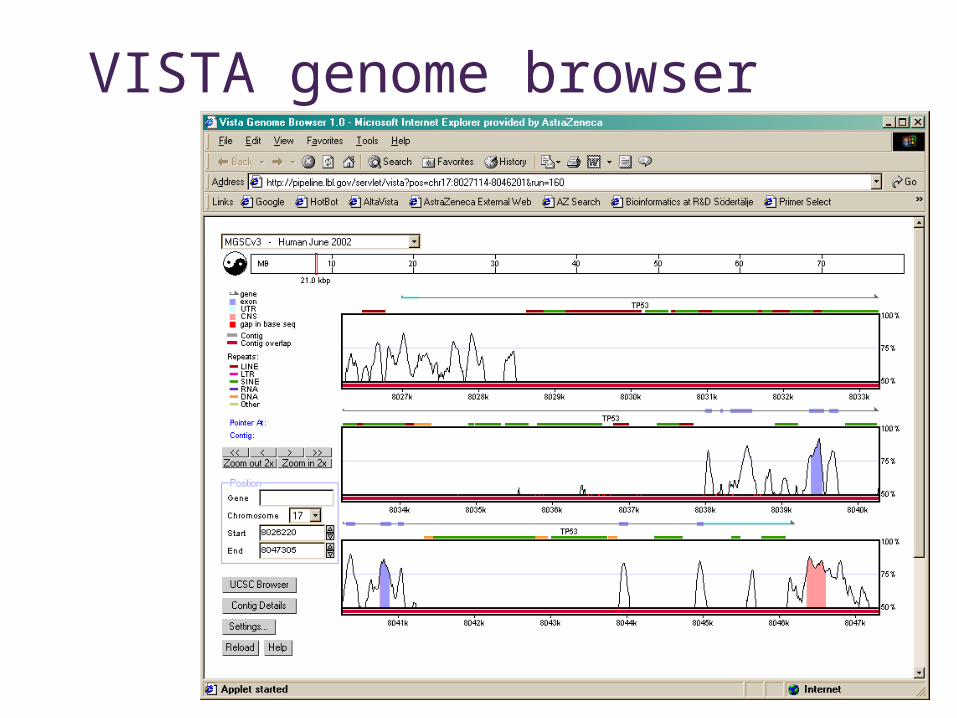
Vista Genome Browser
• Human – Mouse - Rat comparisons• VISTA viewer• http://pipeline.lbl.gov/
VISTA genome browser
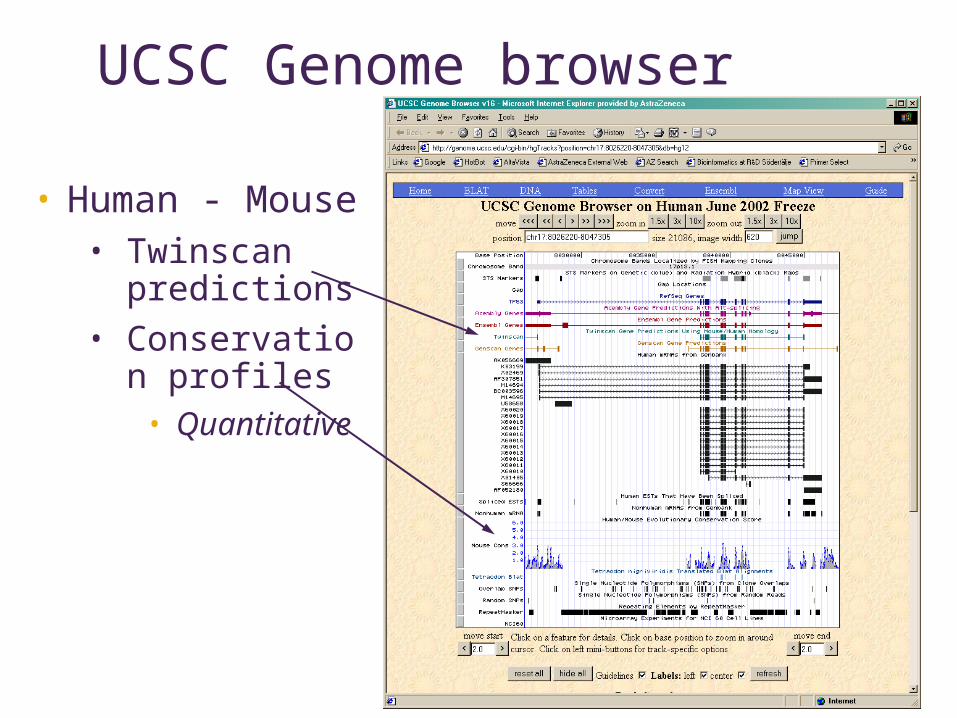
UCSC Genome browser
• Human - Mouse• Twinscan
predictions• Conservation
profiles• Quantitative
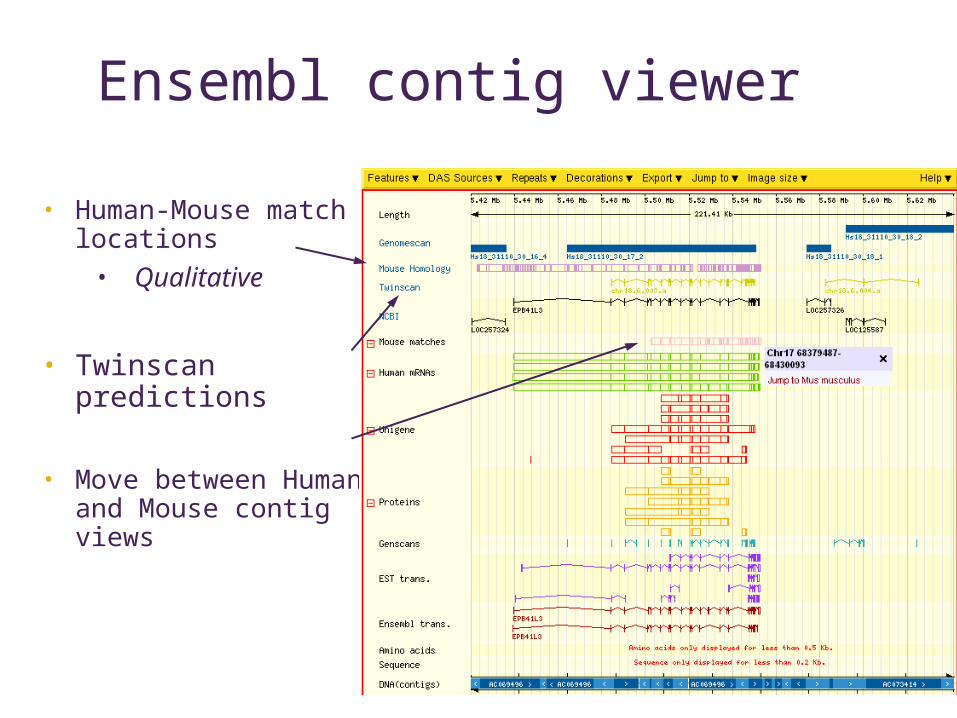
Ensembl contig viewer
• Human-Mouse match locations
• Qualitative
• Twinscan predictions
• Move between Human and Mouse contig views

Comparative Analysis Examples
• Interspecies non-coding regions conservation• Coding region predictions• Regulatory region predictions

Comparative Analysis of Noncoding Regions of 77 Mouse and Human Gene Pairs
Jareborg, Birney, and Durbin.(1999)
Genome Research 9:815
• How conserved are non-coding regions between mouse and human?
• Measure of conservation?• % identity• fraction conserved

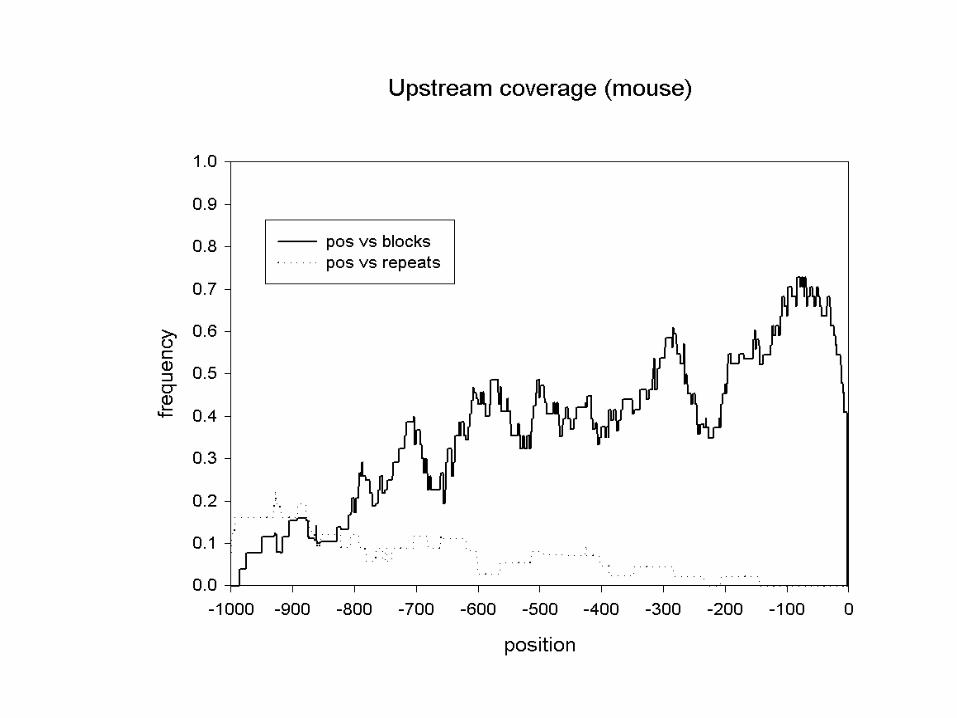
A “typical” intron

mouse/human data set• Genomic sequences from the EMBL database
containing 78 pairs of mouse-human orthologous genes
• Features as defined in feature tables• Corresponding features aligned with DBA:
• Fraction covered by blocks >60 % identical:• Upstream regions: 36 %• 5’ UTRs: 49 %• Introns: 23 %• 3’ UTRs: 56 %
• Sizes:• 20 - 700 bp
Jareborg, Birney & Durbin. Genome Research 9:815-824
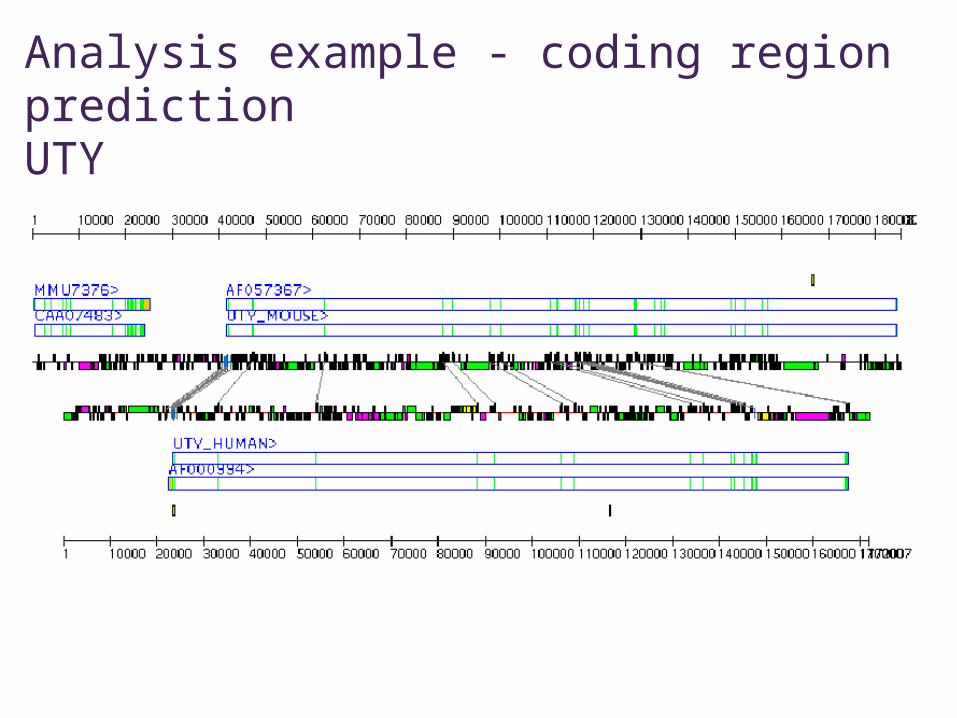
Analysis example - coding region predictionUTY

Analysis example - cont.

Analysis example - Regulatory regions
• BTK - Bruton’s Tyrosine Kinase• agammaglobulinemia• Expression
• early stages of B-cell differentiation• myeloid cell lines• not in T cells
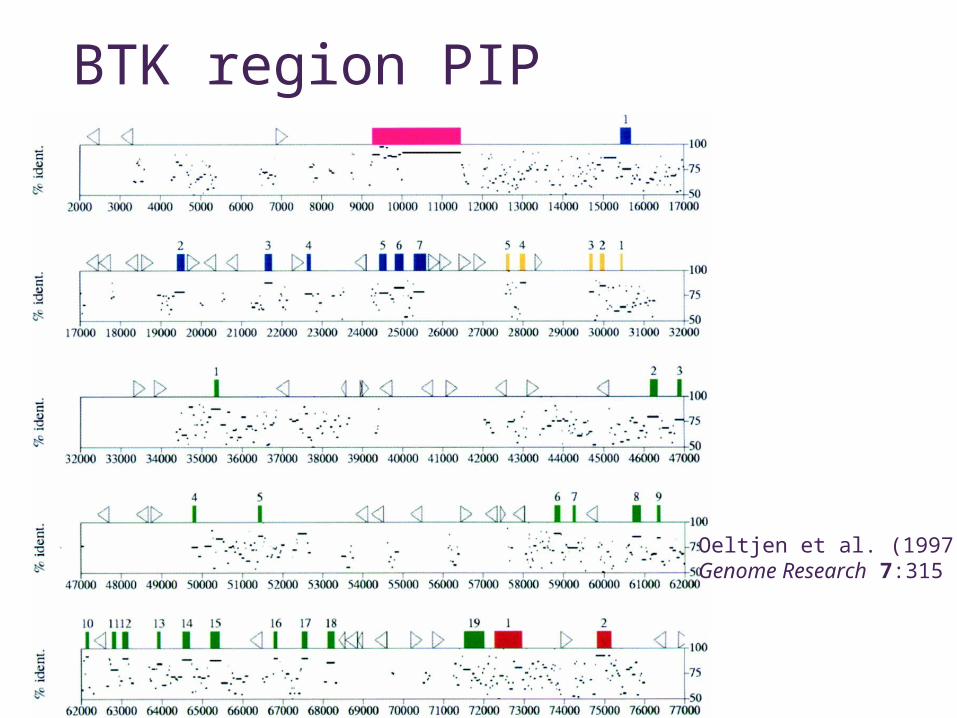
BTK region PIP
Oeltjen et al. (1997)Genome Research 7:315
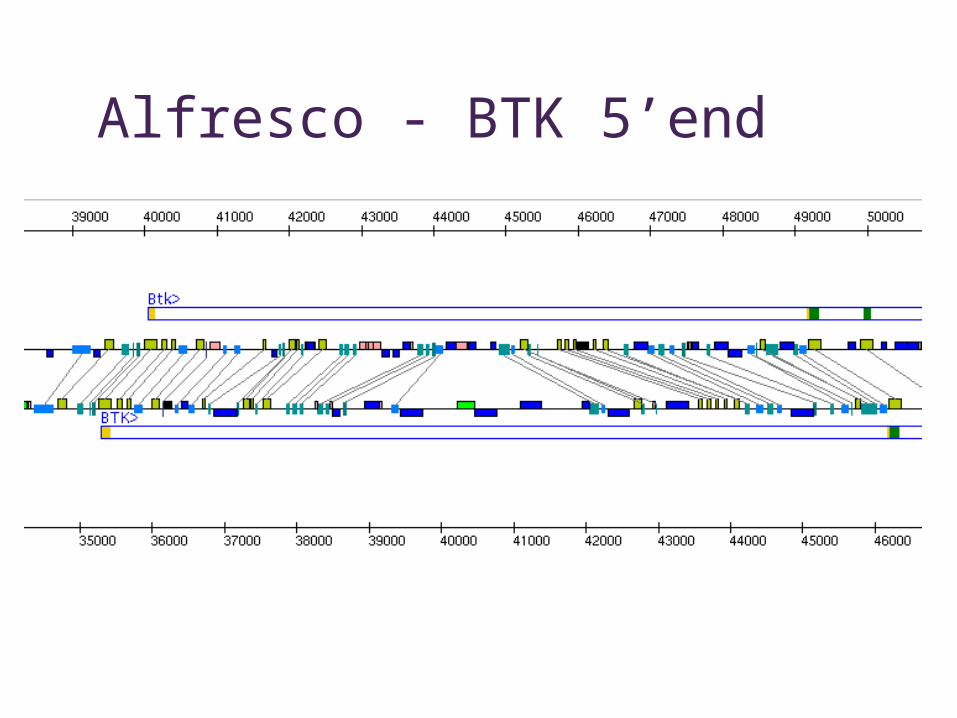
Alfresco - BTK 5’end
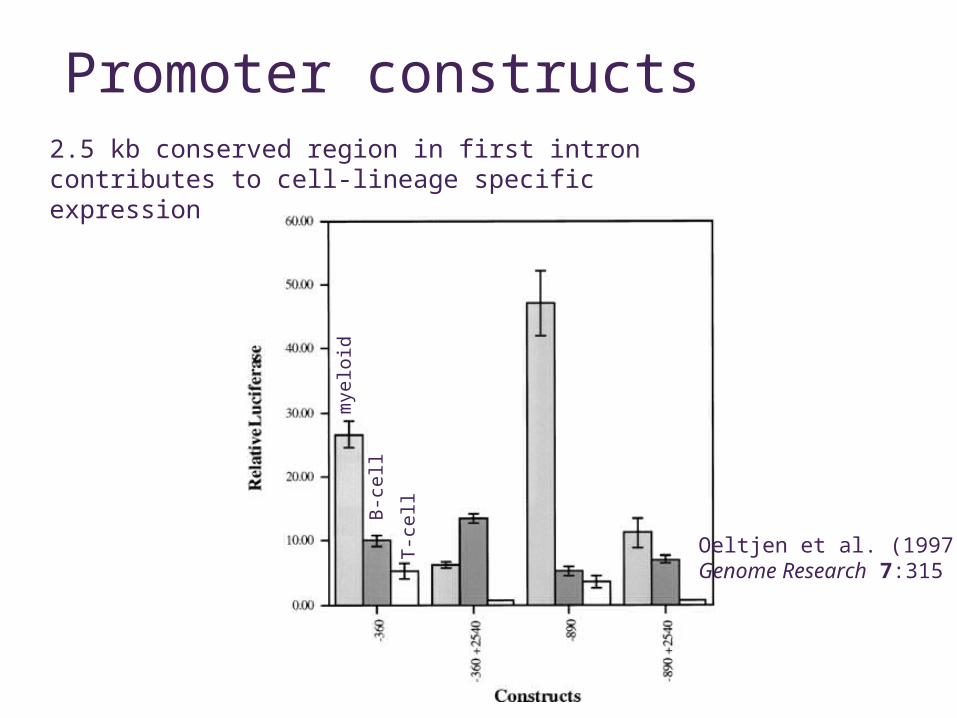
Promoter constructs
mye
loid
B-c
ell
T-c
ell
Oeltjen et al. (1997)Genome Research 7:315
2.5 kb conserved region in first intron contributes to cell-lineage specific expression

Comparative AnalysisIssues for the future
• Faster/better algorithms for aligning vertebrate genomes
• Multiple alignments• Comparing several species can give clues to which
regulatory sequences are of a basic nature, and which are lineage specific
• Cataloguing of comparative data• Better visualisation
• Whole syntenic region <> nucleotide level• Multiple genome sequences

Future Issues - cont.
• Genome evolution • macro scale• molecular evolutionary rates• repeats
• Transcriptional regulatory regions• definition/modelling
• identification of combinations of conserved TFBSs coupled with gene expression data
• prediction

Fin





























