Embed Size (px)
Citation preview

Genome evolution: a sequence-centric approach
Lecture 10: Selection in protein coding genes
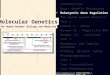
Probabilistic models
Inference
Parameter estimation
Genome structure
Mutations
Population
Inferring Selection
(Probability, Calculus/Matrix theory, some graph theory, some statistics)
Simple Tree ModelsHMMs and variantsPhyloHMM,DBNContext-aware MMFactor Graphs
DPSamplingVariational apx.LBP
EMGeneralized EM (optimize free energy)
Tree of lifeGenome SizeElements of genome structureElements of genomic information
Today refs: See Gruer/Li chapter 3/4, and papers we cite
Models for populationsDriftSelection and fixationDraft

Protein genes: codes and structure
1 2 3 codons
Intron/exons
Domains
Conformation
Degenerate code
Recombination easier?
Epistasis: fitness correlation between two remote loci
5’ utr3’ utr

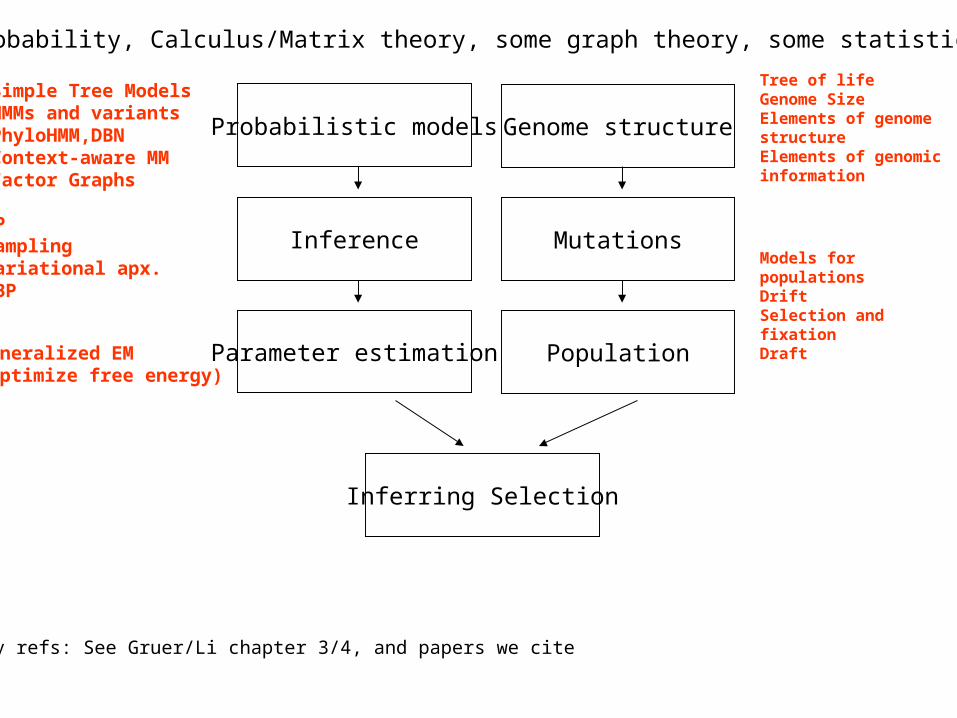
Identifying protein coding genes
From mRNAs
Spliced ESTs : short low quality fragments that are easier to get
Identifying protein coding genes
Computationally identify protein coding genes using probabilistic models is a common genome annotation approach
Comparative/Conservation based methods are becoming increasingly popular

Questions on protein function and evolution
Identification:• Identify protein coding genes
– Not yet resolved for new species, but with new technology this question may be less important than before
Structure/Function:• Define functional domains
– Highly important for understanding protein function• Which parts of the proteins are “important” (e.g., catalytic?)
– Difficult since structural modeling is hard and context dependent
Evolution • Identify places and times where a new protein feature emerged
– Positive selection• Understand mutation/selection through codon degeneracy• Understanding processes of duplication and diversitification

The classical analysis paradigm
BLAT/BLAST
Target sequence
Genbank
Matching sequences CLUSTALW
ACGTACAGAACGT--CAGAACGTTCAGAACGTACGGA
Alignment
PhylogeneticModeling
Analysis: rate, Ka/Ks…
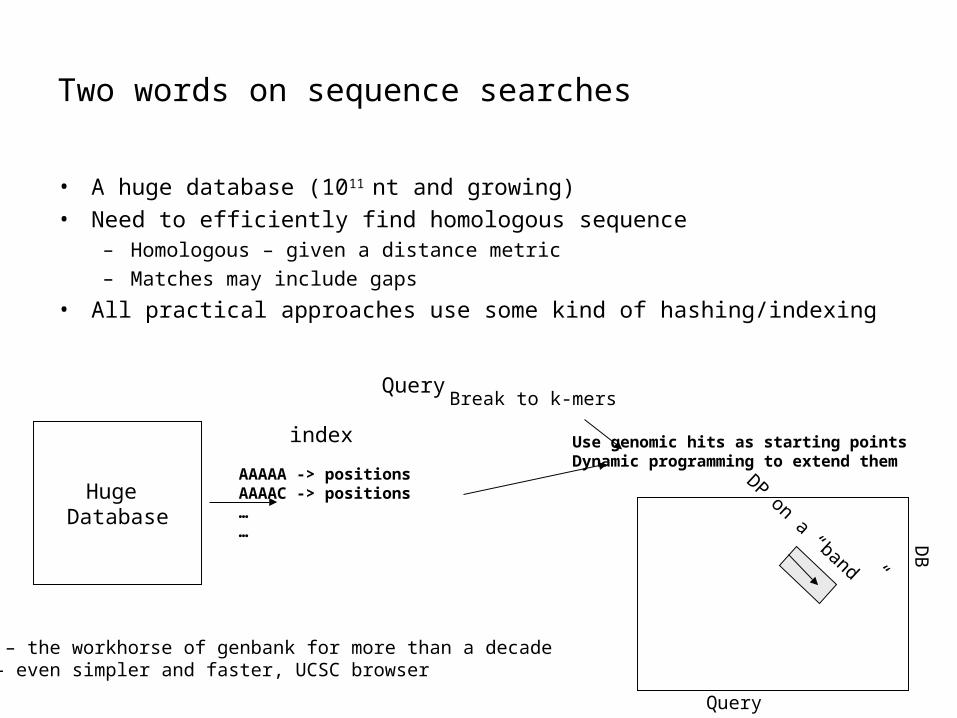
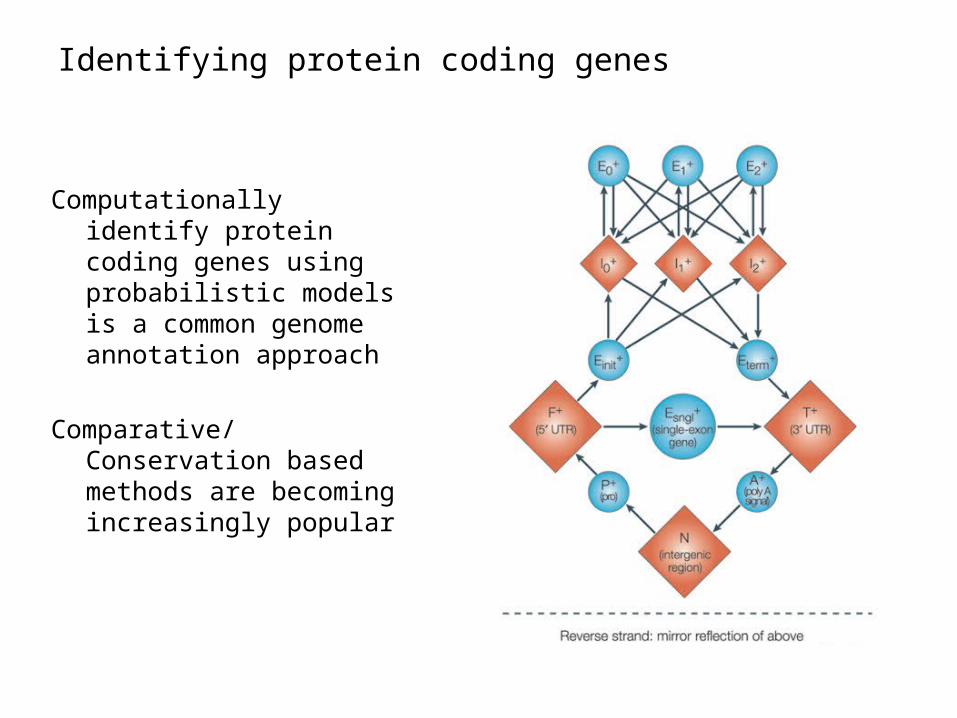
Two words on sequence searches
• A huge database (1011 nt and growing)
• Need to efficiently find homologous sequence– Homologous – given a distance metric– Matches may include gaps
• All practical approaches use some kind of hashing/indexing
Huge Database
AAAAA -> positionsAAAAC -> positions……
index
QueryBreak to k-mers
Use genomic hits as starting pointsDynamic programming to extend them
BLAST – the workhorse of genbank for more than a decadeBLAT – even simpler and faster, UCSC browser
DP on a “band”
Query
DB

Clustalw and multiple alignment
• ClustalW is the semi-standard multiple alignment algorithm when sequences are relatively diverged and there are many of them
ClustalW
Compute pairwise sequence distances (using pairwise alignment)
Build a guide-tree: approximating the phylogenetic relations between the sequences
“Progressive” alignment on the guide tree
S2S1
S4S3
S5 Dist(s1,s2) = best pair align
DistanceMatrix
NeighborJoining
Guide tree is based on pairwise analysis!
From the leafs upwards:Align two children given their “profiles”Several heuristics for gaps
• Other methods are used to multi-align entire genomes, especially when one well annotated model genome is compared to several similar species. Think of using one genome as a “scaffold” for all other sequences.

Nucleotide substitution models
• For nucleotides, fewer parameters are needed:
A
C T
G
A
C T
G
Jukes-Kantor (JK) Kimura
• But this is ignoring all we know on the properties of amino-acids!

Simple phylogenetic modeling: PAM/BLOSSOM62
• Given a multiple alignment (of protein coding DNA) we can convert the DNA to proteins. We can then try to model the phylogenetic relations between the proteins using a fixed rate matrix Q, some phylogeney T and branch lengths ti
When modeling hundreds/thousands amino acid sequences, we cannot learn from the data the rate matrix (20x20 parameters!) AND the branch lengths AND the phylogeny.
Based on surveys of high quality aligned proteins, Margaret Dayhoff and colleuges generated the famous PAM (Point Accepted mutations): PAM1 is for 1% substitution probability.
Using conserved aligned blocks, Henikoff and Henikoff generated the BLOSUM family of matrices. Henikoff approach improved analysis of distantly related proteins, and is based on more sequence (lots of conserved blocks), but filtering away highly conserved positions (BLOSUM62 filter anything that is more than 62% conserved)

Universal amino-acid substitution rates?
Jordan et al., Nature 2005
“We compared sets of orthologous proteins encoded by triplets of closely related genomes from 15 taxa representing all three domains of life (Bacteria, Archaea and Eukaryota), and used phylogenies to polarize amino acid substitutions. Cys, Met, His, Ser and Phe accrue in at least 14 taxa, whereas Pro, Ala, Glu and Gly are consistently lost. The same nine amino acids are currently accrued or lost in human proteins, as shown by analysis of non-synonymous single-nucleotide polymorphisms. All amino acids with declining frequencies are thought to be among the first incorporated into the genetic code; conversely, all amino acids with increasing frequencies, except Ser, were probably recruited late. Thus, expansion of initially under-represented amino acids, which began over 3,400 million years ago, apparently continues to this day. “

Phylogenetic modeling: ML
• Given a rate matrix and an alignment, a phylogenetic tree and branch lengths are estimated to maximize the likelihood.
• Frequently the ML gene tree is NOT the ML/fossil based species tree– due to border line statistics– Or due to real lateral effects/duplications problems
• Adding rate variation may be critical for elucidating protein function!
• Finding the ML model is notoriously difficult:– Estimating the ML branch lengths given a tree can be done using EM– Estimating the optimal tree topology can be done in several ways, some
using sampling techniques we learned

ML Phylogeny using sampling
• Develop a metropolis algorithm to sample from Tree topologies and branch lengths
• See how easy it is to use Sampling:• Define a proposal distribution T,t -> T’,t’
• You accept with P ~ P(T’,t’|S)/P(T,t’|S) = Pr(S|T’,t)/Pr(S|T,t)
• Collect samples after some burn in period to compute relevant features probabilities
• Sampling can be used to ask different question on the tree:• What is the probability of a set of species being mono-phyletic
• Chance of particular branch point
• And of course, what is the optimal (MAP) Tree
• We can tune branch lengths further using EM.
• Critical to the success of the Metropolis algorithm is the proposal distribution we are using– Using something completely random is not a good idea (we will be stuck in
relatively good positions for a long time)
– Too small changes are also problematic

Analysis: rate variation
• If our ML model include rate variation, we can use the inferred rates to annotate the protein
• Same can be done by constructing a conservation profile, even if the model is simplistic.
• Shown here are example from Tal Pupko’s work on the Rate4Site and ConSurf programs

How prevalent are compensatory mutations?
PDB structuresHomology modelling
3-Alignments
Pairs of interacting residues
Rat Mouse Human
Choi et al, Nat Genet 2005
Find pairs of mutations in interacting residues (DRIP)Coupled: occurring in the same lineageUncoupled: occurring in different lineages

Synonymous vs. non synonymous mutations
• The structure of the genetic code is a powerful tool in characterizing evolutionary trends• This is because at degenerate positions of codon we can deterministically distinguish between
mutations that change the protein and mutation that do not• This provide us with a powerful “internal control” – we are comparing two different types of evolutionary
events at the same loci, so all sources of variation in the mutational process are not affecting us.
Given aligned proteins, we can count:
MA – number of non-synonymous changes
Ms – number of synonymous changes
We then want to estimate:• Ka – rate of non-synonymous mutations (per syn site)• Ks – rate of synonymous mutations (per syn site)• Estimate V(Ka), V(Ks)
• Comparing Ka and Ks can provide evolutionary insights:– Ka/Ks<<1: negative selection may be purging protein modifying mutations– Ks/Ka>>1: positive selection may help acquiring a new function– (statistics using, e.g., T-test)
Non-SynSyn
ChangeMAMS
No Change
NA-MANS-MS
Chi-square test
Average number of sites

From dN/dS to Ka and Ks
• Classically, we start by looking at noncoding sequence and a one-parameter substitution model
• In a first approximation K = d/N = p (N being the number of loci), but this ignore multiple mutations
• Taking into account the model:
(L is the number of loci)
If we assume NA,NS non synonymous/synonymous sites:
A more realistic approach:
• classify sites to 0-fold, 2-fold and 4-fold degenerate (consider isoleucine a two fold site) .
• For each type one count the number of syn/non-syn transitions or transversions
• Express the mean KA, and KS as a function of these counts (Kimura, see Graur/Li book)
)3
41ln(
4
3pK
2
2
34
1
)(
pL
ppKV
)3
)(41ln(
4
3
SS N
dSK )
3
)(41ln(
4
3
AA N
dNK
• How would you do it using methods we learned in the course?

Codon bias
• Different codons appears in significantly different frequencies, which is not expected assuming neutrality
• Bias is measured in several ways, most popular is the codon adaptation index:
• Possible sources of bias:
– Selection for translational efficiency given different tRNA abudnances• Highly expressed genes tend to have stronger codon adaptation indices
– Sequence context mutational effects• E.g. CpGs are highly mutable
– Selection for low insertion/deletion potential
• Weak selection for codon bias should be stronger for genomes with larger effective population size. In some cases this is true
L
i
iLi X
XCAI
1
max,1
Codon frequency divided by the frequency of the synonymous codon with maximal frequency

Positive selection in humans vs chimp
Nielsen et al., 2005 PloS Biol
Testis genes: P<0.0001
Immunity genes, Gematogenesis, Olfaction P<1e-5Inhibition of apoptosis P<0.005Sensory perception P<0.02
Kn vs Ks Looking at trends for families of genes
Significantly enriched functions/tissues Example
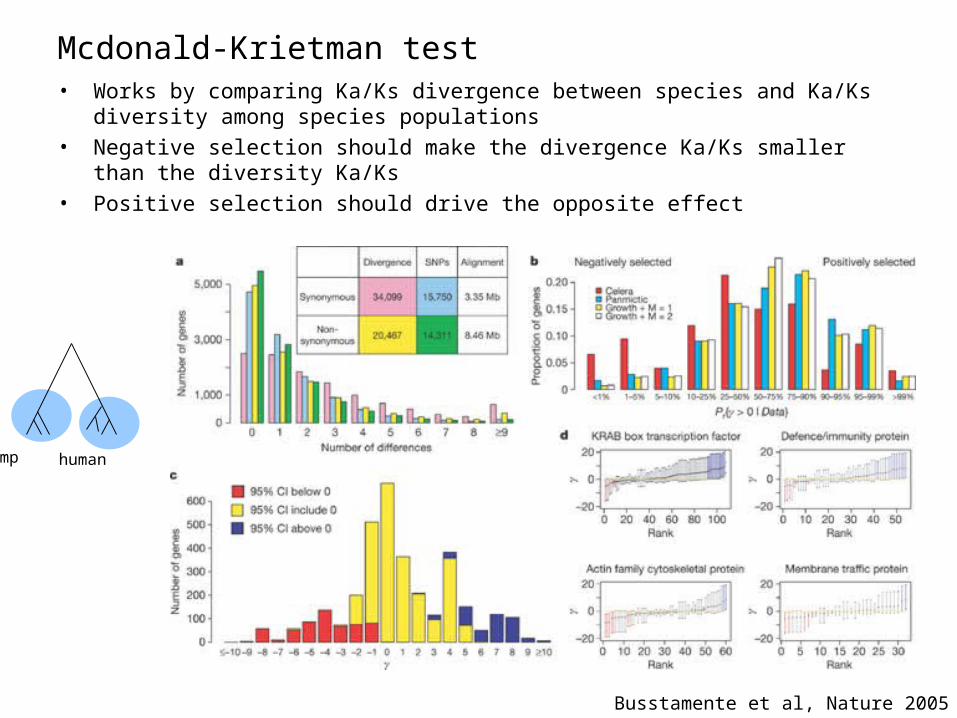
Mcdonald-Krietman test• Works by comparing Ka/Ks divergence between species and Ka/Ks diversity among
species populations• Negative selection should make the divergence Ka/Ks smaller than the diversity
Ka/Ks• Positive selection should drive the opposite effect
Busstamente et al, Nature 2005
humanchimp

Codon volatility
• Volatility is the number/fraction of adjacent non-synonymous codons
• Genes under positive selection may have increased volatility
• Think about the distance from the stationary codon distribution
• No need to align!!
Plotkin et al, Nature 2004
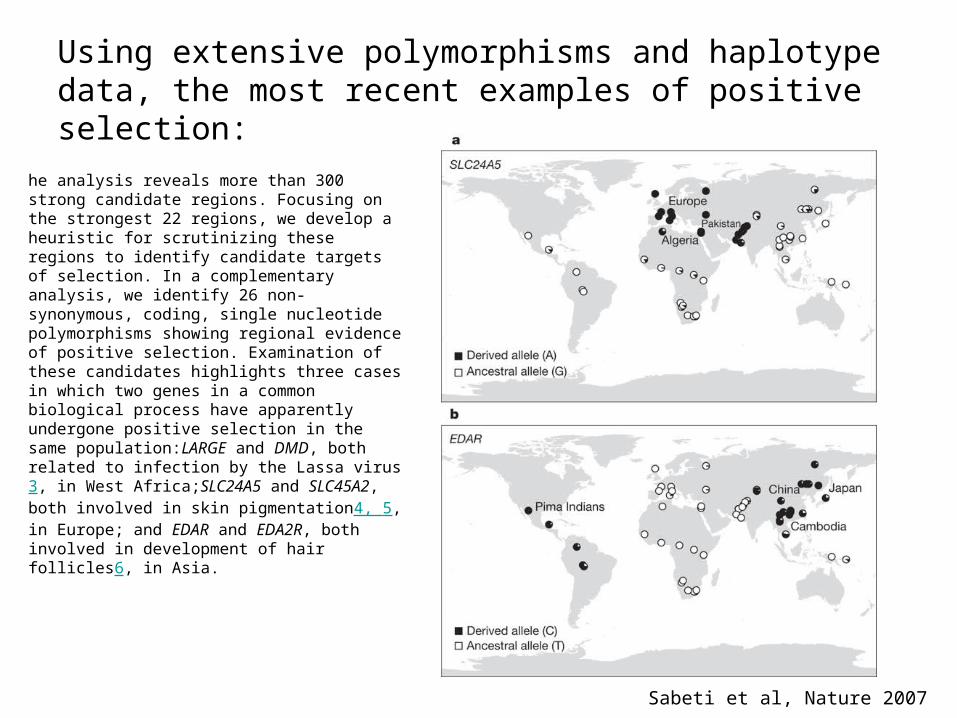
Using extensive polymorphisms and haplotype data, the most recent examples of positive selection:
Sabeti et al, Nature 2007
he analysis reveals more than 300 strong candidate regions. Focusing on the strongest 22 regions, we develop a heuristic for scrutinizing these regions to identify candidate targets of selection. In a complementary analysis, we identify 26 non-synonymous, coding, single nucleotide polymorphisms showing regional evidence of positive selection. Examination of these candidates highlights three cases in which two genes in a common biological process have apparently undergone positive selection in the same population:LARGE and DMD, both related to infection by the Lassa virus3, in West Africa;SLC24A5 and SLC45A2, both involved in skin pigmentation4, 5, in Europe; and EDAR and EDA2R, both involved in development of hair follicles6, in Asia.
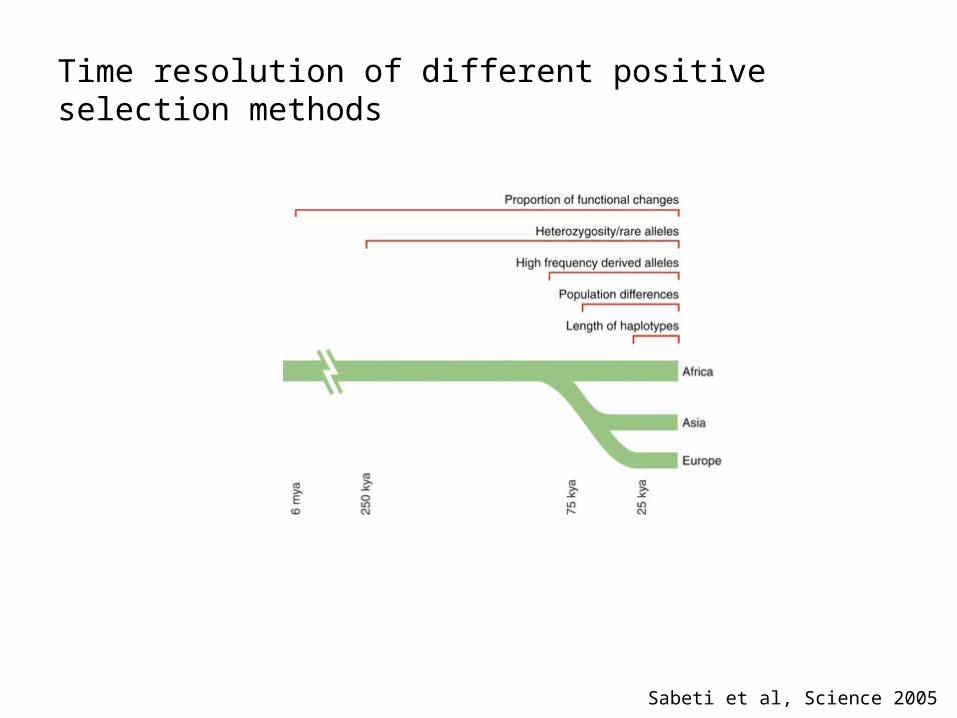
Time resolution of different positive selection methods
Sabeti et al, Science 2005

Molecular clocks and lineage acceleration
• How universal is the rate of the evolutionary process?• Mutations may depend on the number of cell division and thus in the
length of generation• Mutations depends on the genomic machinery to prevent them
(Lynch?)• Mutations may also depend on the environment• The molecular clock (MC) hypothesis state that evolution is working
in a similar rate for all lineages
A B C
O
Relative rate test:
KOA – KOB = 0 ?
Test: KCA – KCB

Different molecular clocks in apes and primates
Kim et al., 2006 PLoS genet

The real challenges ahead
• A model for protein evolution that include multiple selective effects• Context-dependent mutations with different molecular clocks• Selection for codon bias• Epistasis (interaction between positions)• Possible additional roles for exon’s DNA
We did not talk about it today, but:
• Protein coding genes are the outcome of complex duplication/loss process
• how different proteins interact and how it affect their evolution?