Embed Size (px)
DESCRIPTION
Gangguan Hemostasis(Idiopathic Thrombositopenic Purpura, Haemophilia, von Willebrand Disease)
Citation preview

BAB 1
PENDAHULUAN
Sistem hemostasis merupakan mekanisme tubuh dalam mengontrol respon terhadap perdarahan
atau terjadinya trombosis yang berlebihan sehingga proses trombogenesis dan proses fibrinolisis
dalam keadaan seimbang. Proses hemostasis pada keadaan normal membantu menghentikan
perdarahan dan bila berlebihan akan menimbulkan oklusi trombotik dan infark sistemik.
Trombosis terjadi bila ada ketidakseimbangan antara faktor trombogenik dan mekanisme
proteksi.
Yang termasuk dalam faktor-faktor trombogenik adalah kerusakan dinding pembuluh darah,
rangsangan agregasi trombosit, aktivasi koagulasi darah dan stasis, sedangkan keadaan-keadaan
yang berpengaruh dalam mekanisme proteksi adalah endotel yang utuh, inhibitor protease dari
sistem koagulasi, inaktivasi koagulasi oleh hati dan sistem fibrinolitik.
1

BAB 2
DATA PELAKSANAAN TUTORIAL
I. JUDUL BLOK
Hematologi, Limpopoitik dan Immunologi
II. JUDUL SKENARIO
Gangguan Hemostasis
III. NAMA TUTOR
dr. Yolanda
IV. DATA PELAKSANAAN TUTORIAL
Tutorial 1
Hari/tanggal : Senin, 17 Maret 2014
Waktu : 07.30 – 09.00 wib
Tempat : Ruang tutorial SGD 8
Tutorial 2
Hari/tanggal : Kamis, 20 Maret 2014
Waktu : 07.30 – 09.00 wib
Tempat : Ruang tutorial SGD 8
Pleno
Hari/tanggal : Sabtu, 22 Maret 2014
Waktu : 10.20 – 12.00 wib
Tempat : Ruang Kuliah
2

BAB 3
SKENARIO
Seorang laki-laki 15 tahun datang berobat ke RS. Dari anamnesa ditemukan epistaksis (+),
skorbut (+). Pada pemeriksaan laboratorium dijumpai leukosit 10.000/mm3, Hb : 11 gr%,
trombosit 100.000/mm3. Protombin Time 18 detik, Activated Partial Tromboplastin Time 45
detik. Apakah yang terjadi dan bagaimana pengobatannya?
3

BAB 4
PEMBAHASAN SKENARIO
I. KLARIFIKASI ISTILAH
1. Epistaksis
Perdarahan pada mukosa nasal atau mimisan.
2. Skorbut
Penyakit yang disebabkan kekurangan vitamin C atau sariawan.
3. Protombin Time
Masa pembekuan darah.
4. Activated Partial Tromboplastin Time
Masa aktivasi tromboplastin dalam darah.
II. MENETAPKAN MASALAH
1. Seorang laki-laki datang berobat ke RS, dengan hasil anamnesa ditemukan:
- Epistaksis (+)
- Skorbut (+)
Pemeriksaan fisik dijumpai:
- Petechiae
- Ekimosis
2. Pada pemeriksaan laboratorium, dijumpai:
- Leukosit 10.000/mm3
- Hb : 11 gr%
- Trombosit 100.000/mm3
- Protombin Time 18 detik
- Activated Partial Tromboplastin Time 45 detik
4

III. ANALISIS MASALAH
1. Epistaksis : disebabkan karena infeksi dan trauma.
Skorbut : kekurangan vitamin C.
Petechiae dan ekimosis : karena gangguan pembuluh darah akibat menurunnya
leukosit dan trombosit.
2. Trombosit menurun (normal (150.000 – 450.000/mm3)
Leukosit normal (5000 – 10.000/mm3)
Hb normal (11 gr% untuk anak)
PT : memanjang (normal 12-15 detik)
APTT : memanjang (normal 30-40 detik)
IV. KESIMPULAN SEMENTARA
Seorang laki-laki 15 tahun mengalami gangguan hemostasis.
V. TUJUAN PEMBELAJARAN
1. Mekanisme Hemostasis dan Faktor Koagulasi
2. Penyakit-penyakit pada Gangguan Hemostasis
3. Defenisi Gangguan Hemostasis
4. Etiologi Gangguan Hemostasis
5. Epidemiologi Gangguan Hemostasis
6. Patofisiologi Gangguan Hemostasis
7. Gejala Klinis Gangguan Hemostasis
8. Diagnosa dan Diagnosa Banding Gangguan Hemostasis
9. Penatalaksanaan Gangguan Hemostasis
10. Pencegahan Gangguan Hemostasis
11. Komplikasi Gangguan Hemostasis
12. Prognosis Gangguan Hemostasis
5

BAB 5
TINJAUAN TEORI
1. Komponen Penting Dalam System Hemostasis
Sistem Hemostasis pada dasarnya terbentuk dari tiga kompartemen hemostasis yang
sangat penting dan sangat berkaitan yaitu trombosit, protein darah dan jaring-jaring
pembuluh darah. Agar terjadi peristiwa hemostasis yang normal, trombosit harus
mempunyai fungsi dan jumlah yang normal. Sistem protein darah sangat berperan
penting tidak hanya sebagai protein pembekuan akan tetapi sangat berperan dalam dalam
fisiologi perdarahan dan trombosis.
a. Pembuluh darah
Pembuluh darah sangat besar peranannya dalam sistem hemostasis. Dinding
pembuluh darah terdiri dari tiga lapisan morfologis: intima, media, dan adventitia.
Intima terdiri dari (1) selapis sel endotel non trombogenik yang berhubungan
langsung dengan pembuluh darah dan (2) membran elastik interna. Media dibentuk
oleh sel otot polos yang ketebalannya tergantung dari jenis arteri dan vena serta
ukuran pembuluh darah. Adventitia terdiri dari suatu membran elastik eksterna dan
jaringan penyambung yang menyokong pembuluh darah tersebut. Gangguan
pembuluh darah yang terjadi seringkali berupa terkelupasnya sel endotel yang diikuti
dengan pemaparan kolagen subendotel dan membran basalis. Gangguan ini terjadi
akibat asidosis, endotoksin sirkulasi, dan komplek antigen/antibodi sirkulasi.
Fungsi pembuluh darah meliputi permiabilitas yang apabila meningkat akan berakibat
kebocoran pembuluh darah fragilitas yang apabila meningkat menyebabkan pecahnya
pembuluh darah dan vaso konstriksi yang menyebabkan sumbatan vaskuler.
b. Trombosit
Trombosit merupakan komponen sistem hemostasis yang amat penting dan kompleks.
Trombosit adalah kuntum sel yang dihasilkan dari megakariosit. Trombosit tidak
punya inti dan disusun dari suatu zona perifer yang terdiri dari suatu glukokaliks
sebelah luar, membran plasma, dan suatu sistem kanalikuler yang terbuka. Dalam
6

zona perifer terdapat suatu zona "sol-gel" yang tersusun dari mikrotubulus,
mikrofilamen, tubulus yang padat dan trombostenin yaitu protein trombosit yang
dapat berkerut. Zona organel mengandung bahan-bahan padat, granula alfa dan
mitokondria. Trombosit berbentuk bulat kecil atau cakram oval. Diameternya 2-4
mikron. Sel megakariosit yang menghasilkan trombosit merupakan sel yang sangat
besar dalam susunan hemopoitik yang berada dalam sum-sum tuilang dan tidak
meninggalkannya untuk memasuki darah.
Konsentrasi normal trombosit dalam darah adalah antara 150.000-350.000 mm3.
Meskipun tidak mempunyai inti, trombosit mempunyai ciri fungsional sebagai sebuah
sel. Dalam sitoplasma terdapat molekul aktif seperti :
1) aktin dan miosin yang menyebabkan trombosit berkontraksi,
2) sisa retikulum endoplasma dan aparatus golgi yang mensintesis enzim dan
menyimpan besar ion kalsium,
3) sistem enzim yang mampu membentuk ATP dan ADP,
4) sistem enzim yang mensintesis prostaglandin,
5) suatu protein penting yaitu faktor pemantap fibrin, dan
6) faktor pertumbuhan yang dapat menyebabkan penggandaan dan pertumbuhan sel
endotel pembuluh darah.
Pada membran sel trombosit terdapat lapisan glikoprotein yang menyebabkan
trombosit bisa melekat pada pembuluh darah yang luka, terutama pada sel endotel
yang rusak dan jaringan kolagen yang terbuka. Trombosit juga mengandung
fosfolipid yang dapat mengaktifkan salah satu sistem pembekuan darah yang disebut
sistem intrinsik. Pada membran trombosit terdapat enzim adenilat siklase yang bila
diaktifkan dapat menyebabkan pembentukan AMP siklik yang menggiatkan aktifitas
dalam trombosit. Jadi trombosit merupakan struktur yang sangat aktif, waktu
paruhnya 8-12 hari setelah itu mati. Trombosit kemudian diambil dari sirkulasi,
terutama oleh makrofag jaringan. Lebih dari separuh trombosit diambil oleh
makrofag pada waktu darah melewati kisi trabekula yang tepat. (Guyton, 1997)
7

a. Protein darah
Protein darah yang terlibat dalam hemostasis meliputi protein koagulasi, protein
enzim fibrinolitik sistem kinin dan sistem komplemen serta inhibitor yang
terdapat pada sistem-sistem tersebut. Sistem protein koagulasi terpusatkan pada
tiga reaksi yaitu pada reaksi pembentukan faktor Xa, reaksi pembentukan
trombin, dan reaksi pembentukan fibrin. Protease serin adalah faktor pembekuan
yang diaktifkan pada reaksi pembentukan faktor Xa dan bagian yang aktif untuk
aktivitas enzim adalah asam amino serin. Pada ketiga reaksi kunci tersebut
memerlukan komponen-komponen seperti substrat, enzim, kofaktor,
fosfolipoprotein dan kalsium. (Sodeman, 1995)
Mekanisme Hemostasis
Istilah hemostasis berarti pencegahan hilangnya darah. Bila pembuluh darah mengalami
cidera atau pecah, hemostasis akan terjadi. Peristiwa ini terjadi melalui beberapa cara
yaitu : vasokonstriksi pembuluh darah yang cidera, pembentukan sumbat trombosit,
pembekuan darah, dan pertumbuhan jaringan ikat kedalam bekuan darah untuk menutup
pembuluh yang luka secara permanen. Kerja mekanisme pembekuan in vivo ini
diimbangi oleh reaksi-reaksi pembatas yang normalnya mencegah mencegah terjadinya
pembekuan di pembuluh yang tidak mengalami cidera dan mempertahankan darah berada
dalam keadaan selalu cair.
1. Vasokonstriksi pembuluh darah
Segera setelah pembuluh darah terpotong atau pecah, rangsangan dari pembuluh
darah yang rusak menyebabkan dinding pembuluh berkontraksi sehingga aliran darah
dari pembuluh darah yang pecah barkurang. Kontraksi terjadi akibat refleks syaraf
dan spasme miogenik setempat. Refleks saraf dicetuskan oleh rasa nyeri atau lewat
impuls lain dari pembuluh darah yang rusak. Kontraksi miogenik yang sebagian besar
menyebabkan refleks saraf ini, terjadi karena kerusakan pada dinding pembuluh darah
yang menimbulkan transmisi potensial aksi sepanjang pembuluh darah. Konstriksi
suatu arterioul menyebabkan tertutupnya lumen arteri. (Guyton, 1997)
8

2. Pembentukan sumbat trombosit
Perbaikan oleh trombosit terhadap pembuluh darah yang rusak didasarkan pada
fungsi penting dari trombosit itu sendiri. Pada saat trombosit bersinggungan dengan
pembuluh darah yang rusak misalnya dengan serabut kolagen atau dengan sel endotel
yang rusak, trombosit akan berubah sifat secara drastis. Trombosit mulai
membengkak, bentuknya irreguler dengan tonjolan yang mencuat ke permukaan.
Trombosit menjadi lengket dan melekat pada serabut kolagen dan mensekresi ADP.
Enzimnya membentuk tromboksan A, sejenis prostaglandin yang disekresikan
kedalam darah oleh trombosit. ADP dan tromboksan A kemudian mengaktifkan
trombosit yang berdekatan sehingga dapat melekat pada trombosit yang semula aktif.
Dengan demikian pada setiap lubang luka akan terbentuksiklus aktivasi trombosit
yang akan menjadi sumbat trombosit pada dinding pembuluh. (Guyton, 1997)
3. Pembentukan bekuan darah
Bekuan mulai terbentuk dalam 15-20 detik bila trauma pembuluh sangat hebat dan
dalam 1-2 menit bila trauma pembuluh kecil. Banyak sekali zat yang mempengaruhi
proses pembekuan darah salah satunya disebut dengan zat prokoagulan yang
mempermudah terjadinya pembekuan dan sebaliknya zat yang menghambat proses
pembekuan disebut dengan zet antikoagulan. Dalam keadaan normal zat antikoagulan
lebih dominan sehingga darah tidak membeku. Tetapi bila pembuluh darah rusak
aktivitas prokoagulan didaerah yang rusak meningkat dan bekuan akan terbentuk.
Pada dasarnya secara umum proses pembekuan darah melalui tiga langkah utama
yaitu pembentukan aktivator protombin sebagai reaksi terhadap pecahnya pembuluh
darah, perubahan protombin menjadi trombin yang dikatalisa oleh aktivator
protombin, dan perubahan fibrinogen menjadi benang fibrin oleh trombin yang akan
menyaring trombosit, sel darah, dan plasma sehingga terjadi bekuan darah.
a. Pembentukan aktivator protombin
Aktivator protombin dapat dibentuk melalui dua jalur, yaitu jalur ekstrinsik dan
jalur intrinsik. Pada jalur ekstrinsik pembentukan dimulai dengan adanya
peristiwa trauma pada dinding pembuluh darah sedangkan pada jalur intrinsik,
pembentukan aktivator protombin berawal pada darah itu sendiri.
9

Langkah-langkah mekanisme ekstrinsik sebagai awal pembekuan
1. Pelepasan tromboplastin jaringan yang dilepaskan oleh jaringan yang luka.
Yaitu fosfolipid dan satu glikoprotein yang berfungsi sebagai enzim
proteolitik.
2. Pengaktifan faktor X yang dimulai dengan adanya penggabungan glikoprotein
jaringan dengan faktor VII dan bersama fosfolipid bekerja sebagai enzim
membentuk faktor X yang teraktivasi.
3. Terjadinya ikatan dengan fosfolipid sebagai efek dari faktor X yang
teraktivasi yang dilepaskan dari tromboplastin jaringan . Kemudian berikatan
dengan faktor V untuk membentuk suatu senyawa yang disebut aktivator
protombin.
Gambar 1. Mekanisme ekstrinsik sebagai awal pembekuan (Guyton, 1997)
10

Langkah-langkah mekanisme intrinsik sebagai awal pembekuan
1. Pengaktifan faktor XII dan pelepasan fosfolipid trombosit oleh darah yang
terkena trauma. Bila faktor XII terganggu misalnya karena berkontak dengan
kolagen, maka ia akan berubah menjadi bentuk baru sebagai enzim proteolitik
yang disebut dengan faktor XII yang teraktivasi.
2. Pengaktifan faktor XI yang disebabkan oleh karena faktor XII yang
teraktivasi bekerja secara enzimatik terhadap faktor XI. Pada reaksi ini
diperlukan HMW kinogen dan dipercepat oleh prekalikrein.
3. Pengaktifan faktor IX oleh faktor XI yang teraktivasi. Faktor XI yang
teraktivasi bekerja secara enzimatik terhadap faktor IX dan mengaktifkannya.
4. Pengaktifan faktor X oleh faktor IX yang teraktivasi yang bekerja sama
dengan faktor VIII dan fosfolipid trombosit dari trombosit yang rusak untuk
mengaktifkan faktor X.
5. Kerja dari faktor X yang teraktivasi dalam pembentikan aktivator protombin.
Langkah dalam jalur intrinsic ini pada prinsipnya sama dengan langkah
terakhir dalam jalur ekstrinsik. Faktor X yang teraktivasi bergabung dengan
faktor V dan fosfolipid trombosit untuk membentuk suatu kompleks yang
disebut dengan activator protombin. Perbedaannya hanya terletak pada
fosfolipid yang dalam hal ini berasal dari trombosit yang rusak dan bukan dari
jaringan yang rusak. Aktivator protombin dalam beberapa detik mengawali
pemecahan protombin menjadi trombin dan dilanjutkan dengan proses
pembekuan selanjutnya.
11

Gambar 2. Mekanisme instrinsik sebagai awal pembekuan (Guyton, 1997)
b. Perubahan protombin menjadi trombin yang dikatalisis oleh activator protombin.
Setelah activator protombin terbentuk sebagai akibat pecahnya pembuluh darah,
activator protombin akan menyebabkan perubahan protombin menjadi trombin
yang selanjutnya akan menyebabkan polimerisasi molekul-molekul fibrinogen
menjadi benang-benang fibrin dalam 10-15 detik berikutnya. Pembentukan
activator protombin adalah faktor yang membatasi kecepatan pembekuan darah.
Protombin adalah protein plasma, suatu alfa 2 globulin yang dibentuk terus
menerus di hati dan selalu dipakai untuk pembekuan darah. Vitamin K diperlukan
oleh hati untuk pembekuan protombin. Aktivator protombin sangat berpengaruh
terhadap pembentukan trombin dari protombin. Yang kecepatannya berbanding
lurus dangan jumlahnya. Kecepatan pembekuan sebanding dengan trombin yang
terbentuk.
12

c. Perubahan fibrinogen menjadi fibrin.
Trombin merupakan enzim protein yang mempunyai kemampuan proteolitik dan
bekerja terhadap fibrinogen dengan cara melepaskan 4 peptida yang berberat
molekul kecil dari setiap molekul fibrinogen sehingga terbentuk molekul fibrin
monomer yang mempunyai kemampuan otomatis berpolimerisasi dengan molekul
fibrin monomer lain sehingga terbentuk retikulum dari bekuan. Pada tingkat awal
dari polimerisasi, molekul-molekul fibrin monomer saling berikatan melalui
ikatan non kovalen yang lemah sehingga bekuan yang dihasilkan tidaklah kuat
daan mudah diceraiberaikan. Oleh karena itu untuk memperkuat jalinan fibrin
tersebut terdapaat faktor pemantap fibrin dalaam bentuk globulin plasma.
Globulin plasma dilepaskan oleh trombosit yang terperangkap dalam bekuan.
Sebelum faktor pemantap fibrin dapat bekerja terhadap benang fibrin harus
diaktifkan lebih dahulu. Kemudian zat yang telah aktif ini bekerja sebagai enzim
untuk menimbulkan ikatan kovalen diantara molekul fibrin monomer dan
menimbulkan jembatan silang multiple diantara benang-benang fibrin yang
berdekatan sehingga menambah kekuatan jaringan fibrin secara tiga dimensi.
13

Gambar 3. Mekanisme pembekuan darah
14

Gambar 4. Faktor-faktor pembekuan darah
2. Penyakit-penyakit Gangguan Hemostasis
a) Gangguan vaskulus
Perdarahan abnormal yang tidak disebabkan oleh kelainan trombosit dan kelainan
mekanisme pembekuan darah digolongkan kedalam perdarahan karena gangguan
vaskulus.
Faktor yang dapat menimbulkan kelemahan vaskulus umumnya dapat dibagi menjadi
1. Faktor kongenital
a. Telangiektasia hemoragika herediter (oslet-weber-rendu)
b. Hiperelastika kutis (ehler-dan los)
2. Faktor didapat (acquired)
a. Scorbut
b. Defisiensi vitamin c
15

c. Panvaskulitis
d. Purpura anafilaktoid (purpura henoch-schenlein)
e. Dan lain-lain, misalnya uremia.
b) Gangguan trombosit
Gangguan kelainan jumlah trombosit
- Purpura trombositopenik imun (PTI) ialah suatu penyakit perdarahan yang
didapat sebagai akibat dari penghancuran trombosit yang berlebihan, yang
ditandai dengan: trombositopenia (trombosit <100.000 mmk), purpura,
gambaran darah tepi yang umumnya normal, dan tidak ditemukan penyebab
trombositpeni yang lainya.
- Purpura trombositopenia sekunder (PTS ), yang trejadi akibat pengaruh imun
yang menyebabkan umur trombosit memendek dan kelainan ini mempunyai
hubungan dengan jumlah megakariosit normal atau meningkat maupaun
megakariosit kurang sampai tidak ada dalam sumsum tulang.
- Trombositopenia neonatal, terjadi bila jumlah trombosit < 100.000/ml,
umunya ditemukan pada distress neonatal terutama pada bayi prematur yang
sakit atau menderita penyakit seperti bakteriemi dan koagulasi intravaskular
diseminata (DIC).
c) Gangguan Pembekuan darah
Gangguan pembekuan darah yang diturunkan
a. Hemofilia merupakan penyakit atau gangguan perdarahan yang bersifat herediter
akibat kekurangan faktor pembekuan VIII atau IX.
Hemofilia terbagi atas :
Hemofilia A adalah penyakit perdarahan herediter yg disebabkan karena
defisiensi atau penurunan aktivasi faktor koagulasi FVIII.
Hemofilia B adalah penyakit perdarahan herediter yang disebabkan karena
defisiensi atau penurunan aktivasi faktor koagulasi FIX.
Hemofilia C adalah penyakit perdarahan herediter yang disebabkan karena
defisiensi atau penurunan aktivasi faktor koagulasi FXI.
16

b. Penyakit von willbrand
Penyakit von Willebrand (vwd) adalah kelainan yang diwariskan secara
otosomal dengan gejala perdarahan, disebabkan mutasi gen faktor von
Willebrand (vwf) sehingga terjadi defisiensi atau disfungsi vwf.
1. Gangguan Pembekuan darah yang didapat
a. Defesiensi vitamin K
Vitamin K penting untuk sintesis prokoagulan faktro II , VII, IX dan
X serta koagulan C dan S
b. Penyakit hati
c. Hati mensintesis semua faktor koagulasi dalam plasma (kecuali faktor
VIII yang juga dapat disintesis di endothel pembuluh darah).
Gangguan pembekuan timbul bila terdapat kerusakan parenkim hati
yang mengganggu sintesis faktor-faktor tersebut.
d. Disseminated intravaskular coagulation (DIC), merupakan suatu
gangguan pembekuan darah yang di dapat, berupa kelainan
trombohaemorrhagic sistemik yang hampir selalu disertai dengan
adanya penyakit primer yang mendasarinya.
Hemorrhagic Disease of the newborn merupakan penyakit perdarahan
yang terjdi pada hari-hari pertama kehidupan akibat kekurangan
vitamin K yang ditandai dengan menurunnya faktor II , VII, IX, X.
3. Defenisi Gangguan Hemostasis
Hemostasis adalah mekanisme tubuh untuk menghentikan perdarahan secara spontan.
Ada beberapa system yang berperan dalam hemostasis yaitu system vascular, trombosit
dan pembekuan darah.
1. System vaskuler
Peran system vascular dalam mencegah perdarahan meliputi proses kontraksi
pembuluh darah (vasokonstriksi) serta aktivasi trombosit dan pembekuan darah.
17

2. System trombosit
Trombosit mempunyai peran penting dalam hemostasis yaitu pembentukan stabilisasi
sumbat trombosit. Pembentukan sumbat trombosit. Pembentukan sumbat trombosit
terjadi melalui beberapa tahap yaitu adhesi trombosit, agregasi trombosit dan reaksi
pelepasan.
3. System pembekuan darah
Proses pembekuan darah terdiri dari rangkaian reaksi enzimatik yang melibatkan
protein plasma yang disebut sebagai factor pembekuan darah, fosfolipid dan ion
kalsium. Factor pembekuan darah dinyatakan dalam angka romawi yang sesuai
dengan urutan ditemukannya.
4. Etiologi gangguan hemostasis
Hemostasis primer melibatkan pembentukan sumbat platelet yang melibatkan trombosit,
dinding pembuluh darah dan faktor von Willebrand , kelainan dapat mencakup masalah
dalam jumlah trombosit , adhesi atau agregasi
Disorders of Primary Hemostasis
von Willebrand disease
Glanzmann thrombasthenia
Bernard-Soulier syndrome
Platelet storage pool disease
Gray platelet syndrome
Wiskott-Aldrich syndrome
Hemostasis sekunder melibatkan pembentukan fibrin melalui kaskade koagulasi humoral,
termasuk kelainan kekurangan faktor pembekuan atau faktor kontak, kekurangan atau
kelainan fibrinogen atau penyakit jaringan ikat Epidemiologi gangguan hemostasis
18

Disorders of Secondary Hemostasis
Factor I (fibrinogen) abnormalities
● Afibrinogenemia
● Hypofibrinogenemia
● Dysfibrinogenemia
Factor II (prothrombin) deficiency
Factor V deficiency
Factor VII deficiency
Factor VIII deficiency (Hemophilia A)
Factor IX deficiency (Hemophilia B)
Factor X deficiency
Factor XI deficiency
Factor XIII deficiency
Combined factor deficiencies
a2-antiplasmin deficiency
a1-antitrypsin deficiency
Ehlers-Danlos syndrome
Osler-Weber-Rendu syndrome
Scurvy (vitamin C deficiency)
19

5. Epidemiologi gangguan hemostasis
1. Purpura trombositomenik imun (PTI)
PTI diperkirakan merupakan salah satu penyebab kelainan perdarahan didapat yang
banyak ditemukan oleh dokter anak, dengan insidens penyakit simtomatik berkisar 3
sampai 8 per 100000 anak pertahun. Di bagian ilmu kesehatan anak RSU Dr soetomo
terdapat 22 pasien baru pada tahun 2000, 80 – 90% anak dengan PTI menderita
episode perdarahan akut, yang akan pulih dalam beberapa hari atau minggu dan
sesuai dengan namanya (akut) akan sembuh dalam 6 bulan. Pada PTI akut tidak ada
perbedaan insidensi laki maupun perempuan dan akan mencapai puncak pada usia 2-5
tahun. Hampir selalu ada riwayat infeksi bakteri, virus ataupun imunisasi 1-6 minggu
sebelum terjadinya penyakit ini. Perdarahan sering terjadi saat trombosit dibawah
20000/mm3. PTI kronis terjadi pada anak usia >7 tahun, sering terjadi pada anak
perempuan. PTI yang recuren didefenisikan sebagai adanya episode trombositopenia
>3 bulan dan terjadi 1-4% anak dengan PTI.
2. Hemorrhagic disease of the newborn
Angka kejadian HDN berkisar antara 1 tiap 200 sampai 400 kelahiran pada bayi-bayi
yang tidak mendapatkan vitamin K profilaksis. Di amerika serikat, frekwensi HDN
yang dilaporkan bervariasi antara 0.25-1.7%. angka kejadian tersebut ditemukan lebih
tinggi di daerah-daerah yang tidak memberikan profilaksis vitamin K secara rutin
pada bayi baru lahir.
Survei di Jepang menemukan kasus ini pada 1:4.500 bayi, 81% diantaranya
ditemukan komplikasi perdarahan intracranial. Angka kejadian ini juga menurut
setelah diperkenalkannya pemberian profilaksis vitamin K pada semua bayi batu
lahir. Di Thailand angka kesakitan bayi karena perdarahan akibat defisiensi vitamin K
berkisar 1:1.200 sampai 1:1.400 kelahiran hidup. Angka tersebut dapat turun menjadi
10:100.000 kelahiran hidup dengan pemberian profilaksis vitamin K pada bayi baru
lahir. Data PVDK secara nasional di Indonesia belum tersedia.
3. Von Willebrand Disease (VWD)
Dokter sekarang berpendapat bahwa VWD dapat mengenai 1 diantara 100 orang.
Karena banyak orang - orang ini hanya mengalami perdarahan ringan, maka hanya
sejumlah kecil yang tahu bahwa dirinya membawa pernyakit ini. Penyakit Von
20

Willebrand dapat mengenai pria dan wanita. Namun, karena banyak wanita dengan
VWD mengalami perdarahan haid yang banyak dan perdarahan lama setelah
melahirkan, lebih banyak wanita yang mempunyai gejala dibandingkan pria. Anak -
anak juga dapat menderita VWD. Mereka dilahirkan dengan penyakit ini. Hal ini
karena vWD adalah kelainan yang diturunkan.
4. Hemophilia
Penyakit ini bermanifestasi klinik pada laki-laki. Angka kejadian hemophilia A
sekitar 1:10.000 orang dan hemophilia B sekitar 1:250.000-300.000 orang. Belumada
data mengenai angka kekerapan di Indonesia, namun diperkirakan sekitar 20.000
kasus dari 200 jutra penduduk Indonesia saat ini. Kasus hemophilia A lebih sering
dijumpai dibandingkan hemophilia B, yaitu berturut-turut mencapai 80-85% dan 10-
15% tanpa memandang ras, geografi dan keadaan social ekonomi. Mutasi gen secara
spontan diperkirakan mencapai 20-30% yang terjadi pada pasien tanpa riwayat
keluarga.
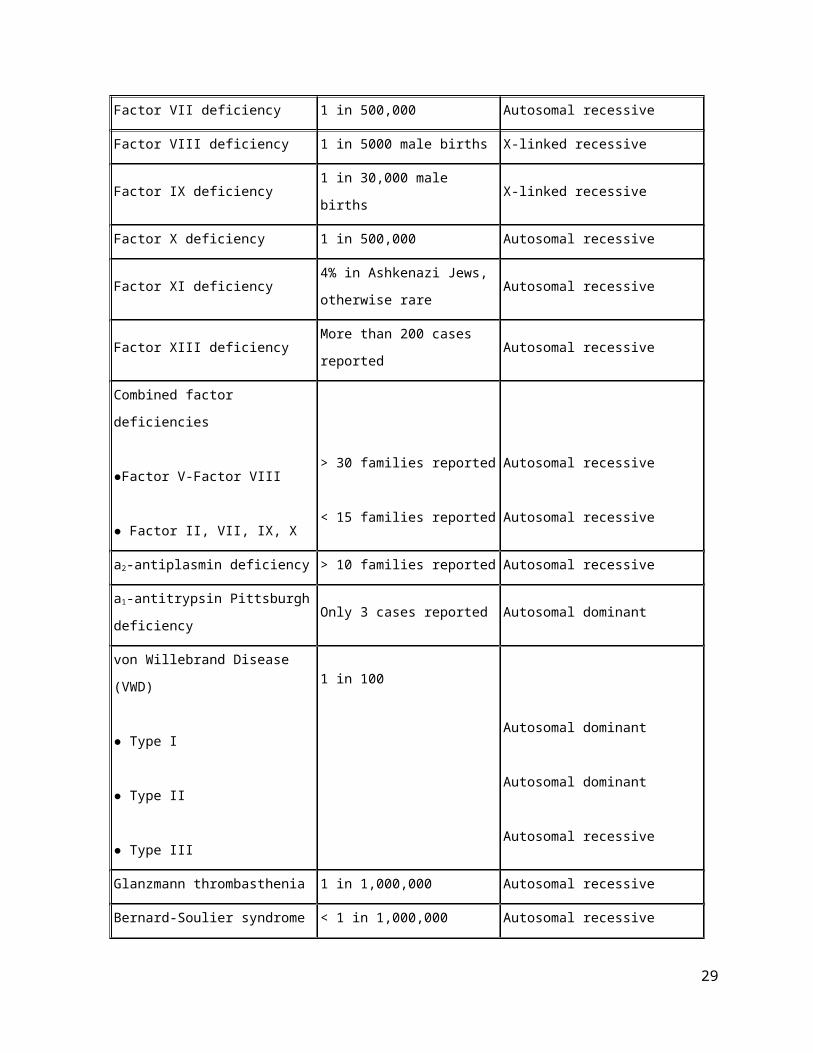
Bleeding disorder Prevalence Inheritance pattern
Factor I (fibrinogen) deficiency
● Afibrinogenemia
● Hypofibrinogenemia
● Dysfibrinogenemia
More than 200 cases reported
Autosomal recessive
Autosomal dominant or recessive
Autosomal dominant or recessive
21

Factor II (prothrombin) deficiency Less than 100 cases reported Autosomal recessive
Factor V deficiency Less than 1 in 1,000,000 Autosomal recessive
Factor VII deficiency 1 in 500,000 Autosomal recessive
Factor VIII deficiency 1 in 5000 male births X-linked recessive
Factor IX deficiency 1 in 30,000 male births X-linked recessive
Factor X deficiency 1 in 500,000 Autosomal recessive
Factor XI deficiency4% in Ashkenazi Jews,
otherwise rareAutosomal recessive
Factor XIII deficiency More than 200 cases reported Autosomal recessive
Combined factor deficiencies
●Factor V-Factor VIII
● Factor II, VII, IX, X
> 30 families reported
< 15 families reported
Autosomal recessive
Autosomal recessive
a2-antiplasmin deficiency > 10 families reported Autosomal recessive
a1-antitrypsin Pittsburgh deficiency Only 3 cases reported Autosomal dominant
von Willebrand Disease (VWD)
● Type I
● Type II
● Type III
1 in 100
Autosomal dominant
Autosomal dominant
Autosomal recessive
Glanzmann thrombasthenia 1 in 1,000,000 Autosomal recessive
Bernard-Soulier syndrome < 1 in 1,000,000 Autosomal recessive
Gray platelet syndrome RareAutosomal dominant, recessive or
X-linked recessive
Wiskott-Aldrich syndrome 1 in 1,000,000 X-linked recessive
6. Patofisiologi dan Gejala Klinis Gangguan Hemostasis
22

a. Von-Willebrand Disease
Sejak tahun 1980, pengetahuan molekul dan seluler telah mendefinisikan hemofilia A
dan penyakit von willebrand secara jelas. mutasi dan subtipe penyakit von willebrand.
Penyakit ini disebabkan oleh abnormalitas (secara kuantitatif dan kualitas) faktor von
Willebrand, dimana merupakan suatu glikoprotein multimerik yang berfungsi sebagai
protein pembawa faktor VIII (FVIII). Faktor von Willebrand juga diperlukan dalam
fungsi normal adesi trombosit. Dengan demikian, faktor ini berfungsi primer (adesi
trombosit) dan sekunder (melibatkan Faktor VIII) dalam proses hemostasis. Pasien
dengan penyakit von willebrand yang berat memiliki gen faktor VIII yang normal
pada kromosom X, sebagian kecil terdapat gen vWF yang abnormal pada kromosom
12. Gen vWF terlokalisasi di dekat ujung lengan pendek kromosom 12, pada 12p13.3.
gen ini memanjang sekitar 178 kb DNA dan mengandung 52 exon. Pada bagian lain,
pseudogen vWF yang belum diproses terletak pada kromosom 22q12.2. pseudogen
ini, memanjang sekitar 25 kb DNA dan berhubungan dengan 23-24 exon. Segmen
gen ini mengkode domain A1A2A3 yang mengikat tempat untuk glikoprotein Ib
(GpIb) trombosit dan kolagen, sebaik pengikatan pada tempat yang dipecah oleh
ADAMTS13. Gen vWF dan pseudogen telah dibedakan dari DNA sekuens.
Pseudogen terdapat pada sel manusia dan kera besar. Pseudogen vWF menyulitkan
deteksi mutasi gen vWF karena polymerase chain reactions (PCRs) tidak dapat
menampilkan amplifikasi segmen dari kedua lokus. Namun demikian, kesulitan ini
dapat diatasi dengan penanganan secara teliti PCR primer gen spesifik. Pseudogen
vWF bertindak sebagai reservoir mutasi sehingga dapat diperkenalkan dengan lokus.
Sebagai contoh, beberapa mutasi yang tidak tampak atau berpotensial patogen
diidentifikasi pada exon 27 dan 28 pada gen vWF pada individu dengan penyakit von
Willbrand. Mutasi penyebab penyakit ini telah diketahui melalui identifikasi gen
vWF. Dalam hal yang bertolak belakang dengan Hemofilia A, dimana re-
arrengement gen mayor tunggal dapat mengakibatkan penyakit yang berat, pada
penyakit von willebrand sering tidak terjadi recurring mutation.
Pada hemostasis primer, faktor von Willebrand menempel pada trombosit melalui
reseptor spesifik glikoprotein Ib pada permukaan trombosit dan bertindak sebagai
jembatan perekat antara trombosit dan subendotelium yang rusak. Sedangkan pada
23

hemostasis sekunder, faktor von Willebrand menjaga agar FVIII dari degradasi dan
mengantarkannya ke lokasi cedera. Faktor von Willebrand terdiri dari sub-unit
dimmer yang terhubung oleh suatu bentuk ikatan disulfide dari kompleks multimers.
Fungsi multimer utamanya untuk membawa FVIII. Sedangkan multimer dengan berat
molekul yang lebih berat memiliki tempat pengikatan trombosit lebih banyak dan
memiliki daya rekat yang lebih besar. Setiap subunit multimers memiliki tempat
untuk reseptor glikoprotein Ib untuk trombosit yang tidak teraktivasi dan reseptor
glikoprotein IIb/IIIa pada trombosit yang teraktivasi. Keadaan ini menjadikan adhesi
dan agregasi trombosit penting untuk fungsi normal trombosit. Daerah pengikatan
FVIII pada faktor von Willebrand telah dilokaliasasi pada NH2-terminal 272 asam
amino bagian dari protein matur. Hubungan antara FVIII dan faktor von willebrand
dapat dihambat oleh antibody monoclonal anti vHF (anti faktor von willebrand), W5-
6A, yang dipetakan oleh epitop antara Thr 78 dan Thr 96 di dalam 272 daerah ikatan
asam amino. Mutasi yang bertenggungjawab terhadap terganggunya pengikatan
faktor VIII dilaporkan berada pada epitop yang sama. Bagian dari domain pengikatan
faktor von willebrand didalam faktor VIII bersifat asam (acidic).
24

Gambar 3. Ilustrasi peran trombosit dan faktor von willebrand
Hemostasis dan koagulasi adalah serangkaian kompleks reaksi yang menyebabkan
pengendalian perdarahan melalui pembentukan trombosit dan bekuan fibrin pada
tempat cedera. Pembekuan diikuti resolusi atau lisis bekuan dan regenerasi endotel.
Pada keadaan hemostatik, hemostasis dan koagulasi melindungi individu dari
perdarahan masif akibat trauma. Trombosit merupakan fragmen sel granuler
berbentuk cakram, tidak berinti yang penting untuk proses hemostasis dan koagulasi.
Trombosit ini berdiameter 1-4 mikrometer dan memiliki siklus hidup 6 hari. Pada
pewarnaan Wright, sel ini akan terlihat biru muda dengan granula berwarna merah
ungu. Pada membran trombosit diabsorbsi faktor V, VIII, IX, protein kontraktil
aktomiosin (trombostenin) dan beberapa protein serta enzim lain. Faktor-faktor
pembekuan, kecuali faktor VIII (tromboplastin jaringan ) dan faktor VI (ion kalsium),
merupakan protein plasma yang berada dalam sirkulasi darah sebagai molekul inaktif.
Prakalikrein (faktor Fletcher) dan kininogen (Faktor Fitzgerald) dengan berat molekul
25

tinggi, bersama faktor XII dan XI disebut factor kontak dan diaktivasi pada saat
cedera dengan berkontak dengan permukaan jaringan.
Aktivasi faktor koagulasi diyakini terjadi karena enzim memecahkan fragmen bentuk
prekursor yang tidak aktif, oleh karena itu disebut prokoagulan. Tiap faktor yang
diaktivasi, kecuali faktor V,VIII,XIII dan I, merupakan enzim pemecah protein yang
mengaktivasi prokoagulan berikutnya. Hepar merupakan tempat sintesis semua faktor
koagulasi kecuali faktor VIII dan mungkin faktor XI dan XIII. Vitamin K penting
sintesis faktor protrombin II, VII, IX dan X. Bukti yang memberi kesan bahwa faktor
VIII merupakan molekul kompleks yang terdiri atas tiga subunit yang berbeda antara
lain :
1. Bagian prokoagulan, yang mengandung antihemofilia (VIIIAHG) yang tidak
dijumpai pada pasien hemofilia klasik.
2. subunit lain yang mengandung tempat antigenik, dan
3. faktor von Willebrand (VIIIVWF) yang diperlukan untuk adhesi trombosit pada
pembuluh darah.
Fase koagulasi diawali dalam keadaan homeostasis dengan adanya cedera vaskuler.
Vasokonstriksi merupakan respon segera terhadap cedera, yang diikuti denagn adhesi
trombosi pada kolagen dinding permbuluh darah. Faktor III (trombosit) juga
mempercepat pembekuan plasma. Dengan cara ini terbentuklah sumbatan trombosit,
kemudian segera diperkuat oleh protein filementosa yang dikenal sebagai fibrin.
Produksi fibrin dimulai dengan perubahan faktor X menjadi faktorXa. Faktor X dapat
diaktivasi melalui dua rangkaian reaksi, yaitu reaksi jalur ekstrinsik dan intrinsik.
Pada jalur ekstrinsik memerlukan faktor jaringan yang dilepaskan oleh endotel
pembuluh darah pada saat cedera, namun faktor jaringan tidak terdapat di dalam
darah maka disebut sebagai faktor ekstrinsik koagulasi. Rangkaian reaksi lainnya
adalah jalur intrinsik. Disebut demikian karena rangkaian reaksi ini menggunakan
faktor yang terdapat di dalam sistem vaskuler plasma. Dalam rangkaian ini terjadi
reaksi kaskade (Cascade), aktivasi satu prokoagulan menyebabkan aktivasi bentuk
pengganti. Jalur reaksi ini dimulai dengan plasma yang keluar terpajan dengan kulit
atau kolagen di dalam pembuluh darah yang rusak. Faktor jaringan tidak diperlukan,
26

tetapi trombosit yang melekat pada endotel cukup berperan. Faktor XIII, XI dan IX
harus diaktivasi secara berurutan, dan faktor VIII harus dilibatkan sebelum faktor X
dapat diaktivasi.
Proses ini berlanjut sehingga masuk dalam jalur bersama. Aktivasi faktor X terjadi
sebagai akibat reaksi jalur ekstrinsik dan intrinsik. Pembentukan fibrin berlangsung
jika faktor Xa yang dibantu fosfolipid dari trombosit yang diaktivasi, memecah
protrombin membentuk trombin. Selanjutnya, trombin memecahkan fibrinogen
membentuk fibrin. Fibrin pada awalnya merupakan jeli yang dapat larut, distabilkan
oleh faktor XIIIa dan mengalami polimerasi menjadi jalinan fibrin yang kuat.
Gambar 5. Kaskade proses pembekuan
Faktor von Willebrand (vWF) disintesis dalam dua bentuk tipe sel. Di dalam endotel
vaskuler, vWF ini disintesis dan disimpan dalam bentuk granul sekretori (Weibel-
Palade bodies) yang dapat dilepaskan bila terjadi stres atau akibat obat seperti
desmopressin (DDAVP, 1-desamino-8-D-arginine vasopressin), suatu obat yang
analog dengan vasopressin. vWF ini juga disintesis di sumsum tulang sebagai
megakaryosit dimana penyimpanannya pada granul trombosit alfa, yang akan
dilepaskan mengikuti aktivasi trombosit. Desmopressin tidak menyebabkan pelepasan
27

faktor vWF trombosit. Faktor von Willebrand merupakan protein yang terbentuk dari
subunit identik menjadi untaian linear dengan ukuran bervariasi menjadi multimers.
Multimers ini dapat mencapai berat molekul lebih dari 20 juta Dalton dan panjang
lebih dari 2 mikrometer. Sel kompleks ini memproses dimerisasi di dalam retikulum
endoplasma, glikosilasi di dalam retikulum endoplasma dan kompleks golgi dan
menyimpannya dalam granul. Dua proses terakhir dikendalikan di bawah propeptida
vWF (vWFpp) yang dipecah dari vWF pada saat penyimpanan. Faktor von willebrand
yang dilepas ke sirkulasi diikuti peningkatan secara paralel FVIII, namun belum jelas
bagaimana hubungan antara protein ini. Kompleks vWF dan FVIII di dalam plasma
bersirkulasi seperti ikatan kompleks protein yang tidak beriteraksi kuat dengan
trombosit atau sel endotel pada kondisi basal. Namun apabila terdapat cedera
vaskuler, vWf akan bersinggungan dengan subendotel. vWF akan menyebabkan
aktivasi dan perlekatan trombosit dan kemudian mengagregasi fosfolipid trombosit.
Hal ini memfasilitasi proses pembekuan yang diperankan FVIII. Dengan karakteristik
spesifik dari hemostasis dan fibrinolisis pada permukaan mukosa, gejala yang muncul
pada vWD lebih berat di jaringan ini.
b. Hemofilia
Proses hemostasis tergantung pada faktor koagulasi, trombosit dan pembuluh darah.
Mekanisme hemostasis terdiri dari respons pembuluh darah, adesi trombosit, agregasi
trombosit, pembentukan bekuan darah, stabilisasi bekuan darah, pembatasan bekuan
darah pada tempat cedera oleh regulasi antikoagulan, dan pemulihan aliran darah
melalui proses fibrinolisis dan penyembuhan pembuluh darah.
Cedera pada pembuluh darah akan menyebabkan vasokonstriksi pembuluh darah dan
terpaparnya darah terhadap matriks subendotelial. Faktor von Willebrand (vWF) akan
teraktifasi dan diikuti adesi trombosit. Setelah proses ini, adenosine diphosphatase,
tromboxane A2 dan protein lain trombosit dilepaskan granul yang berada di dalam
trombosit dan menyebabkan agregasi trombosit dan perekrutan trombosit lebih lanjut.
Cedera pada pembuluh darah juga melepaskan tissue factor dan mengubah
permukaan pembuluh darah, sehingga memulai kaskade pembekuan darah dan
28
menghasilkan fibrin. Selanjutnya bekuan fibrin dan trombosit ini akan distabilkan
oleh faktor XIII.
Pada penderita hemofilia dimana terjadi defisit F VIII atau F IX maka pembentukan
bekuan darah terlambat dan tidak stabil. Oleh karena itu penderita hemofilia tidak
berdarah lebih cepat, hanya perdarahan sulit berhenti. Pada perdarahan dalam ruang
tertutup seperti dalam sendi, proses perdarahan terhenti akibat efek tamponade.
Namun pada luka yang terbuka dimana efek tamponade tidak ada, perdarahan masif
dapat terjadi. Bekuan darah yang terbentuk tidak kuat dan perdarahan ulang dapat
terjadi akibat proses fibrinolisis alami atau trauma ringan.
Gambar 1. Pathway Hemofilia
PK: Prekallikrein, HK: High molecular weight kininogen, TF: Tissue factor, PTT: Partial
Prothrombin time, PT: Prothrombin time
29

Defisit F VIII dan F IX ini disebabkan oleh mutasi pada gen F8 dan F9. Gen F8
terletak di bagian lengan panjang kromosom X di regio Xq28, sedangkan gen F9
terletak di regio Xq27. Terdapat lebih dari 2500 jenis mutasi yang dapat terjadi,
namun inversi 22 dari gen F8 merupakan mutasi yang paling banyak ditemukan yaitu
sekitar 50% penderita hemofilia A yang berat. Mutasi gen F8 dan F9 ini diturunkan
secara x-linked resesif sehingga anak laki-laki atau kaum pria dari pihak ibu yang
menderita kelainan ini. Pada sepertiga kasus mutasi spontan dapat terjadi sehingga
tidak dijumpai adanya riwayat keluarga penderita hemofilia pada kasus demikian.
c. Idiopatic Trombositopenic Purpura
Mekanisme terjadinya trombositopenia pada ITP ternyata lebih kompleks dari yang
semula diduga. Kerusakan trombosit pada ITP melibatkan autoantibody terhadap
glikoprotein yang terdapat pada membrane trombosit. Sehingga terjadi penghancuran
terhadap trombosit yang diselimuti antibodi (antibody-coated platelets) oleh
makrofag yang terdapat pada limpa dan organ retikuloendotelial lainnya.
Megakariosit dalam sumsum tulang bisa normal atau meningkat pada ITP. Sedangkan
kadar trombopoitin dalam plasma yang merupakan progenitor proliferasi dan
maturasi dari trombosit mengalami penurunan yang berarti, terutama pada ITP kronis.
Adanya perbedaan secara klinis maupun epidemiologis antara ITP akut dan kronis,
menimbulkan dugaan adanya perbedaan mekanisme patofisiologi terjadinya
trombositopenia di antara keduanya. Pada ITP akut, telah dipercaya bahwa
penghancuran trombosit meningkat karena adanya antibodi yang dibentuk saat terjadi
respon imun terhadap infeksi bakteri/virus atau pada pemberian imunisasi, yang
bereaksi silang dengan antigen dari trombosit. Mediator-mediator lain yang
meningkat selama terjadinya respon imun terhadap infeksi, dapat berperan dalam
terjadinya penekanan terhadap produksi trombosit. Pada ITP kronis mungkin telah
terjadi gangguan dalam regulasi sistem imun seperti pada penyakit otoimun lainnya,
yang berakibat terbentuknya antibodi spesifik terhadap trombosit. Saat ini telah
diidentifikasi beberapa jenis glikoprotein permukaan trombosit pada ITP, di antaranya
GP IIb-IIa, GP Ib, dan GP V.7-9 Namun bagaimana antibody antitrombosit
30

meningkat pada ITP, perbedaan secara pasti patofisiologi ITP akut dan kronis, serta
komponen yang terlibat dalam regulasinya masih belum diketahui. Hal tersebut di
atas menjelaskan mengapa beberapa cara pengobatan terbaru yang digunakan dalam
penatalaksanaan ITP memiliki efektifitas terbatas, dikarenakan mereka gagal
mencapai target spesifik jalur imunologis yang bertanggung jawab pada perubahan
produksi dan destruksi trombosit.
Gambar 1. Jalur ITP
GEJALA KLINIS GANGGUAN HEMOSTASIS
- epistaksis
- menorrhagia
- bleeding after dental extraction
- ecchymoses
- bleeding from minor cuts or abrasions
- gingival bleeding
- postoperative bleeding
31

- hemarthrosis
- GI bleeding
7. Diagnose dan Diagnosa Banding Gangguan Hemostasis
a. Anamnesis
Pada anamnesis, pasien akan mengeluhkan :
1) Gangguan vascular, trombosit dan koagulasi, seperti :
- Perdarahan pada mukosa hidung (epistaksis) atau mimisan yang berulang
- Biru-biru pada kulit atau di persendian
- Gusi mudah berdarah
- Pembengkakan dan nyeri pada sendi
- Luka lama sembuh
- Mudah memar
2) Riwayat Pengobatan, seperti :
- Mengkonsumsi obat-obatan yang dapan menurunkan produksi, destruksi, dan
perubahan fungsi trombosit seperti (Sulfonamide, Quinidine, Karbamazapine,
Aspirin, Dipiridamol, Kloramfenikol, Estrogen, Heparin, Digoksin).
- Menjalani kemotrapi atau Radiasi
3) Riwayat penyakit keluarga seperti karier Hemofili dari Ibu
b. Pemeriksaan fisik gangguan hemostasis didapatkan :
- Petechiae
- Ekimosis
- Hemartrosis
- Perdarahan pada gusi
- Epistaksis
- Purpura
- Hematoma
- Splenomegali
32

c. Pemeriksaan penunjang
1) Tes penyaring
- Pemeriksaan darah lengkap dan pemeriksaan apusan darah
Trombositopenia sering merupakan penyebab perdarahan abnormal, oleh
karena itu pada pasien yang diduga menderita kelainan perdarahan, pertama
kali harus dilakukan pemeriksaan hitung darah lengkap dan pemeriksaan
apusan darah tepi. Selain untuk memastikan adanya trombositopenia, dari
pemeriksaan darah apus dapat menunjukkan kemungkinan penyebab yang
jelas terlihat (misalnya leukemia).
- Pemeriksaan penyaring sistem koagulasi darah
Pemeriksaan penyaring meliputi penilaian jalur intrinsic dan ekstrinsik dari
sistem koagulasi dan perubahan dari fibrinogen menjadi fibrin.
Waktu protrombin (PT)
Untuk mengukur faktor VII, X, V, protrombin dan fibrinogen. Waktu
normal untuk koagulasi adalah 10-14 detik. Hal ini dapat dinyatakan
sebagai INR (international normalized ratio).
APTT (Activated partial thromboplastin time) mengukur faktor VIII,
IX, XI, dan XII, selain itu tambahan pada faktor V, X, protrombin dan
fibrinogen. Waktu normal untuk pembekuan adalah sekitar 30-40
detik.
perpanjangan dari PT dan APTT yang disebabkan karena defisiensi
faktor koagulasi dapat dikoreksi dengan penambahan plasma normal
kedalam plasma yang diperiksa. Apabila tidak dapat dikoreksi atau
hanya sebagian terkoreksi, dicurigai kemungkinan adanya inhibitor
koagulasi.
Waktu trombin (TT)
Waktu trombin cukup sensitif untuk menilai defisiensi fibrinogen atau
adanya hambatan terhadap thrombin. Nilai normal kurang dari 15-20
detik. Bila thrombin time memanjang berarti terdapat
hipofibrinogenemia.
33

Table 1. tes penyaringan yang digunakan dalam diagnosis gangguan koagulasi
Tes penyaring Kelainan yang diindikasikan oleh
pemanjangan
Kausa tersering penyebab
gangguan koagulasi
Waktu thrombin (TT) Defisiensi atau kelainan fibrinogen atau
inhibisi thrombin oleh heparin atau FDP
DIC
Terapi heparin
Waktu protrombin
(PT)
Defisiensi atau hambatan pada satu atau
lebih faktor koagulasi berikut: VII, X, V, II,
fibrinogen
Penyakit hati
Terapi heparin
DIC
Activated partial
trhomboplastin time
(APTT atau PTTK)
Defisiensi atau hambatan pada satu atau
lebih faktor koagulasi berikut: XII, XI, IX
(penyakit Christmas), VIII (hemophilia), X,
V, II, fibrinogen
Hemophilia, penyakit Christmas
(+kondisi-kondisi diatas)
Kuantitas fibrinogen Defisiensi fibrinogen DIC, penyakit hati
DIC,disseminated intravascular coagulation (koagulasi intravascular diseminata);FDPs, fibrin degradation products (produk penguraian fibrin).
NB, Hitung trombosit dan tes fungsi trombosit juga digunakan sebagai penyaring bagi pasien dengan gangguan perdarahan.
- Pemeriksaan faktor koagulasi khusus
Pemeriksaan faktor koagulasi khusus termasuk disini misalnya fibrinogen,
faktor vW, dan faktor VIII. Pemeriksaan bisa secara kuantitatif atau dengan
cara membandingkan efek koreksi dari plasma yang mengandung kekurangan
substrat tertentu yang mempunyai perpanjangan waktu pembekuan (PT,
aPTT), dengan efek koreksi terhadap plasma normal, yang hasilnya
dinyatakan dengan persentase aktivitas normal.
- Waktu perdarahan
Tes waktu perdarahan berguna untuk pemeriksaan fungsi trombosit abnormal
misalnya pada defisiensi faktor vW. Pada trombositopenia, waktu perdarahan
juga akan memanjang, namun pada perdarahan abnormal yang disebabkan
kelainan pembuluh darah, waktu perdarahan biasanya normal. Pemeriksaan
dilakukan dengan cara memberi tekanan pada lengan atas dengan memasang
manset tekanan darah. Selain itu, dibuat insisi kecil di kulit lengan bawah
34

bagian fleksor. Pada keadaan normal, perdarahan akan berhenti dalam, waktu
3-8 menit.
- Pemeriksaan fungsi trombosit
Tes agregasi trombosit merupakan pemeriksaan yang mempunyai nilai
penting. Tes ini mengukur penurunan penyerapan sinar pada plasma kaya
trombosit sebagai agregat trombosit. Agregasi primer berasal dari rangsangan
agen eksternal, sedangkan respon sekunder berasal dari agen yang dilepas dari
dalam trombosit sendiri. Agen agregasi yang sering digunakan misalnya:
ADP, kolagen, ristosetin, asam arakidonat dan adrenalin.
- Pemeriksaan fibrinolisis
Peningkatan activator plasminogen dalam sirkulasi dapat dideteksi dengan
memendeknya waktu lisis bekuan euglobulin. Beberapa tehnik imunologik
digunakan untuk mendeteksi produk degradasi dari fibrin maupun fibrinogen.
Pada pasien yang mengalami peningkatan fibrinolisis, kadar plasminogen
dalam darah mungkin rendah dapat ditemukan.
2) Tes Khusus
Tes khusus lanjutan, yaitu tes untuk mengetahui penyebab
kelainan faal hemostasis tersebut. Tes ini dikerjakan sesuai
petunjuk tes penyaring :
Tes faal trombosit
Tes Ristocetin
Pengukuran faktor spesifik (faktor pembekuan)
Pengukuran alpha-2 antiplasmin
Biopsi sumsum tulang
35

Tabel 2. Gambaran Laboratorium pada Hemofilia A, B dan von-willebrand
Hemofilia A Hemofilia B Von-Willebrand
Pewarisan X-linked recessive X-linked recessive Autosomal Dominant
Lokasi perdarahan
utama
Sendi, otot,
pascatrauma/operasi
Sendi, otot,
posttrauma/operasi
Mukosa, kulit, post
trauma/ operasi
Jumlah trombosit Normal Normal Normal
Waktu perdarahan Normal Normal Memanjang
PPT Normal Normal Normal
Aptt Memanjang Memanjang Memanjang/normal
F VIII C Rendah Normal Rendah
F VIII AG Normal Normal Rendah
F IX Normal Rendah Normal
Tes ristostetin Normal Normal Terganggu
Tabel 3. Gambaran laboratorium perdarahan akibat Def. Vitamin K, Penyakit hati, dan DIC
Komponen Def. Vitamin K Penyakit Hati DIC
Morfologi eritrosit
PTT
PT
Fibrin Split Product
Trombosit
Faktor koagulasi
yang menurun
Normal
Memanjang
Memanjang
Normal
Normal
Faktor II,VII,IX,X
Sel target
Memanjang
Memanjang
Normal/ naik
Normal / turun
I,II,V,VII,IX,X
Sel target, sel burr.
Fragmentosit, sferosit
Memanjang
Memanjang
Naik
Menurun
I,II,V,VIII,XIII
36

d. Diagnosis banding
1) PTI
Pada pemeriksaan fisik didapatkan bukti adanya perdarahan tipe trombosit, yaitu
petekie, purpura, perdrahan konjungtiva, atau perdarahan mukokutaneus lainnya.
Jika ditemukan pada palpasi adanya pembesaran hati dan atau limpa, meskipun
ujung limpa sedikit teraba pada lebih kurang 10 %. Pada pemeriksaan penunjang
yang dilakukan selain adanya trombositopenia, pemeriksaan darah tepi dengan
PTI umunya normal. Lebih kurang 15% pasien didapatkan anemia ringan karena
perdarahan yang dialaminya. Pada pemeriksaan hapusan darah tepi diperlukan
untuk menyingkirkan kemungkinan pseudotrombositopenia, dan kelainan
hematologi lainnya. Pada pemeriksaan dengan flow cytometry terlihat trombosit
pada PTI lebih aktif secara metabolic. Diagnosis PTI ditegakkan dengan
menyingkirkan kemungkinan penyebab trombositopenia.
2) Hemofilia
Pada pemeriksaan klinis tanda dan gejala hemofili A dan B sulit dibedakan. Pada
pemeriksaan yang didapati adalah perdarahan yang umum dijumpai ialah
hematoma, dapat berupa kebiruan, pada berbagai bagian tubuh dan hemarthrosis
atau perdarahan yang sukar berhenti dan Ada riwayat keluarga. Pada pemeriksaan
laboratorium hasil yang didapat pada umumya hasil pemeriksaan darah rutin
maupun hemostasis sederhana pada hemofili Adan B sama. Pada pemeriksaan
darah rutin biasanya normal sedangkan pada pemeriksaan tes penyaring masa
pembekuan memanjang, masa protrombin normal, masa tromboplastin parsial
memanjang dan masa pembekuan tromboplastin abnormal. Untuk mendiagnosi
pasti ialah dengan memeriksa kadar faktor VIII untuk hemofili A dan kadar faktor
IX untuk hemophilia B.
3) DIC
Pada pemeriksaan inspeksi yang didapat yaitu perdarahan pada bagian kulit
seperti: peteki, ekimosis. Dan perdarahan pada mukosa seperti: epistaksis,
perdarahan gusi, hematemesis, dan lain-lain. Dan biasanya penderita mengalami
perdarahan dari banyak tempat. Perdarahan timbul karena penurunan faktor II, V,
VII dan fibrinogen plasma, trombositopenia berat dan peningkatan kadar fibrin
37

degradation product (FDP). Untuk menegakkan diagnosis pemeriksaan
laboratorium didapatkan: PT, APTT dan TT memanjang, jumlah trombosit
menurun, biasanya <100.000/mm3 , penurunan konsentrasi fibrinogen, aktivitas
protrombin, faktor V dan VIII, adanya fragmentasi dari sel darah merah pada apus
darah tepi, peningkatan kadar FDPs dan peningkatan D-Dimer, diukur dengan
antibody monoclonal yang menunjukkan adanya suatu fibrinolisis. Dan criteria
minimal untuk menegkkan diagnosis DIC adalah didapatkan keadan klinis yang
menyebabkan DIC dengan manifestasi perdarahan, tromboemboli atau keduanya
disertai trombositopenia dan gambaran sel burr pada sel darah merah atau D-
dimer positif.
4) Von Willbrand’s Disease
Pada pemeriksaan gejala klasik pada penyakit von Willebrand ialah terjadinya
perdarahan dari ringan sampai keberat berupa kebiruan dikulit, epistaksis,
perdarahan yang memanjang pada luka kecil, menoragia dan perdarahan yang
berlebihan setelah trauma atau operasi. Dan pemeriksaan laboratorium untuk
menegakkan diagnosis penyakit von Willebrand: hitung trombosit (pemeriksaan
darah lengkap), masa perdarahan, faktor VIII, faktor vW. Dan diagnosis mudah
ditegakkan bila ditemukan jumlah trombosit yang normal, masa perdarahan >10
menit dan kadar faktor VIII, faktor vW semuanya dibawah 40 u/dl.
8. Penatalaksanaan dan pencegahan gangguan hemostasis
a. Hemofilia A (herediter) dan Hemophilia B (herediter)
Tatalaksana penderita hemofilia harus dilakukan secara komprehensif meliputi.
1) Pemberian faktor pengganti yaitu F VIII untuk hemofilia A dan F IX untuk
hemofilia B. Penggantian faktor IX dilakukan dengan infus plasma beku segar
(PBS) atau konsentrat faktor IX . 1 unit faktor IX/mL menaikan faktor IX 1%.
38

2) Bila terjadi perdarahan akut terutama daerah sendi, maka tindakan RICE (rest, ice,
compression, elevation) segera dilakukan. Sendi yang mengalami perdarahan
diistirahatkan dan diimobilisasi. Kompres dengan es atau handuk basah yang
dingin, kemudian dilakukan penekanan atau pembebatan dan meninggikan
daerah perdarahan.
3) Penderita sebaiknya diberikan faktor pengganti dalam 2 jam setelah perdarahan.
4) Untuk hemofilia A diberikan konsentrat F VIII dengan dosis 0.5 x BB (kg) x
kadar yang diinginkan (%). F VIII diberikan tiap 12 jam sedangkan F IX
diberikan tiap 24 jam untuk hemofilia B. Kadar F VIII atau IX yang diinginkan
tergantung pada lokasi perdarahan dimana untuk perdarahan sendi, otot, mukosa
mulut dan hidung kadar 30-50% diperlukan. Perdarahan saluran cerna, saluran
kemih, daerah retroperitoneal dan susunan saraf pusat maupun trauma dan
tindakan operasi dianjurkan kadar 60-100%. Lama pemberian tergantung pada
beratnya perdarahan atau jenis tindakan. Untuk pencabutan gigi atau epistaksis,
diberikan selama 2-5 hari, sedangkan operasi atau laserasi luas diberikan 7-14
hari. Untuk rehabilitasi seperti pada hemarthrosis dapat diberikan lebih lama lagi.
5) Kriopresipitat juga dapat diberikan untuk hemofilia A dimana satu kantung
kriopresipitat mengandung sekitar 80 U F VIII.
39

6) Aspirin dan obat antiinflamasi non steroid harus dihindari karena dapat
mengganggu hemostasis.
7) Profilaksis F VIII atau IX dapat diberikan kepada penderita hemofilia berat
dengan tujuan mengurangi kejadian hemartrosis dan kecacatan sendi. WHO dan
WFH merekomendasikan profilaksis primer dimulai pada usia 1-2 tahun dan
dilanjutkan seumur hidup. Profilaksis diberikan berdasarkan Protokol Malmö
yang pertama kali dikembangkan di Swedia yaitu pemberian F VIII 20-40 U/kg
selang sehari minimal 3 hari per minggu atauF IX 20-40 U/kg dua kali per
minggu.
8) Untuk penderita hemofilia ringan dan sedang, desmopressin (1-deamino-8-
arginine vasopressin, DDAVP) suatu anolog vasopressin dapat digunakan untuk
meningkatkan kadar F VIII endogen ke dalam sirkulasi, namun tidak dianjurkan
untuk hemofilia berat. Mekanisme kerja sampai saat ini masih belum jelas, diduga
obat ini merangsang pengeluaran vWF dari tempatsimpanannya (Weibel-Palade
bodies) sehingga menstabilkan F VIII di plasma. DDAVP dapat diberikan secara
intravena, subkutan atau intranasal.
Pencegahan
- Hindari trauma
- Hindari mengkonsumsi obat-obatan yang mempengaruhi kerja trombosit yang
berfungsi membentuk sumbatan pada pembuluh darah, seperti asam salisilat,
obat antiradang jenis nonsteroid, ataupun pengencer darah seperti heparin.
- Kenakan tanda khusus seperti gelang atau kalung yang menandakan bahwa ia
menderita hemofilia. Hal ini penting dilakukan agar ketika terjadi kecelakaan
atau kondisi darurat lainnya, personel medis dapat menentukan pertolongan
khusus.
- Penderita hemofilia dianjurkan untuk berolah raga rutin, memakai peralatan
pelindung yang sesuai untuk olahraga, menghindari olahraga berat atau kontak
fisik.
- Berat badan harus dijaga terutama bila ada kelainan sendi karena berat badan
yang berlebih memperberat arthritis.
40

- Menjaga kebersihan mulut dan gigi.
- Pihak sekolah sebaiknya diberitahu bila seorang anak menderita hemofilia
supaya dapat membantu penderita bila diperlukan.
- Upaya mengetahui status pembawa sifat hemofilia dan konseling genetik.
- Konseling genetik perlu diberikan kepada penderita dan keluarga.
- Deteksi hemofilia pada janin dapat dilakukan terutama bila jenis mutasi gen
sudah diketahui.
b. Idiopathic Thrombocytopenia Purpura (ITP)
Terapi suportif:
membatasi aktifitas fisik,
mencegah perdarahan akibat trauma,
menghindari obat yang dapat menekan produksi trombosit,
dan yang tidak kalah pentingnya adalah memberi pengertian pada pasien dan
atau orang tua tentang penyakitnya,
pemberian vitamin K dan vitamin C.
ITP Akut
- Tanpa pengobatan, karena bisa sembuh secara spontan.
- Pada keadaan berat dapat diberi kortikosteroid (prednisone) per oral
dengan/tanpa transfuse darah. Bila setelah 2 minggu belum ada kenaikan
trombosit, dapat dianjurkan pemberian kortikosteroid.
- Pada trombositopenia yang disebabkan oleh DIC, dapat diberikan heparin
intravena. Pada pemberian heparin ini sebaiknya selalu disiapkan
antidotumnya yaitu protamin sulfat.
- Bila keadaan sangat gawat hendaknya diberikan transfuse suspense trombosit.
ITP Menahun
- Kortikosteroid minimal 6 bulan.
- Obat imunosupresif (misal: merkaptopurin, azatioprin, siklofosfamid)
41

- Splenektomi, bila tidak diperoleh hasil dengan penambahan obat
imunosupresifvselama 2-3 bulan.
Dosis yang dipakai:
a. Prednisone : 2-5 mg/kgBB/hari secara oral.
b. Merkaptopurin : 2,5-5 mg/kgBB/hari secara oral.
c. Azatioprin : 2-4 mg/kgBB/hari secara oral.
d. Heparin : 1 mg/kgBB secara intravena. Dilanjutkan dengan dosis 1
mg/kgBB per infuse setiap 4 jam sampai tercapai masa pembekuan lebih
dari 30 menit.
e. Protamin sulfat : 1 mg/kgBB secara intravena
f. Transfuse darah : umumnya 10-15 ml/kgBB/hari. Dapat diberikan lebih
banyak pada perdarahan massif.
Pencegahan
- Idiopatik Trombositopeni Purpura (ITP) tidak dapat dicegah, tetapi dapat
dicegah komplikasinya.
- Menghindari obat-obatan seperti aspirin atau ibuprofen yang dapat
mempengaruhi platelet dan meningkatkan risiko pendarahan.
- Lindungi dari luka yang dapat menyebabkan memar atau pendarahan.
- Lakukan terapi yang benar untuk infeksi yang mungkin dapat berkembang.
Konsultasi ke dokter jika ada beberapa gejala infeksi, seperti demam.
4. Disseminated Intravascular Coagulation (DIC)
Penatalakasanaan KID yang utama adalah mengobati penyakit yang mendasari
terjadinya KID. Jika hal ini tidak dilakukan , pengobatan terhadap KID tidak akan
berhasil. Kemudian pengobatan lainnya yang bersifat suportive dapat diberikan.
a. Antikogulan
Secara teoritis pemberian antikoagulan heparin akan menghentikan proses
pembekuan, baik yang disebabkan oleh infeksi maupun oleh penyebab lain.
Meski pemberian heparin juga banyak diperdebatkan akan menimbulkan
42

perdarahan, namun dalam penelitian klinik pada pasien KID, heparin tidak
menunjukkan komplikasi perdarahan yang signifikan.
Dosis heparin yang diberikan:
- Secara intermiten diberi bolus heparin 75-100unit/kg/4jam
- Infus kuntinyu diberi bolus 50-75 unit/kg, diikuti dengan infus kontinyu
10-25unit/kg/jam.
Indikasi:
Penyakit dasar tak dapat diatasi dalam waktu singkat.
Terjadi perdarahan meski penyakit dasar sudah diatasi.
Terdapat tanda-tanda trombosis dalam mikrosirkulasi, gagal ginjal,
gagal hati, sindroma gagal nafas. Dosis:100iu/kgBB bolus dilanjutkan
15-25 iu/kgBB/jam (750-1250 iu/jam) kontinu, dosis selanjutnya
disesuaikan untuk mencapai aPTT 1,5-2 kali. Kontrol Low molecular
weight heparin dapat menggantikan unfractionated heparin.
b. Recombinan activate protein C digunakan untuk pasien sepsis. Dosis 24
mcg/kg/hour.
c. Terapi suportif diberikan pada kondisi pendarahan yang aktif.
d. FFP jika PT memanjang. Dosis: 15 mL/kg.
e. Cryoprecipitate mempertahankan faktor fibrinogen > 100 mg/dL. Dosis: 1-1.5
bags/10 kg.
f. Recombinant VIIa for intractable bleeding or volume overload, dosis: 75
mcg/kg every 2 hours.
Pencegahan
Pengobatan yang tepat untuk salah satu kondisi yang berhubungan dengan DIC
dapat mengurangi risiko Anda untuk DIC.
5. Penyakit Von Willebrand
1. Infuse desmopressin (DDAVP) yang dapat melepaskan vWF dari cadangan
dalam endotel.
2. Terapi ganti dengan single donor cryoprecipitate
43

3. Dapat juga diberikan epsilon aminocaproic acid atau asam traneksamat.
Pencegahan
Tidak ada cara pencegahan khusus untuk Penyakit Von Willebrand saat ini. Untuk
mencegah terjadi komplikasi, hindari penggunaan obat-obatan jenis aspirin,
ibuprofen, dan naproxen tanpa resep dokter. Obat-obat ini dapat mengencerkan
darah dan mencegah darah membeku jika terjadi luka. Beberapa obat anti depresi
juga dapat menghalangi proses pembekuan darah.
Tetap jaga berat badan ideal dan aktif secara fisik. Olahraga yang
direkomendasikan di antaranya berjalan, bersepeda, dan berenang. Olahraga jenis
ini dapat melatih kekuatan otot dan kelenturan sendi. Sementara itu, sebaiknya
hindari olahraga yang rawan benturan. Seperti sepakbola, gulat, atau hoki.
6. Skorbut
Merupakan penyakit akibat kekurangan Vitamin C.
Pengobatannya: memberikan Vitamin C 200 mg/hari selama 1 minggu kemudian
dikurangi perlahan-lahan sampai 1 bulan.
9. Komplikasi dan prognosis gangguan hemostasis
a. Komplikasi1) Von wilebrand
- Perdarahan GI- Nyeri pada persendian
2) Hemofilia- Artropati hemofilia- Sinovitis- Hematom- Kerusakan sendi yang menyebabkan atrofi otot dan dapat menyebabkan
kecacatan.- Perdarahan intrakranial
3) ITP- Perdarahan intrakranial- Perdarahan GI- Perdarahan SSP
44

4) DICKegagalan organ luas seperti :
- Syok- Koma- Gagal ginjal- Gagal napas- Iskemia- Edema pulmoner- Stroke
5) Scurvy disease- Penyakit gusi- Sindrom sjogren
b. Prognosis1) Von wilebrand
Tingkat kematian VWD mendekati nol dinegara-negara Barat karena kemampuan untuk mendiagnosa penyakit dan mengobatinya dengan aman dan efektif.
2) HemofiliaBila penanganannya adekuat dalam medikasi dan psikologis maka umumnya prognosisnya tidak buruk, tetapi bila tidak ditangani dengan tepat dan adekuat dan pasien tidak menjaga diri, maka prognosis akan buruk dan bisa menyebabkan kematian.
3) ITP50–60% penderita berespons dengan kortikosteroid. Penderita ITP dewasa dapat mengalami remisi spontan (2%), menjadi kronis (tidak mengalami remisi komplit setelah kortikosteroid dan splenektomi) sebanyak 43%. Kematian biasanya disebabkan perdarahan serebral (3%), perdarahan berat lain (4%).
4) DICPrognosis untuk pasien DIC biasanya buruk, 10-50% mengalami kematian bergantung pada luas thrombosisnya dan komplikasinya, pasien dengan sepsis/infeksi mempunyai % kematian lebih tinggi yang signifikan.
5) Scurvy diseaseSkorbut yang tidak diobati ini selalu fatal. Namun, kematian akibat skorbut langka dizaman modern. Karena semua yang diperlukan untuk pemulihan penuh adalah dimulainya kembali asupan vitaminC yang normal, mudah untuk mengobati jika diidentifikasi dengan benar. Konsumsi suplemen makanan dan/atau buah jeruk adalah cara yang digunakan untuk mencapai hal ini.
45

BAB 6
KESIMPULAN AKHIR
Nama : -
Umur : 15 tahun
Jenis kelamin : Laki-laki
Hasil anamnesa, dijumpai :
1. Epistaksis (+) :
a. Adanya trauma local
b. Kemungkinan adanya tumor
c. Kemungkinan adanya benda asing
2. Scorbut (+) :
a. Disebabkan karena kekurangan vitamin C
Dari pemeriksaan fisik :
1. Petechiae :
a. Kemungkinan terganggu hemostasis dan factor koagulasi
b. Karena perdarahan yang keluar dari pembuluh-pembuluh darah yang kecil sekali
dibawah kulit
c. Jumlah platelet yang rendah
2. Ekimosis :
a. Terganggunya mekanisme hemostasis dan factor koagulasi
b. Adanya trauma
Dari pemeriksaan laboratorium
1. Leukosit 10.000/mm3 :
a. Karena sel darah putih (leukosit) diserang/infeksi sehingga leukosit mencapai batas
maksimal.
2. Hb menurun (normal Hb 12) :
a. Kekurangan gizi
b. Terjadi perdarahan yang berlebihan
c. Kemungkinan adanya penyakit kronik
46

3. Trombosit menurun :
a. Disebabkan oleh gangguan fungsi trombosit
b. Gangguan produksi trombosit
c. Gangguan penghancuran trombosit
d. Gangguan distriksi trombosit
4. PT memanjang (normal : 11-14 detik): karena defisiensi factor koagulasi.
5. APTT memanjang (normal : 25-35 detik): karena defisiensi factor koagulasi.
Anjuran pemeriksaan penunjang:
1. BMP (Bone Marrow Punction)
2. Immunoglobulin
Diagnosis :
Laki-laki, 15 tahun mengalami gangguan hemostasis suspek ITP.
DD :
1. Gangguan hemostasis
2. ITP
3. DIC
Penatalaksanaan
Untuk sementara menunggu hasil pemeriksaan penunjang maka dapat diberikan terapi suportif.
47

DAFTAR PUSTAKA
1. Guyton, A., & Hall, J. 2006. Buku Ajar Fisiologi Kedokteran. Edisi 11 Jakarta : Penerbit Buku Kedokteran EGC. Hal 480-486
2. Hall E. Jhon. 2007. Buku Saku Fisiologi Kedokteran. Edisi 11 Jakarta : Penerbit Buku Kedokteran EGC. Hal 284-287
3. Sherwood Lauralee. 2009. Buku Ajar Fisiologi Kedokteran. Edisi 6 Jakarta : Penerbit Buku Kedokteran EGC. Hal 433-438
4. Penyunting; Permono H. Bambang, Sutaryo, Ugrasena IDG, Windiastuti Endang, Abdulsalam Maria. 2012. Buku Ajar Hematologi-Onkologi Anak. Catatan keempat. Jakarta. Badan Penerbit IDAI.
5. Widjanarko A, Sudoyo A W, Salonder H. 2009. “Anemia Aplastik”. Dalam : Sudoyo A W, Setiyohadi B, Alwi I, Simabidrata M, Setiati S (Editor). Buku Ajar Penyakit Dalam, Jilid II Edisi V. Jakarta
6. Setiabudi, Rahajuningsih D. 2012. Hemostasis dan Trombosis. Jakarta;Badan Penerbit FKUI
7. Corringan. J. J. 2012. Dalam Buku Nelson Ilmu Kesehatan Anak; Penyakit Perdarahan dan Trombosis. Jakarta: EGC
8. http://repository.usu.ac.id/bitstream/123456789/35611/4/Chapter%20II.pdf 9. http://www.info-kes.com/2013/06/pengobatan-hemofilia-hemophilia.html 10. http://www.hemofilia.or.id/perawatan.php
48





























