Embed Size (px)
Citation preview

24/10/2011 1
Techniques et Stratégies en Biologie Moléculaire
Deuxième partie
Octobre 2011
Vincent Ecochard Didier Fournier Laurence Nieto
Laurent Paquereau
Biotechnologie
Master Protéines
Recombinantes Master Professionnel
Expression Génique et Protéines Recombinantes
e-mail : [email protected] / Tel : +33 (0) 5 61 17 58 59 http://www.m2p-egpr.ups-tlse.fr/

24/10/2011 2
ANALYSE DE LA FONCTION D’UN GENE .................................................................................................................. 6 STRATEGIE BASEES SUR L’HYBRIDATION D’ARN ............................................................................................................... 6
L’interférence d’ARN (RNAi) ......................................................................................................................................... 6 LE KNOCK-OUT OU L’INVALIDATION D’UN GENE PAR RECOMBINAISON HOMOLOGUE ...................................................... 10
La recombinaison homologue : un mécanisme à l’origine de la technologie de Knock-out ........................................ 10 Les souris KO ............................................................................................................................................................... 11 Les cellules ES ............................................................................................................................................................. 11 Fabrication de souris transgéniques ............................................................................................................................ 12 L’interruption d’un exon : knock-out de gène .............................................................................................................. 12 Création de mutation ponctuelle .................................................................................................................................. 13 Le système Cre/Lox ...................................................................................................................................................... 14 Exemple d’application ................................................................................................................................................. 15 Mutation propre ........................................................................................................................................................... 15 Large délétion ou translocation ................................................................................................................................... 15 Les possibilités d’induction de la cre recombinase ...................................................................................................... 16 Le système tet-on .......................................................................................................................................................... 16
DETECTION DES MUTATIONS / POLYMORPHISMES ........................................................................................... 17 I) DETECTION DES MUTATIONS ......................................................................................................................................... 17
Méthodes basées sur la séquence ................................................................................................................................. 17 Méthodes basées sur l’hybridation .............................................................................................................................. 19 Méthodes basées sur la PCR ........................................................................................................................................ 24 Ligation ........................................................................................................................................................................ 26 RFLP ............................................................................................................................................................................ 27 Méthodes basées sur la conformation de l’ADN .......................................................................................................... 28 Hétéroduplex sensibilité à une coupure ....................................................................................................................... 29
II ) ANALYSE DU POLYMORPHISME ................................................................................................................................... 32 Protéines ...................................................................................................................................................................... 32 Analyse des fragments de restriction ........................................................................................................................... 32 Génotypage et cartographie génétique ........................................................................................................................ 34 Les micro et minisatellites ............................................................................................................................................ 35 MAAP (Multiple Arbitrary Amplification profiling) .................................................................................................... 37
III) DETECTION D'UNE SEQUENCE ..................................................................................................................................... 38 EXPRESSION DES PROTEINES RECOMBINANTES .............................................................................................. 39
INTRODUCTION ................................................................................................................................................................. 39 RAPPELS SUR LA TRADUCTION .......................................................................................................................................... 39
Les événements de la traduction .................................................................................................................................. 39 Régulation de la traduction .......................................................................................................................................... 44 Compartimentalisation et modifications post-traductionelles ..................................................................................... 48
EXPRESSION TRANSITOIRE A PARTIR D'UN ARNM OU D'UN ARN IN VITRO (CRNA) ......................................................... 50 Obtention d'un cRNA. ................................................................................................................................................... 50 Traduction en milieu cellulaire. L’ovocyte de Xénope ................................................................................................. 54
EXPRESSION EN SYSTEME PROCARYOTE ........................................................................................................................... 55 I) Escherichia coli ........................................................................................................................................................ 55 II) Autres bactéries utilisées en production de protéines ............................................................................................. 66
EXPRESSION EN SYSTEME EUCARYOTE : LES CHAMPIGNONS ............................................................................................. 70 les levures ..................................................................................................................................................................... 70 Les champignons filamenteux ...................................................................................................................................... 76

24/10/2011 3
Dictyostelium discoideum ............................................................................................................................................ 76 LES CELLULES VEGETALES ............................................................................................................................................... 78 LES CELLULES D'INSECTES ................................................................................................................................................ 80
a) Baculovirus. ............................................................................................................................................................. 80 b) Cellules d’insecte ayant intégré le gène d’intérêt dans leur génome ....................................................................... 85
LES CELLULES DE VERTEBRES ........................................................................................................................................... 87 1) L'ovocyte de Xénope ................................................................................................................................................ 87 2) Cellules de mammifère ............................................................................................................................................. 87
EXPRESSION DANS DES EUCARYOTES UNICELLULAIRES .................................................................................................. 103 LES CAUSES D'ECHECS DE LA PRODUCTION DE PROTEINES. ............................................................................................. 104
1) Il n’y a pas ou peu de protéine ............................................................................................................................... 104 2) La protéine est produite mais est toxique .............................................................................................................. 108 3) La protéine est surexprimée mais est mal repliée .................................................................................................. 110 4) La protéine est produite mais est protéolysée ........................................................................................................ 112 5) Les maturations post-traductionelles ne sont pas correctes .................................................................................. 113 6) La protéine est produite mais on ne sait pas la détecter ........................................................................................ 115 7) La protéine est produite mais on ne sait pas la purifier ........................................................................................ 116 8) La protéine est produite mais il existe aussi des protéines proches produites par la cellule, des endogènes ....... 117 9) Problème particulier lié à l'utilisation de la protéine chez l'homme ..................................................................... 117
TAG, PROTEASES ET ACIDES AMINES MODIFIES ............................................................................................. 118 LES TAG ........................................................................................................................................................................ 118 LES PROTEASES ............................................................................................................................................................... 124 INTEIN ............................................................................................................................................................................. 125 LES ACIDES AMINES MODIFIES ........................................................................................................................................ 127
PURIFICATION DES PROTEINES RECOMBINANTES .......................................................................................... 129 DETERMINATION DE LA CONCENTRATION EN PROTEINE .................................................................................................. 129
La spectrophotométrie UV ......................................................................................................................................... 129 La fluorimétrie ........................................................................................................................................................... 129 Les méthodes colorimétriques .................................................................................................................................... 130
SOLUBILISATION DES PROTEINES .................................................................................................................................... 131 Extraction des protéines cellulaires ........................................................................................................................... 131 Solubilisation et renaturation des corps d’inclusion .................................................................................................. 133
LA CONCENTRATION DES PROTEINES .............................................................................................................................. 136 LES DIFFERENTES METHODES DE SEPARATION DES PROTEINES ....................................................................................... 137
La chromatographie ................................................................................................................................................... 137 L’électrophorèse ........................................................................................................................................................ 138 Séparation selon la taille ........................................................................................................................................... 139 Séparation selon la charge ......................................................................................................................................... 140 Séparation selon la taille et la charge........................................................................................................................ 141 Séparation selon l’hydrophobicité ............................................................................................................................. 141 Séparation selon l’affinité .......................................................................................................................................... 142 Séparation par des colorants ..................................................................................................................................... 144
EVALUATION DE LA PURETE ........................................................................................................................................... 144 CARACTERISATION DES PROTEINES ................................................................................................................................. 145 STABILISATION DES PROTEINES ...................................................................................................................................... 147
Stabilisation des protéines par l’utilisation d’additifs. .............................................................................................. 148 Stabilisation des protéines par mutagenèse dirigée ................................................................................................... 149
ANALYSE DES INTERACTIONS ENTRE MACROMOLECULES ........................................................................ 153 INTERACTIONS ACIDES NUCLEIQUE-PROTEINE ................................................................................................................ 153
Criblage de banque d’expression : ......................................................................................Erreur ! Signet non défini. Simple Hybride .....................................................................................................................Erreur ! Signet non défini. Triple hybride .......................................................................................................................Erreur ! Signet non défini. South-Western ......................................................................................................................Erreur ! Signet non défini.

24/10/2011 4
SELEX ..................................................................................................................................Erreur ! Signet non défini. Immunoprécipitation de chromatine ....................................................................................Erreur ! Signet non défini. Electrophoretic Mobility Shift Assay (EMSA) - gel retard ...................................................Erreur ! Signet non défini. Footprint – empreinte sur l’ADN .........................................................................................Erreur ! Signet non défini. BIAcore ................................................................................................................................Erreur ! Signet non défini.
INTERACTIONS PROTEINE - PROTEINE .............................................................................................................................. 183 Introduction ................................................................................................................................................................ 183 Démarche expérimentale permettant de mettre en œuvre puis de caractériser une interaction protéine – protéine .............................................................................................................................................Erreur ! Signet non défini. Méthodes permettant s’assurer que la protéine appartient à un complexe et de déterminer la taille de ce complexe .............................................................................................................................................Erreur ! Signet non défini. Méthodes permettant d’isoler un complexe protéique .........................................................Erreur ! Signet non défini. Cross-linking (association par des agents pontants .............................................................Erreur ! Signet non défini. Identification des protéines du complexe .............................................................................Erreur ! Signet non défini. Vérification de l’interaction in vivo .....................................................................................Erreur ! Signet non défini. Méthodes permettant d’identifier un partenaire protéique tout en isolant son ADNc .........Erreur ! Signet non défini.
REFERENCES ................................................................................................................................................................... 212 FOURNISSEURS ............................................................................................................................................................ 221
INDEX ............................................................................................................................................................................... 222

24/10/2011 5

24/10/2011 6
Conservartion de l’interférence chez les eucaryotes
Analyse de la fonction d’un gène
Plusieurs approches peuvent être envisagées pour aborder la fonction d’un gène. Les
techniques de biologie moléculaire et de biochimie permettant par exemple de localiser l’expression
d’un gène ou son produit (Northern, western, hybridation in situ, immunofluorescence …), de
déterminer la structure de la protéine (RMN, cristallographie …) ou de trouver des partenaires
protéiques (double hybride, immunoprécipitation …) donnent des informations importantes sur la
fonction d’un gène. D’autres stratégies basées sur des modifications de l’expression d’un gène ou sur
une modification de l’activité de la protéine permettent d’approfondir cette étude.
Un gène dans une cellule ou un organisme peut être soit surexprimé soit supprimé et, dans ce
dernier cas, on regroupe les stratégies sous le nom d’interruption de gène. L’analyse (difficile
certaines fois) du phénotype qu’engendrent ces modifications devrait permettre d’accéder à la (ou
les) fonction(s) du gène.
Stratégie basées sur l’hybridation d’ARN
L’interférence d’ARN (RNAi)
Le phénomène d’interférence d’ARN (RNAi) découvert par Fire et al. (1998) chez le nématode
Caenorhabditis elegans désigne l’inhibition
génique post-transcriptionnelle induite par de
l’ARN double brins (ARNdb). C’est à dire
que la présence dans une cellule d’un ARN
db conduit dans certaines conditions à la
dégradation spécifique de l’ARN messager
de séquence identique (à l’un des brins). Ce
mécanisme, qui semble être un moyen de
résistance vis-à-vis de virus et des
transposons chez les eucaryotes (hormis la levure), est à la base d’une stratégie actuellement
largement utilisé pour cibler la dégradation d’un ARNm dont on cherche la fonction. De par sa

24/10/2011 7
Complexe enzymatique Dicer
siRNA
Clivage de l ’ARNdb
Ciblage de l ’ARNm
Complexe RISCBrin antisens du siRNA
Clivage de l ’ARNm
ARNm dégradé
Poly(A)
grande spécificité d’action et sa relativement bonne efficacité d’inhibition la stratégie de RNAi a
supplanté l’utilisation d’ARN antisens.
Le RNAi est un processus en 2 étapes qui commence à être caractérisé. Dans la première
étape les longues molécules d’ARNdb sont clivées par la protéine Dicer (une nucléase de la famille
des RNases-III) en petits fragments d’ARNdb de 21 à 25
nucléotides de long : les siRNA (small interfering
RNAs). Dans la seconde étape, les siRNA sont
incorporés dans un complexe ribonucléasique, le
complexe RISC (RNA-induced silencing complex), qui
va guider les siRNA sur l’ARNm de séquence
homologue. Le brin antisens des siRNA s’apparie à
l’ARNm cible ce qui provoque son clivage (par Dicer)
au milieu de la zone d’appariement, et donc
l’impossibilité de traduire la protéine.
Amplification des siRNA
Chez C. elegans, il n’y a besoin d’une très faible quantité de siRNA pour oberver une
inhibition de la traduction d’un gène. Par exemple, on peut inhiber un gène du nématode en le
nourissant avec des bactéries qui expriment des RNAdb, et 2 molécules d’ARNdb suffisent à éteindre
un gène fortement exprimé tel que unc-22 gène codant pour une protéine musculaire. Cette puissance
d’action est retrouvée chez les plantes et le champignon Neurospora mais pas chez la drosophile ni
les cellules de mammifères. Ce faible besoin en siRNA pour observer un effet, rend leur action
transmissible à la descendance, les siRNA se retrouve dans les cellules germinales.
Pour isoler la protéine responsable de
l’amplification des siRNA, une approche génétique a
été faite chez Neurospora par une une analyse de
mutants déficients. Ceci a permis d’identifier un
gène codant pour une enzyme responsable du
phénomène d’amplification observé. Il s’agit d’une
RNA polymérase RNA dépendante (RdRP) présente
chez les plantes et chez le nématode mais absente
ARNdb
Dicer
Clivage de la cible
RISCComplexe siRNA actif
siRNA secondaire
Synthèse d ’ARNpar une RdRP

24/10/2011 8
U5’3’U(N19)
3’ U 5’U (N’19)
chez la drosophile ou les vertébrés. Cette enzyme est capable de synthétiser un ARN complémentaire
à partir d’un ARN matrice et d’une amorce provenant d’un siRNA. Ceci entraîne la production
d’ARNdb qui sera à son tour clivé par l’enzyme dicer pour donner des siRNA secondaires qui eux
même pouront servir d’amorce à une nouvelle polymérisation d’ARNdb. Ce système d’amplification
rends l’utilisation d’ARNdb très efficace pour inhiber un gène chez C. elegans.
Les longs ARNdb sont aussi couramment utilisés aussi chez la drosophile ou dans les cellules
de drosophile en culture (cellule S2). Cependant, chez les mammifères, l’utilisation de longs ARNdb
entraîne des effets non spécifiques tels que l’activation de la production d’interferon avec l’induction
d’une protéine kinase ARN dépendante (PKR) conduisant à la dégradation non spécifique de tous les
ARNm, puis à l’apoptose des cellules. Pour remédier à ce problème, le groupe de T. Tuschl en 1999,
a montré que l’on pouvait l’utiliser directement des siRNA et non de longs ARNdb.
Pratiquement, les siRNA peuvent être obtenus de différentes manières :
- par synthèse chimique
- par clivage in vitro de long ARNdb (à l’aide de l’enzyme Dicer recombinante)
- par l’utilisation de vecteurs codant des ARN tige boucle
Synthèse de siRNA
Les siRNA peuvent s’acheter chez de nombreux fournisseurs ; il suffit de donner les
séquences sens et antisens. La figure montre la
structure d’un siRNA synthétique, composé de 21
nucléotides de longueur hybridant sur 19 nucléotides
avec 2 U sortant en 3’. Generallement on remplace les
2 U par 2 T ce qui permet une meilleure stabilité des siRNA pour un coup moindre.
Il a été montré de façon empirique que le choix de la séquence cible était très important pour
l’efficacité de l’inhibition. Le siRNA doit être choisi dans l’ORF (cadre ouvert de lecture)
préférentiellement au milieu de la séquence codante, 100 nucléotides après l’AUG et 100 avant le
stop. Actuellement il se développe des logiciels pour dessiner les siRNA à partir de la séquence d’un
ARNm.
Les paramètres à prendre en compte lors du choix d’un siRNA
- Pourcentage en GC

24/10/2011 9
- Recherche dans les banques de données afin de s’assurer de la spécificité d’inhibition
- Choix d’une méthode de transfection efficace en fonction du type cellulaire.
Clivage in vitro de longs ARNdb :
Une stratégie utilisée avec succès permet de synthétiser in vitro
des siRNA en ultilisant l’enzyme Dicer qui est produite par
génie génetique (protéine recombinante).
Principe : l’ADNc du gène à éteindre (ou uniquement son
ORF) est cloné dans un vecteur comprenant deux promoteurs
phagiques. Les ARN sens et antisens sont transcrits in vitro et
hybridés en solution. L’enzyme Dicer est rajoutée à l’ARN
puis le siRNA sont purifiés et transfectés.
L’avantage de cette approche est que l’on synthétise en
quelque sorte des siRNA correspondant à toute la séquence de
l’ARN.

24/10/2011 10
Insertion au hasard Recombinaison homologue
Le Knock-out ou l’invalidation d’un gène par recombinaison homologue
Le knock-out d’un gène (ou K.O.) signifie la perte physique de la séquence (ou d’une partie
de la séquence) de ce gène conduisant à l’absence de l’ARN messager et donc de la protéine. Le KO
est donc transmissible à la descendance. Attention cependant : chez les eucaryotes, le KO d’un gène
signifie que la séquence des deux allèles est affectée, la descendance respecte les lois de Mendel.
Le KO revient à réaliser de la mutagenèse dirigée (par délétion). Ceci est rendu possible par
l’existence d’un mécanisme fondamental d’échange de séquence d’ADN homologue au niveau
chromosomique : la recombinaison homologue.
La recombinaison homologue : un mécanisme à l’origine de la technologie de Knock-out
La recombinaison homologue est un échange de fragments d’ADN entre deux molécules
(d’ADN) au niveau des séquences nucléotidiques homologues. C’est ce qu’il se passe lors de
crossing over à la méiose entre les chromatides des paires de chromosomes. Le KO est basé sur de la
recombinaison homologue entre de l’ADN exogène et de l’ADN génomique. Ce mécanisme connu
depuis longtemps chez la levure a été mis en évidence en 1980 chez les mammifères par injection
directe d’ADN dans des cellules de mammifères.
Lorsque de l’ADN (plasmidique par exemple) est transfecté dans des cellules eukaryotes, il peut : - soit rester à l’état épisomique (transfection transitoire)
- soit s’intégrer dans le génome (transfection stable)
Dans ce dernier cas, l’ADN s’intègre au hasard (généralement en plusieurs copies inversées),
mais dans un nombre de cas rares, il peut
s’intégrer par recombinaison homologue (RH)
si une séquence identique existe dans le
génome.
Les facteurs qui favorisent la recombinaison homologue :
- La taille de la région homologue (de 4 à 9 kb, on multiplie par 20 la RH)
- Le fort pourcentage d’homologie

24/10/2011 11
- L’ADN sous forme linéaire (par rapport à circulaire)
- Le mode de transfection
Il semble en revanche que ni le nombre de copies présentes dans la cellule, ni la localisation
chromosomique du gène à invalider, n’influe sur la RH.
En tout état de cause, il est indispensable de sélectionner les événements de recombinaison.
Les différents types de sélection utilisés chez les eucaryotes supérieurs ( pour l’établissement de
lignées stables) reposent sur l’utilisation de gènes de résistance à des drogues telles que la néomycine
(ou G418), la bléomycine, l’hygromycine, la puromycine.
D’autres marqueurs de sélection sont également utilisés :
Le gène codant pour la DHFR (dihydrofolate réductase) résiste au méthotrexate.
Le gène HPRT : Hypoxantine phosphoribosyl transferase. Ce gène permet une double sélection,
positive et négative. Les cellules possédant ce gène poussent en milieu HAT (hypoxanthine,
aminoptérine, thymidine) mais sont sensible au 6-thioguanine, inversement l’absence de ce gène rend
les cellules sensibles au milieu HAT et résistantes à la 6-thioguanine.
Le gène HSV-TK : la thymidine kinase du virus herpès simplex. Cette enzyme est capable de
phosphoryler le gencyclovir qui devient ainsi toxique pour les cellules.
Les souris KO Nous venons de voir qu’il est possible de créer et sélectionner des cellules stables déficientes
pour un gène. Nous allons voir comment il est possible de créer à partir de ces cellules un organisme
entier KO. Ceci est devenu possible grâce à l’isolement de cellules souches embryonnaires de souris,
les cellules ES (embryonic stem cell)
Les cellules ES
Ce sont des cellules isolées à partir d’embryons de souris et possédant des propriétés
remarquables :
- Elles sont totipotentes, c’est-à-dire que ce sont des cellules indifférenciées possédant le potentiel de devenir n’importe quel type cellulaire
- Leur croissance est illimitée en culture
- Elles sont facilement transfectables (et sélectionnables)
- Il est possible de les réimplanter (après modification génétique) dans un blastocyste de
souris et elles pourront alors coloniser tous les types cellulaires (donc aussi la lignée germinale) et,

24/10/2011 12
ATG
ATG
Stop
Stop
HSV-TK
Néo RGanc R
Néo
Néo
1 2 3 4 5
2 3 3 4
Exons :
Vecteur
Chromosome
dans ce cas, donneront naissance à des souris chimères pouvant donner des descendants
hétérozygotes pour la modification génétique.
Fabrication de souris transgéniques
Une fois les cellules ES sélectionnées,
elles vont être injectées dans le blastocoele
(cavité) d’embryons au stade blastocyste prélevé
sur une femelle donneuse (dont la couleur du
pelage est plus claire que la femelle donneuse de
cellules ES). Quelques blastocystes ainsi
modifiés vont alors être réimplantés dans une
femelle receveuse. Les souriceaux dérivant des
blastocystes modifiés seront mosaïques :
certains tissus proviennent des cellules ES et
d’autres dérivent du blastocyste hôte. En
fonction de l’étendu du pelage foncé provenant
des cellules ES modifiées, on peut se rendre
compte de l’importance de la colonisation
tissulaire de ces cellules, l’espoir étant que les
cellules modifiées aient colonisé la lignée germinale. Seuls ces individus seront capables de
transmettre le gène invalidé à leur descendance. Le croisement de ces souris avec des souris sauvages
donnera 50% d’hétérozygotes (+/-) pour la mutation et 50% de souris sauvages (+/+) . Le croisement
des hétérozygotes entre eux permet d’obtenir 25% d’homozygotes possédant les deux allèles mutés (-
/-) : ce sont les souris KO pour le gène considéré.
Les différentes constructions utilisées
L’interruption d’un exon : knock-out de gène La figure ci-contre montre un exemple de construction simple aboutissant, après
recombinaison homologue, à la perte de
l’expression d’un gène. Le gène de
résistance à la néomycine a été introduit
dans la séquence de l’exon 3 d’un gène

24/10/2011 13
HSV-TK
ATG Stop
StopATG
Néo
Néo
Transcription - Epissage
StopATG
*
*
*
*
Néo RGanc R
ATG Stop
hprt
ATG StophprtHAT R6-TG S
*
ATG Stop*HAT S6-TG R
1er évènement de recombinaison
2ème évènement de recombinaison
quelconque. Le gène de la thymidine kinase du virus de l’herpes est introduit en aval de la séquence
du gène. Les événements de recombinaison homologue sont sélectionnés par la double sélection :
positive par le néomycine et négative par la thymidine kinase. Les cellules recombinées au niveau du
gène sont néomycine résistantes et gancyclovir résistantes, alors que les intégrations au hasard seront
néomycine résistantes et gancyclovir sensibles.
Création de mutation ponctuelle
Si maintenant le gène de résistance
à la néomycine se situe dans un intron et
que l’on a créé une mutation dans l’exon
3 du gène, la recombinaison homologue
de cette construction aboutira à
l’introduction de la mutation au niveau
du génome. La présence de la cassette
de résistance dans un intron n’empêche pas la traduction ni l’épissage, les modifications qu’entraîne
cette construction sont théoriquement dues uniquement à la mutation. Il à cependant été montré que
dans certains cas la présence d’une cassette de sélection dans un intron peut avoir un effet sur le
niveau de transcription du gène. Ce qui a conduit le chercheurs à imaginer des stratégies pour
éliminer le gène de sélection afin d’aboutir à des mutations « propres »
Mutation propre par double remplacement
Dans cette exemple on se base sur la
propriété du gène HPRT permettant une double
sélection (voir plus haut). Le premier événement
de recombinaison apporte gène hprt dans l’intron
(cellules HAT résistantes et 6-thioguanine
sensibles). La seconde RH amène la mutation et
élimine le gène hprt, les cellules deviennent
HATS et 6-TGR .

24/10/2011 14
Le Knock-in
Cette stratégie permet de créer un KO, et en même temps de remplacer le gène par un autre gène qui s’exprimera ainsi comme le gène interrompu. Si le nouveau gène est un gène rapporteur, type LacZ ou GFP, ceci permet de localiser l’expression du gène délété dans la souris au cours du développement ou chez l’adulte (ce qui n’est pas toujours facile autrement).
Cette stratégie peut aussi être utilisée pour montrer qu’un autre gène peut remplacer la fonction de
celui qui est interrompu. Par exemple, la cycline E peut sauver le phénotype du KO de la cycline D.
Les systèmes inductibles
Il se peut que des souris KO pour un gène ne soient jamais obtenues. C’est le cas lorsque la mutation est lethale embryonnaire ; les homozygotes mutants ne sont jamais mis au monde, ou bien l’expression du gène étudié est très large, donc le phénotype mutant très complexe. Dans ces cas, on va vouloir contrôler la mutation dans le temps et dans l’espace.
Le système Cre/Lox
La Cre est une recombinase (de la famille des intégrases) du bactériophage P1. C’est une protéine de 38 KDa qui catalyse la recombinaison entre deux site de reconnaissance, les sites LoxP. LoxP est une séquence d’ADN de 34 pb comprenant aux extrémités 13 nucléotides palindromiques (il est donc impossible de trouver cette séquence dans un gènome eucaryote). Les deux sites peuvent être très éloignés l’un de l’autre et être quand même recombinés par la Cre. Cette recombinase fonctionne dans les cellules eucaryotes génétiquement modifiées comportant deux sites loxP. En fonction de l’orientation des sites loxP, il peut se
produire une délétion ou une inversion. Il est à noter que ce
mécanisme est réversible.
Dans les exemples qui suivent la RH permet d’apporter les

24/10/2011 15
HSV-TKNéo
StopATG *
*
+ Cre
Néo
Néo
Puro
Hyg
ro
Hyg
Hyg Puro ro
Hyg ro
Néo R
Néo
Hygro R
+ Cre
sites loxP sans altérer le gène étudié. La cre recombinase permettra d’éliminer la région entre les
deux sites. L’induction de la cre entraîne l’induction de la déletion.
Exemple d’application La délétion d’exon Deux sites loxP sont insérés de part et d’autre d’un exon du gène à invalider. On dit que
l’exon est « floxé ». Cette construction peut être
utilisée pour remplacer le gène sauvage par
recombinaison homologue dans des cellules ES. On
vérifie que des souris transgéniques homozygotes
(pour l’exon floxé) sont parfaitement saines. Ces
souris peuvent alors être croisées avec des souris
transgéniques exprimant la Cre recombinase dans un
tissu particulier (le génome de ces souris contient le
gène Cre sous la dépendance d’un promoteur tissu spécifique).
Mutation propre Il a été montré que la présence d’un gène de résistance dans un intron, par exemple dans le cas
de mutations ponctuelles, pouvait influer sur la régulation de l’expression du gène (voir plus haut). Et
donc que le phénotype observé n’était pas seulement dû à la mutation. Dans ce cas, on peut utiliser le
système Cre/lox pour éliminer le gène de
résistance. Il suffit, dans la construction, de floxer
la cassette de résistance qui sera éliminée par la
cre.
Large délétion ou translocation
Certaines maladies génétiques sont dues à
de grandes délétions dans le génome (ou à
des translocations). Il est possible de créer
des souris porteuses de ce type de maladie
(afin par exemple d’analyser l’effet de

24/10/2011 16
Cre Cre
Cre
Cre
Cre Cre
P (ubiquitaire ou spécifique)
Cre-ind
Cre-ind
Séquence « floxée »
Injection de l’agonisteà un temps donné
Agoniste(tamoxifène, RU486)
Domaine de fixation au ligand(récepteur des stéroïdes)
InactiveActive
X
certaines thérapies). La figure montre une stratégie basée sur le système cre/lox qui à été utilisée avec
succès. Deux événements de recombinaison homologue sont nécessaires pour introduire les deux
sites lox P et deux parties de la cassette hygromycine. Le premier événement est sélectionné par la
cassette néomycine et le second par puromycine. La cre recombinase permet de rapprocher les deux
parties non fonctionnelles de l’hygromycine devenant ainsi fonctionnelle. Si les deux sites loxP sont
sur le même chromosome il s’en suit une large délétion (plusieurs centaines de Kb). Si ils sont sur
deux chromosomes différents cela conduit à une translocation.
Les possibilités d’induction de la cre recombinase
Un système très sophistiqué d’induction de la cre recombinase à été utilisé récemment (ref). Il
repose sur l’expression d’une protéine de fusion entre la cre et un récepteur tronqué aux
glucocorticoïdes. L’expression de cette
fusion peut être contrôlée par un
promoteur quelconque (ubiquitaire ou
tissu spécifique). La cre ainsi fusionnée
est inactive et ne devient active qu’en
présence d’un agoniste de
glucocorticoïde (et non pas de
glucorticoïdes endogènes) comme le
tamoxifène et le RU486. La figure
montre que le croisement de souris transgénique pour la cre inactive (commerciale) avec de souris
floxé pour un gène donné permet d’obtenir des souris contenant à la fois la cre inactive et un gène
floxé. L’injection de l’agoniste dans un tissu quelconque provoquera la délétion du gène (ou d’une
partie du gène) au moment et au lieu choisi.
Le système tet-on

24/10/2011 17
Détection des mutations / polymorphismes
I) Détection des mutations Les mutations ponctuelles affectent un seul nucléotide et sont souvent appelées SNP (single
nucléotide polymorphism). Elles peuvent être silencieuses, mais peuvent aussi causer des mutations
non-sens, modifier la séquence en acide aminé d’une protéine ou interférer avec l’épissage de l’ARN.
Ces modifications peuvent alors être responsables de maladies génétiques, ou modifier le phénotype
du porteur.
Méthodes basées sur la séquence
La méthode de séquencage de Sanger permet
d'identifier la mutation d'une manière sûre.
Toutefois cette méthode est lourde et génère plus
d’information qu’il n’en faut. Aussi elle n'est
souvent utilisée que pour la mise en évidence
pour la première fois d'une mutation.
Une fois la séquence connue ce n'est pas la peine de séquencer toutes les pistes. Ce n'est pas non plus
la peine de séquencer en aval de la mutation. On peut
utiliser la méthode de séquençage de Sanger en
utilisant uniquement trois desoxynucléotides et un
didesoxynucléotide et une amorce proche de la
mutation de telle sorte que la polymérisation soit plus
ou moins longue en fonction de la présence de la
mutation. Les produits de la réaction sont séparés sur
gel et la présence d'un produit permet de connaître la présence de la mutation.
Exemple de mise en évidence d’une mutation ponctuelle par séquençage
32P NNNNNNNNNNNNNNNNNTGCAdd32P NNNNNNNNNNNNNNNNNTCGGGCAdd
NNNNNNNNTGC GCANNNNNNNAG
Mutation à détecter
Produits de la réaction en présence de ddATP

24/10/2011 18
Cette méthode présente un avantage, si la mutation est présente dans une partie de la population de
départ, en quantifiant la présence des deux produits on peut estimer sa fréquence. Elle a par exemple
été utilisée pour estimer le taux d'édition de certains ARN messagers (Schiffer et Heinemann, 1999).
On peut aussi faire une extension d’amorce en utilisant uniquement le nucléotide à détecter. Dans de
cas le nucléotide sera marqué de façon à déterminer l’incorporation. On peut par exemple utiliser
uniquement un didesoxynuclétotide fluorescent et détecter l’incorporation par electrophorèse
capillaire. Dans ce cas on fait les deux réactions, avec l’extension sauvage et l’extension mutée, et on
fait migrer les produits de la réaction. Pour éviter la migration des nucléotides non incorporés on les
dégrade à l’aide d’une phosphatase alcaline.
La révélation peut aussi se faire à l’aide d’anticorps.
Différentes étapes :
- on amplifie la région de la mutation,
- on fixe l’amorce à un support,
- on hybride le produit de l’amplification avec l’amorce,
A G
Hybridation
A G
Polymérisation en présencede dTTP lié à un antigène reconnu par un anticoprs lié à une phosphatase alcalinede dCTP lié à un antigène reconnu un anticoprs lié à une péroxidase
AT
GC
a b
AT
GC
a bAP HRP

24/10/2011 19
- on polymérise en présence des deux oligonuclétides correspondant au type sauvage et au
type mutant,
- on lave pour éliminer les nucléotides non incorporés
- et on révèle le nucléotide incorporé à l’aide d’un anticorps associé à une activité
enzymatique.
Pour améliorer l’hybridation on peut détruire un des deux brins de l’amplification en le digérant par
une exonucléase, le brin à hybrider sera alors protégé en utilisant un oligonucléotide modifié non
sensible à l’exonucléase.
Cette méthode est commercialisée par Invitrogen. La méthode de séquence en utilisant le pyroséquençage peut être utilisée pour détecter des mutations
(Ahmadian et al., 2000). L’avantage de cette méthode est de faire passer le nucléotide « sauvage » et
« mutant » l’un après l’autre la hauteur relative des deux pics permet d’estimer la proportion de
mutants dans une population.
Méthodes basées sur l’hybridation
Southern
L'hybridation de deux chaînes d'ADN est due à l'appariement de bases complémentaires A-T et G-C.
Elle est donc diminuée lors de mésappariement. Le Tm d'un oligonucléotide de 10 à 20 bases sera
donc abaissé s'il y a un mésappariement.

24/10/2011 20
La méthode de Southern (1975) est particulièrement adaptée pour détecter les grandes altérations
telles que les insertions, les délétions ou les
réarrangements. Mais cette méthode peut aussi être
utilisée pour détecter les mutations ponctuelles si on
utilise un oligonucléotide comme sonde.
Il n’est pas toujours nécessaire de digérer l’ADN par
une enzyme de restriction et de séparer les fragments
par electrophorèse. On peut directement spotter
l’ADN à analyser sur une feuille de nitrocellulose
(« dot-blot »).
Dans certains cas si l'altération se retrouve dans
l'ARN, un northern peut être employé pour la
détecter.
Il existe plusieurs méthodes pour détecter l’hybridation. La plus ancienne est l’utilisation d’un
marquage radioactif. On utilise comme sonde un oligonucléotide marqué en 5' par une kinase et du
α-32P ATP. Comme contrôle, on peut utiliser une sonde plus grande dont l’hybridation n’est pas
sensible à la présence de la mutation (ADNc sur la figure) pour vérifier que la cible était bien en
quantité égale dans les différentes pistes.
L’hybridation peut se faire sur puce à ADN. Un premier spot contient un oligonucléotide sauvage et
un deuxième spot contient un oligonucléotide muté. L’ADN de la région à analyser est amplifié et
marqué avec un fluorophore. Il est hybridé avec les oligonucléotides fixés à la puce, s’il y a
hyburidation il y aura fluorescence, si la mutation diminue le Tm, il n’y aura pas de fluorescence.
Comme on peut disposer d’autant de spots qu’on le désire, on peut cribler en une seule fois la
présence de nombreuses mutations.
sondecDNAoligonucléotide
ADN
mutant témoin mutant témoin
Exemple de mise en évidence d’unemutation ponctuelle par Southern avecun oligonucléotide portant la mutation

24/10/2011 21
On peut suivre l’hybridation en FRET en utilisant deux oligonucléotides, chacun sera porteur d’un
fluorophore. Lorsque l’oligonucléotide n’est pas hybridé, on n’a pas de fluorescence, par contre
lorsqu’il est hybridé on obtient une fluorescence.
Si on augmente peu à peu la température, à faible
température, l’oligonucléotide se fixera d’une
manière non spécifique et on aura pas de
fluorescence. Lorsque la température atteindra le
Tm, on observera une fluorescence. Au dessus de
Tm on perdra la fluorescence. Comme la
présence d’une mutation modifie le Tm, les
courbes de fluorescence en fonction de la température permettent de détecter la présence d’une
mutation.
Hybridation partielle
Dans cette technique on va détecter le fait que
l’oligonucléotide soit ou ne soit pas
complètement hybridé lorsque l’hybridation est
totale ou lorsque l’hybridation présente un
mésappariement.
Le fragment portant la mutation est amplifiée
par PCR, puis on hybride deux oligonuléotides
qui portent à leurs extrémités des fluorophores
tels que le pyrène. Un pyrène est positionné en
3' d'un oligonucléotide et un autre en 5' de
l'autre oligonucléotide. Lorsque les deux
pyrènes sont proches l'un de l'autre, lorsque les
deux oligonucléotides s'hybrident parfaitement
on obtient une émission de fluorescence
(excitation à 350 nm et émission à 490 nm) par
O
O
O
PP
PP
cible
oligonucléotidesfluorescence
pas de fluorescence
fluoresceineémission dansle rouge
mutation
ADN
excitationémission
Principe

24/10/2011 22
contre lorsqu'il y a un mésappariement, dans le cas d'une mutation, les deux pyrènes ne sont plus
proches l'un de l'autre et on n'obtient pas de fluorescence (Paris et al., 1998).
Comme amélioration de la méthode on a l'utilisation de d'appareil PCR permettant de suivre
l'émission de la fluorescence au cours des amplifications successives.
Utilisation d’un anticorps
On peut suivre l’hybridation en utilisant un anticorps qui reconnaît l’ADN double brin (Viazov et al.,
1994) Cette méthode est souvent appelée « DNA enzyme immunoassay » (DEIA). Dans une
première étape, une région contenant la mutation à détecter est amplifiée par PCR. Ce produit
d’amplification est alors hybridé à un nucléotide spécifique fixé sur un support solide via un pont
streptavidine-biotine. S’il y a hybridation, elle est détectée par ELISA en utilisant un anticorps se
fixant sur l’ADN double brin.
DGGE = Denaturing Gradient Gel Electrophoresis Principe : Le Tm dépend de la séquence, de la température, de la
concentration en certaines molécules telles que l'urée ou la
formamide. La dénaturation est un phénomène coopératif. La
migration d'un fragment d'ADN change avec la dénaturation, un
ADN double brin partiellement ouvert migre moins vite qu'un
fragment double brin (Myers et coll. 1985)
Dans cette technique, on fait migrer un fragment d'ADN dans un
gel gradient allant de conditions natives ou l'ADN est sous
forme double brin à des conditions dénaturantes ou l'ADN est
sous forme simple brin (7M urée, 40% formamide). La
température est maintenue constante.
L'ADN est extrait puis digéré par une enzyme de restriction qui coupe souvent, tous les 200-400 pb.
Le produit de la digestion est chargé sur un gel gradient linéaire, il migre sous la forme de double
brin mais à un moment il atteint son Tm, est partiellement dénaturé et migre moins vite. Ainsi des
fragments de séquences différentes, même d'une seule base sur 100 pb migrent à des endroits
**
sauvage mutant
gel
sauvage mutant
**
sauvage mutant
gel
sauvage mutant

24/10/2011 23
différents. Pour détecter les bandes, L'ADN est transféré et hybridé avec une sonde spécifique d'une
région ou on analyse des fragments de PCR après les avoir marqués
Au lieu d’effectuer un Southern, on peut amplifier la région de la mutation par PCR, faire migrer les
deux produits de PCR dans un gel à concentration croissante d’un agent dénaturant.
Un des problèmes rencontrés avec cette technique : les fragments se séparent trop tôt dans le gel.
Pour remédier à de problème, on effectue la PCR avec des amorces riches en GC dans la région 5'
(non importante pour la réaction de PCR). Ces extrémités GC stabilisent le duplex et permettent une
meilleure séparation des homoduplex et des hétéroduplex. DHPLC: Denaturing high-performance liquid chromatography
Cette méthode est proche de la DGGE mais ici la détection des hétéroduplex se fait par HPLC en
utilisant une colonne de paire d’ions en phase reverse dans les condition partiellement dénaturantes
(Oefner et Underhill, 1995).

24/10/2011 24
Méthodes basées sur la PCR
Les altérations du génome peuvent être détectées par PCR en utilisant plusieurs amorces, ce qui
permet l'amplification simultanée de plusieurs régions (multiplex PCR). Cette méthode a été
employée pour la première fois par Chamberlain et coll. (1988) pour détecter les mutations de la
dystrophine responsable de la maladie de Duchenne.
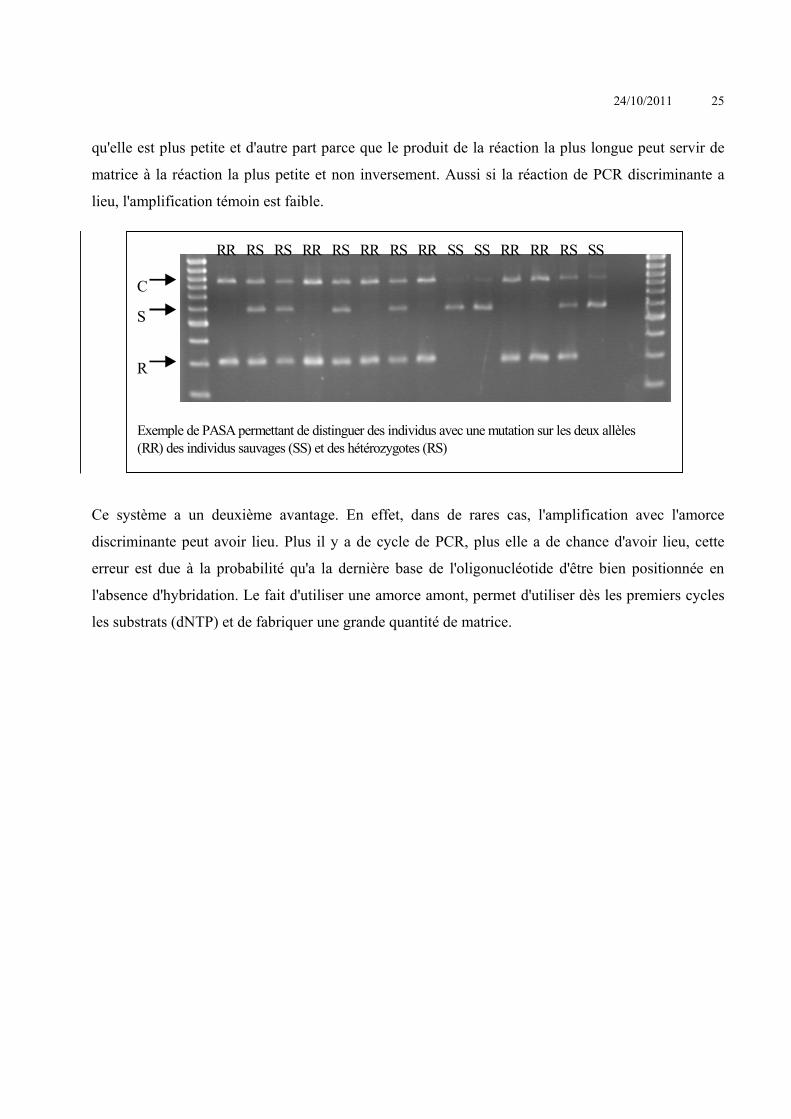
PCR oligonucléotide allèle spécifique, PASA (PCR Amplification of Specific Alleles)
En PCR, la polymérase ne peut polymériser que si la dernière base en 3' est stabilisée par des liaisons
hydrogènes, si la dernière base est hybridée. La plupart des polymérases ont une activité 3'-
5'exonucléase, mais certaines d'entre elles comme l’ADN polymérase Taq en est dépourvue. En
prenant deux oligonucléotides, un spécifique de la mutation et un autre spécifique du témoin
(sauvage) on peut faire deux PCR à l'aide d'un troisième oligonucléotide situé en 3' (en aval) à
environ 1 kb. On aura amplification uniquement qu’avec un des deux couples (Sommer. 1992)
L'absence de bande ne signifie pas toujours l'absence d'hybridation en 3' mais peut résulter d'un
défaut lors de PCR, d'une erreur de manipulation. Un témoin peut être ajouté, c'est un oligonucléotide
qui hybride sur tous les allèles (connus) en amont de l'hybridation permettant de voir la mutation. On
obtient donc deux bandes, une bande haute permettant de voir si la polymérisation a bien eu lieu et
une bande basse permettant de voir la présence ou non de la mutation, suivant l'oligonucléotide qui a
été utilisé.
Les deux réactions de PCR (entre l'amorce amont et l'amorce aval et entre l'amorce discriminante et
l'amorce aval) entrent en compétition, et l'amplification la plus petite est favorisée, d'une part parce
matrice
amorce aval
amorces discriminantes
amorce amontou
24/10/2011 25
qu'elle est plus petite et d'autre part parce que le produit de la réaction la plus longue peut servir de
matrice à la réaction la plus petite et non inversement. Aussi si la réaction de PCR discriminante a
lieu, l'amplification témoin est faible.
Ce système a un deuxième avantage. En effet, dans de rares cas, l'amplification avec l'amorce
discriminante peut avoir lieu. Plus il y a de cycle de PCR, plus elle a de chance d'avoir lieu, cette
erreur est due à la probabilité qu'a la dernière base de l'oligonucléotide d'être bien positionnée en
l'absence d'hybridation. Le fait d'utiliser une amorce amont, permet d'utiliser dès les premiers cycles
les substrats (dNTP) et de fabriquer une grande quantité de matrice.
RR RS RS RR RS RR RS RR SS SS RR RR RS SS
R
S
C
Exemple de PASA permettant de distinguer des individus avec une mutation sur les deux allèles(RR) des individus sauvages (SS) et des hétérozygotes (RS)

24/10/2011 26
Ligation
Les ADN ligases catalysent l'estérification entre un nucléotide 5' phosphate et un nucléotide 3' OH. Il
faut toutefois que ces deux fonctions soient maintenues proches l'une de l'autre.
Pour détecter l'événement de mutation, on peut marquer un oligonucléotide. Une méthode pour
augmenter le signal consiste à répéter plusieurs fois l'expérience en faisant des cycles successifs de
dénaturation et de ligation. Dans ce dernier cas on utilisera de préférence une ligase thermostable
(dans ce cas l'amplification ne suit pas une progression exponentielle).
Ligation
Gel, conditionsdénaturantesautoradiographie
Hybridation

24/10/2011 27
RFLP
Dans certains cas la mutation recherchée coïncide avec un site de restriction, toutefois c'est assez
rare. Dans d'autre cas, la mutation est proche d'une autre mutation qui, elle, coïncide avec un site de
restriction. La coupure est alors détectée par Southern ou par coupure d'un fragment de PCR.
Sandwich: L'ADN cible est fixé sur une membrane de nitrocellulose puis hybridé avec une sonde.
Après formation du duplex il y a coupure avec une enzyme de restriction. S'il y a homoduplex
l'enzyme coupe et l'oligonucléotide est deshybridé, s'il n'y a pas coupure l'hétéroduplex reste sur la
membrane.

24/10/2011 28
Méthodes basées sur la conformation de l’ADN SSCP (Single Strand Conformation Polymorphism)
Cette méthode est basée sur l'influence de la séquence primaire sur la migration en gel natif de
fragment d'ADN simple brin (Orita et coll., 1989). En condition non dénaturante, les brins d'ADN
forment des structures secondaires en tiges/boucles qui sont uniquement dépendantes de la séquence
primaire, de plus il existe une structure tertiaire provenant de liaisons entre les sucres et les bases.
Donc une variation dans une séquence primaire s'accompagne d'une différence de conformation de
l’ADN simple brin correspondant.
Le fragment obtenu par PCR ou le génome entier est coupé par une enzyme de restriction. L'ADN
est dénaturé soit par la soude soit par la chaleur et déposé sur un gel d'acrylamide. L’ADN est ensuite
mis dans des conditions permettant la formation de structures secondaires en abaissant la
températurure ou en neutralisant le pH. La migration est détectée soit par coloration à l'argent
(produit de PCR) soit par Southern (ADN génomique), soit après marquage du produit PCR
(radioactif ou fluorescent).
Cette méthode permet de repérer des mutations dans une région, même si elles ne sont pas connues.
R
S
SS SS RS SS RS RS SS SS RR RS RR SS
Exemple de SSCP pour determiner le génotype des individus d’une population pour une
mutation conférant la résistance : homozygote sensible (SS), homozygote resistant (RR) et
hétérozygotes (RS).

24/10/2011 29
ddF (Dideoxyfingerprinting)
Cette méthode (Sakar et coll. 1992) est un hybride
entre la séquence selon la méthode de séquençage à
l'aide des dideoxynucléotides et la SSCP. Le fragment
d'ADN où se trouve la mutation est amplifiée puis une
réaction de séquence est faite en employant un seul
dideoxynucléotide et une amorce marquée en 5'. Le
produit de la réaction est dénaturée puis chargée sur un
gel d'acrylamide non dénaturant.
Hétéroduplex sensibilité à une coupure
Dans cette technique les deux fragments d'acide nucléique sont hybridés et on détecte les
mésappariements en les coupant avec des enzymes ou chimiquement.
Utilisation de la nucléase S1
Cette nucléase d'Aspergillus oryzae est spécifique de
l'ADN simple brin et ne digère pas l'ADN double brin. On
fait une sonde ADN simple brin marqué radioactivement
et on hybride avec les différentes populations. On fait
ensuite un gel en condition dénaturante pour voir la taille.
Une autre possibilité est d'amplifier la région portant la
mutation avec des oligonucléotides portants des
fluorophores. Les fragments amplifiés de l'ADN témoin et
de l'ADN testé sont réunis dans le même tube, dénaturés et
hybridés. La présence d'une mutation est ensuite mise en
évidence en incubant le produit de l'hybridation avec la
nucléase puis en faisant migrer les produits de la digestion sur un gel en conditions dénaturantes.
types1 1 2 1 3 3 3 3 4 3 4
exemple de ddF (Langemeier et coll., 1994)
ADN 1 ADN 2
PCR
dénaturation / hybridation
digestion par une nucléasedénaturation

24/10/2011 30
D'autres nucléases peuvent être employées, la nucléase P1 de Penicillium citrinum ou la mung bean
nucléase de Vigna radiata ou la nucléase CEL 1 du céleri. Les trois premières nucléases sont des
protéines à Zinc et sont principalement actives à pH 5. Cette action à pH acide pose quelques
problèmes pour les régions riches en AT en effet à ce pH on peut avoir une dénaturation partielle de
l'ADN. Dans ce cas on peut utiliser une nucléase de plante comme la nucléase CEL 1 du céleri qui
coupe l'ADN simple aux pH neutres (Oleykowski et coll. 1998)
Certaines nucléases comme la nucléase S1 digèrent très difficilement lorsque le mésappariement est
limité à une seule base, mais au-dessus de deux bases la vitesse de réaction augmente énormément.
Dans ce cas, la technique n'est donc valable pour détecter des délétions de petites tailles.
Utilisation des RNAse (Myers et coll., 1985)
Les RNAse A, T1 ou T2 coupent les ARN sous forme
simple brin et non lorsqu'ils sont hybridés. Lors d'une
hybridation entre deux clones de deux individus
différents, les mésappariements seront digérés et seuls
les régions hybridées resteront. La technique est donc
équivalente à la digestion par la nucléase S1 mais ici
on utilise une sonde ARN.
Cependant les RNAse ne digèrent pas tous les
mésappariements, elles digèrent uniquement C-A, C-
C, C-T et U-T, c'est à dire 4 sur les 12 possibles.
Pour augmenter ce nombre on peut faire deux ARNs
marqués pour les deux espèces et dans ce cas on voit
60% des mésappariements.
Un fragment de PCR est synthétisé, une des deux amorces comporte la séquence du promoteur de la
T7 et l'autre la séquence SP6.
Utilisation de la nucléase ABC
Cette nucléase ne reconnaît pas les mésappariements à moins qu'ils n’aient auparavant réagi avec la
carbodiimide. L'hétéroduplex est modifié par la carbodiimide et ensuite digéré par la nucléase ABC.
ARN marquéARN messager
Hybridation
coupure à la RNAse A + T1
gel
sauvage mutant

24/10/2011 31
Cette méthode permet de voir toutes les mutations mais la nucléase ABC n'est pas actuellement
disponible.
Coupure par les méthodes chimiques (CCM= Chemical Cleavage of Mismatch)
Les méthodes chimiques ont été développées par les auteurs qui ont étudié les structures secondaires
des ARN. Les produits chimiques présentent un certain nombre d'avantage sur les enzymes, ils
agissent sur une gamme de pH et de force ionique beaucoup plus large, il n'y a pas de problème
d'instabilité et ils ne nécessitent pas d'avoir un acide nucléique purifié.
De nombreux produits chimiques réagissent avec les bases des acides nucléiques, ils produisent des
lésions qui peuvent être ensuite coupées par un traitement alcalin, c'est le principe de base de la
méthode de séquence de Maxam et Gilbert.
Parmi ces produits l'hydroxylamine (H) réagit avec les C mésappariés, le tetraoxide d'osmium réagit
avec les T mésappariés (OT). La combinaison des deux (HOT) permet de détecter les C et T. (Cotton
et coll., 1988).

24/10/2011 32
II ) Analyse du polymorphisme
On peut analyser le polymorphisme en analysant plusieurs mutations mais souvent on s'intéresse à
plusieurs locus.
Protéines Les premières méthodes d’analyse du polymorphisme utilisaient les différences de migration des
protéines d’un individu sur gel à l’état natif. Quelques protéines telles que les estérases étaient
révélées par leurs activités.
Ces méthodes ont été très utilisées par les généticiens des populations mais le nombre de variants
était assez faible et pouvait résulter d’un effet transcriptionnel.
Analyse des fragments de restriction RFLP
On peut déterminer l'identité d'un plasmide en effectuant sa carte de
restriction. Cette méthode peut s'appliquer à tous les fragments d'ADN
à la condition qu'ils ne soient pas trop grand, à condition que la
digestion ne génère qu'un nombre limité de fragment. Chez les
eucaryotes, les petits génomes sont représentés par les génomes
mitochondriaux et chloroplastiques. On purifie donc les mitochondries
ou les chloroplastes puis on isole leur ADN. La carte de restriction
permet ainsi de les identifier et en conséquence d'identifier leur porteur.
Cette méthode n'ayant pas besoin d'information de départ, elle a été une
des premières utilisées pour différentier des individus ou des
populations.
On peut aussi analyser les sites de coupures par des enzymes de restriction sur l’ADN génomique.
Cependant la digestion donne trop de fragments pour pouvoir être analysé. Pour rféduire le nombre
de fragment on peut soit faire un Southern avec un mélange de sonde, soit amplifié certaines régions
Hae III EcoRI
RFLP sur l’ADNchloroplasmique detrois individus(Mariac et coll. 2000)

24/10/2011 33
du génome par PCR puis ensuite analyser le polymorphisme de restriction sur les fragments
amplifiés.
AFLP (Amplified Fragment Length Polymorphism) On digère l'ADN avec deux enzymes, une qui coupe
rarement comme par exemple Eco RI (G/AATTC) et une
qui coupe souvent comme par exemple Mse I (T/TAA).
On ajoute deux adaptateurs aux deux extrémités à l'aide
d'une ligase. Un adapteur différent est utilisé pour chaque
site et l'enzyme de restriction ne coupe pas le produit de la
ligation. Les adaptateurs sont à une concentration
supérieure à celle des fragments d'ADN si bien que la
ligation se fait préférentiellement avec l'adaptateur.
L'adaptateur de site rare (Eco RI) est lié à la biotine ce qui permet de sélectionner les fragments
EcoRI/ MseI et Eco RI / Eco RI sur une colonne de streptavidine agarose.
Ces fragments sont amplifiés par PCR avec des oligonucléotides spécifiques des deux adaptateurs
utilisés, celui hybridant sur l'adaptateur du site rare (Eco RI) est marqué radioactivement pour ne pas
voir les amplifications des fragments Mse I/ MseI qui n’auraient pas été éliminés lors de la sélection
par la biotine.
Le résultat de la PCR est analysé sur gel de séquence par radioautographie. S’il y a trop de bandes,
un site coupant plus rarement est utilisé.
Adaptateur MSE I (T/TAA)
TANNNNNNNNNNNNNNNNNNNNNN
Adaptateur Eco RI (G/AATTC)
AATTNNNNNNNNNNNNNNNNNNNNNNNNNNNN-biotine

24/10/2011 34
Génotypage et cartographie génétique
Parmi les variations de séquences entre individus, l'identification d'une mutation ponctuelle (SNP,
single-Nucleotide Polymorphism) est une information utile dans de nombreux domaines et plus
particulièrement comme marqueur de maladie génétique.
Il existe plusieurs techniques permettant de détecter une mutation ponctuelle, mais récemment une
équipe a développé une méthode qui permet de mettre en évidence des milliers de mutations
ponctuelles dans le génome humain en une seule expérience (Wang et al., 1998).
Il fallait au préalable identifier les SNP, ils ont donc séquencé un grand nombre de STS (Sequence-
Tagged Site) qui sont des séquences réparties au hasard sur l'ensemble du génome. Mais cette
méthode est laborieuse. Une deuxième méthode a alors été mise en œuvre, à l'aide de puces à ADN.
Des oligonucléotides correspondant à des STS ou des EST ont été synthétisés sur une puce, et des
oligonucléotides comportant des mutations ont été synthétisées sur les spots voisins. En hybridant
l'ADN d'individus différents sur ces puces, ils ont pu détecter des différences d'hybridation et donc
mette en évidence des SNP. Ils ont ainsi utilisé 149 puces comportant chacune 15000 à 30000
oligonucléotides. Ce qui a permis de mettre en évidence environ 3000 SNP.
La position des SNP sur la carte génomique a pu ensuite être déterminé en utilisant une série de
lignées cellulaires comportant chacune une fraction du génome humain (radiation-hybrid mapping).
Une fois les SNP déterminés et éventuellement cartographiés, il a suffit de synthétiser des petits
oligonucléotides variants hybridant sur un allèle ou sur l'autre puis à les déposer sur une puce à ADN.

24/10/2011 35
Les micro et minisatellites
Les différentes parties du génome sont plus ou moins variables, les parties codantes et les promoteurs
présentent des séquences très conservées par contre, les parties non codantes sont plus variables.
Mais il existe des éléments qui sont encore plus variables, ce sont les micro et minisatellites. Ce sont
des éléments hautement répétés qui forment des répétitions de 1000 ou de million de copies
similaires. Leur nom vient du fait qu'au départ ils ont été identifiés parce qu'ils étaient présents dans
une bande différentiée en centrifugation isopycnique (en gradient de CsCl). Cela est du au fait que
certains d’entre eux avaient une composition en base différente de l'ADN total. Ils sont généralement
présents dans l'hétérochromatine des chromosomes, région ou la fréquence de recombinaison est
faible comme par exemple le centromère.
Ils ont la propriété d'être très variables, ceci étant du
- à des mutations, comme pour le reste de l'ADN
- à des crossing over inégaux
- à un glissement de polymérase
- à une réplication en "rolling circle".
La variabilité est fonction du satellite analysé, le plus variable dans l'individu ne sera pas utilisable,
un moins variable permettra de voir des différences entre les individus d'une même population
(utilisable par exemple en criminologie), un moins variable permettra de voir des différences entre les
populations.
Microsatellites ou STR (Short Tandem Repeat) : répétitions de 2 à 4 bases, ils sont très variables,
On les clone en criblant une banque génomique avec un oligonucléotide composé de la répétition. On
séquence les clones pour connaître les séquences adjacentes, qui elles sont présentes en simples
copies dans le génome. On synthétise des oligonucléotides spécifiques qui hybrident de chaque coté
de la répétition. On amplifie par PCR en présence de nucléotides marqués et on fait un gel dénaturant
de type séquence. On peut ainsi déterminer la taille de la répétition.

24/10/2011 36
Minisatellites (ou VNTR : Variable Number Tandem Repeat) répétitions de 8-15 bases, variabilité
qui proviendrait de crossing over inégaux.
Comment les clone t’on? En prenant une sonde au hasard, au début on prenait M13 comme sonde et
on criblait une banque d'ADN génomique. L'hybridation se fait mal mais comme elle se fait
fréquemment on obtient une hybridation décelable. Une autre méthode fait appel au hasard, en
criblant des banques avec un oligonucléotide pour trouver un gène, par le même effet, on clone des
minisatellites. Le clone est séquencé et on obtient la séquence du minisatellite qui peut servir pour
l'espèce et pour des espèces proches.
Une troisième méthode consiste à digérer l'ADN avec une ou plusieurs enzymes en qui coupent
fréquemment (tous les 400 pb) comme Sau3A, GATC. Puis à faire un gel qui est coloré au bromure
d'éthidium. On voit des bandes de haut poids moléculaire. Ces bandes sont extraites du gel et clonées,
au besoin après coupure au hasard (ultrason), puis séquencées.
Utilisation des minisatellites : L'ADN des populations analysées est digéré par une enzyme de
restriction qui coupe souvent mais pas dans la répétition. On obtient ainsi des fragments qui
correspondent à la taille des répétitions. Ces fragments sont séparés sur un gel d'agarose, transférés
sur nitrocellulose ou nylon et hybridés avec le minisatellite. On obtient un fingerprint.
Utilisation: typage au niveau de l'individu : criminologie, recherche de paternité
Détection du nombre de répétition d’un élément répété
Il existe plusieurs méthodes pour quantifier le nombre de répétitions :
- On peut faire une PCR quantitative en utilisant deux amorces hybridant dans l’unité
répétée. On estime ainsi le nombre de répétition dans le génome.
- On peut faire un Southern pour estimer la longueur de chaque répétition.
- On peut faire une PCR avec des amorces externes. Dans ce cas on estime le nombre de
répétitions à un seul locus.
- Une méthode de détection a été développée par Schalling et al. (1993). On utilise un
oligonucléotide complémentaire à la séquence répétée. Correspondant par exemple à 5
répétitions. On incube l’ADN à analyser avec l’oligonuléotide et une ADN ligase
thermostable. A chaque cycle, il y a hybridation puis ligation jusqu’à ce que le produit de
la ligation corresponde à la taille de la répétition. Le produit de la réaction est ensuite

24/10/2011 37
chargé sur un gel dénaturant et la taille des produits de la ligation est déterminé, soit par
Southern, soit en ayant marqué les oligonucléotides au préalable.
MAAP (Multiple Arbitrary Amplification profiling)
Cette méthode inclus (Caetano-Anollis, 1994) :
DAF: DNA amplification fingerprinting
RAPD : random amplified polymorphism DNA
AP-PCR : Arbitrary primed PCR
Ces trois méthodes impliquent une amplification entre des amorces prises au hasard pour obtenir un
pattern variant.
La principale différence entre les trois
techniques est principalement la longueur des
amorces utilisées par la PCR (5-15 nt pour le
DAF, 9-10 pour le RAPD et 18-32 pour le
AP-PCR)
Dans les deux premiers la température
d'hybridation est proche du Tm de chaque
oligonucléotide de telle façon que les sites
d'hybridation correspondent à la séquence.
Dans le dernier cas la température
d'hybridation est en dessous de Tm de telle
sorte que des oligonucléotides de 11-15 hybrident. L'AP-PCR revient en fait à utiliser un mélange de
différents oligonucléotides plus petit. Cette méthode présente l’avantage de ne pas nécessiter
d’informations au préalable, on peut l’appliquer sur des espèces dont on ne connaît aucune séquence.
Dans les MAAP on peut inclure l’IMA (Inter Microsatellite Amplification). On amplifie l’ADN
génomique avec des amorces microsatellites (Zietkievwicz et al., 1994) comportant en 3’ quelques
nucléotides au hasard pour diminuer le nombre de bandes.
Distinction de deux espèces d’insectes par RAPD(les deux espèces ne sont que très difficilementdifférentiables par des critères morphologiques)
Espèce 1 Espèce 2

24/10/2011 38
III) Détection d'une séquence
Dans ce cas on veut détecter une séquence particulière par exemple pour détecter un parasite dans un
organisme. La technique la plus sensible est le PCR associé à une technique de détection de
l'amplification (voir chapitre sur la PCR) ou le NASBA si on veut détecter un ARN.
Dans certains cas on veut détecter la fréquence d’une séquence, par exemple dans l’union
européenne, la proportion d’organisme génétiquement modifié ne doit pas dépasser 3% dans la
nourriture. On utilise alors la PCR quantitative en prenant comme témoin un gène de l’organisme qui
est donc présent aussi bien dans les organismes modifiés que dans les organismes sauvages. Le
rapport des deux amplifications donne la proportion d’OGM dans l’échantillon.

24/10/2011 39
Expression des protéines recombinantes
Introduction
Une fois qu'un gène a été cloné, on désire souvent obtenir une protéine à partir d'un fragment
d'ADN. La technique utilisée dépendra tout d'abord de l'objectif final. Par exemple, si on a besoin de
la protéine pour faire un anticorps polyclonal, on n'a généralement pas besoin d'avoir une protéine
active mais par contre la protéine doit être facilement purifiable. Dans ce cas on s'orientera vers la
production en bactérie, en fabriquant des protéines de fusion ou la protéine d'intérêt est fusionnée
avec une autre protéine facilement purifiable. Si le but est d'obtenir une protéine pour des études de
biochimie ou de biologie cellulaire, la purification n'est pas toujours le problème prédominant par
contre l'activité de la protéine (enzyme, récepteur) devient plus important. Enfin, pour des études de
biochimie ou de biophysique, on peut avoir besoin de grande quantité de protéines purifiées comme
par exemple pour les études de biochimie structurale.
On va pouvoir exprimer des protéines soit dans des systèmes procaryotes, soit dans des systèmes
eucaryotes. Il faudra donc tenir compte des différences entre ces deux systèmes. Par exemple chez les
procaryotes, l'initiation s'effectue par reconnaissance d'une séquence particulière (RBS). Donc, si on
veut exprimer un clone provenant d'une cellule eucaryote dans une bactérie, il faudra incorporer cette
séquence en amont de l'ATG d'initiation. Rappels sur la traduction
Les événements de la traduction
Couplage de l'acide aminé sur l'ARNt
L'acide aminé se fixe à l'extrémité 3' OH ou en 2'OH des ARN de transfert par une liaison ester. Cette
réaction est catalysée par une enzyme, l'aminoacyl-tRNA synthétase. Il y a tout d’abord accrochage
de l'acide aminé sur l'ATP (aa + ATP ----> aa-AMP + 2 Pi). Le pyrophosphate est hydrolysé, la

24/10/2011 40
réaction est donc irréversible. Il y ensuite transfert de l'acide aminé sur l’ARNt (aa - AMP + ARNt ---
> aa - ARNt + AMP). Sur l'enzyme il y a donc trois sites (ARNt, acide aminé et ATP) et toutes les
réactions se passent sur l'enzyme sans que les constituants intermédiaires quittent l'enzyme.
La liaison ester est riche en énergie (provenant de l’énergie de la liaison anhydride aa-AMP). Cette
énergie sera utilisée pour la formation des liaisons peptidiques, pour l’accrochage des acides aminés
les uns aux autres.
La réaction est spécifique, à un ARNt correspond un seul
acide aminé (le code génétique est du à cette spécificité de
l'aminoacyl-tRNA synthétase). Pour le vérifier on peut
faire l’expérience suivante : une cystéine liée à son ARNt
est chimiquement transformée en alanine. Dans la
protéine, les cystéines seront remplacées par des alanines.
L'aminoacyl tRNA synthétase reconnaît l’ARNt et l’acide
aminé, pour que la réaction soit spécifique, il faut que les
reconnaissances présentent une très bonne affinité.
Mais la reconnaissance de l'ARN de transfert et de l'acide
aminé n'est pas parfaite. La réaction se fait quelquefois
avec un mauvais partenaire, et il y a dans ce cas des
possibilités de correction.
sitetRNA
Site acideaminé
SiteATP
PO
O O
O
PO O
O
PO O
OAdénosine
-
-
-
-
C
0 0
NH2
RH
-
C
0
NH2
RH
O
PO O
OAdénosine
-OH
C
C
NH2
RH
O O

24/10/2011 41
L'initiation
Il y a 3 phases de lecture possibles: l'initiation permet de déterminer l'endroit exact où commence la
traduction. L’initiation s’effectue pour ainsi dire toujours au niveau d’un codon AUG qui code pour
une méthionine. Comme il y a plusieurs AUG dans une séquence, Il faut que le bon AUG soit
reconnu comme codon de départ.
Chez les procaryotes:
Dans la bactérie il y a équilibre entre les formes 70S et
30S + 50 S. Au 30S se lie le facteur d'initiation IF3 ce qui
déplace l'équilibre précédent. Il y a liaison de l'ARN
messager avec la sous unité 30S. La liaison s'effectue à
une séquence particulière sur l'ARN (Shine Dalgarno, site
de liaison du ribosome) juste en amont, en 5' de l'ATG
initiateur. Cette liaison s'effectue par hybridation avec une
séquence complémentaire en 3' de l'ARN 16S. l’initiation
est donc interne à l’ARN, il peut y avoir plusieurs sites
d’initiation sur l’ARN.
IF3 a deux fonctions : une fonction d’anti-association des
deux sous unités 30S et 50S contrôlant ainsi le nombre de
30S disponibles pour la traduction et une fonction de
liaison de l'ARNm sur le 30S.
Un autre facteur d'initiation (IF1) se lie sur le complexe, il
aide à la dissociation entre les deux sous unités du ribosome en stabilisant le complexe IF3-30S.
Un autre complexe se forme entre IF2 et l'ARN de transfert d'initiation portant une
formylméthionine.
Les deux complexes se reconnaissent au niveau d'un site appelé P. Ce nouveau complexe est capable
de se lier au 50S, il y a hydrolyse du GTP en GDP + pi (probablement par une protéine du ribosome
activée par IF2 et relargage des facteurs d'initiation (IF1, IF2 et IF3).
met
+
70 S 50 S 30 S
+
30 S IF3
+
IF3 30 S mRNA
IF2
met
+
methionyl tRNA
+
metmet
met
+
met
met met
+ +

24/10/2011 42
Chez les eucaryotes, l'initiation est différente
Le ribosome reconnaît le cap à l’extrémité 5’ de l’ARNm. De ce fait l'ARN ne peut pas être
polycistronique. Il n’y a pas comme chez les
procaryotes de reconnaissance d'une séquence
particulière en amont de l'ATG.
La reconnaissance de l’ARNm par le ribosome est
presque identique. Un complexe contenant une
protéine eIF2, du GTP et l’ARNt se lie au 40S libre,
c'est ce nouveau complexe qui reconnaît l’ARNm :
la différence par rapport aux bactéries est que
l’ARNt doit être présent. Comme pour les
procaryotes, la traduction nécessite des facteurs
d'initiation et d'élongation.
Dans certains cas particuliers, il existe des initiations internes chez les eucaryotes : des séquences
appelées IRES sont reconnues par le ribosome et permettent également l’initiation de la traduction.
met
eIF2
met
+
methionyl tRNA
+
metmet
met
+
metmet
+
40 S
met

24/10/2011 43
L’élongation
Il y a ensuite reconnaissance d'un aminoacyl tRNA sur le site A
Il y a transfert de la formylméthionine de l'aminoacyl tRNA
d'initiation sur l'aminoacyl tRNA du site A. Il y a alors dans le
site A, un peptidyl tRNA et sur le site A un ARNt deacétylé
L’ARNt désacetylé va dans un autre site, le site E, le peptidyl
tRNA va dans le site P (avec l'ARN messager). L’ARNt
désacetylé part du site E ce qui permet l'accessibilité du site A à
un autre aminoacyl tRNA Cette élongation nécessite la présence
de plusieurs facteurs appelés EF (facteurs d’élongation)
Bilan énergétique de l'élongation : l'addition d'un acide aminé à la
chaîne nécessite l'hydrolyse d'une molécule d'ATP et de deux
molécules de GTP. L'ATP permet l'accrochage de l'acide aminé
sur l’ARNt, une molécule de GTP permet le positionnement de
l'aminoacyl-tRNA sur le site A, une deuxième molécule de GTP
permet la translocation du peptidyl tRNA du site A vers le site P.
Si on regarde le nombre de liaisons riches en énergie, il en a fallu
4 (1ATP en AMP et 2 GTP en GDP), ce qui est important pour
faire juste une liaison amide. Mais ici, l'énergie est surtout
employée pour assurer la fidélité de la traduction.
La terminaison
Il y a trois codons STOP = UAA, UAG et UGA. Des protéines de libération se lient aux
codons STOP au niveau du site A. On les appelle RF (Releasing Factor). Ces protéines modifient
l'activité de la peptidyl transférase: il y a addition d'une molécule d'eau à la place d'un acide aminé. Il
y a trois releasing factors chez les procaryotes (RF1 reconnaît UAA et UAG; RF2 reconnaît UAA et
UGA, RF3 permet la dissociation de RF1 ou de RF2 du site A en hydrolysant une molécule de GTP)
et une seule chez les eucaryotes. Il y a ensuite libération de la chaîne polypeptidique en croissance du
ribosome, dissociation des deux sous-unités ribosomales et libération de l'ARNm. Ainsi, au codon
met - a.a2
AUG N1N1N1
met
site P site A
a.a2
AUG N1N1N1
site P site A
met - a.a2
AUG N1N1N1 N2N2N2
site P site A
a.a3

24/10/2011 44
stop, le ribosome se dissocie, et en aucun cas ne continu à traduire l’ARN, même s’il y a une phase
de lecture ouverte en 3’.
Régulation de la traduction
Elle peut se faire à deux niveaux, au niveau transcriptionnel ou au niveau traductionnel.
Exemples de régulation au niveau transcriptionnel
L'opéron lactose chez E. coli
En l'absence de lactose, la bactérie ne
fabrique pas les enzymes nécessaires à
sa métabolisation (β-galactosidase,
galactoside perméase et galactoside
acétylase). L'expression de ces gènes est
gouvernée par le même système de
régulation, ils sont sous la dépendance
du même promoteur. En présence de
lactose, l'allolactose se forme dans la
cellule grâce au peu de β-galactosidase
présente dans la cellule, l'allolactose se
fixe à un répresseur, induit un changement allostérique de la protéine qui se détache de l'opérateur,
l'ARN polymérase peut se fixer sur le promoteur qui synthétise un ARN polycistronique codant pour
les trois protéines.
C'est une régulation négative, le répresseur fixé empêche la transcription Le répresseur est codé par le
gène i qui est transcrit sans arrêt à un taux faible.
répresseur
opérateur
β−galactosidase perméase acetylaserépresseur
opérateur
pas de transcription
lactose allolactose
transcription
ARN

24/10/2011 45
La protéine d'activation catabolique (CAP).
A coté de la régulation négative, il existe aussi des régulations positives, dues à des activateurs :
l'activateur favorise localement l'action de
l'ARN polymérase. La CAP (protéine
d’activation catabolique) est un activateur qui
permet de n'utiliser d'autres sources de
carbone qu'en l'absence de glucose chez E.
coli. En l'absence de glucose, l'AMP cyclique
induit un changement de conformation, la
CAP se fixe sur l'ADN, l'opéron lactose est
transcrit. Ce phénomène est connu sous le
nom de répression catabolique
Régulation du métabolisme du galactose chez la levure Saccharomyces cerevisiae
La régulation des gènes utilisés pour la métabolisation du galactose a été très étudiée chez la
levure comme source de promoteurs inductibles dans les systèmes d’expression de protéines. Il y a
deux protéines régulatrices principales (GAL4 et GAL80) qui contrôlent l'expression des gènes :
GAL1 (une kinase), GAL2 (une perméase) GAL7 (une transférase), GAL10 (une epimérase) et MEL1
(une galactosidase). Lorsque le galactose est présent dans le milieu, GAL4 s'accroche sur un UAS
(Upstream Activation Site, équivalent d'un enhancer pour les autres eucaryotes) et, ainsi, favorise la
transcription du gène en aval. En l'absence de galactose, GAL80 s'accroche sur l'extrémité C
terminale de GAL4, l'empêchant de s'accrocher sur l'UAS. A coté de ce système il y a comme pour E.
Coli un système de répression en présence de glucose dans le milieu.
- glucose + lactose
CAP transcription
- glucose - lactose
CAP répresseur
+ glucose + lactose
+ glucose - lactose
répresseur
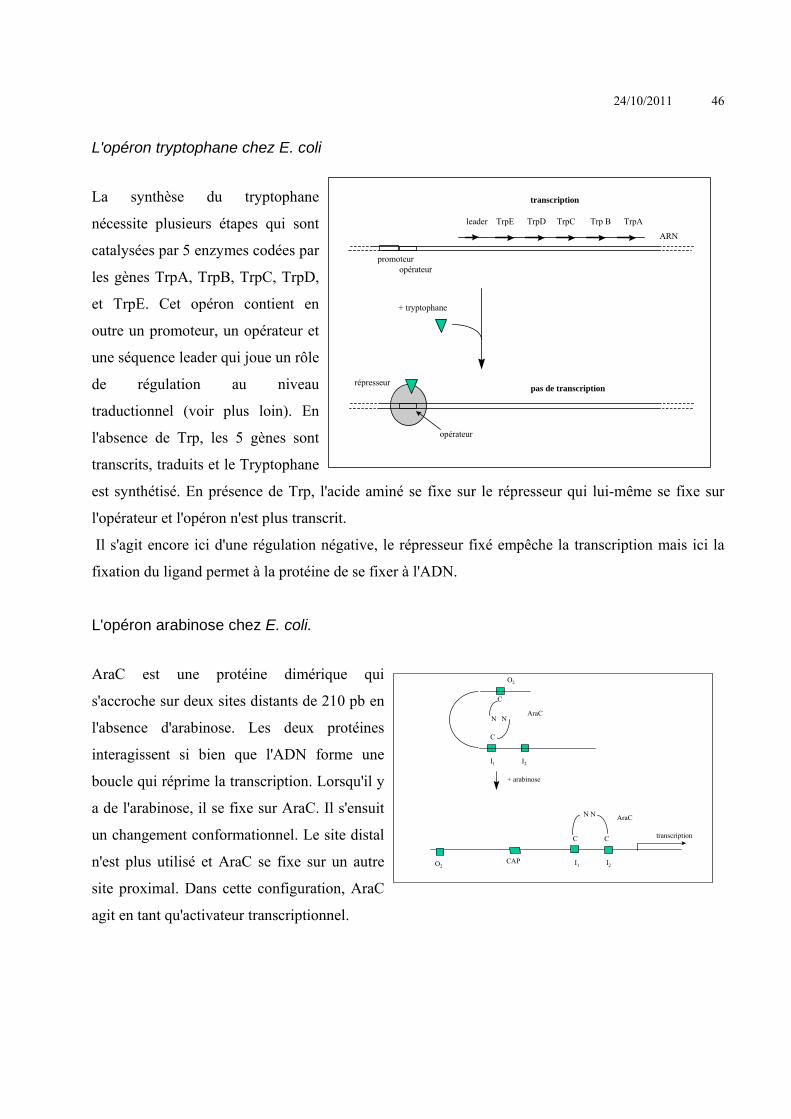
24/10/2011 46
L'opéron tryptophane chez E. coli
La synthèse du tryptophane
nécessite plusieurs étapes qui sont
catalysées par 5 enzymes codées par
les gènes TrpA, TrpB, TrpC, TrpD,
et TrpE. Cet opéron contient en
outre un promoteur, un opérateur et
une séquence leader qui joue un rôle
de régulation au niveau
traductionnel (voir plus loin). En
l'absence de Trp, les 5 gènes sont
transcrits, traduits et le Tryptophane
est synthétisé. En présence de Trp, l'acide aminé se fixe sur le répresseur qui lui-même se fixe sur
l'opérateur et l'opéron n'est plus transcrit.
Il s'agit encore ici d'une régulation négative, le répresseur fixé empêche la transcription mais ici la
fixation du ligand permet à la protéine de se fixer à l'ADN.
L'opéron arabinose chez E. coli.
AraC est une protéine dimérique qui
s'accroche sur deux sites distants de 210 pb en
l'absence d'arabinose. Les deux protéines
interagissent si bien que l'ADN forme une
boucle qui réprime la transcription. Lorsqu'il y
a de l'arabinose, il se fixe sur AraC. Il s'ensuit
un changement conformationnel. Le site distal
n'est plus utilisé et AraC se fixe sur un autre
site proximal. Dans cette configuration, AraC
agit en tant qu'activateur transcriptionnel.
répresseur
opérateur
pas de transcription
transcription
leader TrpE TrpD TrpC Trp B TrpA
promoteur opérateur
+ tryptophane
ARN
O2
I1 I2
C
N N
C
+ arabinose
CAP I1 I2O2
C C
N N
transcription
AraC
AraC

24/10/2011 47
Exemple de régulation au niveau traductionnel : l’opéron tryptophane chez E. coli.
En parallèle à la régulation au niveau transcriptionelle, il existe une régulation au niveau de la
traduction, appelée atténuation
rappel:
- chez les procaryotes la transcription et la traduction sont simultanées
- les ARNs sont polycistroniques
- l'arrêt de la transcription : l’ARN polymérase s'arrête quand elle synthétise une série de résidus U
dans la mesure où elle a d'abord synthétisé une séquence auto-complémentaire capable de se replier.
L'ARN, dans la région du leader-attenuateur, peut prendre deux structures secondaires distinctes:
1) Dans le cas ou la partie codante ne
forme pas de structure secondaire, les
parties 2 et 3 suivantes forment une
structure secondaire. Il y a continuation
de la transcription.
2) Dans le cas ou la partie codante forme
une structure secondaire avec la partie
suivante (2), il se forme une deuxième
boucle présentant une structure
secondaire d'arrêt de la transcription.
Ainsi, selon les structures secondaires au niveau de la séquence d’atténuation, il y a soit transcription
des 5 phases de lecture soit arrêt prématuré de la transcription et donc pas de traduction des 5
protéines nécessaires à la synthèse du tryptophane.
La séquence leader est traduite en un peptide de 14 acides aminés comportant du tryptophane dans sa
séquence, lorsqu'il y a du trp dans le milieu le ribosome passe à travers et on a la structure 1 et donc
arrêt de la transcription.
En absence de Trp, le ribosome reste bloqué, la transcription continu, la partie 1 ne peut pas
s'hybrider avec la partie 2 du fait de la présence du ribosome, la transcription continue.
répresseur
transcription
leader TrpE TrpD TrpC Trp B TrpA
promoteur opérateur
ARN
tryptophane
12 3
4
ribosome bloqué sur la séquence leader
transcription
UUUUU
12
3 4
arrêt de la transcription

24/10/2011 48
Ainsi en l’absence de tryptophane, le ribosome reste bloqué sur l’ARN en cours d’élongation et
provoque un changement conformationnel de l’ARN induisant un arrêt de transcription.
Exemple de régulation au niveau traductionnel : contrôle de la traduction du gène ompF.
La régulation est due à la synthèse d'un petit ARN contrôlant directement la traduction du gène
ompF.
Lorsque la pression osmotique augmente,
il y a activation de la protéine ompR qui
active la transcription de micF. Il y a
synthèse d’un petit ARN de 174 bases
appelé micRNA (mRNA interfering
complementary RNA). Cet ARN est
antisens complémentaire d'une région de
ompF qui inclue le site de liaison au
ribosome : il empêche la traduction de
ompF et il devient sensible à des RNases qui digèrent l’ARN double brin. OmpF code pour une
protéine de la membrane externe de E. Coli qui ne doit plus être traduite lorsque la pression
osmotique augmente.
Utilisation de ce système : cet exemple a servi de modèle pour étudier l'expression de certains gènes.
On peut soit transcrire un ARN complémentaire à celui du gène à éteindre soit utiliser des
oligonucléotides qui s'hybrident au niveau de l'AUG. Le problème est qu'il faut une grande quantité
d'antisens.
Compartimentalisation et modifications post-traductionelles
Chez les procaryotes, transcription et traduction sont simultanées, il n ‘y a pas de modification post-
transcriptionelle. Il y a un seul compartiment, et il existe peu de modifications des protéines après
leurs synthèses (modifications post-traductionelles). Par exemple, les protéines ne sont pas
glycosylées.
OmpR
transcription
gene micF
ARN micF 5’3’ 5’ 3’
ARN de ompF
traduction
OmpF
inhibition de la traduction
hybridation

24/10/2011 49
Chez les eucaryotes, transcription et traduction ont lieu dans deux compartiments différents, le noyau
et le cytoplasme. Les ARN sont maturés avant leur exportation dans le cytoplasme, ils sont cappés en
5’, épissés et pourvus d’une queue polyA à l’extrémité 3’. Une fois dans le cytoplasme, ils sont
traduits. Il y a alors deux possibilités. Soit la protéine se replie dans le cytoplasme dans un milieu
réducteur soit la protéine est exportée dans le réticulum endoplasmique, cet événement est
cotraductionel. L’exportation dans le réticulum endoplasmique est due à un peptide signal en N-
terminal de la protéine, ce peptide est reconnu par la particule de reconnaissance du signal, et le
peptide en cours de synthèse est dirigé à l’intérieur du réticulum endoplasmique. Le milieu est
oxydant, il va y avoir formation de ponts disulfure, la protéine peut être glycosylée et son repliement
est aidé par des protéines spécialisées, les foldases. Ces protéines passent ensuite dans les différents
compartiments de l’appreil de Golgi et sont soit secrétées soit insérées dans la membrane.

24/10/2011 50
Expression transitoire à partir d'un ARNm ou d'un ARN in vitro (cRNA)
Il est possible de fabriquer un ARN in vitro à partir d'un gène et ensuite de le faire traduire
soit in vitro dans un lysat cellulaire, soit dans des cellules. Dans ce dernier cas on utilise par
exemple l'ovocyte de Xénope.
Obtention d'un cRNA.
La méthode classique consiste à cloner le fragment à exprimer dans un plasmide en aval
d'un promoteur reconnu par ARN polymérase puis à polymériser l’ARN in vitro avec l’ARN
polymérase. On utilise généralement les ARN polymérases des phages SP6, T3 ou T7. Le
plasmide est ensuite coupé avec une enzyme de restriction dont le site se situe en aval de la
séquence codante pour éviter des brins 3' trop longs qui peuvent affecter la traduction. Le
produit de cette digestion, une fois purifié, est incubé avec l’ARN polymérase correspondante
au promoteur et des ribonucléotides.
On peut faire directement une réaction de RT-PCR sur des ARN extraits de cellules
exprimant le gène d’intérêt. L’amorce sens (en 5’) comportera en amont de la séquence
hybridant sur le gène, la séquence reconnue par la RNA polymérase et éventuellement les
séquences nécessaires à la traduction.
Enfin si le clone n’est pas disponible, si on ne dispose pas de cellule exprimant la protéine,
on peut toujours faire un gène syntétique par assembalage d’oligonucléotides.
L’initiation de la traduction dans les systèmes eucaryotes nécessite la reconnaissance d’un
Cap, puis le démarrage de la traduction au premier ATG. L’ARN néoformé est dépourvu de
Cap. Les réactions de capping étant difficiles à faire car elles nécessitent plusieurs étapes, on
utilise des astuces. Une astuce consiste à remplacer 90% du GTP fourni à la cellule par du
diguanosine triphosphate (m7G5'ppp5'G). Ce produit doit être enlevé avant la traduction in
vitro, il est en effet un inhibiteur fort de la traduction (il rentre en compétition avec l’ARNc).
Une deuxième astuce vient d’une observation, la traduction du gène de la globine ne dépend
pas de la présence du cap, on peut donc utiliser le leader de la globine en fusion avec le gène
destiné à être traduit. De plus il est important que l'ATG initiateur soit le premier ATG suivant
l’initiation de la transcription.

24/10/2011 51
Traduction in vitro
Des extraits de nombreuses cellules peuvent être utilisés pour traduire des ARNm. Il faut
que tous les éléments de la machinerie nécessaires à la traduction (ribosome, ARNt, acide
aminé, aminoacyl-tRNA synthetase, facteurs d'initiation, d'élongation et de terminaison) soient
présents. Par contre le noyau, les polysomes et les inhibiteurs de la traduction doivent être
enlevés pour éviter la transcription (noyau) et la traduction d’ARNm endogènes (polysomes).
De plus l'extrait doit avoir peu d'activité RNAsique pour ne pas dégrader les ARNm.
La préparation d'un tel extrait cellulaire consiste le plus souvent à lyser les cellules par
choc osmotique, et à retirer les polysomes et les noyaux par centrifugation. Le plus souvent les
acides aminés sont aussi retirés par tamisage moléculaire par exemple sur G25. Les acides
aminés froids et un acide aminé marqué sont rajoutés.
La synthèse est simple il suffit de rajouter l’ARNc au lysat cellulaire. Quelquefois une
étape de digestion des ARN par une nucléase a été rajoutée en fin d'incubation mais
généralement cette digestion n'est pas utile.
Plusieurs extraits sont utilisés en système eucaryote mais le lysat de réticulocyte et l'extrait
de germe de blé sont les plus populaires car ils sont les plus efficaces et sont actuellement
commercialement disponibles. Mais on peut effectuer des traductions in vitro avec un d'autres
d'extraits cellulaires tels que :
- les ascites. Ces cellules sont prélevées à partir de la peau abdominale de souris, lysées par
choc osmotique et les ribosomes sont préparés par centrifugation.
- la levure : l'avantage de ce système est qu'on dispose d'un grand nombre de mutants de
régulation de la traduction.
Un système est utilisé pour les procaryotes, un lysat d'E. coli S30 (par exemple
commercialisé par Promega). L'avantage de ce système est qu'il peut être couplé à la
transcription in vitro: pendant la transcription l'extrémité 5' devient disponible pour la
traduction. Le système de trascription comporte tous les éléments nécessaires aux deux étapes :
NTPs, RNA polymérase, tRNAs, acides aminés, un système de régénération de l’ATP, des
ribosomes…

24/10/2011 52
Dans l'utilisation de systèmes couplés, il faudra faire attention au promoteur utilisé, certain
promoteur de E. Coli tel que lac ne sont efficaces que si l'ADN est surenroulé alors que d'autres
ne sont pas sensibles au surenroulement (lacUV5, tac, λPL ou λPL ). Seuls ces derniers seront
efficaces sur un fragment linéaire.
La traduction in vitro synthétise des protéines pendant environ 30 minutes à température
ambiante. Mais des systèmes en flux continu ont été développés. Les composants de la
traduction sont maintenus dans un boudin de dialyse dans lequel on injecte des acides aminés,
la protéine synthétisée sort du boudin de dialyse alors que les composants de la traduction
formant des complexes ARN-protéines sont trop gros pour passer à travers la membrane de
dialyse. De tels systèmes peuvent fonctionner jusqu'à 45 heures.
Marquage de la protéine : les extraits sont riches en protéine, la protéine néosynthétisée
n'est donc pas biochimiquement pure
mais comme on traduit un seul ARN, on peut
la marquer. Le plus communément on
utilise un acide aminé radioactif. La [35S]
méthionine est le plus souvent utilisée,
Mais d’autre marquage sont possibles en
l’absence de méthionine tel que la [35S]
cystéine ou la [14C] leucine ou l’[3H]
leucine. Dans ce cas la protéine
néosynthétisée est radiochimiquement
pure. Pour éviter l’utilisation d’éléments
radioactifs on peut utiliser des marquages froids.
On peut utiliser des Lysinyl-tRNA modifiés avec une lysine marquée à la position epsilon
par de la biotine ou par un fluorophore. L’ARNt est ajouté à la solution de traduction et la
lysine modifiée est incorporée à la protéine. Dans le cas du marquage à la biotine, la protéine
est révélée à l’aide de streptavidine conjuguée à une enzyme (péroxydase ou phosphatase
alcaline), dans le cas d’un marquage par un fluorophore, la protéine est révélée directement par
fluorescence.
CCO
NNB
OAO
NH2
N
OF F
CH3
CH3
H
O OO OO OO OO OO OO OO O
O OO
O OO
OO
OO
OO
OO
OOOO
OOOO
OOO
OOOO
OO
OO
OOOO
OOOO
OOO
OO O
OO
OOO
OOO
O OO OO OO O
OOO
OOOO
OO
O
Lysine Fluorophore

24/10/2011 53
Avantage de la méthode
La traduction in vitro présente principalement l'avantage de permettre l'expression de
protéines très toxiques pour la cellule. De plus, lors de la traduction d'un ARNc, on obtient une
protéine marquée. On n'a donc pas besoin de purifier la protéine. Dans les systèmes à flux
continu, la protéine qui sort de la dialyse est pure et non uniquement marquée, les acteurs de la
traduction restant dans le compartiment intérieur.
Inconvénients de la méthode
Les protéines exprimées in vitro ne sont généralement pas actives. Toutefois, on peut dans
certains cas obtenir des protéines actives. Ce sera le plus souvent le cas des protéines exprimées
dans le cytosol. Par contre les protéines exprimées dans le réticulum endoplasmique puis
maturées dans le golgi seront le plus souvent inactives.
Utilisation
- Cette méthode est utilisée pour vérifier qu'un ARN messager est bien présent. Par
exemple si on veut cloner un gène et qu'on a choisi comme moyen le criblage avec un
anticorps. On commence par purifier les ARN messagers pour faire une banque d’ADNc,
l'anticorps servira alors à cribler la banque. Mais on ne sait pas toujours si l'ARN est présent au
départ si le gène n'a pas encore été cloné, on ne connaît dans le meilleur des cas que la
répartition spatio-temporelle de la protéine. Un des moyens de vérification consiste à traduire in
vitro les ARN messagers puis à sélectionner les protéines réagissant avec l'anticorps, si on
sélectionne une protéine de taille attendue, l'ARN est bien présent dans le pool de départ.
- Cette méthode permet d'incorporer des acides aminés modifiés contenant par exemple des
groupes photoactivables ou fluorescents. De même on peut incorporer des acides aminés
modifiés tels que de la lysine biotinylée, on obtient alors une protéine biotinylée sur les lysines.
- Cette méthode permet d’obtenir des précurseurs utiles par exemple pour étudier
l'adressage des protéines dans la mitochondrie. Lors de la translocation dans la mitochondrie, la
protéine est dépliée, puis à nouveau repliée à l'intérieur de la mitochondrie. Pour étudier le

24/10/2011 54
passage, on utilise une protéine fabriquée in vitro et marquée radioactivement. De même si on
étudie le repliement des protéines in vitro à partir du précurseur néosynthétisé et non à partir
d’une protéine dénaturée, la traduction in vitro permet de suivre les différentes étapes.
L’obtention du précurseur permet d’étudier certaines étapes de maturation des protéines comme
la N-glycosylation en ajoutant une source de réticulum endoplasmique au lysat de réticulocyte
sous forme de microsomes.
Traduction en milieu cellulaire. L’ovocyte de Xénope
Les ovocytes de Xénope sont des grosses cellules dans lesquelles on peut injecter une
solution d’ARN. Le Xénope est un batracien anoure aquatique vivant en Afrique du Sud et qui
se maintient aisément en aquarium. Il pond plusieurs centaines d’œufs. Si on traite les femelles
par des hormones, on peut synchroniser la maturation des ovocytes. Environ deux mois après
on dissèque les femelles et on prélève les ovaires après avoir anesthésié la femelle en la mettant
au froid, sur de la glace. Le Xénope est relativement robuste et on peut facilement recoudre la
femelle et la réutiliser après. On traite les fragments d’ovaire à la collagènase pour retirer les
cellules folliculaires et on obtient des ovocytes qui font quelques millimètres de diamètre. La
solution d’ARNc, de l'ordre de 100 nl, est injectée dans le cytoplasme. Les ovocytes sont mis à
incuber dans un tampon contenant au besoin un acide aminé marqué. Un à deux jours après, la
protéine a été synthétisée.
Ce système est particulièrement performant pour étudier les récepteurs situés sur la
membrane externe de l'ovocyte. Il est alors facile de faire les expériences d'électrophysiologie
pour étudier le récepteur. L'avantage est que l'ovocyte est dépourvu de récepteur. Plus
rarement, ce système est utilisé pour étudier d'autres protéines. Mais il s'est avéré très utile pour
étudier des protéines sécrétées. En effet l'ovocyte de Xénope ne secrète pas de protéines, si bien
que la seule protéine sortant dans le tampon d’incubation des ovocytes est la protéine dont
l’ARNc a été injecté. On obtient donc directement une protéine pure. L'inconvénient du
système est qu'on en n'obtient que très peu.

24/10/2011 55
Expression en système procaryote
I) Escherichia coli
Escherichia coli a été très étudié depuis les années 60 si bien que c'est un des organismes
actuellement les mieux connus. L'ensemble de ces connaissances en biochimie, génétique et
biologie moléculaire a été exploitée pour exprimer des protéines en grande quantité. Cette
bactérie a plusieurs propriétés intéressantes lui permettant d’être utilisée pour exprimer des
protéines : elle est facile à manipuler, elle pousse vite dans des milieux relativement peu cher et
les souches de laboratoires sont inoffensives. De plus, tous les laboratoires de biologie
moléculaire utilisent E. coli pour d’autres expériences (clonage, séquence…). Cette facilité en a
fait le système d'expression le plus populaire.
Ce qui est indispensable pour produire une protéine dans E. coli.
Pour exprimer un gène dans E. coli, il doit être inséré dans un vecteur qui contient
plusieurs éléments : 1) une origine de réplication, un marqueur sélectionnable pour trier et
maintenir les bactéries ayant incorporé le vecteur et un polylinker, comme tous les plasmides
servant de vecteur, 2) un promoteur, si possible contrôlable, dont l'induction produira une
grande quantité d’ARNm à partir du gène cloné, 3) les séquences responsables de la traduction
telle qu'une séquence de liaison au ribosome, ces séquences doivent être bien positionnées.
a) L’origine de réplication
Le gène doit être répliqué dans la bactérie, autrement, on va le perdre très rapidement. La
méthode générale consiste à l’insérer dans un plasmide qui porte une origine de réplication.
Il existe plusieurs origines de réplication (ori) disponibles. Celle de bluescript (500-700
copies par chromosomes), celle de pBR322 (15-20 copies) et celle de P15A (10-12 copies). Si
le produit du gène est toxique on utilisera une origine de réplication qui donne peut de copies,
au contraire si le gène n’est pas toxique et s’il est bien régulé, on utilisera une origine à fort
nombre de copies

24/10/2011 56
On peut positionner l’origine de réplication pour favoriser la production de protéines.
- Si on veut exprimer un gène non toxique pour la bactérie, l'expression sera simultanée
à la croissance de la population bactérienne. La fourche de réplication doit alors plutôt
progresser dans le même sens que la transcription du gène de sélection, ainsi la
transcription ne gênera pas la réplication du plasmide. Ce phénomène est le même que
celui qui gouverne réplication et transcription dans le chromosome bactérien.
- Si on veut exprimer un gène toxique pour la bactérie, l'expression s'effectuera en phase
stationnaire, lorsqu'il n'y a plus de réplication. La fourche de réplication doit progresser
en sens inverse de la transcription pour avorter toute transcription non désirée.
L’origine de réplication la plus utilisée est ColE1, elle donne de 15 à 20 copies par cellule.
L’origine M15 est parfois utilisée (10-15 copies). Elle est plus particulièrement utilisée avec
ColE1 lorsqu’on veut produire deux protéines simultanément dans la même bactérie (Kholod et
Mustelin, 2001). En effet ColE1 et M15 sont compatibles.
L’utilisation de plasmides pour exprimer un gène dans E. coli est breveté, ceci a amené
l’utilisation d’une autre origine de réplication (Olson et al., 1998). Le gène d’intérêt est ici
incorporé au chromosome bactérien. Pour ce faire, on transforme les bactéries avec un ADN
comportant le site d’attachement du phage λ, aatP, mais ne comportant pas d’origine de
réplication. En présence d’une intégrase codée par un plasmide, l’ADN va s’intégrer dans le
site attB du chromosome bactérien. Pour sélectionner les intégrants, on ajoute un gène de
résistance à la kanamycine à l’ADN. Dans un tel système, on n’obtient qu’une copie du gène
d’intérêt. Pour en avoir plusieurs copies et ainsi augmenter la quantité de protéine produite, on
peut ajouter à l’ADN des séquences permettant la duplication médiée par rec-A et un gène de
résistance à la tétracycline pour sélectionner les bactéries contenant un grand nombre de copies
du gène en tandem. Une augmentation progressive de la dose de tétracycline permettra
d’augmenter le nombre de copies.

24/10/2011 57
b. Les promoteurs
Deux domaines en amont du site d'initiation de la transcription sont importants dans les
promoteurs procaryotes. Le domaine à -10 (Pribnow box, 5' T-A-T-A-A 3') et un domaine à -35
(5' T-T-G-A-C-A 3'). Ces deux domaines sont en contact avec l’ARN polymérase lors de
l'initiation de la transcription.
Deux caractéristiques des promoteurs sont importantes pour l’expression des protéines, la
force du promoteur et son inductibilité.
- La force du promoteur : les promoteurs bactériens sont relativement faibles du moins
pour les besoins de production. Pour les améliorer, des mutations ponctuelles ou des
petites délétions ont été effectuées au sein de ces promoteurs, ce qui a permis
d'augmenter la transcription.
- L’inductibilité, c’est à dire son contrôle : le plus souvent on désire utiliser un
promoteur inductible par exemple lorsque la protéine est très toxique pour la bactérie.
Dans ce cas, la protéine est produite en début de la culture et inhibe la croissance
gène d’intéret
promoteur
attP
kanR
tetR
attBP attPB
intégration dans le chromosome bactérien
attBP
amplification génique
(sélection pour la résistance à la tétracycline)

24/10/2011 58
cellulaire. En utilisant un promoteur inductible, on cherchera à inhiber la production au
début de la phase de croissance des bactéries puis à l’approche de la phase stationnaire,
on induira la production.
La force des promoteurs :
Plusieurs techniques ont été utilisées pour augmenter la force des promoteurs. On peut
éliminer des répresseurs. Ainsi Le promoteur Plac du gène lacZ dépends de l'activation par la
CRP/cAMP. Pour pouvoir être utilisé, ce promoteur doit être muté (mutation UV5) afin que la
répression catabolique ne puisse pas s'exercer (le rôle de la répression catabolique est de ne pas
utiliser le galactose en présence d'autres sources de carbone, principalement en présence de
glucose). Ainsi on pourra avoir une transcription en présence de glucose (Hirshel et al., 1980).
Le remplacement des séquences du promoteur du gène lacZ en amont de la position -20
par les séquences du promoteur de l'opéron tryptophane a permis de faire un promoteur hybride
(Ptac). Cette construction donne une augmentation de la transcription de 10 fois (De Boer et al.,
1983).
La technique de choix pour augmenter la force d’un promoteur est d’utiliser un promoteur
spécifique d’une ARN polymérase qu’on surproduit dans la cellule. Le plus utilisé est le promoteur
de la protéine 10 du phage T7 (Studier et Moffatt, 1986). La transcription utilisant l’ARN polymérase
T7 est très efficace. En effet,
- La polymérase utilise la plupart des nucléotides triphosphate de la cellule ce qui inhibe
la transcription des gènes de l'hôte. Elle est 5 fois plus rapide que les polymérases
bactériennes. Cette polymérase reste la plupart du temps sur l'ADN. Il n'y a que peu
Boite – 35 Boite de Pribnow Ptrp GAGCTGTTGACAATTAATCATCGAACTAGTTAACTAGTACGCA PlacUV5 CCAGGCTTTACACTTTATGCTTCCGGCTCGTATAATGTGTGGA Ptac GAGCTGTTGACAATTAATCATCGGCTCGTATAATGTGTGGA

24/10/2011 59
d'arrêt prématuré de la transcription. La transcription peut en fait, faire plusieurs fois le
tour du plasmide donnant un ARN très grand.
- L’ARN polymérase T7 est très spécifique, elle ne reconnaît pas les promoteurs de la
bactérie, si bien qu'elle ne transcrit que le gène d'intérêt.
- L’ARN polymérase T7 n’est pas sensible aux inhibiteurs des ARN polymérases bactériennes
tels que la rifampicine. Ainsi, l'ajout de rifampicine dans le milieu bloque toutes les
polymérases sauf celle de T7, seul le plasmide est transcrit, tous les ribonucléotides sont utilisés
pour la transcription du gène d’intérêt.
L’ARN polymérase T7 n'est pas
présente dans la bactérie, il faut donc
l'ajouter par exemple sur un plasmide. Ce
deuxième plasmide doit avoir une autre
origine de réplication compatible avec celle
utilisée dans le vecteur portant le gène
d'intérêt. Il doit contenir un gène de
résistance à un autre antibiotique et enfin il
doit contenir le gène de l’ARN polymérase
T7 lui-même inductible. On peut aussi
produire l’ARN polymérase T7 à partir du
génome de la bactérie. On utilise alors une
souche qui a intégré le gène de l’ARN
polymérase T7 sur le chromosome via un lambda lysogène (DE3).
Plac
cI-857
KanR
PL
T7 RNA polymérase
p15A (origine de réplication)
AmpR
Col E1origine deréplication
Promoteur P10RBS
ATG
site de clonage

24/10/2011 60
L’inductibilité des promoteurs :
-> On peut utiliser des répresseurs :
Le promoteur/opérateur du gène lac Z. Une des inductions les plus utilisées est celle du
gène lacZ. (Plac). La protéine LacI est un répresseur qui se positionne en aval du promoteur et
inhibe la transcription. La transcription est déréprimée (ou induite) par un analogue du
galactose, l'isopropyl β-D thiogalactoside (IPTG) qui se fixe sur lacI, induit un changement de
conformation de lacI qui ne se fixe plus sur l’opérateur. Pour augmenter l’efficacité du
répresseur, on peut d’une part surproduire le répresseur en ajoutant un gène le codant dans le
plasmide et d’autre part augmenter le nombre d’opérateurs (lacO) en aval du promoteur du
gène d’intérêt.
Pour que la régulation soit bonne il faut utiliser un souche qui surproduit LacI (lacIq),
L’induction peut se faire par l’IPTG ou en utilisant un LacI thermosensible (LacIts), Dand ce
dernier cas à basse température (30°C) lac I est actif et réprime l’expression du gène d’intérêt
alors qu’à haute température (37 – 42°C) lac I est inactif, et le gène d’intérêt est exprimé
(Hasan et Szybalski, 1995).
Le systéme de régulation LacI/LacO peut être utilisé pour agir directement sur la
transcription du gène d’intérêt ou indirectement en contrôlant la transcription de l’ARN
polymérase T7.
-> On peut utiliser des inhibiteurs de l’ARN polymérase. L’ARN polymérase T7 est
inhibée par le lysozyme. Si on introduit le gène codant pour le lysozyme sur un plasmide, la
.....GTGAGCGGATAAC....
.....CACTCGCCTATTG....
Séquence de l’opérateur LacO reconnue par le répresseur LacI

24/10/2011 61
production de lysozyme inhibera le peu d’ARN polymérase T7 produite en l’absence
d’induction. Par contre, en présence d’induction, l’inhibiteur sera en trop faible quantité
comparativement à l’ARN polymérase T7 qui est surproduite.
On peut ajouter l’ARN polymérase au moment de la production. Dans certains cas la
protéine à exprimer est toxique pour la bactérie. Or tous les promoteurs inductibles ont une
activité basale, même faible, si bien que la protéine est produite en faible quantité en l'absence
d'induction (on dit que le promoteur fuit). Si la protéine est très toxique, on peut utiliser une
autre stratégie pour produire l’ARN polymérase T7 en utilisant un phage M13 la produisant.
On fait pousser les bactéries puis, avant de produire, on infecte les bactéries par le virus. Le
virus produit l’ARN polymérase T7 puis le gène d'intérêt est transcrit.
Le promoteur PL du bactériophage λ: Ce promoteur est fort et peut être presque
complètement éteint par le répresseur cI du phage. Ce répresseur est présent dans le génome
des bactéries appelées "lysogènes défectives", ce qui revient à dire que le génome de ces
bactéries contient une partie du phage λ intégrée. Cette partie du génome de λ produit la
protéine CI réprimant la synthèse de l'ARN en aval de PL. Une telle bactérie peut donc pousser
à haute densité sans produire l'ARN sous la dépendance du promoteur PL. Pour pouvoir induire
une synthèse de protéine, on utilise un répresseur mutant (cI 857), thermosensible. Dans ce cas,
on cultive les bactéries à 32°C, le répresseur est produit, il n'y a pas de production de l'ARN
sous la dépendance du promoteur PL. Lorsqu'elles sont arrivées à haute densité, on élève la
température à 42°C, le répresseur n'est plus produit, l'ARN est transcrit et donc la protéine est
traduite.
L'induction peut aussi se faire chimiquement avec un répresseur sauvage en induisant une
protéase qui dégrade le répresseur CI. Pour produire la protéase, on ajoute de l'acide nalidixique
dans le milieu ce qui inhibe la DNA gyrase, cette inhibition amène des lésions sur l'ADN et il y
a donc déclenchement la réponse SOS qui induit la RecA protéase. Cette protéase coupe le
répresseur CI et le gène est transcrit.
Le promoteur tétracycline. Ce promoteur est inductible par l'anhydrotétracycline. Pour
obtenir une bonne régulation de ce promoteur, on inclu souvent dans le vecteur un gène codant

24/10/2011 62
pour un répresseur (TetR) (Skerra et al., 1994). C'est donc un promoteur qui d’une part doit être
activé et d’autre part peut être réprimé.
Le promoteur arabinose.
Ara C est une protéine dimérique
qui, en l’absence d'arabinose,
s'accroche sur deux sites distants
de 210 pb (I1 et O2) par leurs
extrémités C-terminales. L'opéron
forme alors une boucle qui réprime
la transcription du gène.
En présence d'arabinose, la
liaison sur Ara C induit un
changement conformationnel du dimère qui s'accroche alors sur un deuxième site (I2). Dans
cette configuration, Ara C agit comme un activateur de transcription.
De plus on trouve un site de liaison de la CAP (protéine d'activation catabolique) si bien
qu'en l'absence de glucose, la transcription est stimulée.
Source de carbone Facteur d'induction
glucose 1
glucose + arabinose 70
glycérol + arabinose 820
Les promoteurs hybrides
Lorsqu’un répresseur interfère directement avec la liaison de l’ARN polymérase, le
paramètre important pour une répression efficace du promoteur est la vitesse de formation du
complexe ARN polymérase*promoteur (kON). Ainsi, d'une manière générale, les promoteurs
O2
I1 I2
CAP
C
N N
C
+ arabinose
CAP I1 I2O2
C C
N N
transcription
AraC
AraC

24/10/2011 63
qui lient l’ARN polymérase à faible vitesse sont bien réprimés mais ils restent faibles après
l'induction. A l'inverse, les promoteurs qui lient l’ARN polymérase rapidement, sont mal
réprimés. Plusieurs promoteurs hybrides ont été fabriqués pour obtenir d'une part une
transcription forte et d'autre part une bonne régulation (Lutz et Bujard, 1997).
Par exemple, le promoteur PL est un promoteur fort et on peut remplacer la répression CI
dépendante par une régulation par l'opérateur de l'opéron tétracycline. On obtient un promoteur
PLtetO-1 qui est éteint par le répresseur tetR et qui peut être activé jusqu'à 5000 fois par
l'anhydrotétracycline. Ainsi on peut combiner différents éléments provenant de différents
promoteurs pour obtenir la régualtion souhaitée.
- On peut rajouter d'autres éléments pour éteindre la transcription du gène, on peut en
particulier adopter une stratégie antisens. Dans la stratégie antisens, on clone un gène à
proximité d'un promoteur fort de E. coli de telle sorte que c'est l'antisens qui est produit. Cet
antisens inhibe la traduction du gène pendant la croissance de la cellule. L'ajout de rifampicine
dans le milieu arrête la synthèse de l'antisens lorsque le gène d’intérêt est en aval d’un
promoteur de l’ARN polymérase T7. Pour la même raison, la transcription du gène de
résistance doit se faire dans l'autre sens de transcription du gène d'intérêt. Ainsi lorsqu'il n'y a
pas d'arrêt de transcription ou lorsque l'arrêt n'est pas total, le gène d'intérêt n'est pas lui-même
transcrit en l'absence d'induction. On aura un effet d'antisens, le transcrit initié au niveau du
gène de résistance s'hybridera avec les quelques transcrits du gène d'intérêt présent en l'absence
d'induction.
c. Les séquences responsables de la traduction
Chez les procaryotes, l'initiation s'effectue par reconnaissance d'une séquence particulière
(RBS, ribosome binding site). Cette séquence est composée d’une séquence riche en purine
(Shine-Dalgarno, SD, AGGAGG) et du codon d'initiation qui doit être proche, idéalement,
l'extrémité 3' de la boite de Shine-Dalgarno doit être à 6 bases de l'ATG.
Il y a plusieurs possibilité de boite de Shine-Dalgarno : UAAGGAGG donne une meilleure
traduction que AAGGA (Ringquist et coll. 1992)

24/10/2011 64
Donc, si on veut exprimer un clone provenant d'une cellule eucaryote dans une bactérie, il
faudra incorporer cette séquence en amont de l'ATG d'initiation. Généralement, on mutagenise
la séquence du gène d'intérêt au niveau de l'ATG en y incorporant un site de restriction tel que
NdeI (CATATG). Ce site est présent sur le vecteur 8 nucléotides en aval d'un site de liaison au
ribosome.
d. Les séquences responsables de l’arrêt de la transcription
On peut dans certains cas ajouter en 3' du gène des séquences responsables de l'arrêt de la
transcription. Au niveau d'un signal de terminaison, la polymérase libère la matrice et l'ARN
monocaténaire néosynthétisé.
Chez E. coli, il y a deux sortes de terminaison, une
terminaison intrinsèque et une terminaison
dépendante d’une protéine, la protéine ρ. Dans la
terminaison intrinsèque, la polymérase s'arrête
quand elle synthétise une série de résidus U dans
la mesure où elle a d'abord synthétisée une
séquence autocomplémentaire capable de se
replier. C'est le repliement, l'hélice en épingle à cheveux, qui se forme rapidement dans cette région,
qui est crucial pour l'arrêt de la transcription. La séquence de la région autocomplémentaire peut
varier. Lors de la terminaison ρ dépendante, la protéine ρ reconnaît une séquence sur l’ARN
néosynthétisé et catalyse l’arrêt de la transcription.
Dans certains cas, on ajoute une séquence de terminaison ρ indépendante en 3’ du gène, il y a
formation d’une boucle qui termine la transcription. Cette arrêt de la transcription a peu d’avantage
pour la production de protéines recombinantes si ce n’est qu’elle stabilise l’ARN en inhibant
l’activité des 3’ 5’ exonucléases qui ne dégradent que l’ARN simple brin.
Toutefois ces séquences ne sont indispensables et sont inefficaces pour l’arrêt de transcription dans
le cas d’une production utilisant l’ARN polymérase T7.
extrémité 3’ de l’ARN
UUUUUU
arrêt de la transcription

24/10/2011 65
Il peut y avoir des signaux de terminaison dans le gène à traduire. Dans ce cas on peut utiliser une
bactérie qui produit la protéine N du phage λ qui a une fonction d'antiterminaison. On utilise des
bactéries lysogènes défectives qui produisent la protéine N ce qui permet d'éviter une terminaison
précoce du message en interagissant avec l’ARN polymérase seulement au site Nut. On utilisera alors
cette séquence Nut (terminateur) en aval du gène cloné pour terminer la transcription au bon endroit.
Les signaux de terminaison de la transcription sont parfois utilisés en aval du promoteurs pour éviter
une transcription venant du gène recombinant. Dans ce cas, la transcription en sens inverse pourrait
inhiber l’initiation de la transcription du gène d’intérêt. De même, on peut utiliser un arrêt de
transcription en amont du promoteur pour diminuer le bruit de fond, due à une transcription initiée
par un promoteur plus en amont.
e. Les séquences responsables de l’arrêt de la traduction
La présence d’un codon stop est indispensable pour assurer la terminaison de la traduction. Il en
existe trois UAA, UAG et UGA. Le plus souvent on met plusieurs codons stop à la suite à la fin de la
séquence codante pour éviter les dérapages et donc les protéines plus longues.
Il faut éviter de produire les protéines dans des bactéries comportant des supprésseurs qui comportent
des tRNA reconnaissant les codons stop. Toutefois cette stratégie est quelquefois utilisée pour obtenir
des protéines de fusion d’une manière conditionelle. Lorsqu’on veut la protéine de fusion on
transforme des bactéries ayant le suppresseur, par contre lorsqu’on veut la protéine seule, on
transforme des bactéries dépourvues de répresseur.
La base suivante le codon stop a une importance sur l’efficacité de la terminaison qui peut descendre
à 7% pour le cas de UGAC. Pour éviter ce problème, on utilise UAAU qui est le plus efficace chez E.
coli.
f. Les séquences permettant l’inversion
Un des moyens de controler l’expression du gène est de le cloner à l’envers puis à le retourner au
dernier moment lorsque la culture arrive à la phase stationaire. Deux systèmes ont été utilisés, le
système Int/att et le système Flp/FRT. Les recombinases sont présentes dans la bactéries et leur

24/10/2011 66
expression est régulée. Suite à l’induction, elles tournent le gène d’intérêt qui est traduit (Sektas et
al., 2001).
II) Autres bactéries utilisées en production de protéines
D'autres protéines Gram négatives ont été
utilisées pour exprimer des protéines. les techniques
et concepts ne sont pas différents de ceux développés
pour E. Coli. Caulobacter crescentus est une bactérie
gram négative non toxique qui est très répendue dans
l’environnement principalement dans les biofilms. C.
crescentus est couverte à sa surface par une proteine
qui s’arrange selon un réseau en deux dimensions
régulièrement structuré. Cette protéine sert
vraissemblablement à se protéger contre les virus, les
bactéries pathogènes ou les enzymes lytiques
susceptibles d’attaquer la protéine. Le système de secrétion de cette protéine (RsaA) a été utilisée
pour produire des protéines recombinantes en faisant une fusion entre la protéine RsaA et la protéine
recherchée. Les protéines secrétées se retrouvene tà la surface de la bactérie ou sur des liposomes. Le
vecteur doit comporter deux origines de réplication, une pour E. coli et une pour C. crescentus, le
signal de sécrétion en C terminal de la protéine produite et un promoteur pour l’expression.
Les Bacillus
Parmi les protéines Gram positives, les Bacillus ont été et sont toujours très utilisés pour
produire des protéines pour plusieurs raisons :
- Ils sont depuis longtemps utilisés par l'industrie pour produire des protéines. La
subtilisine, protéase incorporée aux lessives, est produite à partir de Bacillus. Il existe
donc de nombreuses souches modifiées pour les contingences industrielles. Toutefois
ces souches ne sont généralement pas disponibles.
Exemple de vecteur utilisé pour exprimer une
protéine chez Caulobacter crescentus.

24/10/2011 67
- On les connaît bien au niveau génétique et leur manipulation par les techniques de
biologie moléculaire est bien développée. En effet, si à priori on peut prendre n'importe
qu'elle bactérie, il faut quand même que l'on ai isolé une origine de réplication efficace,
un promoteur, etc. De plus on doit avoir mis au point un système de transformation
efficace. Des promoteurs forts sont connus et des peptides signaux responsables de la
sécrétion sont disponibles. Par exemple le peptide signal d'une subtilisine de Bacillus
amyloliquefasciens de 30 acides aminés a été utilisé avec succès.
- Le Bacillus ne sont pas pathogènes et ne produisent pas d'endotoxine.
- Ils sécrètent un grand nombre de protéines, leur système de sécrétion est donc bien
développé. Ce sont des bactéries Gram-positives, si bien que les protéines n'ont que la
membrane cytoplasmique à traverser.
Un cas intéressant est l'expression avec Bacillus brevis. La souche 47 produit de 12 à 25
grammes de deux protéines sécrétées appelées MWP et OWP par litre de milieu. Durant la phase
logarithmique, les deux protéines sont insérées dans la membrane mais elles continuent d'être
produites pendant la phase stationnaire et elles sont alors sécrétées dans le milieu. Les deux protéines
appartiennent au même opéron (cpw), elles sont donc sous la dépendance d'un seul promoteur, et
présentent un peptide signal. La production est énorme, on a donc à faire à un promoteur très fort
qu'on peut utiliser pour la surproduction de la protéine recombinante (cette notion d'utiliser des
promoteurs extrêmement forts est la même que celle qui a donné lieu à l'utilisation de certains
promoteurs en système eucaryote comme par exemple le promoteur de la polyédrine du Baculovirus).
Un plasmide contenant ce promoteur, un site de liaison au ribosome et le peptide signal a donc été
construit. La production d'EGF humain (epidermal growth factor) ou d'α-amylase a donné des
productions de plusieurs centaines de mg de protéine par litre de milieu.
Bacillus megaterium. Un des éléments régulateurs de l’utilisation du carbone est l’opéron xylose
ce qui permet une inductibilité de 350 fois. L’expression des protéines est relativemnt stable du fait
de l’absence de protéases alkaline qui sont par exemple présentes chez B. subtilis.
Ralstonia eutropha

24/10/2011 68
Cette bactérie n’utilise pas la même voie de métblisation des hexoses que E. coli ou que les Bacillus
et synthétisent moins d’acides organiques losque l’oxygène devient limitant ce qui affecte la
fermentation. De ce fait Ralstonia peut pousser à haute densité, jusqu’à 230 g par litre et il semble
que les protéines qui font des corps d’inclusion chez E. coli reste solubles. Un promoteur fort,
inductible par une limitation en phosphate a été isolé en comparant les protéomes de cellules
cultivées en milieu pauvre en phosphate, du protéome de cellules cultivées en milieu riche en
phosphate (Srinivasan et al., 2002).
Le système qui a été développé utilise une souche qui produit la T7 RNA polymérase sous le control
du promoteur fort et inductible phaP. Le gène d’intérêt est cloné en aval du promoteur T7 puis
introduit dans le génome de la bactérie (Barnard et al., 2004)
La production d’une protéine eucaryote dans un système bactérien n'est souvent
envisageable que
- Pour des protéines dont l'activité n'est pas requise (pour faire un anticorps par
exemple),
- Pour les protéines dont les modifications post-traductionnelle ne sont pas importantes.
En effet, les procaryotes sont incapables de réaliser la plupart des modifications
catalysées par des enzymes.
- Pour les protéines dont le repliement n'est pas dû à des protéines comme c'est le cas
pour la plupart des protéines synthétisées dans le réticulum endoplasmique.
Dans les autres cas on utilisera un système eucaryote

24/10/2011 69

24/10/2011 70
Expression en système eucaryote : les champignons
les levures
Plusieurs levures sont utilisées pour exprimer les protéines recombinantes. Les plus
souvent utilisées sont Saccharomyces cerivisiae et Pichia pastoris.
a) Saccharomyces cerevisiae (levure de boulangerie)
Cette levure présente deux avantages : d'une part, on dispose de vecteurs qui peuvent se
maintenir dans les cellules comme des plasmides bactériens, et, d'autre part, on peut facilement
intégrer un gène par recombinaison dans le chromosome.
Origine de réplication ou ARS
L'origine de réplication de l'épisome 2-μm est le plus souvent utilisé, il permet d'avoir
entre 10 et 40 copies du plasmide par cellule. Ce nombre est très variable et on peut perdre le
plasmide au cours des divisions cellulaires. Aussi, pour obtenir des copies plus nombreuses, on
utilise un marqueur de sélection faible et on augmente la sélection. Toutefois il n'est pas aussi
évident que l'augmentation du nombre de copies augmente la production de protéine. En effet
on a souvent remarqué qu'un grand nombre de copies affecte la croissance des levures et donc
la production. Les levures utilisables avec cette origine de réplication doivent être dépourvues
de l'épisome 2-μm.
Le problème est que ces vecteurs sont peu stables et, lorsqu'ils contiennent uniquement une
origine de réplication, ils sont vite perdus en l'absence de sélection, ce qui est différent des
plasmides d'E. coli. En effet, il n'y a pas comme dans la bactérie de découplage entre la
réplication du plasmide et du chromosome. Pour palier à cette perte on peut ajouter une
séquence centromérique (CEN) qui permet la ségrégation des plasmides dans les cellules filles.
L'inconvénient est que dans ce cas, si on gagne en stabilité on perd en nombre de copies, il y a
en effet 1 ou 2 copies du plasmide par cellule.

24/10/2011 71
Un autre moyen pour augmenter la stabilité consiste à intégrer le plasmide dans le génome,
on profite alors de la séquence centromérique du chromosome. On transforme alors un ADN
linéaire contenant le gène d'intérêt et un marqueur de sélection flanqué des séquences d'un gène
non essentiel. Il y a intégration dans ce gène non essentiel.
Les marqueurs de sélection
De nombreuses souches de laboratoire sont auxotrophes pour des acides aminés ou des
nucléotides, c'est à dire qu'elles ne peuvent pas pousser en l'absence du composé dans le milieu.
On peut donc utiliser la complémentation des enzymes entrant dans les voies de biosynthèse.
Les plus utilisés sont les gènes URA3 ou LEU2 responsables de la synthèse de l'uracile ou de la
leucine. Mais il existe d'autres systèmes de complémentation. L'inconvénient de ce système est
qu'il faut utiliser des milieux biochimiquement définis or, dans ces milieux, les levures
poussent moins bien et, de ce fait, la production de protéine est moins bonne. De plus, les
milieux définis sont plus chers à fabriquer que les milieux non définis comportant par exemple
de l'extrait de levure, de la bactone peptone comme les milieux bactériens.
Aussi, on préfère souvent utiliser des marqueurs de sélection qui donnent la résistance à
des drogues, la tunicamycine (TUN) ou l'hygromycine (Hgm), mais il y a toujours un risque
d'apparition de résistances spontanées dans la souche utilisée et donc de perte du plasmide.
Le promoteur
On peut utiliser des promoteurs "constitutifs". Les gènes codant pour l'alcool
déshydrogénase (ADH1) ou pour 3-phosphoglycerate kinase (PGK) sont exprimés à très haut
niveau, en présence de glucose dans le milieu, leurs ARNm représentent jusqu’à 1% des ARN
totaux. Ils ne sont pas tout à fait constitutifs, en effet l'ADH1 est transcrit 2 à 10 fois moins en
absence de glucose et la PGK 20 à 30 fois moins. Mais ces différences sont faibles par rapport
à celles observées pour d'autres promoteurs tels que GAL1 ou PHO5.
La régulation des gènes utilisés pour la métabolisation du galactose est très utilisée pour
réguler la production de protéines recombinantes. Ce promoteur est en effet très fort : en

24/10/2011 72
présence de galactose, la production de la kinase GAL1 est augmentée de 1000 fois et
représente 0.8% des protéines totales de la levure.
Un autre gène inductible très utilisé est celui codant pour la phosphatase acide (PHO5).
Cette phosphatase acide a pour rôle de déphosphoryler les protéines du milieu extérieur et ainsi
de procurer une source de phosphate à la cellule. Lorsque le phosphate inorganique dans le
milieu est faible, le gène est transcrit, la phosphatase acide est sécrétée, elle déphosphoryle des
protéines, le phosphate libéré est incorporé dans la levure. A l'inverse, quand il y a du
phosphate inorganique dans le milieu, la phosphatase devient inutile, le gène la codant n'est pas
transcrit. On peut utiliser ce système en employant un milieu riche en phosphate inorganique
pour éviter la transcription et un milieu dépourvu pour l'augmenter. Toute une série de
répresseurs et d'activateurs ont été mis en évidence comme pour le système de la galactosidase.
Parmi ceux-ci, PHO2 est nécessaire pour l'activation et PHO80 a un rôle dans la répression. On
utilise une souche qui a une mutation de perte de fonction du répresseur PHO80, il n'y a donc
pas de répression par PHO80. De plus on utilise une mutation thermosensible de l'activateur
PHO2 de façon à contrôler l’activation par des changements de température.
Les deux exemples précédents viennent des études de génétique de la levure, mais on peut
emprunter des systèmes chez les autres eucaryotes. Les récepteurs des stéroïdes de mammifères
gardent leurs activités en tant que régulateurs conditionnels de la transcription chez la levure.
Dans ce cas, en amont d'un promoteur de levure on clone plusieurs éléments de réponse aux
glucocorticoïdes (GRE, Glucocorticoïdes Response Elements) qui sont des éléments de 26 pb.
Le récepteur est lui cloné dans un deuxième plasmide. L'addition de glucocorticoïdes favorise
la liaison du récepteur sur les GRE et le gène est transcrit.
Sécrétion des protéines
Les peptides signaux hétérologues fonctionnent pour la plupart dans la levure, il n'est donc
pas nécessaire d'en changer lorsqu'on veut exprimer une protéine habituellement sécrétée. Dans
le cas où on désirerait faire secréter une protéine cytoplasmique, il suffirait de faire une fusion
avec un peptide signal d'une protéine de la levure tel que celui de l'invertase.
Toutefois, il est difficile de produire des protéines sécrétées dans Saccharomyces
cerevisiae.

24/10/2011 73
- En étudiant la glycosylation des protéines, il est apparu qu'il existe une étape limitante
dans cette maturation quelque part entre le réticulum endoplasmique et le golgi. Une première
technique consiste à changer d'espèce et utiliser Pichia pastoris. Cette dernière permet
l'expression de protéines membranaires qui sont difficiles à obtenir avec S. cerevisiae.
- La sécrétion ne s'effectue que pendant la phase de croissance exponentielle, pour une
protéine sécrétée, il est donc préférable d'utiliser un promoteur constitutif.
- La plupart des modifications post-traductionnelle sont effectuées par S. cerevisiae.
Toutefois la N-glycosylation est très différente de celle observée pour les autres eucaryotes. Les
sites sont les mêmes (N-X-S/T), mais les sucres ajoutés sont différents et présents en très grand
nombre. On compte par exemple de 50 à 150 résidus mannose dans chaque chaîne.
Transformation
On dispose de plusieurs méthodes pour faire rentrer un ADN dans la levure. On peut
utiliser soit une transformation chimique avec le l'acétate de lithium, soit une transformation
avec du CaCl2 sur des sphéroblastes (les sphéroblastes sont obtenus par une lyse de la paroi des
levures par une lyticase), soit par électroporation.
Avantage de la levure. On a quelques millénaires d’expérience et on sait qu’il n'y a pas de
toxicité pour l'homme si bien qu'on peut produire dans la levure des protéines destinées à être
utilisées comme vaccin. Par exemple, le vaccin contre l'hépatite B a été fait dans la levure.
b) Schizosaccharomyces pombe
L'expression dans cette levure est similaire à l'expression dans S. cerevisiae. Le vecteur
peut se répliquer d’une manière autonome et peut être sélectionné par auxotrophie en utilisant
Leu2p. Parmi les promoteurs utilisés on peut citer nmt1 qui est inductible par la thiamine, en
présence de thiamine nmt1 est réprimé et l’induction se fait en retirant las thiamine du milieu.
La TATA box de ce promoteur a été modifiée pour avoir des promoteurs plus ou moins fort et
donc avoir des quantités de protéines produites plus ou moins importantes.

24/10/2011 74
c) Pichia pastoris
P. pastoris est une levure qui a été développée dans les années 70 pour utiliser le méthanol
qui était alors un sous produit de l'industrie pétrolière. Elle est en effet capable de se développer
sur du méthanol comme seule source de carbone. Ce méthanol était converti par P. pastoris en
protéines et donc en aliment pour bétail. Avec la crise pétrolière, la difficulté de concurrencer
les aliments tels que le soja a amené à l'abandon de P. pastoris. De plus d'autres utilisations du
méthanol ont été trouvées si bien que l'utilisation de P. pastoris a été arrêtée au début des
années 80.
La première étape de la métabolisation du méthanol est une oxydation par le produit du
gène AOX1 (le produit d'un deuxième gène, AOX2 est aussi impliqué, mais AOX1 est
responsable de la majeure part de l’oxydation). L'expression de AOX1 est régulée au niveau
transcriptionnel, en présence de méthanol dans le milieu de culture, l'ARN codant pour
l'oxydase représente 5% des ARN polyA+.
Le vecteur typique contient un gène de
résistance à un antibiotique et une origine de
réplication pour la propagation dans E. coli.
Il contient, en outre, le promoteur de AOX1
et la partie terminale (3') de AOX1. Ces deux
parties sont interrompues par un site de
clonage multiple suivi au besoin d'une
séquence codant pour un peptide signal pour
insérer le gène d'intérêt. Comme moyen de
sélection, on utilise soit un gène codant pour
une histidinol déshydrogénase permettant la
synthèse d'histidine (HIS4) soit un gène
bactérien procurant la résistance à la kanamycine et procurant chez la levure la résistance au
G418, soit un gène de résistance à la zéocine.
Ce plasmide est introduit dans P. Pastoris soit par électroporation soit par transformation
de sphéroblastes comme pour S. Cerivisae.
Vecteur d’expression dans Pichia pastoris

24/10/2011 75
Il y a plusieurs possibilités d'intégration dans le chromosome:
Le remplacement de gène ou
insertion oméga. Dans ce cas il y a
deux crossing over entre les
promoteurs de AOX1 et la partie 3'
de AOX1, les recombinants ont le
phénotype Muts. (Muts signifie que
la levure n'est plus capable
d'oxyder le méthanol et donc plus
capable d'utiliser cet alcool comme seule source de carbone). Dans ce cas, le plasmide est
coupé en deux endroits avant la transformation.
L'insertion de gène s'effectue
par un seul crossing over entre le
locus AOX1 du chromosome et
une des séquences d'AOX1
présente sur le vecteur (région 5'
ou région 3'). On a dans ce cas
insertion d'un plasmide en amont
ou en aval de AOX1 qui peut
demeurer fonctionnel. Le
phénotype de tels transformant est
Mut+.
Plusieurs copies peuvent ainsi s'intégrer en tandem. De telles insertions multiples peuvent
être sélectionnées en utilisant la résistance au G418 ou à la zéomycine. Chaque gène entraîne
une faible résistance, en augmentant la dose d‘antibiotique, on sélectionne les recombinants qui
ont le plus de copies.
5’AOX1
gène d’intérêt
3’AOX1 plasmide linéarisé
génome de Pichia
plasmide intégré dans le génome de pichia
gène AOX15’ 3’
PAOX1 gène d’intérêt
gène de sélection
3’AOX1
gène desélection
3’AOX1 5’ AOX1(promoteur)
gène d’intérêt
génome de pichiagène AOX15’ 3’
ori

24/10/2011 76
L'insertion de gène peut aussi se faire au locus his4. Dans ce cas, le phénotype devient
His+ et Mut+ comme précédemment. Ici aussi on peut avoir des insertions multiples. Dans ce
cas, on utilise un mutant his portant une mutation ponctuelle pour pouvoir avoir un sauvetage
dans une des deux copies générées par la recombinaison.
Avantages de la production de protéines dans P. pastoris
- La production de protéine est de 10 à 100 fois plus importante qu'avec S. cerevisiae
- Elle permet l'expression de protéines membranaires qui sont difficiles à obtenir avec S.
cerivisiae
- Comparé à S. Cerevisiae, les protéines sécrétées sont moins glycosylées, il y a en effet
moins de résidus mannose ajoutés dans chaque N glycosylation (8 à 14 par chaîne alors que S.
cerevisiae en comporte de 50 à 150).
- On peut utiliser un marquage des protéines au 13C, utile pour les études en RMN. On
utilise du 13C méthanol qui est métabolisé et le 13C est incorporé dans les protéines.
Les champignons filamenteux
On utilise principalement deux espèces, Trichoderma reesei et Aspergillus niger. Les
productions sont très importantes, la cellulase est produite à 30 g/l, l'interleukine à 300 mg/l, la
chymosine bovine à 1 g/l et la lactoferrine humaine à 3 g/l. Cette production au stade industriel
est due à des mutagenèses aléatoires visant à obtenir des superproducteurs.
Généralement on effectue une fusion avec une protéine du champignon pour obtenir la
sécrétion de la protéine d'intérêt.
Il y a toutefois plusieurs problèmes :
- Les souches surproductrices sont industrielles et donc ne sont pas disponibles.
- Comme pour Saccharomyces cerevisiae, il y a une hyperglycosylation des protéines.
- Les méthodes de transformations sont difficiles.
Dictyostelium discoideum

24/10/2011 77
Eucaryote monocellulaire qui forme des agrégats et, en condition défavorable, il y a
formation d'un sporocarpe. Cet eucaryote a l'avantage de posséder des plasmides et donc un
vecteur potentiel de transformation.
Plasmide utilisable chez Dictyostelium discoideum
(Manstein et al., 1995)
Pact15: promoteur constitutif
Dpd2 : origine de réplication
AmpR et ori : résistance à l’ampicilline et origine
de réplication pour les constructions dans E. coli.
Ce système est peu utilisé mais il a été montré qu'une protéine recombinante pouvait représenter 1%
des protéines totales de la cellule.
Pact15MCS
terminateur
géne de sélection(G418)
oriAmpR
Dpd2

24/10/2011 78
Les cellules végétales
Cette technologie vient de la transformation des plantes, la seule différence est qu’au lieu
de régénérer une plante entière, on cultive les cellules. Pour transformer une cellule végétale, le
plus facile est d’utiliser Agrobacterium tumefasciens. Cette bactérie infecte certaines plantes, et
les cellules infectées ont acquis la capacité de croître de façon indépendante et non régulées
(elles sont transformées). Ces cellules sont capables de se développer en culture même sans
hormone et même en l'absence de la bactérie.
Les plasmides Ti sont grands 200 kb, une partie (ADN-T) porte les gènes responsables de
la synthèse d'acide aminés peu courants appelé les opines qui ne peuvent pas être utilisés par la
plante. Elles sont utilisées uniquement par la bactérie qui a sur le plasmide Ti des gènes dont
les produits sont capables de dégrader les opines comme source énergétique. Cette utilisation
des opines est vraisemblablement un moyen de se maintenir. Une autre partie de l'ADN-T code
pour des hormones qui induisent la prolifération cellulaire. Enfin le plasmide Ti porte aussi des
gènes de virulence.
Les cellules végétales endommagées produisent des substances chimiques qui déclenchent
l’expression des gènes de virulence (vir) situé sur le plasmide Ti. L'élément T est excisé de
l'ADN plasmidique au niveau de séquence répétée de 25 bp sous la forme simple brin (un seul
des deux brins est excisé comme dans le cas de la conjugaison bactérienne). Cet ADN-T simple
brin est transféré à la cellule végétale, rentre dans le noyau et s'intègre dans l'ADN.
Ce système a été utilisé pour introduire des gènes via l'ADN-T. Comme le plasmide Ti est
trop grand (200 kb) pour pouvoir être utilisé in vitro, L'ADN-T a été introduit dans un petit
plasmide utilisé couramment en biologie moléculaire. Il a été raccourci, les gènes codant pour
les hormones ont été éliminés pour éviter la croissance incontrôlée des cellules infectées. Dans
cet ADN-T on introduit le gène à exprimer, un gène de sélection dans l'agrobactérium et un
gène de sélection dans la plante.
Le plasmide est ensuite introduit dans agrobactérium par conjugaison. Il y a alors des
événements de recombinaison et ceux là seul sont sélectionnés car le plasmide venant de E. coli
n'est pas capable de se répliquer dans Agrobactérium. La sélection des recombinant (par rapport
au Ti sauvage s'effectue par un gène de résistance). Le plasmide Ti recombinant est alors
transféré dans les cellules végétale en les infectant avec l'agrobactérium recombinant.

24/10/2011 79
Un deuxième système a été mis au point. Le plasmide propagé dans E. Coli ne contient que
les deux séquences répétées bordant l'ADN-T (LB et RB). Entre ces deux fragments, on a un
site de clonage et un gène de sélection pour sélectionner les recombinants dans la plante (NPTII
qui confère la résistance à la kanamycine). On dispose par ailleurs d'un plasmide Ti modifié qui
ne contient plus l'ADN-T mais toujours les genes vir. Le plasmide de E.coli est transféré dans
agrobactérium par conjugaison mais ici il n'y a pas de recombinaison. La bactérie contenant les
deux plasmides est utilisée pour infecter des cellules végétales. Il y a intégration de l'insert
entre les deux extrémités répétés du plasmide construit dans E. coli.
Les cellules les plus utilisées sont les cellules de tabac et en particulier la souche BY2
(Bright Yellow 2) qui se multiplie rapidement.
Avantage de l’expression dans les cellules végétales. Ces cellules se multiplient
facilement.

24/10/2011 80
Les cellules d'insectes
a) Baculovirus.
Les Baculovirus sont responsables de polyédroses nucléaires des insectes en particulier des
lépidoptères. Ce sont de gros virus dont le génome est constitué d'un ADN d'environ 100 kb. A
la fin de l'infection, ces virus forment des corps d'inclusion (appelés polyèdres) dans la cellule.
Ces polyèdres sont principalement constitués d'une protéine d'environ 30 kDa, la polyédrine,
qui protège les virions. Dans la nature, ces polyèdres sont ingérés par les insectes, dégradés par
les protéases du tube digestif, les virions sont libérés et vont se fixer sur les microvillosités des
cellules épithéliales de l'intestin moyen et ils traversent les cellules intestinales. Ils se
multiplient dans les hémocytes et dans le tissu adipeux. Les virions sont transmis de cellules à
cellules par exocytose. En fin de cycle, les noyaux renferment de très nombreux polyèdres, ces
polyèdres constituent donc le mode de transmission d'insecte à insecte.
Depuis quelques années on sait utiliser le Baculovirus pour produire des protéines in vitro.
En fin de cycle le virus produit une très grande quantité de polyédrine. L'astuce a donc été de
remplacer la partie codante du gène codant
pour la polyédrine par celui qui nous intéresse.
On transfecte des cellules. Le virus, au lieu de
produire une très grande quantité de
polyédrine, produit la protéine voulue. A coté
de ce promoteur, il existe un autre promoteur
tardif qui est très fort, le promoteur de P10.
On peut cloner notre gène derrière ce
promoteur. Dans ce cas, la lyse des cellules est retardée ce qui a pour effet d'augmenter la
production. L'utilisation des deux promoteurs, P10 et polyédrine permet en outre de coproduire
deux protéines, une protéine exprimée à partir du promoteur de la polyédrine et une autre
Exemple de vecteur de transfert à deux promoteurs : p10 et polyédrine
AmpR
promoteur P10
promoteur polyédrine
ori

24/10/2011 81
exprimée à partir de P10. En utilisant un vecteur comportant deux promoteurs P10 tête-bèche,
on peut même produire trois protéines simultanément.
Comment remplacer le gène de la polyédrine par le gène d'intérêt ? Comme il n'est pas aisé
de manipuler de grands fragments d'ADN (100 kb) in vitro on peut utiliser deux méthodes,
utilisant soit la recombinaison dans la cellule d’insecte, soit la transposition dans la bactérie.
La recombinaison consiste à utiliser la "plasticité" du génome des Baculovirus qui est très
importante. En effet il arrive qu'ils échangent une partie de leur information génétique. Les
recombinants peuvent donc acquérir des propriétés nouvelles, en associant dans le même virus
des propriétés des deux "parents". On profite donc de la facilité d'obtention de recombinant en
manipulant en bactérie une petite partie du génome du Baculovirus comportant le promoteur de
la polyédrine, une région d'ADN en amont du promoteur et une région en aval pour avoir des
régions ou peuvent s'effectuer les recombinaisons. Ce plasmide bactérien est appelé vecteur de
transfert. On co-transfecte ensuite des cellules avec le virus sauvage et le plasmide bactérien
comportant une petite partie de l'ADN viral et on cherche les événements de recombinaison, les
plages de lyse qui ne produisent pas de polyédrine.
Il existe une solution pour éviter de passer par le vecteur de transfert. Le gène d'intérêt est
amplifié avec des amorces spécifiques, mais en amont de ces amorces, on ajoute 50 nucléotides
correspondant aux séquences amont et aval de la polydédrine sur la séquence du virus. Ces
nucléotides vont permettre la recombinaison comme dans le cas des séquences présentes sur le
vecteur de transfert (Gritsun et al., 1997).
La recombinaison est un phénomène relativement rare et l'isolement des recombinant était
jusqu'à récemment relativement fastidieux.
Une technique améliorant ce processus a été
développée. Elle consiste à couper l'ADN
viral dans la région qui doit être remplacée
par le gène d’intérêt. Pour ce faire, on coupe
le virus par une enzyme de restriction qui ne
DNA viral (100 kb)
polyédrine p10
ORF 603 LacZ ORF1629
promoteur polyédrine
Bsu36I Bsu36I Bsu36I

24/10/2011 82
coupe qu'à cet endroit (Bsu36I). On cotransfecte l'ADN viral et le vecteur de transfert et on
attend la recombinaison sur les ORF 603 et 1629. Seuls les recombinants vont se développer, il
restera toutefois quelques virus sauvages si la coupure n'a pas été totale.
Enfin on peut fabriquer les recombinants directement dans la bactérie si on arrive à cloner
directement le gène d'intérêt derrière le promoteur de la polyédrine en bactérie. Il faudra alors
simplement extraire l'ADN et transfecter des cellules et la recombinaison dans les cellules
d'insecte sera alors évitée. Cette construction est difficile à effectuer in vitro, en effet les sites
de restriction uniques sur
100 kb sont rares et la
manipulation de grand
fragments d'ADN sans
les casser est difficile.
Mais on peut effectuer la
construction dans la
bactérie soit par
recombinaison soit par
transposition. C'est cette
dernière méthode qui est
la plus utilisée. L'ADN
viral a été cloné dans son
ensemble dans E. coli.
Pour ce faire, l'origine de réplication du facteur F et un gène de résistance à un antibiotique ont
été ajoutés au génome de baculovirus. On a ainsi obtenu un génome du baculovirus capable de
se répliquer à faible nombre de copies chez E. coli. Cette construction a pris le nom de bacmide
(Luckow et al., 1993).
Dans le bacmide, on a rajouté un site cible du transposon bactérien Tn7 (mini-att Tn7) en
phase avec le gène codant pour le peptide α de la β-galactosidase (LacZY). Ainsi un événement
de transposition peut se repérer par l’arrêt de synthèse de la β-galactosidase dans une souche
déficiente en peptide α. Le transposon vient du plasmide donneur. Il contient, entre les deux
bacmide100 kbmini-F-ori
LacZa
mini-att Tn7
kanamycine R
Helper
tetracycline R
ori
tnsA-E
donneur5000 bps
ampiciline R
Tn7Lgentamicine R
gèneTn7RPpol
LacZα
pSC101 ts ori

24/10/2011 83
LTR de Tn7, le promoteur de la polyédrine cloné en amont du gène d’intérêt et un gène de
sélection.
On fabrique donc in vitro le plasmide donneur, avec le gène d’intérêt en aval de son
promoteur. Ce plasmide est alors utilisé pour transformer des bactéries contenant le bacmide et
un plasmide helper. Ce dernier fourni les différents composants nécessaires à la transposition
comme la transposase. Les colonies où un évènement de transposition a eu lieu sont blanches et
sensibles à l’antibiotique présent dans le plasmide donneur en dehors du transposon. Il faut
alors éliminer le plasmide donneur et, pour ce faire, on a plusieurs moyens : utiliser le lacZα en
dehors du transposon, les bactéries exprimant la β-galactosidase ne seront pas prises, ou utiliser
une origine de réplication thermosensible. Après la transposition, les bactéries seront incubées à
42°C (Leush et al., 1995).
Pour isoler le virus on a deux méthodes. La première ("plaque assay") consiste à étaler des
cellules sur une boite puis à les infecter avec la population de virus. On coule ensuite de
l'agarose pour limiter le passage des virus aux cellules voisines. On utilise de l'agarose à bas
point de fusion pour éviter de cuire les cellules. On obtient ainsi des plaques de lyse comme
losqu'on isole des clones de lambda recombinants dans une banque. La deuxième méthode est
appelée "end point dilution". On utilise une plaque de microtitration de 96 puits qui sont
inoculés par des cellules. Dans chacun des puits d'une rangée, on infecte les cellules par 1
microlitre de la solution virale, dans la seconde par 100 nl soit 10 fois moins, dans la troisième
par 10 nl, etc... Cette quantité est obtenue par dilution successive de la solution primordiale. A
partir d'une certaine dilution, les virus sont isolés. Par exemple si la solution virale au début est
de 1 virus par nl, à la dilution de 1 nl et de 0.1 nl par puits on aura des clones isolés. Cette
méthode permet donc d'une part d'isoler des virus d'une solution et d'autre part de la titrer. Dans
les deux méthodes, il faut ensuite reconnaître les virus "sauvages" des virus recombinants. Les
premiers essais utilisaient un virus sauvage produisant la polyhédrine. Ces virus étaient
reconnaissables au polyèdre présent dans le noyau des cellules infectées. Maintenant on utilise
plutôt comme virus "sauvage" un virus exprimant la β-galactosidase, repérable par une réaction
enzymatique avec le X-gal donnant une couleur bleue, les recombinants ne donnant pas de
réaction colorée avec le X-gal.

24/10/2011 84
La production de protéine est le plus souvent effectuée à l’aide de culture de cellule
d’insectes. Quelques cellules d'insectes ont été immortalisées d'une manière assez empirique,
généralement en dissociant des cellules non différentiées et en les mettant en culture (les larves
d'insectes hétérométaboles possèdent des amas de cellules non différentiées qui seront utilisées
pour générer les tissus spécifiques de l'adulte). Des cellules dissociées ont été mises en culture,
certaines d'entre elles se sont maintenues. On dispose ainsi de trois souches de cellules, des Sf9
et Sf21 provenant d'ovaires de Spodoptera frugiperda et Tn5 provenant de Trichoplusia ni. Les
cellules sont infectées avec un virus recombinant, le virus se développe, produit la protéine et
lyse les cellules.
Une autre possibilité est d’infecter des larves de lépidoptère avec le virus recombinant. Les
chrysalide du vers à soie (Bombyx mori) sont actuellement utilisé en Asie, parce que c’est un
élevage mis au point depuis longtemps pour fabriquer de la soie naturelle, parce que c’est un
insecte assez gros et parce que l’insecte est « non toxique » en effet les chrysalides sont
consommées en chine. La technique consiste à injecter une solution virale dans les chrysalides
et à les récolter 4-5 jours après.
Avantage de l'expression en baculovirus:
Ce système de production en infectant soit des insectes soit des cellules d'insecte présente
plusieurs avantages:
Fonctionnalité des protéines recombinantes : Les insectes sont des eucaryotes et ils sont
capables d'effectuer tous les repliements des protéines avec réalisation des bons ponts
disulfures et les bonnes oligomérisations, ce qui n'est pas le cas des bactéries ou des levures.
Cependant, lorsque la protéine fonctionne sous la forme d’hétérodimère, elle ne sera pas
fonctionnelle à moins que la deuxième protéine ne soit co-exprimée.
Modifications post-traductionnelles : La plupart des modifications post-traductionelles sont
effectuées en baculovirus et ce aux sites où on les trouve dans les protéines natives. Toutefois,

24/10/2011 85
ce système produit une très grande quantité de protéine et on observe parfois une saturation du
système. Par exemple on observe parfois un plus faible niveau de glycosylation, de
phosphorylation ou de glypiation (ajout d’un ancrage GPI) de la protéine. Dans certains cas, la
modification post-traductionnelle est espèce ou tissus spécifique, cette modification post-
traductionnelle est due à des enzymes particuliers et on ne les observe pas chez les cellules
d'insectes à moins que ces enzymes ne soient coexprimées.
Haut niveau d'expression : Le record de l'expression mentionnée avec baculovirus est de 1
gramme de protéine recombinante pour 109 cellules soit plus d'un gramme de protéine par litre
de culture. Cependant il s'agit d'un record et il est difficile de prévoir la quantité de protéine que
l'on obtiendra. Par exemple une protéine cytotoxique tuera précocement la cellule et la
production sera plutôt de l'ordre du microgramme par litre.
Capacité de grande insertion : jusqu'à maintenant on ne connaît pas de taille maximale
pouvant être intégré dans le baculovirus. La seule limitation est donc la manipulation de grands
fragments d'ADN dans le vecteur de transfert.
Capacité d'épisser les gènes : La plupart des sites d'épissage seront utilisés par la cellule.
Cependant l'épissage des sites alternatifs, qui sont tissus spécifiques seront plus aléatoires.
Expression simultanée de plusieurs gènes : Des vecteurs comportant jusqu'à trois
promoteurs (deux P10 et un polyédrine) sont actuellement disponibles. La seule limitation est
simplement la difficulté de construire les vecteurs de transfert très grands, comportant plusieurs
inserts.
b) Cellules d’insecte ayant intégré le gène d’intérêt dans leur génome
Toutes les lignées cellulaires ont la propriété intéressante de pouvoir intégrer de l'ADN
dans leurs chromosomes. Si on transfecte une cellule avec un plasmide on a tout d'abord
expression transitoire à partir du plasmide puis dans un deuxième temps établissement d'une

24/10/2011 86
culture cellulaire exprimant la protéine d'une manière stable puisque le plasmide s'est intégré
dans le génome. Des centaines de copies du plasmide se retrouvent dans le génome.
Le promoteur. Pour obtenir ces lignées stables on a besoin d’un promoteur reconnu par la
machinerie transcriptionnelle de la cellule et d’un agent de sélection. On connaît un très grand
nombre de promoteurs chez les insectes. On utilise soit un promoteur constitutif soit un
promoteur inductible. Dans ce dernier cas, on peut utiliser le promoteur de la metallotionéine
qui est inductible par le sulfate de cuivre. Une autre alternative est d'utiliser un promoteur viral
précoce. Ainsi le promoteur OpIE2 du baculovirus de Orgyia pseudotsugata est utilisé car il est
fonctionnel dans un grand nombre de cellules d'insecte différentes.
La sélection : le facteur limitant est la transfection, si une cellule incorpore de l'ADN, elle
incorpore un grand nombre de molécules présentes dans le milieu. Une première technique
consiste à faire une cotransfection avec un plasmide comportant un gène de résistance. Les
cellules ayant incorporé des plasmides dans leur génome seront hygromycine résistante, les
autres seront sensibles et seront éliminées. Une deuxième technique consiste à intégrer le gène
de sélection au plasmide. On augmente la pression de sélection pour sélectionner les cellules
qui sont les plus résistantes, ces cellules comportent plus de plasmide, ainsi plus de copies du
gène d’intérêt et donc produiront plus de protéines. En augmentant graduellement la sélection,
on peut espérer sélectionner des lignées produisant un grand nombre de copies par
recombinaison.
Ce système offre l’avantage de pouvoir utiliser un grand nombre de cellules différentes, et
donc de choisir celle qui sera le plus adaptée à produire la protéine d’intérêt. En effet, les
capacités à effectuer les différentes modifications post-traductionnellles varient en fonction des
cellules.
Par contre de système est peu efficace si on veut exprimer une protéine présentant une
toxicité pour la cellule. En effet, plus on exprime la protéine toxique, moins on a de chance de
pouvoir établir la lignée stable.

24/10/2011 87
Les cellules de vertébrés
1) L'ovocyte de Xénope
On peut faire exprimer une protéine par l'ovocyte de Xénope en injectant de l’ADN dans le
noyau. Le noyau est situé au 2/3 du pôle animal et n'est pas visible sur l'ovocyte vivant. Pour
pouvoir injecter l'ADN on centrifuge doucement (200-400 g pendant 5 min. Le noyau plus
dense que le cytoplasme se déplace vers le pôle animal jusqu'à le toucher. On observe alors une
décoloration au pôle animal qui nous permet de le localiser. On injecte environ 10 nl d'ADN.
Le gène d'intérêt est cloné derrière un promoteur eucaryote fort. Il y a alors transcription puis
traduction.
Les avantages et inconvénients du système sont les mêmes que ceux énoncés pour
l'injection d'un ARNc, c'est à dire peu de protéines sont produites mais cette protéine est active.
On va donc l'utiliser principalement pour exprimer des récepteurs.
Par rapport à l'injection d'un ARNc, il y a plusieurs avantages: c'est plus facile à réaliser
puisque la transcription in vitro n'est plus nécessaire de plus, on obtient généralement plus de
protéines. Toutefois il y a aussi des désavantages : la production est très variable selon les
ovocytes, certains produisent beaucoup de protéines alors que d'autres n'en produisent que très
peu. Cette variabilité est sans doute due au moins en partie à la centrifugation et à l'injection
dans les noyaux qui peut les abîmer.
2) Cellules de mammifère
On peut distinguer trois grands types d'expression, l'expression à partir d'une infection
virale, l'expression transitoire et l'établissement d’une lignée stable. Dans les deux premiers cas,
les cellules sont infectées ou transfectées et analysées quelques jours après, dans le troisième
cas l'ADN injecté est incorporé au chromosome.

24/10/2011 88
a) Expression à l'aide d'un virus
De nombreux virus qui infectent les cellules de mammifère ont développé des mécanismes
pour détourner la machinerie de synthèse des protéines. Il a alors été tentant d'utiliser ces virus
pour produire des protéines.
Les premiers vecteurs viraux ont été fabriqués à partir du virus tumorigène simien SV40.
Ce virus est petit (5 kb) et ne peut pas s'accommoder de séquence supplémentaire importante. Il
faut donc remplacer quelques gènes viraux par le gène d'intérêt. Mais ces gènes sont
indispensables pour la viabilité du virus. On transfecte donc la cellule avec deux virus SV40 :
le virus recombinant portant le gène d'intérêt et un virus muté à un autre endroit du génome.
Les deux virus se complémentent et on obtient un stock viral comportant les deux virus. (N.B.
ce système d'helper suit la même stratégie que la formation de simple brin en utilisant un
plasmide portant l'origine de réplication des phages f1 ou M13).
Le BPV (Bovine Papilloma Virus) est un papovavirus de 8 kb, ADN double brin circulaire.
In vitro, il peut infecter un faible nombre de cellules et 30% de son génome peut être remplacé
par un ADN étranger ce qui permet d'insérer des séquences de plasmide permettant la
propagation dans E. coli ainsi que le gène d'intérêt. Son avantage est d'être présent en grande
quantité dans les cellules (50 à 200 copies), la transfection des cellules de souris (C127 ou 3T3)
est aisée et on peut même établir des lignées stables où le virus se maintient comme un épisome
(équivalent d'un plasmide).
Les lentivirus ont l’avantage de pouvoir infecter toutes les lignées cellulaires. Un système
de production est commercialisé par Invitrogen sous le nom de ViraPower®. On transforme le
plasmide contenant le gène d’intérêt avec deux autres plasmides dans une souche de cellules
pour reconstituer le virus portant le gène en aval d’un promoteur eucaryote fort. Le surnageant
de culture contient les virus et peut être utilisé pour infecter d’autres types cellulaires.
Le virus de la vaccine est un poxvirus contenant un ADN de grande taille (185 kb) dont la
réplication s'effectue entièrement dans le cytoplasme. Le gène d'intérêt est placé en aval d'un

24/10/2011 89
promoteur de poxvirus, les promoteurs eucaryotes n'étant pas reconnus par la machinerie
transcriptionnelle du virus. Comme pour Baculovirus, l'ADN du virus est trop grand pour être
manipulé aisément in vitro. La construction sera donc effectuée par recombinaison homologue
avec un vecteur navette portant des séquences du virus. Ici aussi, la recombinaison est assez
rare, on obtient environ un recombinant pour 1000 parents. Pour améliorer ce pourcentage et
ainsi faciliter la sélection des recombinants plusieurs systèmes ont été mis au point:
- On utilise le gène viral codant pour la thymidine kinase (TK) comme séquence
flanquante permettant la recombinaison, de telle façon que la recombinaison l'inactive.
On utilise des cellules mutées sur leur gène cellulaire de la thymidine kinase donc TK-.
Lorsqu’il y a le virus, les cellules sont TK+ du fait de la présence d’une thymidine
kinase virale. Les recombinant seront TK- due à l’inactivation de la thymidine kinase
virale. Or, on sait trier les cellules TK- des cellules TK+ : les cellules TK- sont
résistantes à la 5-bromodésoxyuridine. En effet, pour être active, cette drogue doit être
phosphorylée par la thymidine kinase.
- Le problème est que la mutation de TK+ vers TK- a une fréquence de 10-4, aussi on
associe souvent cette sélection à une autre méthode. Cette deuxième méthode utilise la
co-expression d'un gène de résistance à un antibiotique codant pour la guanine
phosphorybosyltransférase (gpt) qui procure la résistance au G418.
- Une troisième méthode utilise, comme pour le Baculovirus, un virus parent exprimant
la β-galactosidase pour pouvoir trier visuellement les virus recombinés des virus
parents. L'avantage du virus de la vaccine est qu'il éteint les productions d'ARNm de
l'hôte si bien que l'ARN du gène cloné peut représenter jusqu'à 30% de l'ARN de la
cellule.
Les adenovirus sont des virus à ADN double brins linéaires de 36 kb. Ils causent chez
l'homme des infections des voies respiratoires ou des conjonctivites. On peut utiliser des virus
atténués comme vecteur en clonant l'ADN d'intérêt derrière un promoteur fort. L'introduction
de l'ADN se fait par un premier clonage dans un plasmide puis par une recombinaison in vivo
comme pour le cas du baculovirus ou du virus de la vaccine. L'avantage de ce virus est que
l'expression peut se faire dans un grand nombre de cellules humaines telles que la lignée 293.

24/10/2011 90
Ainsi, les modifications post-traductionnelles caractéristiques des cellules humaines sont
respectées. L'expression est souvent élevée, jusqu'à 10-20% des protéines cellulaires.
Une autre catégorie de virus peut être utilisée : les rétrovirus. Ce sont des virus à ARN, une
fois l'ARN entré dans le cytoplasme, il est rétrotranscrit en ADN et cet ADN est intégré dans le
génome avec à chaque extrémité les LTR (Long Terminal Repeat). Il produit de l'ARN viral à
partir d'une séquence proche de l'extrémité du virus. Cet ARN sert à produire les protéines
virales et donc à la création de nouveaux virus. Ces virus sont assemblés dans le cytoplasme et
sortent de la cellule par bourgeonnement. Ainsi tout gène inséré dans un virus recombinant sera
intégré dans le génome.
Pour utiliser un tel système
- on fabrique un plasmide dans lequel on a ajouté le gène d’intérêt derrière son
promoteur et le signal d’empaquetage de l’ARN (ψ+), le tout est inséré entre les deux
LTR. Ce virus est incapable de s'intégrer dans le génome et est dépourvu de gag
(protéine de structure), de pol (reverse transcriptase, intégrase) et d’env (protéine de
l’enveloppe).
- Dans un deuxième plasmide (ou intégré dans le génome), on a un virus complet mais
qui manque de séquences nécessaires à l'encapsidation de l’ARN. Le plasmide portant
le gène d’intérêt est transfecté dans la cellule qui produit les protéines virales (gag, pol,
env), ce qui permet d'obtenir un virus à ARN. Ce système a simplement permis de
passer d’un plasmide à un virus, incapable de se répliquer mais pouvant s’intégrer dans
un génome.
Ces virus sont purifiés et infectent d'autres cellules qui ne comportent pas de virus. L'ARN
est rétrotranscrit en ADN, circularisé et intégré dans le génome. Le nouveau virus est incapable
de s'encapsider, l'intégration est stable.

24/10/2011 91
Les alfavirus tels que le SFV (Semliki Forest Virus) sont des petits virus à ARN simple
brin qui peuvent se répliquer dans toutes les cellules animales. A la suite de l'infection, l'ARN
code pour quatre protéines qui produisent l'autre brin appelé brin -. Ces derniers servent alors
de matrice pour produire des brins + et des petits génomes qui servent à faire les protéines de
structure. Plusieurs alfavirus ont été clonés sous la forme d’ADNc pour produire des ARN in
vitro, infectieux dans lesquels les gènes codant pour les protéines de structures ont été
remplacés par le gène d'intérêt.
Ce système a plusieurs avantages : on obtient une grande quantité de protéine, la
construction du virus s'effectue entièrement dans E. coli, il n'y a pas besoin d'helper. La
principale difficulté provient de la transfection de l'ARN dans les cellules qui est faite par
électroporation. Ce système est proche de l'injection d’ARN dans l'ovocyte de Xénope mais,
ici, il y a réplication de l'ARN par une réplicase.
4) Reconnaissance de Ψ par les protéines virales
vecteur retroviral
1) Transfection
2) intégration
3) transcriptiontraduction
gene X, Ψ, resistanceARN
gag, pol, env
6) Virus infectantsmais incapables de se répliquer
5) Empaquetage
génome

24/10/2011 92
Il y a quelques dangers à utiliser des virus pour exprimer des protéines. Par exemple, le
virus de la vaccine n'est pas toxique et on s'en sert pour les vaccinations contre la variole.
Toutefois, il faut faire attention, il est en effet capable d'attaquer les cellules humaines.

24/10/2011 93
b) Expression transitoire
Les vecteurs
Les vecteurs utilisés ici sont constitués de deux parties, une permettant la manipulation
dans les bactéries, avec au minimum une origine de réplication (pBR322 ori ou pUC ori ou col
E1 ori) et un gène de résistance aux antibiotiques. A ces deux parties, on peut ajouter des
parties accessoires : une origine d'un phage à ADN simple brin (M13 ou f1) pour faire certains
protocoles de mutagenèse ou pour séquencer, les promoteurs des phages T3, T7 ou Sp6 si on
veut faire de l'ARN in vitro. Une deuxième partie servira à la manipulation dans les cellules
eucaryotes. Ces deux parties ne sont pas forcement spatialement séparées et on peut ajouter ou
retirer les parties que l'on souhaite.
Les marqueurs de sélection
Un des principes de base des techniques de biologie moléculaire est d'utiliser un marqueur
biologique pour identifier ou sélectionner les cellules contenant des molécules d'ADN
recombinant. Le gène de sélection doit lui-même être en aval d'un promoteur fonctionnant dans
la cellule transfectée
Une première technique consiste à complémenter une déficience de la cellule. Exemple de
la thymidine kinase déjà vue dans l'utilisation du virus de la vaccine. Cette enzyme est codée
par le gène tk qui a été isolé chez le virus de l'herpès simplex (HSV). Elle est impliquée dans
une des voies de synthèse de la thymidine monophosphate à partir de la thymidine et donc de la
thymidine triphosphate, nucléotide indispensable pour la synthèse de l'ADN et donc pour la
réplication et la viabilité de la cellule.
Une deuxième méthode consiste à utiliser des enzymes résistants à une drogue comme par
exemple :

24/10/2011 94
- Une dihydrofolate réductase (DHFR) résistante au méthotrexate qui peut alors être ajouté
au milieu de culture (la DHFR résistante a été trouvée dans les cellules cancéreuses
résistante au méthotrexate).
- Une hygromycine phosphotransférase (HPH) inactive l'hygromycine qui inhibe aussi la
synthèse protéique.
- Une aminoglycosyl phosphotransférase (APH) donne la résistance au G418, proche de la
néomycine. Le G418 bloque la synthèse protéique et est détruit par l'APH. C'est le
marqueur le plus utilisé car le gène bactérien de résistance à la kanamycine procure la
résistance au G418 chez les eucaryotes. Dans ce cas, le même gène marqueur peut être
utilisé pour les étapes de construction dans E. coli comme pour les étapes d’expression
dans les cellules eucaryotes.
Il faudra alors que le gène soit fonctionnel dans les deux systèmes. Il faudra deux
promoteurs en amont du gène, un procaryote et un eucaryote, un site de liaison au
ribosome en amont de l’ATG d’initiation de la traduction pour l’expression en procaryote
et un site de polyadénylation en 3’ pour l’expression en eucaryote.
Il est aussi possible de trier les cellules transfectées par coexpression d'un anticorps sur la
surface externe de la cellule. Les cellules transfectées expriment l'anticorps à leur surface, ainsi
que la protéine d'intérêt, ces anticorps réagissent avec un haptène liée à une bille magnétique,
ces billes peuvent être triées avec un aimant.
Une autre méthode consiste à exprimer une protéine qui colore les cellules. On utilise le
plus souvent la GFP (green fluorescent protein) qui colore les cellules en vert. Les cellules
promoteur procaryote
promoteur eucaryote
neomyciner signal de polyadénylation
RBS

24/10/2011 95
transfectées sont vertes. Cette méthode peut être en plus employée en combinaison avec la
sélection à une drogue. On peut en effet fabriquer une protéine de fusion entre la GFP et
l’hygromycine phosphotransférase. Les cellules qui se développent en présence d’hygromycine
doivent être vertes. On peut ainsi vérifier que la sélection à l’hygromycine est efficace.
Les promoteurs
Les promoteurs et enhancers consistent en des séquences qui interagissent avec les
protéines impliquées dans la transcription. La combinaison de ces séquences détermine
l'efficacité avec laquelle un gène donné est transcrit dans une cellule donnée.
On distingue grossièrement deux types de séquence dans un promoteur, la "TATA box" et
les éléments en amont. La "TATA box" est localisée 25 à 30 bp en amont du site d'initiation de
la transcription. Ce site est directement impliqué dans le positionnement de l’ARN polymérase
II qui commencera la synthèse au bon site. Les éléments en amont, eux, sont responsables de la
vitesse de l'initiation de la transcription. L'action de ces éléments est indépendante de leurs
orientations, ils sont localisés 100 à 200 bp en amont de la TATA box.
Les enhancers stimulent la transcription, même à grande distance du promoteur (plus d'un
kb), ils peuvent se situer en amont ou en aval du promoteur. La plupart d'entre eux fonctionnent
uniquement dans un type de cellule et, de plus, sont actifs seulement dans des conditions
spécifiques telles que la présence d'un inducteur comme une hormone ou un ion métallique.
Ainsi le choix d'un promoteur et d'un enhancer va principalement dépendre de la lignée
cellulaire utilisée.
Les promoteurs et enhancer les plus utilisés proviennent de virus qui ont une faible
spécificité cellulaire. Par exemple l'enhancer précoce de SV40 est actif dans presque toutes les
cellules. De même, le promoteur précoce du cytomégalovirus (CMV) humain permet
l'expression dans un grand nombre de lignées cellulaires. Mais on peut aussi utiliser des
promoteurs de gène codant pour des protéines ubiquitaires. Ainsi, UbC le promoteur de
l’ubiquitine qui est une protéine très conservée chez les eucaryotes est utilisé parce qu’il
fonctionne dans un très grand nombre de cellules et que la transcription est équivalente à celle
obtenue avec le promoteur CMV.

24/10/2011 96
Un système hybride utilise le virus de la vaccine et l’ARN polymérase T7. On infecte des
cellules avec un virus recombinant codant pour l’ARN polymérase T7. On transfecte ensuite les
cellules avec un plasmide où le gène d'intérêt est sous le contrôle d'un promoteur de T7. Ce
système permet d'avoir une grande quantité de protéine en profitant d'une part de la forte
transcription et traduction de la polymérase puis de sa forte capacité de synthèse de l'ARN
d'intérêt.
Pour avoir un bon contrôle de l'expression, on peut utiliser des promoteurs inductibles par
exemple par les métaux lourds, un choc thermique ou une hormone stéroïdienne. Toutefois tous
ces systèmes fuient, il y a toujours une expression faible en absence d’induction. De plus les
inducteurs sont souvent toxiques et l’induction n’est généralement pas spécifique.
Pour résoudre ce problème, on utilise une régulation hétérologue.
Par exemple, on peut utiliser une séquence régulatrice d'insecte dans le promoteur.
L’ecdysone est une hormone de mue des insectes, absente chez les vertébrés. On fabrique des
cellules (CHO ou 3T3) qui synthétisent d'une manière constitutive un récepteur
hétérodimérique de l'ecdysone. On utilise dans le plasmide, un promoteur minimal comportant
en amont la séquence de réponse à l'ecdysone. On induit alors l'expression avec de la
muristerone A, un analogue de l'ecdysone qui n'a aucun effet sur les cellules de mammifère, et
qui n'induit aucun autre gène.
Un deuxième exemple est l’utilisation d’un système de régulation bactérien comme
l’opéron tetracycline de E. coli (Gossen et Bujard, 1992 ; Gossen et al., 1995). Le gène
d’intérêt est cloné en aval d’un promoteur minimal du cytomegalovirus (PminCMV) en aval d’un
élément de réponse à la tétracycline (TRE) qui contient plusieurs copies de l’opérateur. Le
plasmide doit coder pour le répresseur qui n’existe pas dans la cellule eucaryote (TetR). En
l’absence de tetracycline, TetR se fixe sur l’opérateur (TRE). Pour convertir TetR, un
répresseur procaryote en transactivateur eucaryote, il a été fusionné avec un domaine

24/10/2011 97
d’activation du virus de l’herpes (VP16). En présence de tétracycline, il n’y a pas transcription
alors que l’induction se fait en retirant la tétracycline.
Mais il n’est pas toujours simple d'éliminer la tétracycline du milieu pour avoir une
induction. TetR a été muté par mutagenèse au hasard et le mutant appelé rTetR, pour reverse
TetR se lie à l’ADN en présence de tétracycline uniquement. Comme la tétracycline a des effets
indésirables, des effecteurs plus efficaces et donc moins toxiques ont été recherchés, la
doxycycline est le plus utilisé actuellement.
Le polylinker
Noté soit avec tous les sites d'enzymes de restriction, soit MCS (multiple cloning site), il
est généralement optimisé pour éviter les structures secondaires de l'ARN pouvant gêner la
traduction. Il y a généralement plusieurs sites de coupures avec différentes localisations ce qui
permet d'orienter l'insert, en utilisant des enzymes qui ne coupent pas à l'intérieur de la
séquence codante. Comme dans le cas des procaryotes, un site de restriction est souvent
positionné sur l'insert par mutagenèse dirigée.
La terminaison
gène d’intérêt Pcmv TREPsv40 tetR VP16
TetR
VP16
TetR
VP16
Régulation de l’expression d’un gène dans une cellule eucaryote par système de régulation procaryote.
+ Tetracycline
-Tetracycline

24/10/2011 98
Lors de la transcription, l’ARN polymérase II passe à travers le site de polyadenylation.
L'extrémité 3' est donc formée lors de la maturation de l'ARN par clivage et ajout de la queue
poly-A. On ne connaît pas bien les arrêts de la transcription chez les eucaryotes. La maturation
est due à deux sites : une séquence riche en GU ou en U située en aval du site de coupure et une
séquence très conservée AAUAA située 11 à 30 nucléotides en amont du site de coupure.
Pratiquement ces séquences doivent être présentes pour assurer une bonne maturation de
l'ARN. Ces éléments sont présents lorsqu'on veut exprimer un ADNc complet mais souvent ils
ne sont pas suffisant pour assurer une bonne maturation. Aussi, on en rajoute une dans la
plupart des vecteurs. Le plus souvent on utilise les sites de polyadenylation des transcrits de
SV40.
Un intron
Chez les eucaryotes l'ARN prémessager (hnRNA) est souvent épissé. Après la
polyadénylation, les introns sont retirés pour générer l'ARN mature. Les séquences consensus
de l'épissage ont été déterminées en comparant un grand nombre de séquences d’ADNc et
d’ADN génomique. On a AG/GU(A)AGU....(UC)nN11CAG/G, le GU au site donneur et le AG
au site accepteur étant invariable. Or, des séquences proches de la séquence consensus peuvent
se trouver dans la séquence codante de l’ADNc et ces sites peuvent être utilisés quand il n’y a
pas d’intron à épisser. Pour éviter ce problème, un intron est généralement ajouté en aval du
site de clonage. De plus, il semble que l'épissage augmente l'efficacité du transport de l'ARN
vers le cytoplasme où il est traduit.
On a remarqué que l’expression chez les mammifères était augmentée lorsqu’un intron est
inclus dans la séquence (Brenner et al., 1994). Comme la présence d’un intron n’est par ailleurs
généralement pas préjudiciable, on préfère l'ajouter.
Une séquence responsable de la traduction
L'ATG d'initiation doit être dans un environnement favorable et respecter la séquence
consensus de Kozac pour une bonne traduction. Cette séquence est (G/A)NNATGG. Bien que

24/10/2011 99
d'autres séquences soient possibles, il est préférable d'avoir une purine en position -3 et une
guanine en position +4.
Des séquences d'adressage
Lorsqu'on souhaite que la protéine soit adressée dans un région particulière, on ajoute en
fusion une séquence codante permettant son adressage. Le plus courant est la sécrétion en
ajoutant un peptide signal, la protéine sera dirigée dans le réticulum endoplasmique puis
sécrétée. Mais on peut aussi utiliser un signal de sécrétion dans les mitochondries ou un signal
de sécrétion dans le noyau.
Dans ce dernier cas, on peut utiliser une fusion avec VP22. Cette protéine de 38 kDa du
virus de l’herpès a la particularité d’être transloquée dans le noyau d’une cellule à l’autre.
Après la traduction dans le cytoplasme, VP22 est importée dans le noyau des cellules
adjacentes, non transfectées et cette faculté de translocation est en même temps conférée aux
protéines de fusion (O’Hare et Elliot, 1997). Un des avantage de cette fusion est de pouvoir
utiliser cette fusion en expression transitoire, dans ce cas, l’efficacité de la transfection n’a plus
grande importance puisque toute les cellules auront la protéine.
Des sites interne d'initiation de la traduction.
Bien que les ARN eucaryotes soient monocistroniques, il existe une exception : le virus de
la polio (picornaviridae) produit un ARN polycistronique pour lequel il existe des sites
d'initiation internes (Peletier et Sonenberg, 1988). Ces sites de liaison du ribosome (IRES,
Internal ribosomal entry site) peuvent être utilisés en amont du gène d’intérêt, ils ont alors la
même fonction que le RBS. Ces IRES seront donc utilisés lorsqu'on veut avoir deux protéines
différentes provenant du même ARN. Une protéine peut ainsi servir de marqueur de
transcription pour la protéine d'intérêt.
c) L'établissement de lignées stable

24/10/2011 100
Même dans les meilleurs cas, 5% à 50% des cellules seulement sont transfectées, on
analyse donc des cellules transfectées et des cellules non transfectées. L'origine de réplication
de E. coli ne fonctionne pas dans les cellules eucaryotes , on a donc trois solutions : soit en
ajouter une, soit intégrer le gène dans le génome et, dans ce cas, on a établissement d'une lignée
stable soit ne pas en rajouter, dans ce dernier cas, on a une expression transitoire, uniquement
dans les cellules transfectées.
Le problème de l'établissement de lignées stables est que l'on ne connaît pas d'origine de
réplication chez les eucaryotes utilisables pour faire répliquer un plasmide.
Utilisation d'un virus
On s'est donc tout d'abord adressé au virus. La méthode utilisant le virus SV40 a peu à peu
évolué, uniquement l'origine de réplication de SV40 a été introduite dans le plasmide et le virus
auxiliaire (helper) a été introduit dans le génome de certaines cellules. Ainsi les cellules COS
ont un virus intégré dans le génome, cette séquence produit l'antigène viral T qui déclenche la
réplication du plasmide. L'antigène T se fixe sur l'origine de SV40, écarte les deux brins d'ADN
et joue le rôle d'une hélicase. De plus l'antigène T se lie à des protooncogènes tels que P53 ce
qui favorise la multiplication des cellules. Après transfection, le plasmide est répliqué en un
grand nombre de copies (entre 10.000 et 100.000 par cellules) 48 heures après l'infection. Il est
possible de transfecter plusieurs plasmides simultanément, et donc d'obtenir plusieurs protéines
ou des complexes multiprotéiques. Ce système a servi au clonage d'un grand nombre de
protéines par expression comme par exemple le clonage de récepteurs.
Il existe certaines limitations à l'utilisation de ce système : ce système ne marche qu'avec
des cellules de singe et il y a souvent des remaniements dans le génome. D'autre part la
production est souvent trop importante si bien que la cellule occupée à faire la protéine au lieu
de ses propres protéines meurt au bout d'une semaine et on se retrouve dans une situation
proche d'une expression transitoire.

24/10/2011 101
Intégration aléatoire
Lorsqu'on introduit un fragment d'ADN dans une cellule, il y a toujours une chance que cet
ADN s'intègre dans le génome. L'intégration se fait de manière aléatoire et on observe assez
souvent une intégration de plusieurs copies en tandem au même locus.
La sélection peut dans certains cas s'effectuer sur un grand nombre de copies. C'est le cas
de la sélection avec la DHFR. Dans les cellules cancéreuses résistante au méthotrexate, le gène
est amplifié. L'amplification n'est pas limitée au gène seul mais les séquences situées à
proximité sont aussi amplifiées. Ce phénomène a été exploité en clonant le gène d'intérêt près
du gène de la DHFR. Après la transfection, les cellules sont soumises à une sélection au
méthotrexate pour sélectionner les cellules ayant incorporé l'ADN puis, progressivement, au
cours de générations, la dose de méthotrexate est augmentée jusqu'à arriver à 80 µM,
concentration pour laquelle l'entrée du méthotrexate dans la cellule devient limitante. A cette
concentration, on a entre 500 et 2000 copies du gène dans le génome. L'inconvénient de cette
méthode est qu'elle peut prendre une durée d’un an.
Un autre méthode de sélection utilise l'amplification de la glutamine synthétase. Ce gène
confère une faible résistance à la methionine sulfoximine (MSX).
Intégration par recombinaison
On peut utiliser la recombinase
Flp si on a les sites FRP dans le
plasmide et dans le génome de la
cellule. On effectue donc une
première transfection stable du site
FRP, sélectionnée avec un gène de
résistance. Puis on transfecte les
cellules avec deux plasmides, un
plasmide portant le gène d'intérêt,
un site FRP et un gène de sélection
et un plasmide codant pour la
gène de
sélection 2FRT
gène d’intérêt
génome
gène desélection 1
gène de sélection 2
gène d’intérêtFRT FRT
FRT
gène desélection 1

24/10/2011 102
recombinase. La recombinaison est alors sélectionnée avec la résistance provenant du plasmide
portant le gène d'intérêt.
L'avantage de ce système est l'absence d'effet de position puisque le plasmide s'intègre
toujours au même endroit.
Choix du système cellulaire.
- Il ne faut pas qu'il y ait de protéine endogène correspondant à celle que l’on recherche ou
d'antigène cross-réagissant avec les anticorps qui seront utilisés pour voir la protéine.
- La température de culture peut avoir une importance.
- Le choix peut avoir des conséquences sur les modifications post-traductionnelles. Les
différents organismes ont une spécificité, par exemple les sucres ajoutés par les levures sont
différents des sucres ajoutés par les cellules de mammifères. A l'intérieur des mammifères
toutes les cellules ne font pas exactement les mêmes modifications post-traductionnelles, par
exemple les cellules CHO (provenant d'ovaire de hamster) ajoutent plus d'acide sialique que les
cellules CV-1 (de singe) ou NIH-3T3 (de souris).
Mais certaines modifications sont tissus spécifique. Uniquement certaines cellules sont
capables d'effectuer certaines modifications post-traductionnelles. Par exemple pour le facteur
IX de coagulation, il y a une γ-carboxylation de certains acides glutamiques qui ne peut être
effectuée que par les cellules du foie. Le problème est d'autant moins facilement résolu qu'une
des caractéristiques des cellules eucaryotes différenciées est de ne pouvoir être cultivées.
Les systèmes hybrides
On ne sait souvent pas quel système choisir au départ et il est souvent nécessaire de faire des
essais en procaryote et en eucaryote. Pour alléger les travaux de construction, on peut cloner le
gène d'intérêt directement dans un vecteur en aval de deux promoteurs, un eucaryote et un
procaryote. L'ensemble des séquences nécessaires aux deux systèmes sera alors présent dans le
vecteur (site de liaison au ribosome procaryote, site de polyadénylation, gène de sélection...).

24/10/2011 103
Expression dans des eucaryotes unicellulaires
Exemple de l’utilisation de Leishmania tarentolae, un Kinetoplastidae non pathogène. Ce système a
été développé par une compagnie, JenaBioScience.
Le gène est introduit par transfection et maintenu soit sous forme épisomale soit par intégration
dans le génome. Les protéines sont produites avec les maturations post-traductionnelles des
eucaryotes.
Avantage du système : la culture est facile dans un milieu peu cher.

24/10/2011 104
Les causes d'échecs de la production de protéines.
Dans de nombreux cas, on n’obtient pas ou très peu de protéine désirée. Les problèmes peuvent
venir de le séquence du gène ou de la séquence de la protéine.
1) Il n’y a pas ou peu de protéine
Le problème peut provenir de la séquence du gène
Dans ce cas il n’y a pas ou peu de traduction du fait de l'hétérogénéité des systèmes : différence
procaryotes/eucaryotes ou eucaryotes/ virus
Les codons
La vitesse de traduction est fonction des codons utilisés. Le code génétique est dégénéré, un
même acide aminé peut être codé par plusieurs codons, mais les concentrations des différents
ARNt ne sont pas identiques selon les espèces. Ils peuvent donc devenir un facteur limitant
dans la vitesse de traduction simplement en la ralentissant. Par exemple le glutamate est codé
par GAG ou GAA mais GAG est utilisé dans 60 % des cas chez les mammifères (et donc GAA
dans 40% des cas) alors que c'est l'inverse chez les bactéries gram- GAG est utilisé dans
uniquement 35% des cas. De même, l'arginine est codé par AGPu ou CGN. Les codons AGPu
sont les plus utilisé chez les mammifères (21% chacun) mais rarement utilisé chez E. coli (4%
chacun).
Comme chez E. coli, la transcription et la traduction sont couplés, un ralentissement de la
traduction affecte la stabilité de l'ARN messager. Si le ribosome est ralenti sur plusieurs codons
rares, l'extrémité 3' du messager est exposée aux RNAses. De plus on observe une terminaison
prématurée de la transcription et donc pas de production de la protéine.
Dans ce cas là, il existe deux solutions : une mutagenèse de certains codons peut être envisagée
pour améliorer la traduction. Si un codon est peu utilisé, c'est généralement parce que l'ARN de

24/10/2011 105
transfert corespondant est rare. Une autre solution consiste alors à coexprimer les ARNt rares
(Sauge-Merle et al., 1999).
Comme il y a généralement de nombreux codons à modifier, un gène synthétique
correspondant à une optimisation des codons peut être construit (Feng et al., 2001). Les codons
choisis correspondent alors à ceux qui sont les plus utilisés pour les protéines qui sont très
exprimées dans l’organisme. Des tables ont été établies pour donner la fréquence relative
d’utilisation des codons,
∑=
= ni
ji
Xijn
XijRCUij
1
1
RCU : relative codon usage ; Xij : nombre de rencontre du codon j pour l’acide aminé i, ni :
nombre de codon pour l’acide aminé i. Un logiciel permet de faire la traduction inverse d’une
séquence de protéine (http://www.entelechon.com/eng/backtranslation.html) pour être
optimisée selon l’organisme.
L’initiation
Chez E. coli, la séquence de liaison du ribosome doit être optimisée lors de l'expression. En
dehors de la séquence de Shine Dalgarno, plusieurs éléments doivent être pris en compte : la
séquence du premier acide aminé après la methionine est importante. On a par exemple 10 fois
plus de protéine produite si on remplace un tryptophane (UGG) par une alanine (GCU)
(Looman et al., 1987). Les codons AGG, CGG, UGG et GGG sont associés à une faible
expression s’il sont localisés entre les positions +2 à +5. Il semble que la présence de ces
codons au début de la traduction entraîne un détachement du peptidyl tRNA (Valdivia et
Isaksson, 2005).
La présence de structures secondaires impliquant des régions amont et aval au site de liaison du
ribosome est défavorable à la traductibilité, (Schauder et McCarthy, 1989), plus ces structures
sont stables, moins on obtient de protéine. Ainsi, si on augmente la proportion de AT dans les
premiers nucléotides en aval de l’ATG, on augmente la production.

24/10/2011 106
Il peut y avoir des initiations internes (séquences de Shine Dalgarno pour les bactéries). Chez
les procaryotes, les ARN sont polycistroniques, si bien qu'il faudra faire attention lors de
l'expression d'un gène eucaryote, qu'il n'y ait pas à l'intérieur de la séquence codante une
séquence susceptible d'être reconnue par un ribosome procaryote.
Il peut y avoir un promoteur procaryote à l’intérieur de la séquence. Ce promoteur peut géner la
production de la protéine soit en initiant la synthèse d’un ARN antisens soit en étant un site de
liaison de la polymérase qui gène le déroulement de la traduction.
Il peut y avoir une initiation prématurée. Chez les eucaryotes, de nombreux gènes comportent
de courtes phases de lecture ouverte en amont de l’ATG initiateur de la protéine d’intérêt. Ces
phases de lecture ont souvent un rôle de régulation de la traduction. Pour obtenir une bonne
surexpression dans un système eucaryote, on enlèvera ces phases de lecture ouverte avant le
codon initiateur.
La terminaison de la transcription
La polymérase bactérienne s'arrête quand elle synthétise une série de résidus U dans la mesure
où elle a d'abord synthétisé une séquence autocomplémentaire capable de se replier.
L'élimination de telles structures par mutagenèse permet une meilleure traduction.
On a le même problème avec l'expression dans le virus de la vaccine, les signaux d'arrêt de
transcription du virus (TTTTTNT) éventuellement présents dans le gène seront enlevés de la
séquence ajoutée par mutagenèse dirigée.
Si on veut exprimer un gène mitochondrial dans une cellule eucaryote, il faudra faire attention
qu’il n’y ait pas de signaux de polyadenylation dans la séquence. En effet les transcrits
5’ TTGACA 17 +/- 1 bp TATAAT
Séquence consensus d’un promoteur chez E. coli(Hawley et MacClure, 1983)

24/10/2011 107
mitochondriaux ne sont pas polyadénylés, si bien que la séquence consensus peut être présente
en amont ou dans la phase de lecture du gène à exprimer.
Les structures secondaires de l'ARN empêchent la traduction
On a montré qu'il existait une corrélation inverse entre le pourcentage de structure secondaire
sur l'ARN messager et l'efficacité de traduction.
Ces structures secondaires sont relativement importantes en 5' où elles ont un rôle dans le
contrôle de la traduction. Pour surexprimer une protéine, il faudra donc éliminer les séquences
inhibitrices de la traduction existant dans la partie non codante en 5' des ARNm, phénomène
connu sous le terme d'atténuation (comme par exemple dans le cas de l'opéron tryptophane
chez E. coli).
Pour l'expression dans le virus de la vaccine, les séquences non traduites en 3' et en 5' seront
courtes pour mimer les ARNm des poxvirus.
Stabilité de l'ARN
Chez les procaryotes, les ARN peuvent être stabilisés en ajoutant en 3’ une structure
secondaire pour inhiber la dégradation par des exonucléases 3’-5’. Bien qu’il n’existe pas de
RNAse 5’-3’ la présence d’une structure secondaire en 5’ stabilise le transcrit en inhibant
l’action d’endonucléases telle que la RNAse E.
Chez les eucaryotes, il y a de grandes différences de stabilité des ARNm. La stabilité peut
dépendre de l’ARN synthétisé. Généralement, les protéines régulatrices dont le taux varie
rapidement dans la cellule sont codées par des ARNs instables. Ainsi, l'ARN de la β-globine a
une durée de demi-vie de plus de 10 heures, alors que l'ARN d'un facteur de croissance a une
durée de demi-vie d'environ 30 min. Cette stabilité dépend souvent des séquences non traduites
en 3'. Pour analyser ce phénomène on transfère les séquences du gène analysé sur un gène
rapporteur. Si on transfère l'extrémité du gène codant la β-globine l'ARN de fusion devient plus

24/10/2011 108
stable. Par contre si on ajoute une séquence riche en A et U présente dans les ARN instables
comme ceux codant pour un facteur de croissance, l'ARN de fusion devient instable.
Lorsqu’on efffectue une surproduction d’une protéine peu synthétisée, on enlève souvent
les séquences en 3' de l’ADNc et, au besoin, on rajoute en 3’ des séquences favorisant la
stabilité de l’ARN. Pour ce faire, on ajoute les séquences 3’ non codantes d’un gène dont
l’ARN est stable. Par exemple, la séquence du gène codant pour l’hormone de croissance
bovine est souvent rajoutée pour les expressions en cellules de mammifère.
Problème inconnu, mutagenèse au hasard
Dans certains cas plutôt que de chercher à mutagenéiser les codons et les structures
secondaires par mutagenèse dirigée, on mutageneise au hasard le plasmide, on transforme des
bactéries et on trie celles qui produisent le plus. Cette expérience a par exemple été réalisée
avec l'α-antitrypsin. Ils ont obtenu une production augmentée de 100 fois. L'analyse de la
séquence exprimant le plus n'a pas permis de comprendre les mutations responsables de cette
surproduction (la surproduction n'a pas été expliquée par une élimination des codons rares ou
des structures secondaires).
Dans de nombreux cas on ne sait pas pourquoi la protéine n’est pas produite. La solution
est alors de changer de système. Si on n’a pas d’expression lors d’une traduction in vitro dans
le système du lysat de réticulocyte, on peut obtenir une bonne expression dans le système du
germe de blé et inversement. Ce phénomène est assez général, la production d’une protéine
dans un système ne permet pas de le valider, la comparaison de deux systèmes de production
ou de deux cellules avec un même système de production ne permet pas de dire qu’un système
est meilleur qu’un autre.
2) La protéine est produite mais est toxique
De nombreuses protéines sont toxiques pour la cellule. La culture ne se développe pas et on
obtient pas ou peu de protéine. Dans certains cas, la toxicité est prévisible. C'est par exemple le

24/10/2011 109
cas des protéines impliquées dans le cycle cellulaire ou la surexpression d’une ADN
polymérase. Par contre, dans certains cas, on ne s'y attend pas. Par exemple l'expression de la
transposase d’un élément transposable de la mouche des fruits est toxique pour E. coli.
Cette toxicité a plusieurs conséquences, elle s'accompagne
- de mutations qui diminuent la transcription et la traduction de la protéine.
- de perte du vecteur (plasmide chez E. coli)
- d'accumulation de mutations dans la protéine la rendant moins toxique.
Il existe plusieurs solutions :
- Utiliser des promoteurs fortement inductibles comme par exemple le promoteur arabinose
avec AraC et un site de liaison de la CAP. Dans ce cas on a une induction de 800 fois dans
un milieu sans glucose et contenant de l'arabinose.
- Augmenter l’inductibilité du promoteur. On peut surexprimer un répresseur tel que LacI et
ajouter des opérateurs tels que LacO en amont du gène d’intérêt. Dans le cas du promoteur
LacZ, le réprimer par le glucose. C’est la répression catabolique qui, bien quelle soit
diminuée en utilisant un mutant (LacUV5), n’est pas totalement abolie. La technique
consiste donc à faire pousser la bactérie dans un milieu riche en glucose puis lors de
l’induction à changer le milieu pour un milieu pauvre (Pan et Malcolm, 2000).
- Produire un inhibiteur de la polymérase. Cette stratégie est utilisée lorsque le gène d’intérêt
est placé en aval d’un promoteur reconnu par une polymérase exogène, produite par la
cellule. C’est le cas de l’utilisation de l’ARN polymérase T7 dans les expressions chez E.
coli. Dans ce cas on coproduit le lysozyme du phage T7 qui est un inhibiteur de l’ARN
polymérase T7 et qui inhibe l’enzyme exprimée à bas niveau avant l’induction (Studier,
1991). Lors de l’induction, l’ARN polymérase T7 est produite en grande quantité, le
lysozyme est en trop faible quantité pour l’inhiber.
- Ajouter l’ARN polymérase au moment de la production par exemple en infectant les
bactéries avec un M13 portant le gène de l’ARN polymérase T7.
- Utiliser une stratégie antisens, en absence d'induction, la fuite du promoteur est contrôlée
par un ARN antisens produit par un promoteur constitutif en aval du gène.
- Utiliser un système de sécrétion de la protéine dans le milieu cellulaire.

24/10/2011 110
Dans le cas ou la protéine est très toxique, on peut toujours se replier sur les systèmes de
traduction in vitro.
3) La protéine est surexprimée mais est mal repliée
Lorsque les protéines sont exprimées en grande quantité dans E. coli, (jusqu'à 50% des
protéines totales), elles sont agglomérées dans des "corps d'inclusion" visibles en microscopie à
contraste de phase aux extrémités de la bactérie. Les protéines ainsi incluses sont généralement
inactives et ne peuvent pas être purifiées par chromatographie. Ces corps d’inclusion sont
fréquents lors des productions dans E. coli.
Plusieurs techniques ont été mises en place pour éviter ou diminuer cette agglomération
- Utilisation de souches particulières. Il n'existe pas de souche éliminant complètement les
corps d'inclusion, mais parfois en changeant de souche on obtient souvent moins de
protéine.
- Diminution de la vitesse de production en faisant varier les conditions de température. On
peut faire varier les températures de 10 à 43°C sans altérer E. coli. Par exemple la
production de subtilisine active est augmentée de 14 fois lorsque la culture est faite à 23°C
au lieu de 37°C. Pour l'interféron, 5% de la protéine est active à 37°C et 73% à 30°C ce qui
a donné une augmentation de 7 fois de protéine active en passant à 30°C. De même, l’ajout
de chloramphénicol au milieu permet d’obtenir plus de protéines solubles. La quantité
d'inducteur a aussi de l'importance pour éviter la formation des corps d'inclusion, il faut
diminuer au maximum la quantité d'inducteur pour obtenir une protéine active. Toutefois,
toutes ces techniques visent à diminuer la production, aussi on observe souvent une baisse
du rendement.
- Utilisation de stabilisants. L’addition de sucres non métabolisables par la bactérie
(saccharose, raffinose ou sorbitol) permet souvent de diminuer le pourcentage de protéine
insoluble. Ce résultat est attribué à l’effet stabilisateur des sucres sur la conformation des
protéines. D’autres méthodes de stabilisation particulières peuvent être efficaces, c'est par

24/10/2011 111
exemple le cas des protéines qui contiennent un métal comme cofacteur soit structural soit
catalytique. Si la protéine est fabriquée plus vite que le métal n'est transporté dans la
cellule, l'apoprotéine sans le métal s'accumulera et ne se repliera pas correctement. Un autre
facteur qui influence la présence d'agrégat est le pH.
- Faire sécréter la protéine dans le milieu extérieur ou dans le périplasme chez E. coli. Le
cytoplasme des bactéries est un milieu réducteur dans lequel les ponts disulfures ne peuvent
pas se former. Par contre le périplasme est un milieu oxydant. Plusieurs protéines de E. coli
sont sécrétées dans le périplasme, par exemple, les β-lactamases conférant la résistance à
certains antibiotiques tels que l'ampicilline ou la pénicilline sont sécrétées dans le
périplasme. L'adressage de ces protéines est dû à un peptide signal qui est coupé lors de la
translocation. Le peptide signal actuellement le plus utilisé est celui du OmpA, la coupure
s'effectue juste au niveau de la jonction si bien que la protéine exprimée se retrouve dans le
périplasme avec son extrémité N-terminale. Problème: la sécrétion est souvent relativement
peu efficace. Il est aussi possible de faire sécréter la protéine dans le milieu extracellulaire
en coexprimant une protéine, la BRP (Bacteriocin Release Protein), qui perméabilise la
membrane externe de la bactérie en activant une phospholipase.
A l’inverse, l’adressage au périplasme peut empêcher le bon repliement de la protéine, c'est
pas exemple le cas de la GFP qui n’est pas fonctionnelle si on essaye de l’exprimer dans le
périplasme.
- Utilisation de foldases. Il y a de nombreuses protéines telles que la thiorédoxine, la protéine
disulfide isomérase ou la prolyl cis-trans isomérase ou les chaperonines, qui aident au
repliement des protéines dans les cellules eucaryotes. Toutes ces protéines n’ont pas le
même rôle, par exemple il semble que la protéine disulfide isomérase participe au
repliement alors que les chaperones maintiennent la protéine bien repliée dans son état. Par
exemple la coexpression de la chaperonine GroEL en même temps que la RuBisCo
(ribulose 1,5 bisphosphate carboxylase/oxygenase) a augmenté le taux de RuBisCo soluble
par 10. Comme autre exemple, on peut citer l’exemple de la thioredoxine qui est une petite
protéine de 11,7 kDa restant soluble même quand elle est exprimée à des taux de 40% dans
la cellule. Une fusion du gène d'intérêt et du gène codant pour la thioredoxine permet de
diminuer la formation de corps d'inclusion.

24/10/2011 112
- Utilisation d’une fusion avec une protéine soluble en amont. Les partenaires les plus
souvent utilisés pour l’expression chez E. coli sont la glutathion-S transférase, la maltose-
binding protein et la thioredoxin. Mais d’autres partenaires ont été décrits tels que Nus An
grpE, la bacterioferritine, la kinase du phage T7 ou une chaperone de E. coli, Skp
(Chatterjee et Esposito, 2005).
Le fait que la protéine s'agrège en corps d'inclusion a toutefois quelques avantages, elle est
protégée de la protéolyse et elle est facile à purifier. Il suffit en effet de purifier les corps
d'inclusion simplement par des séries de centrifugations et de lavage du culot. Il faut ensuite
dénaturer la protéine puis la renaturer en faisant varier les proportions d'agent dénaturant.
Techniquement, on ajoute des agents dénaturants, typiquement de l'urée ou des sel de
guanidinium, des détergents et un agent réducteur pour couper les ponts disulfures (β-
mercaptoethanol ou DTT). Ces agents sont ensuite éliminés soit par dialyse dans une solution
diluée pour éviter les interactions entre les protéines soit en bloquant la protéine sur un gel
d'affinité.
4) La protéine est produite mais est protéolysée
La plupart des protéines hétérologues que l'on fait synthétiser dans la bactérie sont protéolysées
très rapidement (en deux minutes par exemple pour l'insuline humaine). Plusieurs solutions ont
été utilisées pour résoudre ce problème :
- Faire une protéine de fusion constituée de la protéine désirée et en amont une autre
protéine. Cette stratégie n'est valable que si la protéolyse est due à la faible stabilité des
petites protéines ou peptides.
- Utiliser des bactéries mutantes, déficientes pour certaines protéases. Cette stratégies a été
adoptée pour les protéines secrétées dans le périplasme (pour permettre les ponts
disulfures) qui sont souvent peu stables. Une série de souches portant des mutations de
déficience en protéases et une mutation sur le gène rpoH codant pour le facteur sigma de
l’ARN polymérase qui contrôle la transcription de certaines protéases ont été fabriquées

24/10/2011 113
(Meerman et Georghiou, 1994). Ces souches permettent une meilleure expression des
protéines, mais on ne peut pas éliminer toutes les protéases, soit parce qu’elles sont
toujours inconnues, soit parce qu’elles sont indispensables au fonctionnement de la cellule.
- Utiliser des inhibiteurs de protéases, mais comme précédemment, ces inhibiteurs sont
souvent toxiques pour la cellule.
Cette protéolyse existe aussi dans les systèmes eucaryotes. Par exemple, en traduction in vitro
dans le lysat de réticulocyte, on a remarqué que la traduction des protéines de petit poids
moléculaire (< 30 kDa) est souvent difficile. Ceci est du en partie au fait qu’elles sont instables
dans les réticulocytes. Pour palier à ce problème, le plus simple est de changer de système de
traduction et de choisir par exemple le lysat de germe de blé qui traduit aussi bien les petites
que les grosses molécules.
Chez les procaryotes comme chez les eucaryotes, des protéines à longue durée de vie coexistent
avec des protéines à courte durée de vie. Chez les eucaryotes, la dégradation des protéines à
courte durée de vie est sous la dépendance de l’ubiquitination. Dans ce système, les protéines
qui seront dégradées sont tout d’abord liées covalement à l’ubiquitine, une petite protéine de 76
acide aminés. C’est une liaison peptidique entre la fonction carboxilique terminale de
l’ubiquitine et, soit une fonction amine d’une lysine interne, soit l’amine présente à l’extrémité
N-terminale de la protéine. Dans ce dernier cas l’acide aminé présent en N-terminal est
important. Ainsi, les acides aminés M, S, A, G, T et V sont stabilisant (Bachmair et al., 1986).
5) Les maturations post-traductionelles ne sont pas correctes
Ce problème est évident lors de l'expression dans des systèmes hétérologues. La cellule hôte
n'est pas capable de faire une modification post-traductionelle qui normalement se produit dans
le système homologue.
Lorsque la modification post-traductionelle existe dans la cellule pour les protéines endogène,
il arrive que la séquence signal ne soit pas reconnue. C’est le cas par exemple de

24/10/2011 114
l'acétylcholinestérase d'insecte : un peptide hydrophobe en C-terminal est normalement
échangé contre un glycolipide. Cette maturation est co-traductionnelle et a lieu dans le
réticulum endoplasmique. Si on exprime cette enzyme dans l'ovocyte de Xénope, on obtient
une protéine avec le peptide hydrophobe et non le glycolipide à l'extrémité C-terminale.
L'ovocyte est capable de faire cette maturation avec d'autres protéines mais n'est pas capable de
la faire avec le peptide de l'acétylcholinestérase de drosophile. Un moyen d'obtention de la
protéine proche de la protéine native, naturelle, trouvée chez l'insecte, est de modifier la
séquence primaire du peptide hydrophobe pour qu'il soit reconnu.
A l'inverse, la cellule hôte peut faire une modification post-traductionnelle qui n'existe pas dans
la cellule. Par exemple, la MCP-1 humaine (Monocyte Chemoattractant Protein-1) est non N-
glycosylée. Si on l'exprime dans Pichia pastoris ou dans Saccharomyces cerevisiae, on observe
une N-glycosylation. Cette N-glycosylation augmente la masse apparente de la protéine
d'environ 12 kDa et peut être identifiée à l'aide de la reconnaissance de la protéine par une
lectine, la concanavaline-A. Dans ce cas la glycosylation peut être retirée en digérant la
protéine par l'endoglycosidase F qui coupe la liaison asparagine/N-acetylglucosamine.
Toutefois cette solution ne peut être envisagée que pour les protéines qui ne sont normalement
pas glycosylées en effet, si la protéine est normalement glycosylée et si la cellule hôte rajoute
une glycosylation, celle-ci ne pourra pas être spécifiquement retirée.
Le problème peut aussi provenir de la surproduction. Il y a saturation d'un système de
maturation qui ne peut pas fonctionner sur toutes les protéines synthétisées. On obtient alors
une hétérogénéité dans les protéines produites, certaines présentent la maturation et d'autres
non.
- La MCP-1 humaine (Monocyte Chemoattractant Protein-1) présente une modification à
l'extrémité N-terminal, qui est bloquée du fait de la présence d'un pyroglutamate. La
surproduction dans Pichia pastoris donne les deux protéines, une avec un pyroglutatmate et
une autre avec une glutamine.
- L'acétylcholinestérase humaine est glycosylée, mais sa surproduction en cellule de
mammifère entraîne une acetylcholinestérase mal glycosylée, qui manque d'acide syalique.
Ce problème a été résolu en coexprimant l'enzyme limitant, une syalictransférase.

24/10/2011 115
- Le codon d'initiation (ATG) code chez les bactéries pour une N-formyl méthionine. Le
groupe formyl est toujours retiré mais la méthionine reste la plupart du temps. Dans le cas
d'une expression faible, elle est enlevée lorsque le premier acide aminé est par exemple une
glycine, une sérine, mais il n'est jamais enlevé avec d'autres acides aminés tels que
l'arginine. De plus lorsque la surproduction est trop importante il y a saturation de l'enzyme
de maturation (méthionine aminopeptidase) et il n'y a pas de coupure. Ce n'est souvent pas
important mais si ça l'est, on a deux possibilités: soit traiter la protéine avec la méthionine
aminopeptidase, soit coexprimer cette enzyme dans la bactérie.
Par ailleurs, le fait de rester dans un système proche du système homologue ne garanti pas
d’obtenir toutes les modifications post-traductionnelles. Par exemple, certaines maturations de
la protéine observées chez Pseudomonas ne se retrouvent pas ou peu chez E. coli alors qu’il
s’agit de deux bactéries.
Parmi les maturations de la protéine, il y a l'oligomérisation. Lorsqu'on ajoute un tag pour
faciliter la détection ou la purification (voir plus loin), ce tag peut favoriser des
oligomerisations non attendues. C'est le cas par exemple avec les queues d'histidines rajoutées
en N ou C terminal de la protéine. Ces histidines favorisent la formation d'oligomères (Hom et
Volkman, 1998).
6) La protéine est produite mais on ne sait pas la détecter
Dans certains cas on n’a pas d’anticorps, on ne connaît pas l’activité de la protéine. On a donc
des difficultés à contrôler la production ou à sélectionner les cellules qui produisent le plus.
Une première solution est de produire simultanément une protéine marqueur.
On peut lors d’une production en bactérie placer le gène marqueur en aval du gène d’intérêt
avec son propre RBS. Les deux protéines seront traduites à partir du même ARN. La même
stratégie peut se faire dans les systèmes eucaryotes, bien que les ARN eucaryotes soient

24/10/2011 116
monocistroniques, on peut ajouter un IRES qui fait office de RBS chez les eucaryotes.
Une autre technique est de cloner le gène
derrière un promoteur bidirectionnel. D’un
coté le promoteur régule la production d’une
protéine marqueur et de l’autre le promoteur
régule la production de la protéine d’intérêt.
Un tel promoteur bidirectionnel peut être
fabriqué autour de l’élément de réponse à la tétracycline.
Une autre technique est d’utiliser un vecteur avec plusieurs promoteurs, le même promoteur
peut être utilisé deux fois dans le même vecteur mais dans ce cas on positionne les deux copies
tête-bêche pour éviter les recombinaisons.
Une deuxième solution est d’utiliser une protéine de fusion, un « tag » (voir chapitre sur les
tag)
7) La protéine est produite mais on ne sait pas la purifier
On peut utiliser une fusion avec une protéine dont on possède un anticorps. Après la
production, la protéine fusionnée sera purifiée par immunoaffinité. Les anticorps sont
accrochés sur une phase fixe et la protéine de fusion se trouve dans la phase mobile. Il y a
accrochage de la protéine de fusion par réaction antigène-anticorps puis la protéine de fusion
est décrochée soit en faisant passer un tampon acide (pH 2.6) soit un tampon comprenant un
agent dénaturant si la protéine n'a pas besoin d'être active. Parmi les anticorps disponibles, on
peut citer ceux dirigés contre la β-galactosidase.
Plusieurs protéines sont facilement purifiables et peuvent donc être utilisées en fusion pour
faciliter la purification. Cette séquence (tag) va permettre à la protéine de fusion de s'attacher
sur un ligand. De plus, comme les protéines utilisées comme tag sont des protéines qui
s'expriment bien in vitro, la fusion permet souvent d'augmenter l'expression de protéines qui
sont faiblement produites.
TRE PcmvPcmv gène d’intérêtgène marqueur
Contrôle de l’expression du gène d’intérêt par un promoteur bidirectionnel

24/10/2011 117
8) La protéine est produite mais il existe aussi des protéines proches produites par la
cellule, des endogènes
Plus le système de production est proche du système naturel, plus on a de chance que la
protéine soit bien produite. Mais parallèlement, plus on a de chance qu’il y ait des endogènes
dans la production. La première solution consiste à s’éloigner du système naturel, la deuxième
solution est d’utiliser un tag pour purifier la protéine recombinante de l’endogène.
9) Problème particulier lié à l'utilisation de la protéine chez l'homme
Dans certains cas, la protéine est destinée à être injecté à l'homme. Il est alors important que ces
cellules ne soient pas infectées par un virus. C'est arrivé dans le cas de la production de vaccin
contre la polyomélyte préparé dans des cellules de singe. On s'est aperçu plus tard qu'elles
étaient infectées par SV40.

24/10/2011 118
TAG, protéases et acides aminés modifiés
Les TAG
Ce sont des protéines ou des peptides qui permettent la production, la détection, la
stabilité et la purification aisée des protéines recombinantes lorsqu'elles sont ajoutées en
fusion à la protéine d'intérêt.
Elles permettent souvent une meilleure production, en effet l'expression d'ARN messager
eucaryote dans E. coli se heurte à de nombreux problèmes dus en particulier à la
mauvaise initiation de la traduction. Si la protéine de fusion est placée en amont de la
protéine d'intérêt, on aura normalement une bonne initiation de la traduction. La sur-
expression de protéines dans E. coli s'accompagne souvent de formation de corps
d'inclusion dans lesquels la protéine surexprimée est mal repliée. La fusion avec une autre
protéine qui se replie correctement aide souvent à avoir une protéine bien repliée. La
protéine de fusion joue alors le rôle de chaperonne intramoléculaire.
Elles permettent la détection de la protéine de fusion. En effet on dispose d'anticorps pour
les protéines de fusion mais de plus certaines d'entre elles sont des enzymes qui peuvent
être détectées par leur activité.
Elles permettent une stabilisation de la protéine. Les petits peptides sont rapidement
dégradés chez E. coli. Un des moyens pour éviter cette dégradation est donc de les
produire en fusion.
Les TAG les plus utilisés :
- Epitopes: Des petites séquences, d'une dizaine d'acides aminé, sont ajoutées
généralement à l’une des deux extrémités de la protéine, plus rarement à l’intérieur. Ces
séquences sont reconnues par des anticorps monoclonaux. Ceci permet de localiser la
protéine sur des filtres, de la quantifier par ELISA ou de la purifier après immunoaffinité

24/10/2011 119
avec un anticorps spécifique. On utilise des épitopes pour lesquels on dispose d'anticorps
très efficaces et très spécifiques qui n'ont pas de crossréactivité dans le système utilisé. Il
existe plusieurs séquences et les anticorps correspondants commercialisés
- Xpress : DLYDDDDK (invitrogen)
- c-myc : EQKLISEEDL (invitrogen). L’anticorps a une très bonne affinité pour ce
tag, il est donc principalement utilisé pour la détection de la protéine de fusion.
- FLAG : DYDDDDK (sigma). L’anticorps n’a pas une très bonne affinité, il est
principalement utilisé pour la purification de la protéine de fusion, en effet la liaison
anticorps-antigène peut facilement se dissocier (Hopp et al., 1988). Pour certains
anticoprs, la liaison tag-anticops peut être rompue par l’ajout d’EDTA ce qui est
assez pratique pour les purifications.
- V5 : GKPIPNPLLGLDST (In vitrogen)
- HA : YPYDVPDYA de l'hémagglutinine d'Influenza : (Roche Molecular
Biochemicals)
- épitope de la protéine C : 12 acides aminés (Roche Molecular Biochemicals)
L'anticorps présente l'avantage de ne se fixer qu'en présence de calcium, ce
qui permet d'éluer la protéine de fusion en présence d'EDTA.
- T7 : MASMTGGQQMG, anticorps disponible chez Abcam (http://www.abcam.com)
- Staphylococcus protein A (Nilsson et Abrahmsen, 1990).
- S-tag : KETAAAKFERQHMDS ce peptide a une forte interaction avec la protéine S qui
dérive de la RNAse A (Karpeisky et al. 1994). La liaison est très forte et généralement
l’élution est obtenue soit à pH2 ou en coupant la jonction entre les deux proteines.
- Lac Z : Si la séquence est insérée en amont de LacZ en fusion. LacZ garde son activité. Il
est donc possible de faire une protéine bifonctionelle, ayant les deux activités, en amont
la protéine d'intérêt et en C-terminal l'activité β-galactosidase. La fusion dans l'autre sens
n'est pas possible, il faut que l'extrémité C-terminale de la β-galactosidase soit libre pour
garder son activité. Cette fusion permet en outre de suivre la protéine lors de la
purification.

24/10/2011 120
- La calmoduline-binding peptide (Stofko-Hahn et al., 1992). Cette protéine très acide
présente une forte affinité pour certains peptides en présence de calcium.
- La maltose binding protéine (MBP) de E. coli peut être purifiable sur une colonne
d'amylose et éluée avec du maltose. La fusion est généralement effectuée en C-terminal
de la MBP, si bien que le peptide signal de la MBP permet de plus d'adresser la protéine
de fusion dans le périplasme (Guan et al., 1988).
- Les interaction streptavidine-biotine sont très utilisées en biologie. Un peptide de 10
acides-aminés qui interagit fortement avec le site de liaison de la biotine de la
strptavidine a été sélectionné à partir d'une banque de peptides, le "strep-tag" (Schmidt et
Skerra, 1993). Ce peptide a été amélioré pour donner le strep-tag II (WSHPNFEK). La
strpetavidine a une affinité de 3,5 µM pour ce peptide. Pour améliorer cette affinité, un
peptide plus long, le SBP (Streptavidin Binding Peptide,
MDEKTTGWRGGHVVEGLAGELEQLRARLEHHPQGQREP) a été isolé. L’affinité de la
streptavidine pour ce peptide est de 2,5 nM (Keefe et al., 2001). Une deuxième stratégie
a consisté à sélectionner un peptide de 13 acides aminés qui est facilement biotinylé dans
E. coli par BirA, l'enzyme endogène de biotinylation chez E. coli. (Schatz, 1993). Dans
les deux cas les protéines comportant le peptide mimant la biotine ou le peptide biotinylé
peuvent être purifiées par affinité sur la streptavidine.
- La glutathion S-transférase de shistosome. Cette protéine catalyse la réaction entre un
tripeptide, le glutathion (γGlu-Cys-Gly) et un substrat électrophile. Si on utilise un seul
substrat, on n'a pas de métabolisation mais uniquement fixation du glutathion sur la
glutathion S-transférase. Cette propriété a été exploitée pour fabriquer des
chromatographies d'affinité en bloquant du glutathion sur la phase fixe. La glutathion
transférase s'accroche sur le glutathion fixé sur la colonne et est décrochée en ajoutant du
glutathion au tampon. L'avantage de cette protéine est son activité enzymatique qu’il est
possible de suivre. Ainsi, on peut connaître au cours de la production la quantité de
protéine produite, sans avoir besoin de faire de gel (Smith et Jonhson, 1988).

24/10/2011 121
- Thioredoxine de E. coli (LaVallie et al., 1993). La purification des protéines de fusions
avec la thioredoxine utilisent les propriétés de la thiorédoxine qui peut être extraite des
bactéries par choc osmotique et qui est résistante à la chaleur. De plus, un mutant
présentant une série d'histidine à sa surface a été construit ce qui permet une purification
sur colonne de nickel.
- Les queues homopolymériques. Plusieurs essais ont été effectués, une queue poly-
arginine permet la purification de la protéine sur une résine d'échange de cation
(Saasenfeld et Brewer, 1984). Une polyarginine permet en outre d’accrocher la protéine
sur le verre, toutefois ce tag est moins performatnt que le Si-tag. Une queue poly-
aspartate permet la purification sur résine échangeuse d'anions, une queue de
phénylalanine permet la purification sur colonne d'hydrophobicité de type phényl
sépaharose et une queue poly-cystéine permet la purification sur thiopropyl-sépharose.
Dans ce dernier cas, on accroche la protéine par une liaison covalente et on l'élue soit à
l'aide de cystéine soit en ajoutant un agent réducteur tel que le dithiotreitol (Persson et al.,
1988). La queue homopolymérique actuellement la plus populaire est une queue histidine
ou HIS-tag, ce qui crée un site de haute affinité pour les cations divalents tel que le nickel
ou le cobalt. La protéine qui a un tel site s'accroche sur colonne d'affinité de Ni et se
décroche spécifiquement en ajoutant de l'imidazole à la phase mobile. Cette séquence de
6 histidines peut se fusionner aux extrémités N ou C terminal de la protéine ou être
incorporée dans une protéine de fusion. Par exemple dans le cas d'une fusion avec la
thioredoxine, protéine servant à solubiliser les protéines recombinantes produites dans E.
coli, la mutagenèse de deux résidus (acide glutamique et glutamine) en histidine produit
un amas d'histidine conférant à la protéine une bonne affinité pour le nickel. L’avantage
de cette fusion est que la séquence rajoutée est petite. Cette technique est actuellement la
plus populaire principalement parce que ce tag est petit et à priori va moins perturber la
fonction de la protéine (Van Dyke et al., 1992). Les histidines ne sont pas nécessairement
à la suite l’une de l’autre, il faut simplement qu’elles soient à proximité. Le tag peut donc
se modéliser sur la structure tridimentionelle de la protéine. Par ailleurs certains

24/10/2011 122
commerçants proposent des tags hybrides comme le HQ tag (HQHQHQ) qui a les mêmes
propriété qu’un tag HHH, une affinité pour le nickel.
- Le Si-Tag : En sélectionnant les protéines batériennes qui s’accrochent sur le verre puis
en les identifiant par spectrographie de masse, on a trouvé que la protéine ribosomale L2
de E. coli a une forte affinité pour les silicates. Une analyse par deletion a ensuite permis
d’identifier deux domaines non structurés de la protéine qui intérragissent avec le verre,
un domaine N terminal correspondant aux acides aminés 1-60 et un domaine C-terminal
correspondant à la séquence 203-273. En fusion avec la protéine d’intérêt, ce tag permet
de fixer la protéine sur des lames de verres utilisées par exemple lors de l’élaboration de
puces à protéines (Taniguchi et al. 2006).
- Le domaine de liaison à la cellulose, domaine de la cellulase de la bactérie clmostridium
cellulovorans peut être utilisé comme Tag (Shpigel et al., 1998). La protéine de fusion est
purifiée sur colonne de cellulose. Le principal avantage par rapport aux autres protéines
est le faible coût de la colonne d’affinité utilisée.
- Les protéines fluorescentes :
La Green fluorescent protein (GFP) a été isolée de la méduse Aequorea victoria, elle
émet de la lumière verte lorsqu'elle est excitée en UV (Prasher et al., 1992). Différents
variants plus efficace ou émettant à différentes longueur d’onde ont été générés par
mutagenèse.
- GFPuv se replie plus facilement que la GFP et a un rendement de fluorescence
16 fois plus importante que la GFP (Crameri et al., 1996).
- GFPmut1 contient deux mutations (F64L et S65T) son rendement de
fluorescence est 35 fois supérieur à celui de la GFP (Cormack et al., 1996).
- GFP+ combine les mutations de GFPuv et de GFPmut1 ce qui lui donne une
augmentation de 130 fois par rapport à la GFP (Scholz et al., 2000).
- L’EGFP est un variant optimisée pour l’expression en cellules eucaryotes.

24/10/2011 123
La HcRed a été générée par mutagenèse à partir d’une protéine non fluorescente du
corail, Heteractis crispa. Elle émet dans le rouge. Ces protéines gardent leurs propriétés
de fluorescence en fusion N ou C terminale. En fusion, elle permet donc la détection et
la localisation de la protéine dans les cellules.
La DsRed a été isolée de Discosoma sp. (Baird et al., 2000), elle peut être excitée à 488
nm comme la GFP mais émet dans le rouge à 583 nm (la GFP émêt à 500 nm. Son
expression a été améliorée par optimisation des codons pour donner la DsRed1. Elle
présentait un défaut important, sa maturation prenait trop longtemps elle avait une
demie vie supérieure à 24 heures. La vitesse de maturation a été améliorée et la
DsRedT4 ainsi obtenu a une demie vie de maturation inférieure à 45 minutes (Bevis et
Click, 2002)
Les protéines fluorescences sont principalement utilisées pour localiser les protéines de
fusion dans les cellules ou dans les tissus.
- Les protéines colorées : La « Coulour-Tag-protein » absorbe dans le jaune parce qu’elle
contient du FAD. C’est une proteine petite (100 acide aminés) soluble et stable (produit
par X-Zyme).
- Le domaine de liaison de la chitine (Chitin-binding domain, CBD) de la chitinase A1 de
Bacillus citrulans. La forte affinité du domaine de liaison à la chitine pour les billes de
chitine permet de bien purifier la protéine de fusion, en effet la colonne peut être lavée
avec un tampon de forte force ionique (1M NaCl) pour retirer les liaisons non
spécifiques. On décroche la protéine avec des agents dénaturants tels que du SDS
(>0.5%) ou des sels de guanidium (6M).
- Tetracystéine : Les protéines portant le motif CCXXCC à l’intérieur d’une hélice α où
XX est n’importe quel acide aminé peuvent être coloré par un dérivé arsénique de la
fluoresceine (FlAsH-EDT2, 4’,5’-bis(1,3,2-dithioarsolan-2-yl)fluoresceine). Ce composé
rentre facilement dans les cellules et se lie au Tag avec une forte affinité (10 pM) donnant

24/10/2011 124
une fluorescence verte (Griffin et al., 1998). On peut aussi utiliser un dérivé arsenique de
la resorufine (ReAsH-EDT2). Ce composé présente l’avantage d’émettre dans le rouge ce
qui permet de faire des expériences de pulse-chase avec la FlAsH-EDT2. De plus le
ReAsH-EDT2 peut être utilisée pour photoconvertir la diaminobenzidine en un composé
insoluble permetttant de détecter la protéine en microscopie électronique (Graham et
Karnovsky, 1966).
Les protéases
Souvent on veut enlever des fusions entre deux protéines après une expression in vitro et
purification. Dans ce cas, on intercale entre les deux protéines une séquence qui va coder
pour un site de coupure.
La coupure peut être chimique par exemple en utilisant le bromure de cyanogène qui
coupe au niveau des méthionines ou l'acide iodosobenzoique qui oxide les tryptophane.
Ces méthodes marchent bien mais on obtient des coupures à des endroits autres que la
jonction, problème qui empire avec la taille de la protéine exprimée.
On peut utiliser une méthode enzymatique, des protéases spécifiques d'une séquence.
- L'enterokinase reconnaît et coupe la séquence Asp-Asp-Asp-Asp-Lys/
- La thrombine reconnaît et coupe la séquence Leu-Val-Pro-Arg-/-Gly-Ser. Selon le
positionnement de la fusion à l'extrémité N ou en C terminale, on obtiendra deux ou
quatre acide aminés.
- Le facteur Xa coupe en C-terminal de la séquence de reconnaissance Ile-Glu-Gly-Arg/
- Les TEV protéinase (Tobacco Each Virus Proteinase) reconnaît la séquence Glu-X-X-
Tyr-Gln/ avec X symbolisant n'importe quel acide aminé. Cette protéase a l'avantage
d'être très spécifique de son site de coupure et de ne pas être inhibée par les inhibiteurs de
protéase à sérine (Parks et al., 1994).

24/10/2011 125
Il y a plusieurs désavantages à l'utilisation de protéases.
la coupure n'est pas toujours spécifique et on peut couper la protéine d'intérêt.
La température assez haute nécessaire à la coupure peut affecter la stabilité de la protéine
d'intérêt.
La coupure est souvent inefficace du fait de l'inaccessibilité du site de coupure dans la
protéine de fusion.
Après la coupure, il faut retirer la protéine de fusion, qui va s'accrocher sur la colonne
d'affinité ayant permis la purification. La protéase est retirée par exemple en utilisant une
protéase biotinylée qui va s'accrocher sur une colonne de streptavidine agarose.
Intein On peut utiliser des protéines qui sont capables
d'autocoupure. C'est le cas par exemple des sites
d'épissages des protéines pour lesquelles on
trouve des inteins (équivalent aux introns) et des
exteins (équivalent aux exons). Ces épissages
ont été trouvés chez des bactéries mésophyles et
thermophyles ainsi que chez la levure. Chez
cette dernière, les différentes étapes de l'épissage
ont été décrites.
On trouve une cystéine au site donneur et une
asparagine et une cystéine au site accepteur. Il y
a tout d'abord un échange entre l'azote de la
liaison peptidique et le soufre de la cystéine
suivi d'une transestérification entre les deux
cystéines. Il y a ensuite coupure petidique
associée à la formation d'un dérivé
succinimidique et enfin un nouvel échange entre
l'azote et le soufre (Chong et al., 1996).
O
NH
SH
N-extein intein
O
O
NH2
NH
SH
C -exteinH2N
H2N
C -extein
O
O
NH2
NH
SH
intein
N-extein
NH2
S
O
NH2
SHN-extein
intein
O
O
NH2
NH
S
C-extein
H2N O
OH2N
C -extein
O
O
NH
intein
N-extein
NH2
SH
S
NH2
+
NH
SH
N-extein C -exteinH2N
O
NH2
SH
intein
O
O
NH2O
CO2-
CO2-
CO2-
CO2-
CO2-
N-S échange
Transestérification
Clivage
N-S échange
cys cys
Asn
Exemple de mécanisme d’épissage d’une intéine.

24/10/2011 126
Ce mécanisme a été décrit pour les levure, chez les archéobactéries, il est à peu près équivalent, la
cystéine est seulement remplacé par un autre nucléophile, une sérine.
Pour obtenir une coupure, on peut utiliser une
intéine mutée au site accepteur. La cystéine du
site accepteur est alors remplacée par un
nucléophile présent dans le milieu (DTT, β-
mercaptoethanol, cystéine...).
la protéine d'intérêt est alors placée en N du site
donneur et la coupure par addition du
nucléophile sur la colonne d'affinité on obtient
une élution de la protéine désirée. (Chong et al.,
1997)
O
NH
SH
protéine d'intérêt inteinH2N
H2N
inteinNH2
S
O
Tag
Tag
N-S échange
protéine d'intérêt
protéine d'intérêtH2N S
O CH2OH+
TagNH2
HS
intein
HSCH2CH2OH
H2O
H2N protéine d'intérêt
O
OH

24/10/2011 127
De la même façon, on peut utiliser le clivage en N et C terminal de l'intéine en mutant le site
accepteur (Chong et al., 1998). En mutant le site
accepteur His-Asn/Cys en Gln-Asn/Ala,
l’induction de la coupure en N-terminal par des
thiols induit le clivage en C-terminal. Dans un tel
système le tag est positionné dans une boucle de
l’inteine.
Lors de la production on obtient une fusion
protéine 1 - N terminus de l’intéine - tag - C
terminus de l’intéine - protéine d’intérêt. Après
induction la protéine de fusion est accrochée sur
une colonne d’affinité. L’action de thiols
(cystéine, DTT ou b-mercaptoethanol) relarge la
protéine 1 et la protéine dans le milieu. Ce système a principalement pour avantage de profiter de
la haute traductibilité de la protéine 1. Si cette protéine 1 est assez petite, elle peut être ensuite
éliminée par dialyse.
Les acides aminés modifiés
Comment introduire une sonde dans les protéines à un site spécifique ?
- Une première technique consiste à muter l’acide aminé par une cystéine puis à modifier
cette cystéine une fois incorporée dans la protéine.
- On peut substituer un acide aminé par un analogue structural en utilisant une souche
auxotrophe (van Hest et al., 2000).
- On peut utiliser une stratégie « non sens ». Le but est ici d’avoir dans la cellule un
aminoacyl tRNA non sens couplé à l’acide aminé non naturel. Un codon ambre est ajouté à
l’emplacement désirée. Il y a deux possibilités pour fabriquer un aminoacyl-tRNA : on peut
aminoacétylé un tRNA suppresseur in vitro (Noren et al., 1989) ou on peut ajouter dans la
bactérie une aminoacyl tRNA synthetase modifiée qui ajoute un aminoacid non naturel
(Wang et al., 2001).
O
NH
SH
proteine 1
O
O
NH2
NHprotéine d'intérêtH2N
H2N
O
O
NH2
NHintéineNH2
S
O
NH2
SH
intéine
O
O
NH
CO2-
CO2-
CO2-
tag
intéine
proteine 1
tag
protéine d'intérêt
OH
OH
S-
HS
tag
NH2
protéine d'intérêt
+
proteine 1H2N
DTT
O
SHS
OH
OH

24/10/2011 128

24/10/2011 129
Purification des protéines recombinantes
Détermination de la concentration en protéine
Pour suivre une purification, on a besoin de connaître la concentration des protéines dans
l’échantillon et d’évaluer la pureté. Toutes les méthodes de détermination de la concentration d’une
solution en protéine sont relatives, en effet elles dépendent de la composition en acide aminé qui
n’est pas connue dans le cas d’un mélange de protéines. Traditionellement, on utilise comme
référence l’albumine de serum de veau (BSA), on fait une courbe étalon avec cette protéine puis on
évalue la concentration de la solution. La BSA sera préparée dans le même solvant, le même tampon
que l’échantillon pour détecter d’éventuelles interférences.
Les méthodes les plus utilisées sont l’absorption en UV et l’absorption dans le visible après réaction
avec un colorant.
La spectrophotométrie UV
Mesure à 280 nm.
Les tryptophanes et la tyrosine absorbent à 280. La mesure est donc fortement dépendante de la
présence de ces deux acides aminés dans la protéine.
Mesure à 205 nm.
La liaison peptidique absorbe à 181-194 nm, mais cette longueur d’onde ne peut pas être utilisée du
fait de l’aborption de l’oxygène. Aussi, on effectue les mesures à 205 nm mais cette méthode est
assez délicate à utiliser, il faut en effet avoir des cuvettes de quartz très propres.
La fluorimétrie
On peut utiliser la fluorescence intrinséque de la protéine en utilisant la fluorescence des
tryptophanes avec une excitation à 280 nm et une émission à 340 nm.

24/10/2011 130
Les méthodes colorimétriques
Le cuivre
Dans des conditions alcalines, les ions cuivre (II) se lient à l’azote des protéines pour donner une
coloration qui absorbe à 540-560 nm (violet). La première méthode dite Biuret est peu spécifique et
peu sensible (on détecte des concentrations de l’ordre de 1mg/ml). Elle a été améliorée en y ajoutant
le réactif de Folin-Ciocalteau (méthode de Lowry) qui colore la protéine en bleu. La sensibilité est de
l’ordre de 100µg/ml. Un autre additif a été l’acide bicinchoninic.
Les colorants
Le coomassie (colorant de Bradford) est le plus utilisé, la liaison du colorant sur les arginines et les
acides aminés aromatiques entraîne un déplacement de l’absorption de 465 nm (rouge) vers 595
(bleu). Ce colorant est un des plus populaire du fait de la facilité de sa mise en œuvre. D’autres
colorants peuvent être utilisés, le rouge ponceau, l’orange G ou l’amino black principalement parce
leur liaison est peu forte.
L’argent
Les protéines se lient à l’argent, cet essai est très sensible mais en parallèle est aussi très sensible à de
nombreux autres composés (les anions, l’EDTA, le SDS, les agents réducteurs tels que le β-
mercapéthanol) aussi, il n’est utilisé que lorsque la protéine est en très faible quantité et qu’il ne faut
pas la « gâcher » uniquement pour déterminer sa concentration.

24/10/2011 131
Solubilisation des protéines
Extraction des protéines cellulaires
Pour aboutir à la purification d'une protéine, il faut tout d'abord préparer l'extrait qui contient cette
protéine. Si la protéine est intracellulaire, on doit passer par une étape de solubilisation : on casse les
cellules et on libère les protéines en solution. Pour casser les cellules, on a à notre disposition
différentes méthodes :
- choc osmotique
- lyse des parois par méthode enzymatique, on utilise par exemple le lysosyme pour
dissoudre la paroi des bactéries Gram+.
- broyage à l’aide d’un homogénéiseur à lames ou d’un potter.
- choc par pression (French Press)
- ultrasons
- billes de verre (pour les levures)
Après séparation par ultrafiltration ou centrifugation, on obtient un extrait clarifié. Parfois les
protéines sont membranaires et il est difficile de les solubiliser. On peut alors utiliser un détergent
qui, par interactions hydrophobes, va solubiliser la protéine. Mais il faut faire attention car un
détergeant trop fort peut inactiver la protéine
Le solvant
Les protéines ne sont solubles que dans une gamme de pH plus ou moins grande selon les protéines.
De même les protéines ne sont souvent pas solubles à faible force ionique. Aussi, le pH et la force
ionique doivent être maintenus constants en utilisant une solution tamponnée. On caractérise un
tampon par son pKa (pKa = -log Ka)
AH+ <-->A- + H+ Ka = [A-][H+] / [AH]
BH+ <--> B + H+ Ka = [B][H+] / [BH+]

24/10/2011 132
Relation Handerson - Hassenbach
pH = pKa + log [base] / [acide]
Le pouvoir tampon d'une solution est maximum au pKa. Le tampon est efficace dans la zone pKa-1/
pKa+1.
Quelques pKa : Acétate : 4,76; Succinate : 5,64; Glycine : 9,8.
Les protéines s'inactivent rapidement par dénaturation (perte de stucture tertiaire) par réaction
chimique (oxydation)ou par protéolyse (coupure des liaisons peptidiques).
Pour résoudre cela :
- on travaille à basse température,
- on élimine les catalyseurs permettant l'oxydation en ajoutant de l'EDTA qui complexe les
métaux qui sont nécessaires à l'oxydation et on peut ajouter un agent inhibiteur comme le ß
Mercaptoéthanol (s’il n’y a pas de ponts disulfures dans la protéine)
- on ajoute des inhibiteurs des protéases
- on ajoute des stabilisants comme le polyol.
- On travaille loin du pI de la protéine
Cas particulier des protéines thermostables : Les protéines thermostables comme par exemple la taq DNA polymérase sont stables à haute
température, au dessus de 80°C. Si on exprime une protéine thermostable dans un organisme
mésophyle comme E. coli, la protéine thermostable se trouve mélangée à des protéines
thermosensibles. Pour purifier la protéine thermostable il suffit de chauffer l’extrait à 80°C, les
protéines thermosensibles se dénaturent et précipitent. A la suite d’une centrifugation, on obtient la
protéine thermostable pure.
On peut utiliser cette technique pour purifier des petites protéines recombinantes en les fusionnant à
une protéine thermostable. Comme elles sont petites, la fusion n’affecte pas la stabilité thermique de
la protéine thermostable et la fusion peut se purifier en dénaturant les autres protéines.

24/10/2011 133
Solubilisation et renaturation des corps d’inclusion
Un cas particulier est la purification des corps d’inclusion lors de la production de protéine dans E.
coli. En effet dans certains cas, on observe l’accumulation de protéine dans des corps non solubles.
Ces agrégats sont visibles en morphologie, leur réfringence permet de les voir en microscopie de
contraste e phase. Ils ont été appelés corps d’inclusion.
Les protéines composant les corps d’inclusion sont généralement mal repliées et présentent un excès
de feuillet β. Ce sont généralement des intermédiaires de repliements qui se sont agrégés avant que le
repliement soit fini (Mitraki et King, 1989). Lorsqu’il y a des cystéines, des ponts disulfure non
correct se sont formés entre les chaînes peptidiques.
La technique de purification consiste alors à purifier ces corps d’inclusion par centrifugation ou par
filtration sur filtre de 0,45 µm. Les corps d’inclusions sont alors lavés en ajoutant de l’EDTA, des
détergents non dénaturants tels que le Triton X-100 ou le desoxycholate ou des détergents
dénaturants à faible concentration. Cette opération de lavage des corps d’inclusion a pour but
d’éliminer les protéines solubles, présentes dans le surnageant qui sont éliminées par centrifugation
ou filtration.
Les corps d’inclusion sont alors solubilisés en utilisant généralement soit l’urée soit le chlorure de
guanidinium, cette solubilisation correspond à leur dénaturation.
- On peut utiliser une concentration de dénaturant qui dénature que partiellement la protéine,
la renaturation sera souvent plus facile. Dans ce dernier cas, on recherche la concentration
minimale qui permet la solubilisation.
- Pour purifier la protéine d’intérêt on peut effectuer une solubilisation différentielle des
protéines. En effet, une première solubilisation à une faible concentration d’agent
dénaturant ne solubilisant pas la protéine recombinante permet d’éliminer les protéines non
désirées.
- Il faut le plus souvent ajouter un agent réducteur tel que le dithiotreitol (DTT) ou le β-
mercaptoethanol pour réduire les ponts disulfures non natifs ou éviter qu’ils ne se forment
lors de la solubilisation.

24/10/2011 134
- Dans certains cas on peut remplacer la solubilisation par l’urée ou les sels de guanidium
par une solubilisation à l’aide pH extrêmes. Ainsi des protéines de fusion de la maltose
binding protein peuvent être dissous à l’aide de 20% acide acétique (Reddy et al., 1998).
Cependant, les pH extrêmes peuvent entraîner des coupures de la protéine ou des
modifications chimiques des résidus. Les détergents comme le sodium dodecylsulfate
(SDS) ou le bromure de n-cetyl trimetylammonium (CTAB) peuvent parfois être utilisés à
faible concentration. L’avantage dans ce cas est que le plus souvent, la solubilisation ne
s’accompagne pas de dénaturation de la protéine et donc d’une perte de son activité. Les
ultrasons sont parfois efficaces pour solubiliser les corps d’inclusion, ici encore, la protéine
n’est pas dénaturée.
Le repliement de la protéine est ensuite effectué par élimination de l’agent dénaturant soit par
dialyse, par diafiltration soit en accrochant la protéine sur une colonne d’affinité. Ce repliement de la
protéine s’avère souvent difficile voir impossible.
Il y a plusieurs recettes pour réussir la renaturation.
- Il faut diluer la solution pour éviter les relations entre les protéines partiellement non
renaturées et donc exposant des surfaces hydrophobes.
- On ajoute généralement du glutathion pour échanger les oxydo-réductions à une
concentration avoisinant les 10 mM. Il est utilisé sous forme réduite (GSH) et oxydé (GS-
SG). Le glutathion a pour rôle de favoriser les échanges de ponts disulfures et ainsi d’éviter
la stabilisation de ponts disulfures mal placés.
- l’ajout de ligand (cofacteurs, métaux, hèmes, inhibiteurs…) dans certains cas favorise la
renaturation
- De nombreux additifs sont utilisés pour faciliter la renaturation sans toujours savoir leur
mode d’action. Ainsi la L-arginine (0,5-1 M) est souvent utilisée. Des détergents (SDS,
CTAB, Triton X-100) amèliorent parfois le repliement des protéines. On peut ajouter des
molécules qui vont favoriser le repliement tels que des molécules switterioniques portant
un groupement sulfobetaine comme groupement hydrophile et un groupement hydrophobe
(Chong et Chen, 20000). Des chaperones et des foldases sont parfois utilisées, elles sont

24/10/2011 135
dans ce cas utilisées sous forme insolubles, liées à des billes d’agarose pour pouvoir les
éliminer facilement à la fin de la renaturation.

24/10/2011 136
La concentration des protéines
Par précipitation :
Pour concentrer les protéines on utilise souvent la précipitation par augmentation de la force
ionique (salting out). Les protéines sont solubles dans l'eau car leurs surfaces hydrophiles présentent
des interactions avec les molécules d'eau. Le sel interagit avec l'eau, si la concentration en sels
augmente, on pompe l'eau de la solution et il y a moins d'eau disponible pour les protéines. Elles
forment des agrégats en interagissant par leur partie hydrophobe. Quand la taille de l'agrégat est
suffisante, il y a précipitation. Le sel le plus utilisé est le sulfate d’ammonium. A 70% de la
saturation, on précipite pour ainsi dire toutes les protéines (La saturation est la solubilité dans l'eau à
20°C. 100% correspond environ à une concentration de 4 moles/litre). D’une manière générale, les
protéines ne sont pas dénaturées par cette précipitation.
Comme toutes les protéines ne précipitent pas avec la même facilité, on peut faire des
précipitations avec différentes concentrations de sulfate d’ammonium. Toutefois comme cette
méthode délicate et peu résolutive n’est plus que rarement utilisée.
Pour quelques protéines, on peut utiliser des solvants organiques tels que l’acétone ou
l’éthanol. Mais l’utilisation de ces solvants organiques est limitée, en effet ils occasionnent souvent
une dénaturation irréversible de la protéine.
Par filtration :
Pour réduire le volume de la solution, la méthode la plus utilisée est l'ultrafiltration. On
soumet la solution protéique à l'action d'une filtration; la taille des pores du filtre doit être telle que
les protéines doivent être retenues. Il existe plusieurs systèmes, le plus simple utilise un boudin de
dialyse, la solution est à l’intérieur du boudin et à l’extérieur on place un polymère absorbant tel que
l’ « aquacide ». L’eau comme les petites molécules passent par les pores du boudin, les protéines
restent à l’intérieur.
On utilise souvent des filtres pour concentrer les protéines, pour les volumes importants, on
crée un courant sur le filtre pour éviter les colmatages, on parle alors d’ultrafiltration tangentielle.

24/10/2011 137
Les différentes méthodes de séparation des protéines
On peut utiliser deux grands types de méthodes pour séparer les protéines, la chromatographie et
l’électrophorèse. Certaines méthodes seront utilisées pour purifier alors que d’autres seront plutôt
utiliser pour vérifier la pureté de la solution.
La chromatographie
La chromatographie est une technique de séparation qui utilise une colonne contenant un support qui
possède plusieurs propriétés.
On distingue plusieurs étapes
1 Régénération et équilibrage de la colonne avec le tampon.
2 Charge de la solution protéique
3 Lavage avec le tampon. On utilise tout d’abord le tampon de charge et au besoin successivement
plusieurs tampons qui vont décrocher des protéines contaminantes.
Pinceserrée
pompe
membrane porée de 10 kDa
échantillon
Ajout de tampon
(pour la diafiltration)
Ultrafiltration tangentielle

24/10/2011 138
4 Elution : elle peut se faire en ajoutant directement le tampon d’élution, en faisant un gradient entre
le tampon de lavage et le tampon d’élution. Ce gradient peut être continu ou discret.
Types de matrice et de pression
Il existe différents types de supports : cellulose, sephadex.
Si on applique une pression élevée on augmente la résolution. On effectue alors une
Chromatographie Liquide Haute Performance (HPLC).
L’électrophorèse
La technique consiste à faire migrer les protéines dans un gel. Le gel le plus utilisé est un gel
d’acrylamide.
L'acrylamide est un monomère, en présence de
radicaux libres (en général persulfate d'ammonium
stabilisé par le TEMED), une réaction
en chaîne est initialisée et on obtient un
polymère.
Si la réaction est faite en présence de N.
N'méthylenebisacrylamide, on a
formation d'un réseau de mailles plus
ou moins serrées en fonction de la
concentration de l'acrylamide.
On obtient donc un gel dont les largeur des mailles dépend des concentrations en acrylamide et
bisacrylamide de départ.
CH2 CH
C O
NH2
C O
NH2
CHCH2 CH2 CH CH2 CH
C O
NH2
C O
NH2
+ persulfate d'ammonium+ temed
CH2 CH
C O
NHCH2NH
C OCHCH2
Bisacrylamide

24/10/2011 139
Séparation selon la taille
Dialyse
Son principe est de mettre la solution dans un boudin de dialyse qui lui-même se trouve dans un
tampon dépourvu de sels. Le boudin de dialyse est constitué d’une membrane qui laisse passer les
petites molécules. Les boudins les plus couramment utilisés laissent passer les molécules qui
présentent un poids moléculaire inférieur à 10 kDa, mais il existe des boudins avec d’autres seuils de
coupure, soit plus faibles (3 kDa) soit plus importants (30, 50 ou 100 kDa). Par un phénomène de
diffusion, les sels vont sortir du boudin jusqu'à atteindre l'équilibre entre la concentration dans le
boyau et la concentration dans le tampon. On effectue plutôt plusieurs dialyses successives qu’une
dialyse dans un grand volume.
Filtration sur gel
Le principe est de faire passer la solution de protéine dans un gel constitué de billes creuses, les
petites molécules passent par l’intérieur des billes et font donc un chemin plus long que les grosses
molécules qui ne passent pas par l’intérieur des billes.
Cette méthode est aussi utilisée comme dessalage pour éliminer les sels d’une solution ou pour
changer le tampon.
Ultrafiltration
La solution est filtrée sur une membrane ayant un seuil de coupure correspondant à des poids
moléculaires de 1 à 100 kDa. Le filtrat ne contient donc que des petites molécules et, si la solution ne
passant pas par la membrane est diluée plusieurs fois avec le tampon, elle ne contient que des
molécules de grande taille. Cette méthode est plutôt utilisée pour les grands volumes.
Ultracentrifugation
Les protéines sont séparées en fonction de leur vitesse de sédimentation dans un milieu visqueux,
généralement une solution concentrée de saccharose. Les protéines les plus grosses sédimentent plus

24/10/2011 140
vite que les molécules plus petites. L’ultracentrifugation ne permet de séparer que de petits
échantillons, c’est donc une méthode analytique.
Séparation selon la charge
On sépare les protéines selon leur charge. Les protéines sont des polyélectrolytes, ainsi, à un pH
donné, chacun des groupements ionisables sera dans un état d'ionisation donné (en fonction du pKa).
Le pI est le pH auquel la charge globale de la protéine est nulle. Si pH < pI, la molécule est positive.
Si pH › pI, la molécule est négative. Ainsi, selon la charge du support et sa propre charge, la protéine
va interagir ou pas avec le support.
On utilise la chromatographie d’échange d’ion.
Si on est à pH 7, les protéines dont le pI > 7 ne seront par retenues sur une échangeuse d’anion
mais seront retenues par une résine échangeuse de cation
Types de groupements échangeurs d’ions Echangeurs d’anions - Carboxy-méthyl (CM)
- Phosphate (P)
- Sulfoéthyl (SE)
- Sulfopropyl (SP)
CH2
COO-
O PO3H2- CH2
CH2
SO3-
CH2
CH2
SO3-CH2
Echanges de cations - Diéthyl-amino-éthyl (DEAE)
- Triéthyl-amino-éthyl (TEAE)
- Quaternary-amino-éthyl (DEAE)
N (C2H5)2(CH2)2
N+
(C2H5)3(CH2)2
N+
(C2H5)3(CH2)2
CH2. CH. CH3OH

24/10/2011 141
L’élution s’effectue généralement en augmentant la force ionique, à l’aide d’un gradient de sel. Elle
peut aussi se faire en faisant varier le pH de la solution, mais dans ce cas on passe souvent par un pH
dénaturant pour la protéine.
On peut utiliser l’électrofocalisation. Il s’agit d’une électrophorèse qui permet de focaliser les
protéines dans une région du gel qui correspond à leurs points isoélectriques. Pour avoir un gel avec
un gradient de pH, on ajoute des ampholines qui sont des polymères avec des groupements
ionisables. La protéine va migrer jusqu'au niveau de pH qui correspond à son pI.
Cette méthode est très utilisée car elle est très résolutive : elle sépare les isoenzymes qui sont très
proches en séquence d'acides aminés.
L’électrofocalisation ne permet pas de séparer une grande quantité de protéine. Cette méthode a été
adaptée à la chromatographie et a pris le nom de chromatofocalisation.
Séparation selon la taille et la charge
On utilise une électrophorèse sur gel de polyacrylamide en condition native. Etant donné que l'on
applique un courant électrique sur le gel, les protéines vont migrer en fonction de leur taille et de leur
charge. Le gel agit comme un tamis qui va plus retenir les grosses molécules que les petites.
La majorité des protéines est chargée négativement et vont donc migrer vers le pôle positif. Ces
gels sont appelés natifs car la protéine garde son activité.
Séparation selon l’hydrophobicité
Chromatographie: On utilise un support qui contient un greffage hydrophobe et qui peut donc
interagir avec les protéines hydrophobes. On connaît deux types de support :
- groupement aliphatique, la longueur des chaînes amine peut varier (C4->C10). Ex : le butyl
sepharose est un greffage en C4.
- noyau aromatique : phenyl sepharose.

24/10/2011 142
Ces supports retiennent les protéines par interaction hydrophobe. On charge la colonne en
présence de sel car il favorise les interactions hydrophobes ((NH4)2 SO4 à 1M). L'élution peut se
faire soit en diminuant la concentration en sels soit en ajoutant un détergent qui solubilise les
interactions hydrophobes.
Séparation de phase. Le triton X-114 est soluble dans l’eau à basse température (4°C) mais la
solubilité est fortement diminuée à plus haute température (35°C). Les protéines hydrophobes ayant
plus d’affinité pour le détergent que pour l’eau, on les retrouvera préférentiellement dans la phase
détergente. La méthode consiste donc à préparer une solution de TX-114 saturée en eau à haute
température, à ajouter l’échantillon, abaisser la température pour favoriser les contacts entre les
protéines et le TX-114. La solution est alors chauffée à 37°C, et centrifugé pour séparer les deux
phases.
Séparation selon l’affinité
Site actif
On fixe sur le support un ligand dont on sait qu'il a une forte interaction avec la protéine que l'on
cherche à purifier.
L'élution peut être non spécifique (augmentation de la force ionique) ou spécifique (élution avec le
ligand libre - il y a alors compétition et fixation de la protéine sur ce ligand libre; le ligand libre est
enfin éliminé par dialyse). L'avantage de cette dernière méthode est qu'elle ne fait pas appel aux
propriétés physico-chimiques qui peuvent être communes à plusieurs protéines mais à la spécificité :
en une seule étape, on a une meilleure purification qu'avec l'échange d'ions ou la chromatographie
hydrophobe. Il faut évidemment avoir une certaine connaissance de l'enzyme.
Exemple de chromatographie d’affinité avec des tag :
- Glutathion-S-transférase : l’élution se fait en rajoutant du glutathion dans le tampon
- Strep-tag : l’élution est obtenue en ajoutant de la desthiobiotine
Modification post traductionnelle.

24/10/2011 143
On peut trier des protéines glycosylées des protéines non glycosylées en utilisant une lectine fixée sur
une colonne. Une lectine est une protéine qui a une forte affinité pour les sucres. En utilisant une
lectine qui reconnaît un sucre composant les structures glycosylées des protéines, on peut accrocher
les protéines glycosylées sur la colonne et les éluer en utilisant le sucre dans la phase mobile.
Une des lectines les plus utilisée est la concanavaline A qui reconnaît le mannose.
Séquence primaire
Certaines séquences peuvent être
reconnues par des ligands. Par exemple,
les queues homopolymériques
d’histidine peuvent être purifiées avec
des colonnes de Nickel ou de Cobalt.
L’élution est obtenue avec une solution
contenant de l’imidazole ou avec une
solution d’EDTA. Cette méthode
proposée en 1975 par Porath et al., est
actuellement une la méthode de purification la plus populaire. Le cobalt est quelquefois préféré au
nickel, parce que la liaison est moins sensible au β-mercaptoethanol et parce que le cobalt forme
quatre liaisons avec les histidines au lieu de deux pour le Nickel ce qui rend l’affinité plus spécifique.
Structure tertiaire
On peut immobiliser un anticorps spécifique de la protéine à purifier sur une colonne en utilisant soit
la protéine A soit un ligand composé de sulfone thioeher (thiophilic resin). Lorsque l’on fait passer
un extrait sur cette colonne, les autres protéines ne seront pas retenues : cette technique est donc
spécifique. Cependant, l'interaction antigène/anticorps est tellement forte qu'il est ensuite difficile de
les séparer : les constantes de dissociation sont comprises entre 10-6 à 10-12. L'élution est donc
problématique. La seule manière de les dissocier serait de baisser fortement le pH (jusqu'à 2) mais à
ce pH là, l'enzyme peut être dénaturée.
A faible pH on dissocie la liaison Anticorps/protéine A si bien qu’on obtient les anticorps avec la
protéine désirée. Pour éviter cette coélution de l’anticorps, il faut fixer de manière covalente
l’anticorps sur la protéine A.
Protéine Nickel Matrice

24/10/2011 144
Séparation par des colorants
Elles ressemblent à une chromatographie d'affinité mais sont moins spécifiques.
Bleu de cybacron : on l’a tout d’abord utilisé comme chromatographie d’affinité pour purifier les
protéines ayant un site de reconnaissance du NAD. Comme de nombreuses protéines sont retenues
par le bleu de cybacron, on s’en est servi pour purifier un grand nombre de protéines. L'élution
s'effectue grâce à un gradient de sels.
Evaluation de la pureté
Si la protéine à purifier a une activité enzymatique, on peut la suivre dans les étapes de purification
grâce à cette activité, et quantifier le rendement de chaque étape en calculant l’activité spécifique,
activité observée/ concentration en protéine.
Mais ce n’est généralement pas possible, et on estime le plus souvent la pureté d’une protéine par un
gel dénaturant. On utilise les propriétés du sodium duodecyl sulfate (SDS), c’est un détergent
anionique qui dénature les protéines en se fixant sur les zones hydrophobes. Il se fixe en moyenne 1.4
g de SDS par gramme de protéine. Les protéines sont dénaturées. Etant donné que les protéines
deviennent toutes chargées positivement à cause de cette interaction, elles ne vont plus migrer en
fonction de leur quantité de charge mais seulement en fonction de leur taille.
Il existe deux types de gel :
- Le gel de séparation qui permet la séparation selon la masse moléculaire.
- Le gel de concentration qui a une teneur plus faible en polyacrylamide, ce qui va permettre
une migration plus rapide. On aura donc des bandes plus fines et donc une meilleure
résolution. Le gel de concentration est généralement coulé au-dessus du gel de séparation.
Avant d’être chargé sur le gel les protéines sont dénaturées en rajoutant du SDS à 1% avec du ß
Mercaptoéthanol (10mM pour couper les ponts disulfure), puis par chauffage 1 à 3 minutes à 100°C.
C'est le test de pureté le plus utilisé car il est fiable et simple.

24/10/2011 145
Comme plusieurs protéines peuvent migrer à la même place dans un gel SDS, on peut augmenter la
séparation en effectuant une électrophorèse bidimensionnelle. C'est une association des méthodes
d'électrofocalisation et d'électrophorèse en conditions dénaturantes. On réalise une première étape
d'électrofocalisation, puis, à 90°, une étape en gel SDS, ainsi, deux protéines qui ont le même pI vont
pouvoir être séparées selon leur poids moléculaire.
Pour colorer les protéines sur le gel, il faut les fixer sur le gel. Pour cela on les traite à l'acide
acétique. La coloration se fait ensuite par :
le bleu de Coomassie : seuil de détection : 1 à 10 µg;
le nitrate d'argent : seuil de détection : 10 à 100 ng.
Caractérisation des protéines Taille et masse : Il existe plusieurs méthodes pour estimer la taille ou la masse d’une protéine, ces deux notions sont souvent prises l’une pour l’autre puisqu’il existe une relation linéaire entre la masse moléculaire d’une protéine et sa taille, du moins pour les protéines globulaires. . - Electrophorèse en condition dénaturante : La solution contenant la protéine est chauffée en présence de SDS ce qui a pour effet de dénaturer la protéine et en présence de 2-mercaptoethanol pour réduire les ponts disulfures. On effectue ensuite un électrophorèse en présence de SDS, a coté de marqueurs, c’est à dire de protéines dont on connaît la masse. Cette méthode permet de séparer des peptides de 5 à 200 kDa. - Electrophorèse en grandient d’acrylamide en condition native. On prépare un gel d’acrylamide à concentration variable, par exemple de 4 à 40%.. dans un tampon éloigné du point isoélectrique de la protéine de façon à ce qu’elle soit chargée. En appliquant le courrant électrique, la protéine migre jusqu’à ce que la maille du gel soit petite. La migration est donc longue, de l’ordre de 16 heures et la protéine n’est pas dénaturée. Comme pour l’electrophorèse en condition dénaturante, des marqueurs sont déposés en parallèle pour avoir un calibration. Cette méthode permet de séparer des protéines de 50 kDa à 1000 kDa. - La centrifugation zonale utilise le fait qu’un protéine de gros poids moléculaire sédimente plus rapidement qu’une protéine de petit poids moléculaire. On dépose la solution de protéine sur solution de saccharose puis on centrifuge plusieurs heures a grande vitesse pour avoir une acceleration de l’ordre de 100 000 g. On récupère ensuite les fractions le long du tube de centrifugation. La migration correspond à une vitesse de sédimentation et est exprimé en Svedberg (S). La calibration

24/10/2011 146
est effectuée en ajoutant dans l’échantillon des protéines qui sont révélées par leurs activités enzymatiques. La centrifugation analytique est une amélioration de la centrifugation zonale en effet, la migration de la protéine dans le tube est suivie au cours de la centrifugation grâce à leur absorbtion à 280 nm. - La chromatographie d’exclusion (SEC, size exclusion chromatographie) utilise le fait qu’une protéine de grosse taille ne passe pas à travers les pores d’une bille, alors qu’une protéine de petite taille passe par les pores et est donc ralentie. Les grosses molécules passent les premières et les petites sont chromatographiées. Il existe plusieurs tailles de pores pour pouvoir analyser des protéines de 10 kDa à 1000 kDa. Selon la pression utilisée en tant que vecteur de migration, on parlera de chromatographie basse pression de FPLC ou d’HPLC. Plus la pression est importante, moins il y ade diffusion et plus la résolution est bonne. . - Diffusion de la lumière statique (Static light scattering, classical light scattering, total intensity scattering) : La lumière reçue par un objet est diffusé selon un angle qui dépend de sa taille. Pour cette technique, on envoie un faisceau lumineux sur un écahntillon et on mesure sa diffusion à plusieurs angles. - Diffusion de la lumière dynanique (Dynamic Light Scattering, DLS ; Correlation photon spectroscopy). Plus un objet est petit plus sa vitesse de diffusion est importante. Si on mesure la diffusion de la lumière au cours du temps on peut estimer la vitesse de diffusion d’un objet et ainsi estimer sa taille ou plus exactement son rayon hydrodyanique. - La microspcopie permet de « voir » des objers de tailles supérieurs à 10 nm. Pour les grosses protéines ou pour les complexes de protéines, on peut utiliser la microspcopie électronique ou la microscopie de champ proche (AFM, atomic force microspcopy) - La spectrométrie de masse permet d’estimer la masse de la protéine. Cette technique est utilisée pour des protéines jusqu’à 50 kDa. - La structure trimentionelle des protéines peut être résolue par diffraction des rayons X dans un cristal ou par RMN en solution. Cette structure, lorsquelle est entièrement résolue nous donne la taille et la masse de la protéine.

24/10/2011 147
Stabilisation des protéines
Trois raisons principales sont généralement mises en avant pour expliquer la dénaturation des
protéines :
1- La protéine perd son activité via un mécanisme monomoléculaire de dénaturation physique. Des
interactions non-covalentes qui la maintiennent sous sa forme native sont remplacées par les
liaisons non covalentes qui la maintiennent sous une forme dénaturée. Une chaîne polypeptidique
peut prendre de nombreuses conformations et une seule d’entre elle correspond à l’état natif.
Dans certains, la conformation native est la seule, la dénaturation est alors réversible, par contre
dans les autres cas, il existe plusieurs conformations possibles qui correspondent à des états
inactifs, la dénaturation est alors irréversible.
2- Cette mauvaise conformation expose des régions hydrophobes à la surface de la protéine. Les
régions hydrophobes de différentes molécules réagissent entre elles et forment des agrégats.
Environ la moitié des résidus présents dans une protéine ne sont pas polaires, ils sont soit enfouis
dans le cœur de la protéine ou alors ils forment à la surface de la protéine des régions qui jouent
un rôle dans les interactions avec les autres protéines, avec les lipides constituants les membranes
ou avec les polysaccharides. Cependant, le contact des résidus hydrophobes avec les molécules
d’eau est thermodinamiquement désavantageux et est préjudiciable pour la stabilité des protéines
in vitro.
3- De nombreux groupes fonctionnels sur les protéines sont réactifs et donc susceptibles
chimiquement modifiés. L’oxidation est la voie majeure de dénaturation des protéines lors de leur
stockage. Les sites d’oxidation potentiels dans les protéines sont les chaînes latérales des
méthionines, cystéines, histidines, tryptophane et tyrosines. De plus il peut y avoir hydrolyse des
liaisons peptidiques Asp-X, destruction des ponts disulfures, et désamidation (Ahern et klibanov,
1986).

24/10/2011 148
Stabilisation des protéines par l’utilisation d’additifs.
L’ajout de stabilisateurs dans les formulations de protéines est la méthode la plus commune pour
augmenter la demi-vie des protéines. Ces composés sont souvent choisis d’une manière empirique,
l’effet obtenu est variable et dépend des caractéristiques de chaque protéine.
- On peut augmenter la différence d’énergie libre entre l’état natif et l’état dénaturé. Pour
cela on utilise des osmolytes. Les osmolytes agiraient en augmentant plus le potentiel
chimique de la forme dénaturée que celui de la forme native, Ainsi les osmolytes
augmentent la différence d’énergie de Gibbs (ΔG) entre les formes natives et dénaturées
(Arakawa et al., 1990). Dans le diagramme
d’énergie, ΔG1 est la différence d’énergie entre
la forme native (Naq) et la forme dénaturée
(Uaq) en solution aqueuse et ΔG3 est la même
différence en présence d’osmolyte. Les
osmolytes augmentent plus le potentiel
chimique de la forme dénaturée (ΔG2) que celui
de la forme native ((ΔG4). Il y a trois grande classes d’osmolytes, i) les polyols tels que le
sucrose, ii) les methylamines telles que la sarcosine iii) certains acides aminés tels que la
proline. On utilise par exemple la L-arginine (Lin et al., 2001), la L-proline, la L-serine, la
glycine, l’acide gamma amino-butyrique, la bétaine. La trimethylamine N-oxide (TMAO)
est un osmolyte naturel qui compense l’effet délétère de l’urée qui est présent à la
concentration de 400 à 600 nM dans les cellules des élasmobranches (Baskakov et al.,
1998).
- On peut stabiliser la conformation des protéines par un mécanisme d’exclusion préférentiel
ou d’hydratation préférentielle due au réseau de liaisons hydrogènes dans la couche
d’hydratation de la protéine. Les molécules génèrent une cage autour de la protéine en
excluant la couche d’hydratation et ainsi en éliminant les molécules d’eau actives à
proximité de la protéine. On utilise des polymères comme la BSA (albumine), le Poly-L-
Histidine, le Poly-L-Arginine, le dextran, le Ficoll et polyethylene glycol. Des sucres tels
que le saccharose, le trehalose (dimère de glucose liés en alpha 1.1) ou le glucose (Krest et
Uaq
Naq
Nos
Uos
ΔG2
ΔG1
ΔG3
ΔG4

24/10/2011 149
al., 1999 ; Felix et al., 1999). Des composé naturel comme l’ectoine qu’on trouve souvent
dans les bactéries halophyles (Lippert et Galinski, 1992). Les sels peuvent être caractérisés
par leur pouvoir de structuration de l’eau, effet Hofmeister. Les sels kosmotropes qui
strucurent l’eau, stablisent les protéines en favorisant une structure plus compacte. Ainsi, le
chlorure de sodium, le phosphate de sodium, le borate de sodium ou le formate de sodium
ou Na2SO4 sont stabilisant du fait de leur pouvoir d’hydratation des protéines (Bowie et
Sauver, 1989 ; Weijers et Van’t Riet, 1992 ; Ahmad et al., 2001 ; Ebel et al. 1999). D’un
autre coté, les sels chaotropes tels que KSCN, CaCl2 ou MgCl2 augmentent la tension de
surface et ainsi déstabilisent la structure des protéines (Arakawa et Timasheff, 1982).
- On peut stabiliser les protéines en diminuant les modifications chimiques des résidus. Le
saccharose peut diminuer la vitesse d’oxydation des résidus méthionine. Si la méthionine
est impliquée dans le site actif, le saccharose protègera l’activité de la protéine (De Paz et
al., 2000). Le poly(ethyleneimine) (PEI) est un polycation qui est un des meilleur
protectant contre l’oxidation. L’EDTA est un agent chélateur qui protège contre l’oxidation
médiée par des traces d’ions métallique (Andersson et al., 2000). Le sorbitol inhibe
complètement la deamidation des résidus de la protéine.
Stabilisation des protéines par mutagenèse dirigée
Il existe plusieurs méthodes pour stabiliser une protéine en modification de sa structure primaire. Une
première méthode consiste à effectuer de la mutagène aléatoire puis à cribler les mutants les plus
stables (Morawski et al., 2001). Une deuxième méthode consiste à effectuer des mutagenèses
dirigées. Pour trouver les résidus à muter, deux informations peuvent être utilisées, les données
structurales et les données phylogénétiques. Dans ce deuxième cas, la comparaison des séquences de
plusieurs espèces peut être une source d’information pour la mutagenèse. Il est par exemple habituel
de comparer des protéines d’espèces mésophile et thermophiles, le problème est qu’elles diffèrent à
de nombreuses positions (Molk et al., 2001 ; Jiang et al., 2001, Perl et al., 2000).
Stabilisation des hélices α
Il existe plusieurs méthodes pour stabiliser les hélices alpha dans une protéine.

24/10/2011 150
Une solution consiste à ajouter une triade de résidus chargés Arg(+)-Glu(-)-Arg(+) espacés à des
intervalles i,i+4 ou i,i+3 dans la chaîne peptidique (Olson et al. 2001).
Une autre solution consiste à changer les glycine en alanine. Les résidus glycine sont les moins
fréquents dans les hélices α, juste après les prolines. Cette propriété a été remarquée par des études
statistiques comme par des études thermodynamiques. L’alanine stabilise l’α hélice de plus de 2
kcal/mol par rapport à la glycine et l’alanine a une forte préférence thermodynamique pour un
environnement hélicoïdal (O’Neil and DeGrado, 1990). Ainsi, la substitution de glycine en alanine
dans les hélices augmente la stabilité des protéines (Chakrabartty et al., 1991, 1995; Munoz et al.,
1994; Pace et al., 1998; Hecht et al., 1986; Ganter et al., 1990 ; Margarit et al., 1992, Blaber et al.,
1995 ; Predki et al., 1995).
On peut aussi ajouter un « cap » à l’extrémité N-terminale de la protéine le plus souvent en ajoutant
une serine à l’extrémité et un glutamate à la position 3 pour établir une liaison hydrogène.
Enfin, certains sites peuvent aider à calculer la stabilité d’une hélice α : http://www.embl-
heidelberg.de/Services/serrano/agadir/agadir-start.html
Stabilisation par rigidification du squelette Pour fixer la structure tertiaire on peut ajouter des prolines dans les boucles. Stabilisation par création de ponts
- Ponts disulfures : on peut introduire des liaisons covalentes en créant des ponts disulfures.
Théoriquement, l’introduction d’un lien intramoléculaire décroît l’entropie de la protéine
dénaturée (Flory, 1956), déstabilisant ainsi l’état dénaturé et stabilisant thermodynamiquement la
protéine. De nombreuses protéines telles que le lysozyme (Johnson et al.,1978, Ueda et al., 1985 ;
Wetzel et al., 1988 ; Matsumura et al., 1989), des RNases (Lin et al., 1984, Futami et al. 2000), la
subtilisine (Pantoliano et al., 1987) ont été stabilisées en ajoutant des liaisons covalentes soit par
des moyens chimiques soit par des moyens génétiques. Cependant, l’introduction de liaison
covalentes peut aussi entraîner une inactivité de la protéine.
- Les ponts salins : l’importance des ponts salins a été suggéré par Perutz et Raitz en 1975. Les
protéines thermostables provenant d’organismes thermostables ont généralement plus de ponts
salins que leurs homologues provenant d’organismes mésophiles et de nombreuses expériences

24/10/2011 151
de mutagenèse dirigée ont montré leur importance dans la stabilité des protéines. La force d’un
pont salin est estimée à 3-5 kcal mol-1. Cependant le rôle stabilisant des interactions
electrostatiques est controversé et dépend fortement de la position de l’interaction. Les ponts à
l’intérieur de la protéine sont plus favorables que les ponts à l’extérieur (Dao-pin et al., 1991) et
les interactions ioniques à la surface de la protéine peuvent même avoir un effet déstabilisant.
- Les liaisons hydrogène stabilisent les protéines. Des mutations occasionnant une perte d’une
liaison hydrogène sont souvent trouvées dans les mutants thermosensibles produisant des
protéines à stabilité réduite (Grutter et al., 1987) et une étude récente (Pace et al., 2001) a montré
que le remplacement des tyrosines en phénylalanine déstabilise habituellement les protéines.
Ainsi, les liaisons hydrogène entre groupes polaires à l’intérieur des protéines sont plus
favorables que les interactions similaires avec l’eau dans les protéines dénaturées. La stabilité due
à une liaison hydrogène a été estimée à 1,3 kcal/mol et l’introduction de nouveaux ponts
hydrogène augmente la stabilité de la protéine (Peterson et al., 1999).
Stabilisation par mutagenèse des résidus exposés au solvant :
On peut remplacer les résidus Asp et Glu qui ont des liaisons hydrogènes exposées au solvant par des
résidus isostériques neutre, Asn or Gln (Irun et al., 2001).
L’exposition des chaînes latérales hydrophobes au solvant entraîne une variation d’énergie libre
défavorable en diminuant l’entropie du solvant. Certains résidus hydrophobes peuvent être moins
exposés au solvant dans l’état dénaturé que dans l’état natif. Pour ces résidus, il y a un effet
hydrophobe inverse qui s’oppose au repliement. Ainsi, des mutations qui remplacent des résidus
exposés au solvant par des résidus hydrophobes peuvent augmenter la stabilité de la protéine (Pakula
et Sauer, 1990).
Stabilisation par ajout de sites de glycosylation
La glycosylation est une des modifications post-traductionelles les plus importantes. Les chaînes de
sucre sont liées soit à l’azote d’une asparagine soit à l’oxygène d’une sérine ou d’une thréonine. La
N-glycosylation s’effectue à la séquence Asn-X-Ser or Asn-X-Thr (Kaplan et al., 1987). L’efficacité
de la glycosylation dépends de l’acide aminé à la position X, elle est impossible lorsqu’il y a une

24/10/2011 152
proline et relativement inefficace lorsque X est un tryptophane, un aspartate ou un glutamate (Shakin-
Eshleman et al., 1996). Cette modification est co-traductionelle et la structure de l’oligosaccharide
est modifiée durant la translocation de la protéine du réticulum endoplasmique vers la membrane. La
plupart des protéines sont glycosylées dés que la chaîne polypeptidique entre dans le réticulum
endoplasmique, cependant la glycosylation ne précède pas nécessairement le repliement.
La stabilisation peut être augmentée par addition de site de N-glycosylation (Khanna et al., 2001).
Cette méthode est valable uniquement pour les protéines qui sont produites en cellules eucaryotes. Il
semble que la glycosylation décroît la dénaturation irréversible et non la dénaturation réversible
(Tams et Welinder, 2001).
Stabilisation par mutagenèse des résidus internes
On peut stabiliser les protéines en re-empaquetant leur cœur hydrophobe. Cette stratégie a donné de
bons résultats pour des protéines qui avaient des défauts de repliement évidents (Finucane and
Woolfson,1999), des résidus chargé à l’intérieur (Waldburger et al., 1995) ou des cavités qui peuvent
être remplies par des acides aminés avec des résidus plus encombrants. Cette stratégie a été appelée
«cavity-filling strategy » (Baldwin et al., 1996 ; Ohmura et al., 2001).
Stabilisation par élimination des résidus réactifs
L’inactivation thermique des protéines provient de certains acides aminés : aspartate, asparagine et
cystéine. L’asparagine est sujet à une déamidation, elle peut être remplacé par des résidus qui ont des
propriétées proches (Gln, Ile ou Thr). La présence d’aspartate conduit à la coupure de la liaison
peptidique, il peut être remplacé par un glutamate.

24/10/2011 153
Analyse des interactions entre macromolécules
Interactions acides nucléique-protéine I - Techniques permettant de déterminer la (les) protéine(s) qui se fixe(nt) sur un acide
nucléique cible (ADN ou ARN) :
I – 1 : Criblage de banque d’expression :
Il s’agit d’utiliser la très classique technique de criblage d’une banque d’expression, le criblage se
faisant ici avec un oligonucléotide double brin marqué radioactivement. Les clones exprimant la
protéine interagissant avec cet ADN seront donc repérés après autoradiographie du filtre.
Une alternative à cette technique consiste à utiliser des phages lytiques de type lambda afin d’éviter
les étapes de lyse des bactéries.
I – 2 : Simple Hybride
Cette technique est dérivée de la technique du double hybride. Elle permet
d’identifier de nouvelles protéines interagissant avec un fragment d’ADN connu – et d’accéder
directement à leur ADNc
de vérifier une interaction entre une séquence d’ADN et une protéine
d’étudier les nucléotides et/ou acides aminés impliqués dans l’interaction.
Bien sûr, la stratégie expérimentale choisie variera légèrement en fonction de ce que l’on veut faire.
Comme pour le double hybride, on travaille chez la levure.
Banque d ’ADNc clonée dans un vecteur d’expression
Transformation chez E. coli
Etale sur boîte
Transfère sur filtre (nitrocellulose ou nylon)
Lyse les bactéries -> les protéines de chaque clone bactérien se fixent sur le filtre
Bloque (ADN double brin non spécifique)
Ajoute la sonde (ADN double brin marqué au 32P)
Repère les clones positifs
Récupère ces clones sur la boîte de pétri
Séquence l’ADNc de ces clones -> identification du partenaire

24/10/2011 154
Principe :
Si la protéine X se lie sur la séquence cis régulatrice E, il y aura activation de la transcription du gène
rapporteur. Si on fait pousser les levures ayant reçu ce vecteur sur un milieu en présence de X-gal, les
colonies obtenues seront bleues.
E : Elément cis régulateur - séquence ADN cible à tester X : protéine dont on teste l’interaction avec la séquence E
• soit une banque d ’ADNc • soit un ADNc d’une protéine connu
A : domaine protéique activateur transcriptionnel (par exemple celui de Gal4) Gène rapporteur - par exemple celui de la β galactosidase Afin d’améliorer le rendement de l’activation transcriptionnelle, la séquence cible E est répétée
plusieurs fois.
Tout comme dans les cas du double hybride, un double système de sélection (β galactosidase et un
gène d’auxotrophie) est en général utilisé.
Afin d’améliorer le rendement de l’activation transcriptionnelle, la séquence cible E est répétée
plusieurs fois.
Tout comme dans les cas du double hybride, un double système de sélection (β galactosidase et un
gène d’auxotrophie) est en général utilisé.
Particularité des ARNs
Triple hybride
R t
X A
E E E
Gène rapporteur
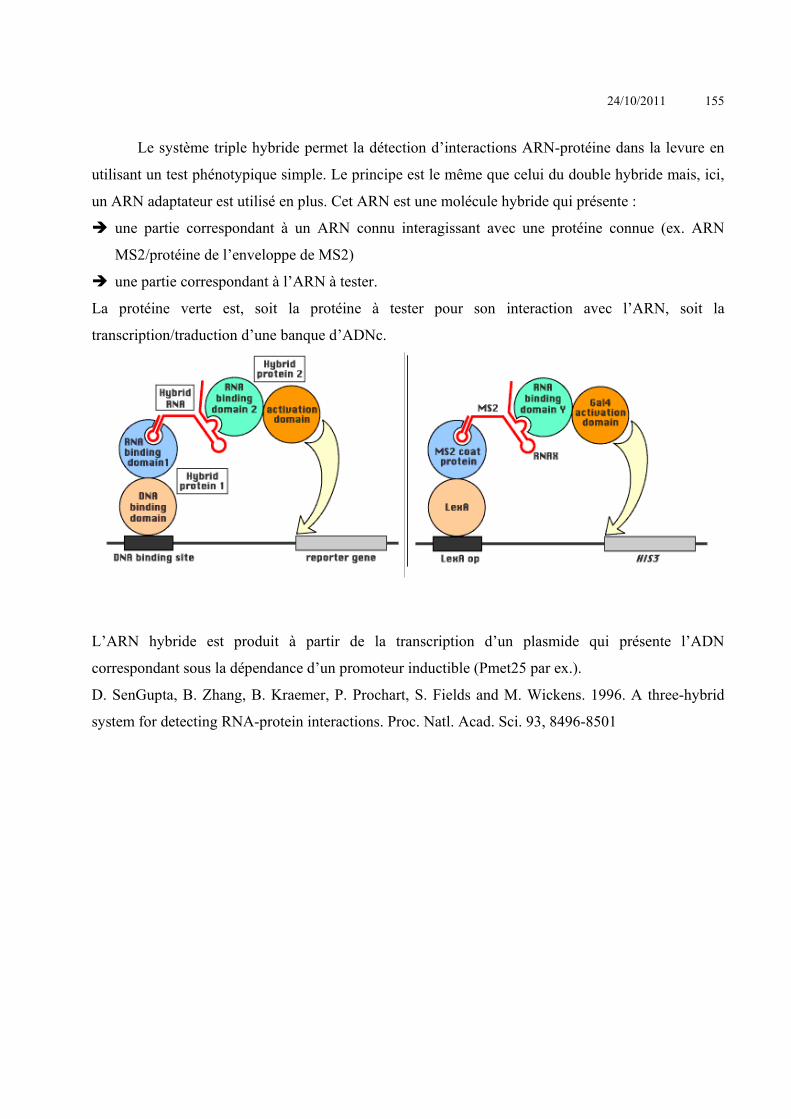
24/10/2011 155
Le système triple hybride permet la détection d’interactions ARN-protéine dans la levure en
utilisant un test phénotypique simple. Le principe est le même que celui du double hybride mais, ici,
un ARN adaptateur est utilisé en plus. Cet ARN est une molécule hybride qui présente :
une partie correspondant à un ARN connu interagissant avec une protéine connue (ex. ARN
MS2/protéine de l’enveloppe de MS2)
une partie correspondant à l’ARN à tester.
La protéine verte est, soit la protéine à tester pour son interaction avec l’ARN, soit la
transcription/traduction d’une banque d’ADNc.
L’ARN hybride est produit à partir de la transcription d’un plasmide qui présente l’ADN
correspondant sous la dépendance d’un promoteur inductible (Pmet25 par ex.).
D. SenGupta, B. Zhang, B. Kraemer, P. Prochart, S. Fields and M. Wickens. 1996. A three-hybrid
system for detecting RNA-protein interactions. Proc. Natl. Acad. Sci. 93, 8496-8501

24/10/2011 156
Méthode de Belasco (Jain et Belasco, 1996)
Le principe est semblable à celui du simple hybride mais les interactions se font chez E. coli (et non
chez la levure) et l’interaction se fait directement avec l’ARN (et non par l’intermédiaire de protéines
comme dans le cas du triple hybride).
La séquence d’ARN à tester est introduite (sous la forme d’un ADN double brin) en amont d’un site
de fixation du ribosome (RBS) lui-même en amont d’un gène β galactosidase. L’ensemble est porté
par un plasmide portant la résistance à l’ampicilline (plasmide reporter). Si l’interaction entre la
protéine à tester et notre ARN cible se fait, l’accès du ribosome au RBS sera empêché et il n’y aura
pas de traduction de la β galactosidase. Si, par contre, l’interaction ARN/protéine ne se fait pas, le
ribosome se fixera sur le RBS et la traduction de la β galactosidase pourra se faire. La présence de β
galactosidase sera vérifiée en présence de X gal.
transcription
traduction
β galactosidase
β galactosidase
ATG
AUG
RBS
RBS ARNm
Séquence à tester
β galactosidaseAUGRBS β galactosidaseAUGRBS
Il y a interactionentre la protéine à tester et l ’ARN
Il n ’y a pas interactionentre la protéine à tester et l ’ARN
ribosome
β galactosidase
X Gal X + Gal
ADN Promoteur

24/10/2011 157
Le plasmide rapporteur est donc cotransformé avec une banque d’ADNc clonée dans un vecteur
d’expression portant la résistance au choramphénicol. Les bactéries transformées sont alors
sélectionnées sur des boites en présence d’ampicilline, de chloramphénicol et de X gal. Si la
bactérie porte un plasmide d’expression avec un ADNc correspondant à une protéine qui fixe
notre séquence d’ARN, la bactérie sera blanche. Le cas échéant, la bactérie sera bleue. On
récupère donc les bactéries blanches, on extrait les plasmides qu’elles portent et on séquence
l’ADNc qu’elles contiennent. Il s’agit de l’ADNc d’une protéine interagissant avec notre ARN. N.B. Il faut utiliser des bactéries ΔLacZ, c’est à dire des bactéries délétées pour le gène codant
pour la β galactosidase.
Jain C. & Belasco G. (1996) A structural model for the HIV-1 Rev-RRE complex deduced from altered-specificity Rev variants isolated by a rapid Genetic Strategy. Cell, 87; 115-125 South-Western Cette méthode permet de caractériser des protéines se liant sur un ADN cible. On peut aussi par cette
méthode cartographier leurs sites de liaison (Lelong et al., 1989). Les protéines sont séparées sur un
gel dénaturant (gel SDS-PAGE ou gel 2D) puis renaturées en présence d’une faible concentration en
urée et transférées sur nitrocellulose par diffusion ou encore directement électrotransférée sur
nitrocellulose. Dans ce dernier cas elle se renaturent durant le transfert. L’ADN à tester est marqué
radioactivement à ses extrémités et incubé avec la membrane de nitrocellulose.
Les fragments d’ADN spécifiquement liés à la protéine testée peuvent être élués de chaque complexe
ADN-protéine, et analysés après amplification par PCR.
L’ADN peut être :
♦ un fragment de restriction
♦ un oligonucléotide (double ou simple brin
♦ un fragment PCR, …
Si au lieu de faire migrer de l’ADN sur le gel, on fait migrer des ARN, la méthode prend le nom de
North-Western (Kwon et al., 1993).

24/10/2011 158
II - Techniques permettant de déterminer la cible (ADN ou ARN) d’une protéine :
II – 1 : Technique du Selex
La technique de SELEX (Systematic Evolution of Ligand by Exponential Enrichment), encore
appelée méthode de sélection in vitro, est une technique qui permet le tri simultané d’un mélange
complexe (plus de 1015 molécules d’ARN ou d’ADN (simple ou double brin) pour une
caractéristique particulière (ici la liaison avec une protéine).
Nous ne présenterons ici que le SELEX réalisé sur des ADN cibles double brin.
Principe :
Tout d’abord, un mélange d’oligonucléotides est synthétisé. Ces oligonucléotides sont composés
d’une vingtaine de nucléotides aléatoires (25 dans l’exemple ci-dessous) encadrés par deux
séquences constantes à partir desquelles se feront les PCR. Au moins 1015 molécules doivent être
synthétisées pour avoir des chances d’avoir un pool suffisamment dégénéré.
5’ TGGGCACTATTTATATCAAC (N25) AATGTCGTTGGTGGCCC-3′
Ces oligonucléotides simples brins sont fabriqués par synthèse chimique. Une fois le premier brin
réalisé, le deuxième est fabriqué en hybridant un oligonucléotide complémentaire de la région 3’
constante : 5’GGGCCACCAACGACATT
5’ TGGGCACTATTTATATCAAC (N25) AATGTCGTTGGTGGCCC-3′ TTACAGCAACCACCGGG-5’
On fabrique alors le deuxième brin à l’aide de polymérase de Klenow en présence de dNTP.
5’ TGGGCACTATTTATATCAAC (N25) AATGTCGTTGGTGGCCC-3′ 3’ ACCCGTGATAAATATAGTTG (N25)TTACAGCAACCACCGGG-5’
La polymérase de Klenow (encore appelée grand fragment de l’ ADN polymerase I d’ E.
coli), est fréquemment utilisé à la place de cette dernière chaque fois que l’ on ne désire

24/10/2011 159
pas avoir l’ activité 5' -> 3' exonuclease de cette dernière. Il a été initialement été produit
par protéolyse à partir de l’ enzyme pleine taille. La protéase utilisée pour cela est la
subtilisine.
Aujourd’hui, ce fragment est exprimé directement dans la bactérie (E. Coli) à partir du gène tronqué
de l’ADN polymérase I
Parmi ce pool d’oligonucléotides dégénéré, la sélection des molécules interagissant avec la protéine à
tester se fait par chromatographie d’affinité, gel retard ou liaison sur filtre.
Chromatographie d’affinité ou techniques dérivées : la protéine cible est tagguée, biotinylée ou on
possède un anticorps dirigé contre cette protéine. On peut alors faire une chromatographie d’affinité,
un pull-down ou une Co-immunoprécipitation
Parce que le pool présente au départ une très faible proportion de molécules d’ADN capables
d’interagir avec la protéine appât, plusieurs cycles de sélection sont nécessaires. Une amplification
par PCR des fragments isolés est réalisée après chaque round de sélection.
Oligonucléotides utilisés dans notre exemple pour la PCR :
5’ TGGGCACTATTTATATCAAC
TTACAGCAACCACCGGG-5’
Plusieurs cycles successifs de sélection/amplification vont nous permettre d’augmenter de façon
exponentielle l’abondance dans le mélange des séquences fonctionnelles, jusqu’à avoir une majorité
de ce type de séquences. La stringence des tampons utilisés pour l’interaction augmente à chaque
cycle.

24/10/2011 160
Clonage et séquençage
Après chaque cycle de PCR, on vérifie par gel retard (EMSA) que l’on a bien augmenté l’affinité du
pool d’oligonucléotides pour notre protéine.
Enfin, on aligne (par bioinformatique les séquences obtenues -> séquence consensus)
II-2 : Immunoprécipitation de chromatine
A – Analyse par gel retard de l’interaction de la protéine X avec les ADN obtenus lors des premiers cycles de SELEX, B : alignement des séquences obtenues par bioinformatique, C : séquence consensus
A
Cycles SELEX 0 1 2 3 4 5
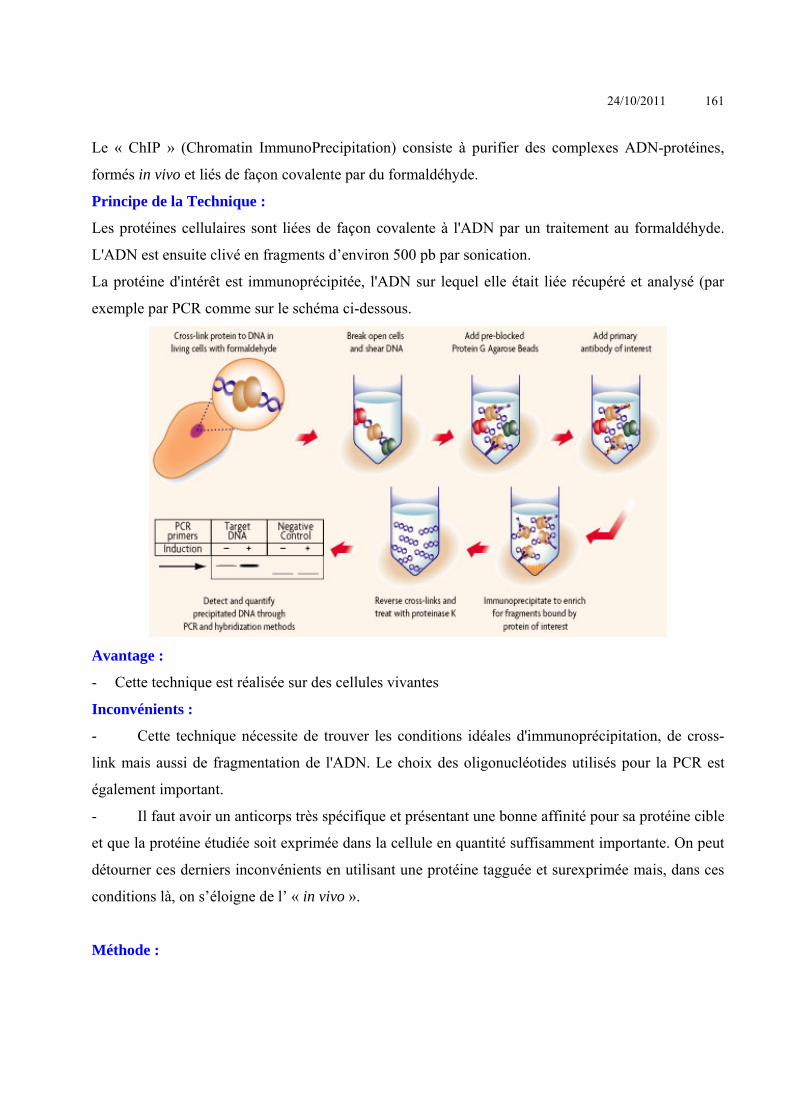
24/10/2011 161
Le « ChIP » (Chromatin ImmunoPrecipitation) consiste à purifier des complexes ADN-protéines,
formés in vivo et liés de façon covalente par du formaldéhyde.
Principe de la Technique :
Les protéines cellulaires sont liées de façon covalente à l'ADN par un traitement au formaldéhyde.
L'ADN est ensuite clivé en fragments d’environ 500 pb par sonication.
La protéine d'intérêt est immunoprécipitée, l'ADN sur lequel elle était liée récupéré et analysé (par
exemple par PCR comme sur le schéma ci-dessous.
Avantage :
- Cette technique est réalisée sur des cellules vivantes
Inconvénients :
- Cette technique nécessite de trouver les conditions idéales d'immunoprécipitation, de cross-
link mais aussi de fragmentation de l'ADN. Le choix des oligonucléotides utilisés pour la PCR est
également important.
- Il faut avoir un anticorps très spécifique et présentant une bonne affinité pour sa protéine cible
et que la protéine étudiée soit exprimée dans la cellule en quantité suffisamment importante. On peut
détourner ces derniers inconvénients en utilisant une protéine tagguée et surexprimée mais, dans ces
conditions là, on s’éloigne de l’ « in vivo ».
Méthode :

24/10/2011 162
Fixation au formaldéhyde, sonication et immunoprécipitation
Le formaldéhyde est une petite molécule de 2 Angström, c’est un agent de liaison qui va permettre de
produire un lien in vivo à la fois « acides nucléiques – protéines » et « protéines – protéines ».
Le formaldéhyde est un composé dipolaire très réactif dans lequel les atomes de carbones agissent
comme des centres nucléophiles. Les groupements « amines » et « imines » des acides aminés
(Lysines, Arginines et Histidines) et de l’ADN (Adénines et Cytosines) réagissent avec le
formaldéhyde pour former une base de Schiff. Cet intermédiaire peut alors réagir avec un second
groupe aminé et se condenser pour donner le complexe ADN-protéine final. Ces réactions se mettent
en place in vivo en quelques minutes après l’ajout de formaldéhyde sur les cellules vivantes. La
réaction est totalement réversible. Cette réaction est réalisée par protonation du groupe « imino » à
faible pH dans un milieu aqueux.
Un autre paramètre qui doit être considéré lors de la mise au point des conditions de fixation est le
fractionnement du matériel fixé. La sonication est une des possibilités (avec la digestion par les
endonucléases) pour solubiliser le matériel fixé, en particulier la chromatine.
Pour l’identification et la caractérisation in vivo des ADNs cibles d’une protéine donnée, après
immunoprécipitation de la chromatine « crosslinkée », l’ADN est purifié puis analysé par les
techniques conventionnelles telles que le southern blot, le séquençage, l’hybridation sur puces et
l’analyse par PCR. De telles analyses demandent l’élimination de toutes les protéines de la fraction
chromatine immunoprécipitée. Pour cela, le cross-link est réversé par chauffage en solution aqueuse
(65°C, au moins 4 heures) – on peut en plus traiter à la protéinase K pour éliminer les protéines puis
l’ADN est purifié par des méthodes standards (par exemple par une partition phénol/chloroforme/eau,
suivi d’une précipitation à l’éthanol).
2) Méthodes d’analyses
- Analyse par PCR :
Si on a une idée des régions chromosomiques sur lesquelles la protéine étudiée a pu se fixer, on peut
réaliser une analyse par PCR. Pour cela, on va dessiner des oligonucléotides qui s’hybrident de par et
d’autre de la séquence à tester (séquence sur laquelle on pense que s’est fixée la protéine étudiée)
puis on réalise une PCR à l’aide de ces amorces afin de tester la présence du fragment d’ADN
correspondant parmi les fragments précipités. Si la protéine était sur cette région de l’ADN, il y aura
amplification par PCR, sinon, on n’aura pas d’amplification.

24/10/2011 163
- Analyse par southern-blot
Les fragments d’ADN immunoprécipités sont marqués (éventuellement après LM-PCR) puis
hybridés par southern blot. Ce n’est bien sûr pas un génome total que l’on a fait migrer sur le blot
mais seulement différents fragments d’ADN que l’on veut tester.
- Hybridation sur « chip » - ChIP on chip ou ChIP to chip
Dans l’expérience appelée « ChIP to chip », tous les fragments d’ADN co-immunoprécipités avec la
protéine d’intérêt sont amplifiés. Ceci est réalisé en ajoutant par ligation des linkers classiques sur les
extrémités des fragments d’ADN réparés (LM PCR). Le matériel amplifié est alors hybridé à une
puce d’ADN (DNA microarray) portant les sondes appropriées – fragments d’ADN génomique ou
correspondant à des séquences promotrices. Ces expériences sont réalisées avec deux marquages, la
deuxième couleur correspondant à l’ADN total comme contrôle (Input). Chaque ADN enrichi par
l’immunoprecipitation est enregistré comme un site de liaison potentiel pour la protéine étudiée.
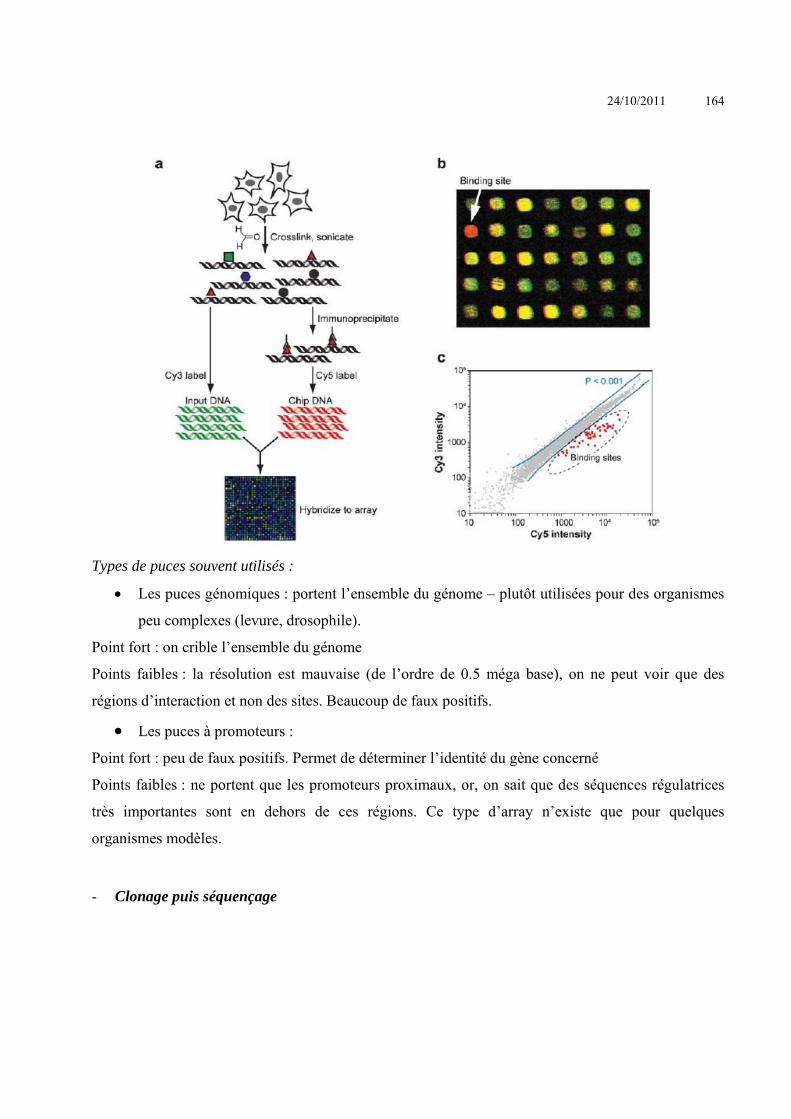
24/10/2011 164
Types de puces souvent utilisés :
• Les puces génomiques : portent l’ensemble du génome – plutôt utilisées pour des organismes
peu complexes (levure, drosophile).
Point fort : on crible l’ensemble du génome
Points faibles : la résolution est mauvaise (de l’ordre de 0.5 méga base), on ne peut voir que des
régions d’interaction et non des sites. Beaucoup de faux positifs.
• Les puces à promoteurs : Point fort : peu de faux positifs. Permet de déterminer l’identité du gène concerné
Points faibles : ne portent que les promoteurs proximaux, or, on sait que des séquences régulatrices
très importantes sont en dehors de ces régions. Ce type d’array n’existe que pour quelques
organismes modèles. - Clonage puis séquençage

24/10/2011 165
L’addition d’un linker permet l’amplification de la banque et le clonage des différents fragments
dans un vecteur d’expression. Clonage
Séquençage
Cette technique est sûrement la plus valable de toutes. Cependant elle est très lourde et onéreuse à
mettre en place. En effet, le nombre de clones à séquencer pour avoir une bonne idée de l’ensemble
des sites de fixation est très important (on estime le nombre de clones à séquencer à environ 100 000
pour l’analyse des sites d’interaction dans un génome humain).
SAGE appliquée au séquençage de fragments d’ADN génomiques
obtenus après immunoprécipitation de chromatine.
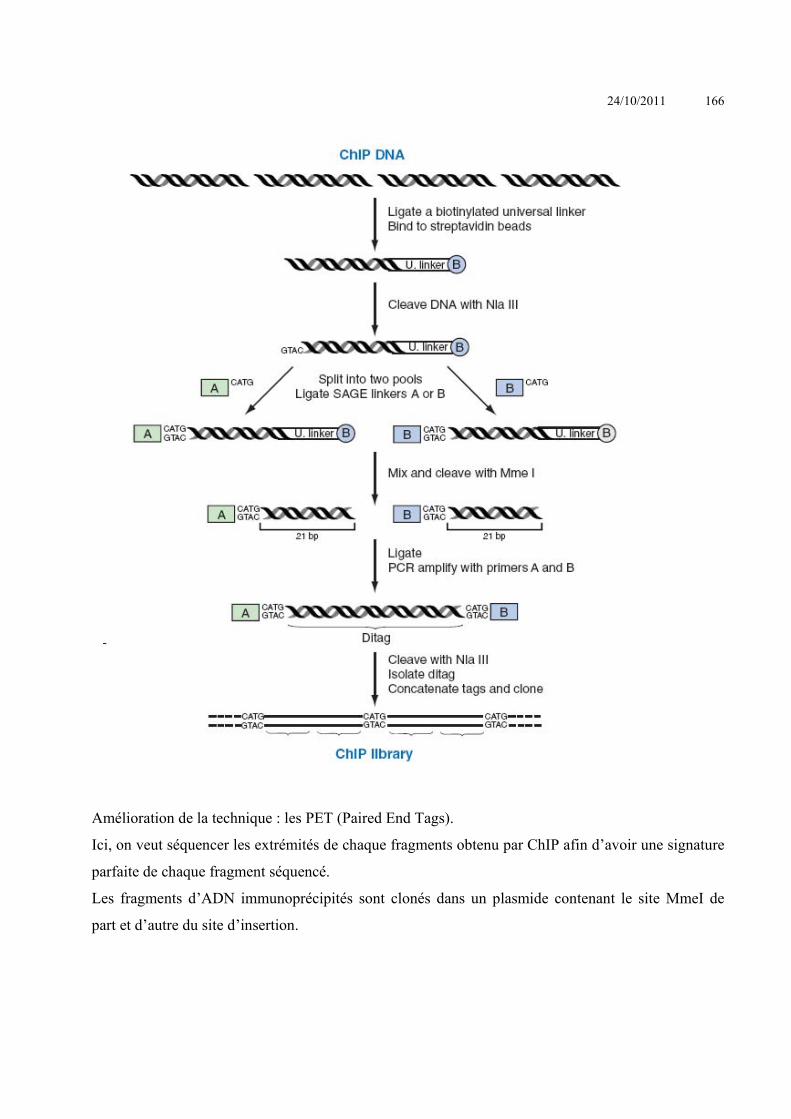
24/10/2011 166
Amélioration de la technique : les PET (Paired End Tags).
Ici, on veut séquencer les extrémités de chaque fragments obtenu par ChIP afin d’avoir une signature
parfaite de chaque fragment séquencé.
Les fragments d’ADN immunoprécipités sont clonés dans un plasmide contenant le site MmeI de
part et d’autre du site d’insertion.
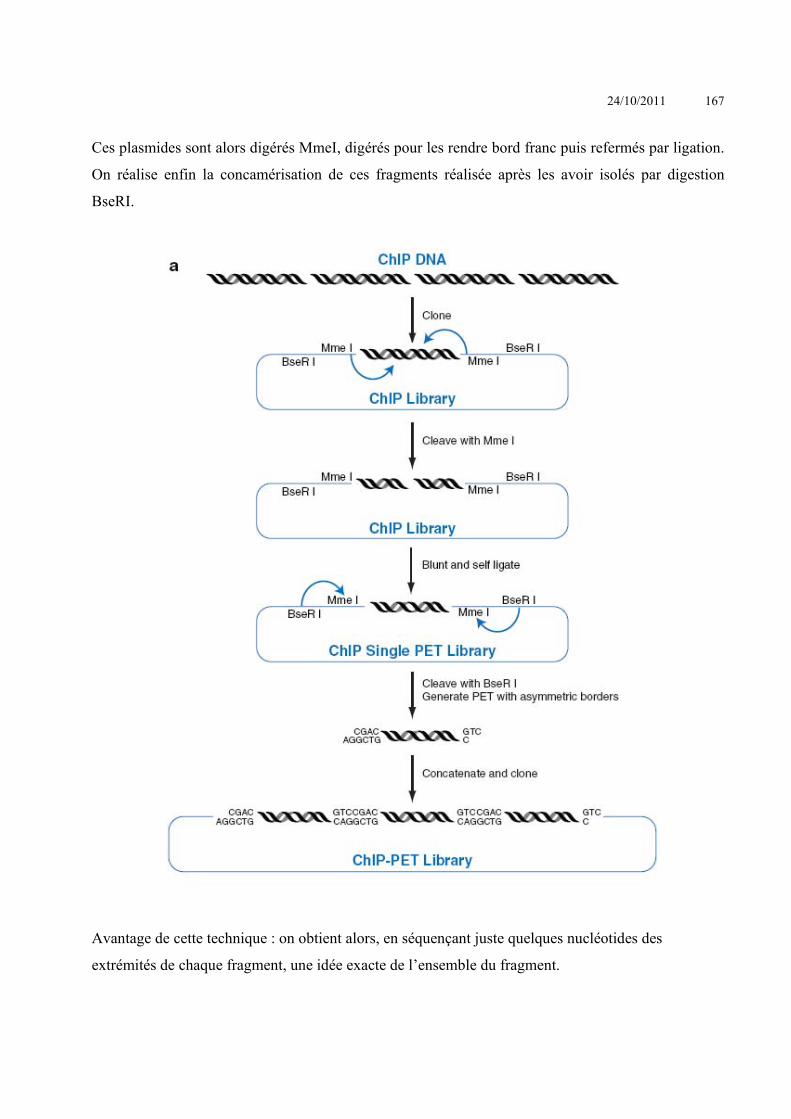
24/10/2011 167
Ces plasmides sont alors digérés MmeI, digérés pour les rendre bord franc puis refermés par ligation.
On réalise enfin la concamérisation de ces fragments réalisée après les avoir isolés par digestion
BseRI.
Avantage de cette technique : on obtient alors, en séquençant juste quelques nucléotides des
extrémités de chaque fragment, une idée exacte de l’ensemble du fragment.
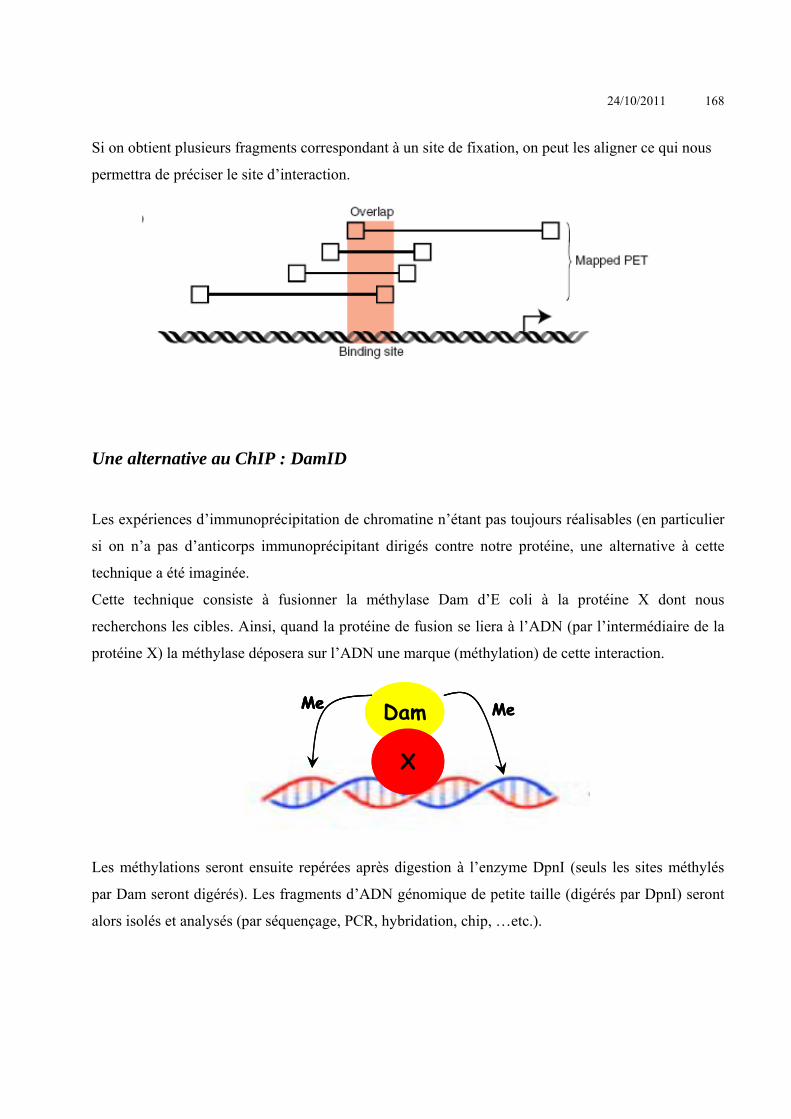
24/10/2011 168
Si on obtient plusieurs fragments correspondant à un site de fixation, on peut les aligner ce qui nous
permettra de préciser le site d’interaction.
Une alternative au ChIP : DamID
Les expériences d’immunoprécipitation de chromatine n’étant pas toujours réalisables (en particulier
si on n’a pas d’anticorps immunoprécipitant dirigés contre notre protéine, une alternative à cette
technique a été imaginée.
Cette technique consiste à fusionner la méthylase Dam d’E coli à la protéine X dont nous
recherchons les cibles. Ainsi, quand la protéine de fusion se liera à l’ADN (par l’intermédiaire de la
protéine X) la méthylase déposera sur l’ADN une marque (méthylation) de cette interaction.
Les méthylations seront ensuite repérées après digestion à l’enzyme DpnI (seuls les sites méthylés
par Dam seront digérés). Les fragments d’ADN génomique de petite taille (digérés par DpnI) seront
alors isolés et analysés (par séquençage, PCR, hybridation, chip, …etc.).
Dam
X
Me MeDam
X
Me MeDam
X
Me Me

24/10/2011 169
Afin d’être sûr que ces méthylation sont bien spécifique de l’interaction de la protéine X avec sa cible
ADN, une transfection de la méthylase Dam seule est réalisée en parallèle.
Le schéma ci-dessous nous montre un exemple d’expérience avec analyse des résultats par chip.
A gauche : cellules transfectées par un plasmide permettant de produire la protéine X fusionnée à la méthylase Dam.
A droite : cellules contrôle transfectées par un plasmide permettant l’expression de la méthylase Dam seule.

24/10/2011 170
Méthodes permettant d’étudier les interactions acide nucléique / protéine
Electrophoretic Mobility Shift Assay (EMSA) - gel retard
Principe de la Technique :
La technique du gel retard est basée sur le retard de migration dans un gel natif de polyacrylamide de
duplexes d'oligonucléotides de séquences courtes en présence de protéines (ou de complexes
protéiques) ayant la propriété de reconnaître spécifiquement la séquence d'intérêt. La variation de
migration des duplexes (ou sondes) complexés aux protéines par rapport aux duplexes libres est
suivie grâce au marquage radioactif au 32P des sondes nucléotidiques.
Le gel retard peut être réalisé à partir de protéines purifiées mais aussi à partir d’un extrait protéique
plus complexe (un extrait nucléaire par exemple).
La spécificité de reconnaissance des sondes marquées peut être testée par l'ajout en excès du même
duplexe non radioactif qui entrent en compétition avec la forme radioactive.
La présence de protéines spécifiques dans le complexe retardé peut être mise en évidence par l'ajout
d'anticorps spécifiques qui permettent un retard plus important sur le gel appelé " supershift ".
La même expérience peut-être réalisée avec de l’ARN.
* *
* ** *
ADN nu marqué en 5 ’
ADN complexé à une protéine
ADN complexé à deux protéines
Concentrations croissantes en protéine

24/10/2011 171
Footprint – empreinte sur l’ADN
Exonucléase A
L’exonucléase A est une enzyme qui a une activité 3’-5’ exonucléase, mais ne s’attaque qu’aux
extrémités proéminentes ou franches. Elle ne coupe ni l’ADN simple brin, ni les extrémités 3’
proéminentes.
Cette activité est utile pour déterminer les limites d’une interaction entre une protéine et un fragment
d’ADN.
Si on marque au 32P une seule des deux extrémités 5’ de notre fragment d’ADN, et si l’on traite celui-
ci à l’exonucléase III pendant qu’une protéine en protège une partie, l’exonucléase III ne pourra
digérer l’ADN que jusqu’à ce qu’elle entre en contact avec cette protéine qui l’empêche de
progresser dans la digestion. La même expérience sera réalisée avec le fragment d’ADN marqué sur
l’autre extrémité.
La taille de l’ADN protégé que l’on détermine sur un gel dénaturant haute résolution nous permettra
de déterminer où est localisée l’interaction ADN-protéine.
Un des avantages de l’empreinte à l’exonucléase III est que l’ADN non-protégé par une protéine est
entièrement dégradé; une interaction partielle ne génère pas de bruit de fond.
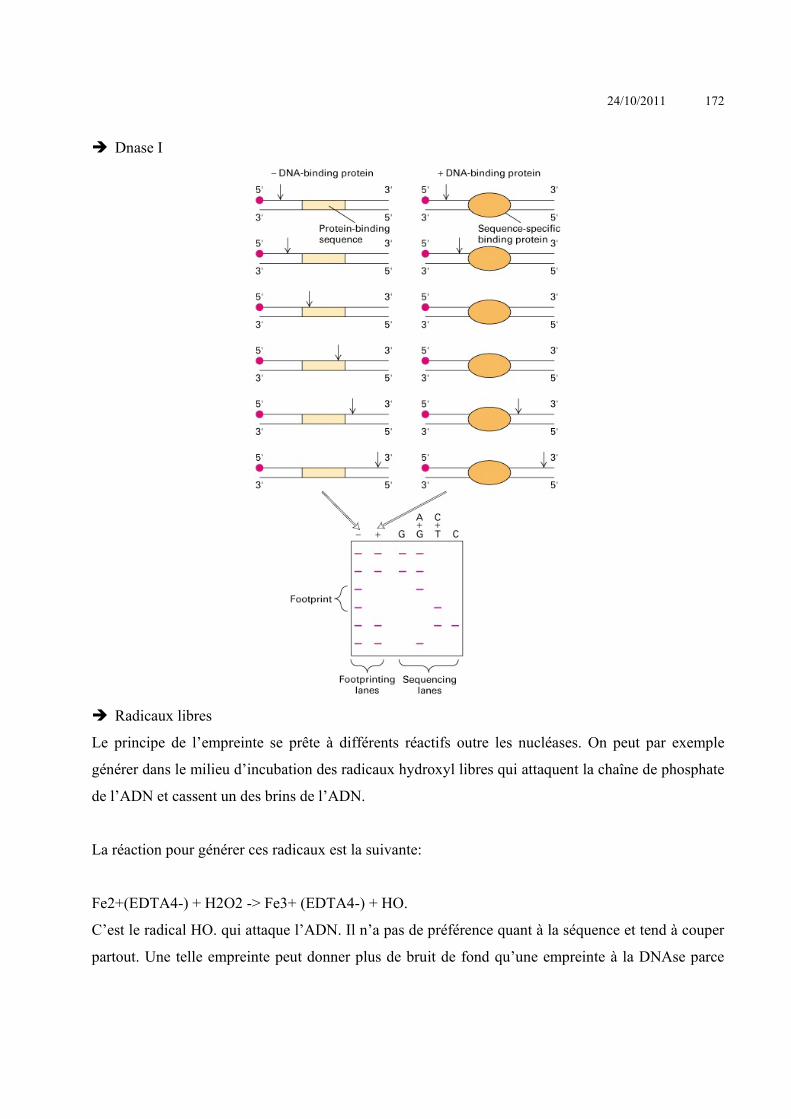
24/10/2011 172
Dnase I
Radicaux libres
Le principe de l’empreinte se prête à différents réactifs outre les nucléases. On peut par exemple
générer dans le milieu d’incubation des radicaux hydroxyl libres qui attaquent la chaîne de phosphate
de l’ADN et cassent un des brins de l’ADN.
La réaction pour générer ces radicaux est la suivante:
Fe2+(EDTA4-) + H2O2 -> Fe3+ (EDTA4-) + HO.
C’est le radical HO. qui attaque l’ADN. Il n’a pas de préférence quant à la séquence et tend à couper
partout. Une telle empreinte peut donner plus de bruit de fond qu’une empreinte à la DNAse parce

24/10/2011 173
que cette dernière, beaucoup plus massive, peut difficilement atteindre des sites situés sous la
protéine qui protège l’ADN, alors qu’une petite molécule comme HO. y arrive plus facilement.
D’autres radicaux existent pour ce type de réaction, comme par exemple le cuivre-phénanthroline.
Le permanganate de potassium (KMnO4) par exemple, est une molécule très réactive qui oxyde
préférentiellement les thymines d’ADN monocaténaire [Rubin et Schmid, 1980; Kahl et Paule,
2001]. Il peut ainsi détecter des portions de séquences d’ADN qui sont monocaténaires. Le KMnO4 a
été utilisé pour déterminer des distorsions dépendantes de la séquence [McCarthy et Rich, 1991], des
structures d’ADN en épingle à cheveux [Balagurumoorthy et Brahmachari, 1994] ou l’ADN de type
B et l’ADN à hélice triple [Jiang et coll, 1991].
Les autres
Presque toutes les techniques utilisées pour étudier les interactions ADN-protéines in vitro peuvent
aussi être utilisées in vivo. Il est juste un peu plus difficile de faire pénétrer les réactifs dans la cellule
et d’en récupérer l’ADN par la suite. Parmi les traitements disponibles pour effectuer des empreintes
in vivo, on trouve les empreintes :
(a) à la DNAse I (il faut alors perméabiliser les cellules pour permettre l’entrée de l’enzyme, et
s’assurer de ce qu’il y ait assez de Mg2+ présent dans le milieu);
(b) au diméthylsulfate ou DMS, qui pénètre spontanément dans les cellules et va causer une
méthylation des guanines non-protégées, une activité particulièrement efficace dans le sillon majeur
de l’ADN; les guanines méthylées peuvent alors être coupées par la piperidine;
(c) l’ UV footprinting, ou formation de dimères de pyrimidines par irradiation aux rayons ultraviolets
UVB (280-320nm) ou UVC (200-280nm). Ces dimères de pyrimidines peuvent être coupés par une
photolyase et la piperidine. Cette empreinte aux UV a l’avantage de ne pas toucher aux cellules du
tout, et permet donc d’étudier une cellule très près de son état naturel.

24/10/2011 174
La technique la plus populaire pour récupérer des fragments d’ADN après une réaction d’empreinte
in vivo est celle du LMPCR, ou ligation-mediated Polymerase chain reaction.

24/10/2011 175
LMPCR et footprint in vivo :
(1) Récupération de l'ADN génomique. Cet ADN contient des coupures simple-brin, là où la DNAse
I ou un autre agent comme le DMS, a endommagé la chaîne d'acides phosphoriques.
(2) Séparation des brins et annélisation de l'un d'eux avec une amorce 1 spécifique à une région qui
nous intéresse. L'autre brin pourra être étudié plus tard lors d'une autre expérience. Notez que les
fragments d'ADN dont nous disposons à cette étape sont de tailles différentes, parce que la coupure à
la DNAse I n'est que partielle.
(2) Extension de l'amorce 1.
(3) Ligation d'un adapteur double brin à l'extrémité franche. Comme l'autre extrémité n'est pas
franche, l'adapteur ne peut pas s'y attacher. Cet adapteur a une extrémité prohéminente, ce qui
l'empêche de se liguer plusieurs fois au bout de notre extrémité franche.
(5) PCR entre une deuxième amorce spécifique (l'amorce 2, à côté du site de l'amorce 1) et une
amorce (l'amorce 3) semblable à l'extrémité prohéminente de l'adapteur que nous avons ajouté à

24/10/2011 176
l'étape précédente. Le premier cycle de ce PCR ne fonctionne que dans un sens, c'est à dire à partir de
l’amorce 2, parce que l'amorce 3 n'a pas encore de séquence complémentaire. Cette séquence sera
synthétisée lors de ce premier cycle.

24/10/2011 177
BIAcore On mesure ici l’interaction entre deux partenaires moléculaires grâce à une machine mesurant
l’intensité d’un rayon de lumière. Cette machine est fabriquée par la compagnie Biacore, qui a
donnée son nom à l’appareil.
La SPR, ou résonance de plasmon, se produit quand un rayon lumineux est réfléchi sous certaines
conditions par un film conducteur à l’interface entre deux milieux d’indices de réfraction différents.
Dans le système Biacore (tm) ces milieux sont (1) la solution dans laquelle se trouvent les molécules
à analyser et (2) le verre d’une lame qui sert de senseur. Le film conducteur à l’interface des deux est
un très fin film d’or à la surface de la lame de verre.
La résonance de plasmon cause une réduction de l’intensité de la lumière réfléchie à un angle
spécifique de réflection. Cet angle varie avec l’indice de réfraction près de la surface du côté opposé
à la lumière réfléchie (c’est donc dire du côté de l’échantillon).
Dans le système Biacore (www.biacore.com) , la molécule appât est attachée à la feuille d’or, du côté
solution. Les molécules avec lesquelles elle peut interagir sont ajoutées en flot continu. Si l’une
d’entre elles interagit avec l’appât, la concentration locale en molécules augmentera dans la région
immédiate de l’appât, ce qui fera changer l’indice de réfraction local.
Le changement d’indice de réfraction aura pour effet de changer l’angle pour lequel la lumière
réfléchie perd un maximum d’énergie par résonance.
Le système détecte un changement et enregistre une interaction. La mesure du niveau d’interaction en
fonction du temps permet d’évaluer le taux d’association.
Une unité de résonance (RU) correspond à un changement de 0.0001° dans l’angle donnant une
intensité minimale de lumière réfléchie, ce qui pour la plupart des protéines correspond à un
changement de concentration de l’ordre de 1 pg/mm2 de surface sur la lame de verre. Le Biacore est
donc extrêmement sensible.

24/10/2011 178
Les puces en général…
ANNEXE :
Les puces à ADN (DNA microarray) permettent un criblage rapide et simultané d’un génome ADN.
Les puces à ADN consistent en un support solide (petite lame de verre comme celles utilisées en
microscopie traditionnelle ou membrane de nylon) sur lequel des milliers de fragment d’ADN sont
déposés de façon géométrique à l’aide d’une micropipette robotisée. Grâce à cette technique, chacun
des fragments d’ADN est représenté par un point sur le support (ou puce). Ils servent de sondes pour
fixer de façon très spécifique les fragments de gènes complémentaires (cibles), présents dans les
échantillons biologiques à tester : leur mise en contact permet de reconstituer la double hélice d’ADN
et ce phénomène (hybridation) peut être mis en évidence par des techniques optiques sous éclairage
fluorescent ou par détection de radioactivité; un système de « marquage » de l’échantillon au moyen
de traceurs fluorescents ou radioactifs ayant été réalisé préalablement. La quantification des signaux
obtenus et l’identification des fragments de gènes reconnus sont ensuite rendues possibles au moyen
d’un système d’acquisition d’image puis d’analyse des données faisant appel à des logiciels
informatiques spécialement conçus à cet effet. Les résultats obtenus sont ensuite validés sur le plan
statistique et interprétés dans un contexte biologique.

24/10/2011 179
Le potentiel de cette méthodologie est énorme, mais la masse de résultats qui résulte de ces
expériences est considérable et leur exploitation par le biais de programmes informatiques n’en est
encore qu’à ses débuts. De nombreux développements sont encore à faire au niveau des logiciels
d’analyse et de représentation afin d’extraire de ces données le maximum de sens sur le plan
biologique.
Le terme « puces à ADN » est un terme générique. Il existe actuellement 2 procédés majeurs de
fabrications de puces à ADN ce qui permet de distinguer différents types de puces : (1) les macro et
microarrays avec un dépôt direct de molécules d’ADN sur leur support (2) les puces à
oligonucléotides avec la synthèse in situ des sondes oligonucléotidiques sur une surface solide.
Les macroarrays ou filtres à haute densité :
Les dépôts (sondes) sont des clones d’ADNc ou des produits de PCR fixés à haute densité sur une
membrane de nylon (8 x 12 cm). Le marquage est le plus souvent radioactif et le criblage est réalisé
en excès de cible, on obtient ainsi une mesure de l’abondance relative de chacun des ARNm présent

24/10/2011 180
dans l’échantillon de départ. On parle de macroarrays jusqu’à une densité d’environ 25 fragments
ADN déposé par cm2.
filtres à haute densité ou macroarrays
Les microarrays :
Ils permettent la miniaturisation des dépôts d'ADN, ce qui permet de fixer plusieurs milliers
de sondes sur des surfaces égales ou inférieures à celle d’une lame de microscope standard. Elles sont
déposées à une densité de 1 000 sondes/cm2 par un robot sur des lames de verre au préalablement
traitées chimiquement, soit jusqu’à 12 000 sondes/lame.
Les sondes sont généralement des ADN double brin de longueur de 200 à 2000 bp amplifiés
par la technique de PCR mais récemment, des oligonucléotides longs (50-70 mers) ont également été
greffés sur la puce après leur synthèse.
Les cibles utilisées sont réalisées par transcription inverse, à partir d’ARN total ou messager
utilisant 2 fluorochromes différents (Cy3 et Cy5) ce qui permet d’hybrider simultanément 2 cibles sur
une même sonde. Les signaux d’hybridation sont analysés grâce à un lecteur capable de discriminer
les 2 fluorochromes et de générer deux images dont le niveau de gris représente l’intensité de la

24/10/2011 181
fluorescence lue. Si on remplace les niveaux de gris par des niveaux de couleur verte pour la
première image et rouge pour la seconde, on obtient en les superposant une image en fausses couleurs
composée de spots allant du vert au rouge en passant par le jaune.
Un excès du gène X dans l’échantillon marqué en rouge donnera un signal rouge ; un excès du
gène Y dans l’échantillon en vert donnera un signal vert ; une expression équivalente du gène Z dans
les deux échantillons donnera un signal jaune. On calcule ensuite le ratio des intensités de
fluorescence rouge/fluorescence verte pour chacun des spots ce qui permet de rechercher une
expression différentielle des gènes dans les 2 échantillons biologiques étudiés. Généralement, on fixe
la limite d’un ratio supérieur à 2 ou inférieur à 0,5 pour considérer qu’un gène est sur- ou sous-
exprimé dans une des cibles par rapport à l’autre.
On peut ensuite essayer de regrouper des gènes ayant le même profil d’expression sur
plusieurs expériences ayant un rapport biologique.

24/10/2011 182
Principe d'une hybridation sur microarrays
Les puces à oligonucléotides :
Elles dérivent à l’origine d’un projet de séquençage par hybridation. Les puces les plus couramment
utilisées sont les puces de la société Affimetrix. Dans ce cas, les sondes sont des oligonucléotides
synthétisés in situ par une technique de photolithographie. On peut synthétiser jusqu’à 300 000
oligonucléotides représentant 30 000 gènes sur une puce d’une surface d’environ 1 cm2. On hybride
une seule sonde par puce et l’intensité de fluorescence mesurée par un scanner permet une mesure de
l’abondance relative de chacun des ARNm présent dans l’échantillon biologique étudié.

24/10/2011 183
Interactions protéine - protéine
1. Avant propos
♦ Liaisons stables – liaisons transitoires
Deux protéines peuvent s’associer indépendamment de tout autre partenaire de deux manières
différentes :
Liaisons stables : toute association donnant naissance à un complexe que l’on peut isoler
(Attention, ne pas confondre avec liaison covalente ! une liaison peut-être stable sans être covalente).
Liaison transitoire : donnent des complexes trop peu stables pour que l’on puisse les isoler. Dans ce
cas là, l’isolement de ces complexes passera souvent par une étape préliminaire de cross-link
(pontage covalent réalisé par un traitement chimique ou physique).
La force de la liaison (affinité) est définie par des paramètres d’équilibre et en particulier par le Kd ou
constante de dissociation.
♦ Notion de Kd Kon Kon : constante de vitesse de formation du complexe A + B <====> AB Koff Koff : constante de vitesse de dissociation du complexe [A] x [B] AB
Kd = [A] [B] / [AB] = koff / kon = 1 / Ka (constante d’association)
Avec [A], [B], [AB] : respectivement concentration en A, B et AB
Quelques exemples de Kd :
streptavidine / biotine : 10-14 M Histone / ADN 10-11 M
Antigène / Anticorps : 10-8 à 10-10 M pour un bon anticorps, 10-6 M pour un anticorps plus faible
♦ Liens moléculaires
Les interactions protéine / protéine sont possibles grâce à la formation de liaisons non covalente. Ces liaisons sont de différente nature : • Liaisons ioniques - interactions qui relient deux atomes de charges opposées- la plus forte des
liaisons non covalentes

24/10/2011 184
• Liaisons hydrogène - se forment à chaque fois qu’un atome d’hydrogène lié à un atome
électronégatif est à proximité d’un autre atome électronégatif (en général oxygène et azote) -
L’énergie de liaison est moyenne
• Liaisons hydrophobes - se rencontrent lorsque deux molécules hydrophobes voisines se
rencontrent (par exemple deux acides aminés hydrophobes) - l’énergie de liaison est faible
• Forces de Van Der Waals – interactions électrostatiques entre deux atomes voisins. Doivent
être très proches car l’énergie de cette liaison est très faible.
2. Introduction
Les génomes de différentes espèces procaryotes mais aussi eucaryotes ayant été séquencés ces
dernières années, la majorité des efforts de la communauté scientifique se porte aujourd’hui sur
l'analyse du produit des gènes, les protéines. En effet, le nombre et la fonction biologique de la
plupart de ces molécules codées par les gènes ne sont pas encore connus. L’étude des protéines est
appelée la protéomique.
Comment accéder à la fonction (aux fonctions) d’une protéine X ?
Analyse in silico :, recherche de protéines présentant des
homologies de séquences, recherche de domaines connus (de
liaison à l’ADN par exemple), …
Etudes biochimiques : recherche d’une activité
enzymatique
Etudes de biologie cellulaire : cellules/compartiments
cellulaires dans lesquels la protéine est présente
Etudes de biologie structurale : recherche de domaines
structuraux de fonction connue
Etudes génétiques : inactivation du gène (KO, RNAi)
suivie d’étude du phénotype obtenu
Etudes biologie moléculaire : facteurs impliqués dans la régulation de ce gène ?
Comme les protéines fonctionnent le plus souvent (toujours ?) en réseaux, l'identification des
interactions entre protéines permet de mieux comprendre leur fonction (si la fonction du (des)

24/10/2011 185
partenaires de la protéine X est connue, cela nous donnera des informations sur sa propre fonction) et
permettra éventuellement de découvrir de nouvelles cibles thérapeutiques.
=> Etude de l’INTERACTOME
3 - Recherche de partenaires protéiques à partir d’une banque d’ADNc
Principe de ces techniques :
Les partenaires protéiques de la protéine d’intérêt (que nous appellerons la protéine appât) seront
recherchés à partir de banques d’ADNc. Ces ADNc seront transcrits puis traduits chez un hôte
particulier (la nature de l’hôte dépend de la technique choisie). L’interaction sera révélée par
différentes méthodologies décrites par la suite.
Intérêt de ces techniques :
une fois le partenaire identifié, on aura directement accès à son ADNc.
Simplicité de mise en œuvre
Inconvénients :
Les interactions binaires pourront essentiellement être étudiées
Les interactions sont réalisées avec des protéines hétérologues ou recombinantes (problème de
quantité relative des partenaires, de compartimentation de la cellule, de modifications post-
traductionnelles, …).
3.1 - Criblage en interaction sur colonies ou sur plages de lyses
Il s’agit d’utiliser dans un premier temps la très classique technique de criblage d’une banque d’expression, la seule différence repose sur le fait que le criblage ne fait pas par hybridation avec une sonde simple brin radioactive mais avec la protéine appât (que nous appellerons protéine X) au préalablement marquée. Les clones exprimant la protéine interagissant avec la protéine X seront donc repérés par sa visualisation (autoradiographie si la protéine X est marquée radioactivement). Les clones positifs, contenant l’ADNc des partenaires de la protéine X, seront identifiés par séquençage.

24/10/2011 186
Pour marquer la protéine appât, on peut la traduire in vitro en présence de méthionine 35S. On peut
aussi la biotinyler ou encore la produire chez E. coli en fusion avec une étiquette facilement
repérable. On peut enfin la repérer grâce à des anticorps.
La banque peut être réalisée dans un plasmide ou dans un phage (lamda par exemple). L’avantage du phage sur le plasmide est qu’il évite l’étape de lyse des bactéries. Avantages de cette technique : ceux déjà cités plus haut pour l’ensemble de ces techniques. Inconvénients de cette technique :
les interactions se faisant à partir de protéines fixées sur des filtres (nitrocellulose ou nylon),
l’efficacité de liaison entre la protéine X et ses partenaires est moins bonne.
Cette technique nécessite d’avoir la protéine X purifiée.
Si on veut tester un grand nombre de partenaires, il faudra cribler un grand nombre de boîtes.
3.2 - Phage display
Une alternative à cette technique consiste à faire du phage display, on peut alors faire un
enrichissement des phages positifs en milieu liquide, ce qui permet de tester un grand nombre de
clones sans augmenter la charge de travail.
Cette technique consiste à faire exprimer la protéine cible à la surface d’un bactériophage non lytique.
Ces phages seront utilisés pour infecter des bactéries E. coli.
Le bactériophage utilisé ici est le bactériophage filamenteux
M13. Pour faire exprimer nos protéines à tester en surface de
ce bactériophage, leur ADNc est cloné en fusion avec le gène
Criblage avec la protéine X marquée

24/10/2011 187
III qui code pour une protéine mineure de la capside qui est
exprimé à la surface du phage.
Le site de clonage se trouve en N terminal de la protéine du gène III afin que le motif protéique
variable soit tourné vers l’extérieur, donc accessible à une sélection. Entre la séquence codante du
gène III et les ADNc, on met une séquence « espaceur » afin que les protéines à tester puissent
adopter un repliement indépendant de celui de la protéine "III". Une banque d’ADNc est insérée dans
l’ADN du phage au niveau du site de clonage.
L’ADN phagique ainsi modifié est transformé dans des bactéries E. coli (avec en théorie un phage
donc une protéine recombinante par bactérie) puis les phages sont obtenus par infection avec le
bactériophage « helper » M13KO7. Ce bactériophage présente un génome modifié qui ne permet pas
sa réplication. Il permettra par contre la formation des bactériophages à partir des ADN phagiques
portant les ADNc. Les phages recombinants sont récupérés dans le surnageant de culture puis
sélectionnés par rapport à leur interaction avec la protéine pour laquelle on recherche des partenaires.
La sélection peut se faire de différentes façons en fonction des caractéristiques de la protéine pour
laquelle on recherche des partenaires (protéine appât) :
• Fixation directement sur un support activé
Les supports ou matrices utilisés pour la chromatographie d'affinité sont généralement des billes d'agarose ("Sepharose™", Pharmacia®). L'immobilisation du ligand sur la matrice nécessite qu'il soit préalablement activé, c'est-à-dire qu'il faut créer des sites réactionnels qui permettent d'établir des liaisons covalentes avec la protéine appât. • Colonne d’affinité
Si la protéine est étiquetée ou biotinylée, ou encore si on lui connait un ligand, on peut sélectionner
les phages positifs en réalisant une colonne d’affinité.
Les phages ainsi sélectionnés sont
utilisés pour réinfecter des souches
bactériennes, les surnageants sont
Particules virales exprimant des fragments d ’ADNc
Fixation surune protéine
cible
lavages
Élutions(augmente
conc. en NaCl par ex.)
Amplifications(infection d ’E. coli) 3 à 4 X

24/10/2011 188
récupérés et resélectionnés contre
la protéine. En répétant cette étape
de sélection plusieurs fois (en
augmentant la stringence de
l’interaction à chaque cycle), on
enrichit notre population en clones
produisant les protéines
recombinantes interagissant le
mieux avec l’appât. Ces candidats
sont alors isolés (par dilutions ou
sur boites – en effet, bien que le
phage ne soit pas
lytique, sa présence dans une bactérie diminue sa vitesse de division, on voit donc apparaître des
« pseudo » plages de lyse sur les tapis bactériens). L’ADNc du phage est alors séquencé, permettant
ainsi l’identification des les partenaires de la protéine X.
Avantages de cette technique : permet de cribler rapidement une grande quantité de clones. Inconvénients de cette technique : seules des interactions de forte affinité peuvent ainsi être étudiées (au moins 10-8M), nécessité d’avoir des quantités importante d’appât purifié.
3.3 - Double hybride ou « piège à interaction ».
Cette technique est basée sur la capacité des domaines de liaison à l’ADN (BD) et d’activation de la
transcription (AD) du facteur de transcription Gal4 à fonctionner de manière indépendante. Dans le
système du double hybride, ces deux domaines sont séparés et chacun est fusionné aux protéines
d’intérêt (X et Y). Ainsi, c’est l’interaction entre les deux protéines X et Y qui permettra de
reconstituer un facteur de transcription actif. Il y aura alors transcription de gènes rapporteurs.
Gène rapporteur : gène non présent dans la souche de levure utilisée et dont l’expression est
facilement repérable. Ex : lacZ : gène d’E. coli, non présent chez S. cerevisiae – facile à repérer
grâce à son activité enzymatique.
La séquence codant la protéine X (l’appât) sera fusionnée à la séquence codant pour le domaine de
liaison à l’ADN de Gal4 -> clonée dans un plasmide
La proie (protéine Y) correspondra soit à l’ADNc de la protéine dont on veut tester son interaction
avec la protéine X, soit une banque d’ADNc si aucune expérience préliminaire n’a encore été

24/10/2011 189
réalisée. Sa séquence sera fusionnée avec le domaine d’activation de la transcription de Gal4. ->
clonée dans un plasmide.
Le gène rapporteur, précédé du promoteur portant les sites de liaison de Gal 4 (répétés 6x), est
inséré dans le génome de la levure. Parmi les sites de liaison de Gal4, l’UAS de Gal1 est souvent
utilisé.
UAS : Upstream Activated Sequence
Les deux plasmides sont co-transformés dans la souche de levure appropriée présentant le gène
rapporteur sous la dépendance d’une UAS de Gal4 et déficiente pour les gènes d’auxotrophie utilisés
comme marqueurs de sélection.
Si le gène rapporteur est LacZ, les colonies positives seront capables de pousser sur milieu galactose
et, en présence de Xgal, les levures seront bleues.
Xgal : Le X-gal, ou 5-bromo-4-chloro-3-indolyl-beta-D-galactopyranoside est un
dérivé du galactose, lié à un noyau indole. Il peut être hydrolysé par la β-
galactosidase, produit du gène LacZ, en formant un composé bleu, ce qui permet de
détecter la présence de cette enzyme.
Le dosage de l’activité β galactosidase peut nous donner une idée de la force de l’interaction.
Les protéines X et Y n’interagissent pas.
Les protéines X et Y interagissent.
Xgal
ExpressionY X
Sites de liaison de Gal 4
Gène rapporteur Gal 4 BD
Gal 4 AD
ARN Pol II
Sites de liaison de Gal 4
X Gal 4 BD
Y Gal 4 AD
ARN Pol II Gène rapporteur
Pas d’expression

24/10/2011 190
Les levures utilisées pour ce double hybride seront donc : Gal4-, déficientes pour les gènes
d’auxotrophie utilisés, et porteront le gène rapporteur (sous la dépendance de Gal4) inséré dans
leur génome
Les limites de la méthode sont essentiellement le très grand nombre de clones artéfactuellement
positifs ou négatifs.
1 - Faux positifs (signaux « positifs » artéfactuels en double hybride).
Pour limiter le nombre de faux positifs, il est nécessaire, en préalable ou suite au criblage, d’effectuer de nombreux contrôles et d’utiliser comme « marqueur » d’interaction plusieurs gènes rapporteurs différents par la séquence de leur promoteur.
On utilisera, chaque fois que cela est possible, des contrôles positifs (deux protéines dont l’interaction a déjà été caractérisée) et des contrôles négatifs (des protéines dont on sait qu’elles ne présentent pas d’affinité l’une pour l’autre, transfection d’un plasmide vide avec l’autre plasmide recombinant). Faux positifs les plus fréquemment rencontrés :
Transactivateurs :
C’est la capacité de X ou Y à activer spontanément la transcription des gènes rapporteurs lorsqu’ils
sont fusionnés à BD ou AD.
1er cas : l’appât X est transactivateur, la transactivation peut être directe (a) ou se faire via une
interaction avec une protéine résidente de la levure (b)
Contrôles: expression indépendante de la protéine de fusion BD-X dans S. cerevisiae (témoin négatif)
2ème cas : la proie Y est transactivatrice. Ceci a lieu quand la protéine AD-Y est recrutée au
promoteur Gal de manière indépendante de la proie. Il y a 2 cas classiques :
1. liaison directe de Y au promoteur (a)
2. liaison à des protéines (b) de l’hôte complexées au promoteur (ex : TATA Binding Protéine)
(a) Y AD (b)
Leu2
Leu2
Leu 2
ZX
BD
X
BD
Y AD
(b)La protéine appât X lie une protéine de la levure capable d’activer la transcription
(a) la protéine appât a une activité transactivatrice

24/10/2011 191
Contrôles à effectuer : expression de AD-Y seul (témoin négatif)
Faux positifs dus à une affinité artéfactuelle des protéines proies et appât par les domaines
BD et AD
2 cas classiques :
1. La protéine proie reconnaît BD
2. La protéine appât reconnaît AD
Intervention d’une troisième protéine Z « adaptatrice » :
2 - Faux négatifs (incapacité à mettre en évidence par double hybride des interactions qu’on
sait exister in vivo).
La (les protéines) n’est (ne sont) pas produite(s) dans la levure
La protéine n’entre pas dans le noyau ⇒ Contrôle : Vérifier la présence et la localisation de nos
protéines de fusion dans les levures (par exemple en immunofluorescence)
Contrôle (témoins négatifs): combinaison AD-Y + BD
Contrôle : combinaison BD-X + AD
Y AD
TB
Contrôle avec techniques d’étude des interactions protéine/protéine in vitro (ex : GST-pull-down, co-immunoprécipitation)
X
BD
Y AD
X
BD
Y
AD
Y AD Z X
BD

24/10/2011 192
Certaines modifications post-traductionnelles sont absentes
Protéines chimériques mal repliées
Les protéines ne sont pas dans leur environnement naturel ⇒ pas d’interaction possible dans un
environnement autre (pH, concentration saline, … non adéquat)
⇒ Contrôle : tester, chaque fois que c’est possible, une proie connue pour interagir avec l’appât
Conclusion
♦ La méthode du double hybride permet de montrer que 2 protéines sont capables d’interagir de
façon binaire lorsqu’elles coexistent dans le noyau de la levure. La technique n’est pas suffisante en
soi pour valider une interaction.
♦ Le résultat doit être validé par d’autres types d’expériences complémentaires (co-
immunoprécipitation, pull-down)
♦ Nécessité d’une validation biologique : les deux protéines coexistent-elles dans l’organisme ?
L’interaction peut-elle avoir une signification biologique ?
Références Brachmann and Boeke (1997) Current opinion in biotechnology 8: 561-568 Vidal M. and Legrain P. (1999) Nucleic Acids Research 27, n° 4, 919-929; Fashena et al. (2000) Gene 250, 1-14 Suter B, Kittanakom S, Stagljar I. (2008) Curr Opin Biotechnol. Aug;19(4):316-23.
3.4 – Complémentation (PCA Protein fragment Complementation Assay).
Cette méthode permet de tester les interactions protéine-protéine à l’intérieur des cellules vivantes,
quelles qu’elles soient. Elle permet soit d’étudier une interaction entre des protéines connues, soit
d’identifier, à partir d’une banque d’ADNc, le (les) partenaires d’une protéine. Dans la stratégie PCA,
une enzyme ou une protéine facilement détectable soit directement par sa présence (protéine
fluorescente) soit par son activité (par exemple avec la β galactosidase). Cette protéine est appelée
« reporteur ». Elle sera alors séparée en deux fragments et les deux fragments seront fusionnés avec
les protéines dont on veut tester l’interaction (protéines A et B dans le schéma ci dessous). L’activité
de la protéine reporteur est alors conditionnelle de l’interaction entre les deux partenaires. Les deux
fragments de la protéine reporteur ne doivent pas à être capables de s’associer indépendamment des
protéines A et B.
Avantages de la stratégie PCA :

24/10/2011 193
Les interactions moléculaires sont visualisées plus directement – et non au travers d’événements
secondaires comme une activation transcriptionnelle -> moins de faux positifs/négatifs.
Ces expériences peuvent être réalisées dans tous organismes et types cellulaires.
La localisation cellulaire peut-être choisie, y compris au niveau des membranes.
Inconvénients de la technique PCA:
peut manquer de sensibilité si les constantes d’affinité entre les protéines à tester sont faibles.
L’interaction entre les protéines A et B doit permettre le rapprochement dans l’espace des deux
parties du reporteur – pour pallier à ce problème, on utilise des espaceurs – séquences de quelques
acides aminés (souvent des glycines) qui permettent une grande flexibilité dans le positionnement des
deux parties des protéines de fusion. On peut aussi tester les fusions en N et C terminal des protéines
reporteurs.
Les protéines reporteurs utilisées peuvent être de différentes natures, l’une d’elles est présentée
ci-dessous.
luciférase (Osawa et al, 2001)
Certains êtres vivants tels que la luciole (firefly) émettent
spontanément de la lumière par un phénomène appelé
bioluminescence.
La luciférase de type Firefly est une protéine monomérique de
61kDa très utilisée en imagerie de bioluminescence. Cette
protéine catalyse l’oxydation de la luciférine selon les réactions
décrites dans la figure ci-contre: l’activation de la luciférine par
la luciférase en présence d’ATP permet la formation d’un
complexe instable qui émettra de la lumière lors de son retour à
un état fondamental.
Réaction d’oxydation de la luciférine par la luciférase Firefly
Ici, la luciférase sera séparée en deux parties : la partie N terminale (acide aminés 1 à 437)
et la partie C terminale (acides aminés 438 à 544).
Ces deux parties ne sont pas capables de s’associer.

24/10/2011 194
L’activité de la luciférase est détectée par la mesure de la bioluminescence émise en présence de
luciférine.
Ozawa T, Kaihara A, Sato M, Tachihara K, Umezawa Y. 2001 Anal Chem. 73(11):2516-21.
4 Recherche de partenaires protéiques à partir d’un extrait protéique : Co-
immunoprécipitation (co IP)
4 – 1 : Principe général : La co immunoprécipitation est une technique qui consiste à isoler un complexe protéique en utilisant un anticorps dirigé contre un des membres du complexe. Si on recherche les partenaires de la protéine X, on va donc utiliser un Ac antiX qui nous permettra de l’immunoprécipiter. Avec elle seront isolés ses partenaires protéiques. Ces protéines seront alors identifiées. L’anticorps utilisé sera de préférence un anticorps polyclonal, afin d’éviter que l’épitope reconnu par l’anticorps ne soit masqué dans le complexe. L’immunoprécipitation se fera alors en utilisant de la protéine A ou de la protéine G, couplée à des billes de sépharose, d’agarose ou des billes magnétiques. Le choix protéine A / protéine G dépend de l’anticorps que l’on va utiliser (ex. protéine A pour un IgG préparé chez la souris).
Les protéines A et G sont des protéines recombinantes d’origine microbienne qui présentent la capacité de fixer les molécules d’immunoglobuline de mammifère. Ces protéines sont couplées de façon covalente à des billes de sépharose. L’interaction entre ces protéines et les immunoglobulines n’est pas équivalente pour toutes les catégories d’anticorps : Antibody Protein A Protein G Human IgG S S W = interaction faible Goat IgG W S S = interaction forte Chicken IgG NB NB NB = pas d’interaction; — non testé Si les Ig dont nous disposons pour faire l’immunoprécipitation ne se fixent ni sur la protéine A, ni sur la protéine G, il est toujours possible de faire un « sandwich » : 4 – 2 : le matériel de départ :
Le matériel de départ correspond à un extrait protéique qui sera réalisé par une lyse des cellules,
suivie d’une sonication (pour casser l’ADN, ce qui permet de diminuer la viscosité de la solution)
puis d’une clarification par centrifugation (afin d’éliminer les débrits cellulaires). Si le nombre de
protéines ainsi isolées est trop important et si on connaît la localisation de la protéine appât dans la
Protéine A
Anticorps
Protéinecible
PartenaireBill
e de
séph
aros
e

24/10/2011 195
cellule, on peut simplifier le système en faisant du fractionnement cellulaire. Le même protocole
précédemment décrit sera alors appliqué après fractionnement.
4 – 3 : Méthodologie : Pratiquement, on va dans un premier temps réaliser l’interaction extrait protéique/protéineA ou G
fixée sur les billes. Cette étape va nous permettre d’éliminer toutes les protéines de l’extrait qui se
fixent sur les billes de façon non spécifique (clearing).
On va alors centrifuger et récupérer le surnageant. Puis on va ajouter l’anticorps dirigé contre notre protéine puis, enfin, un nouveau lot de billes protéine A/ G. On va à nouveau centrifuger (ou passer sur le portoir aimanté) et récupérer les billes que l’on va laver afin de se débarrasser de toutes les interactions non spécifiques.
Enfin, il faudra éluer les complexes des billes. Différentes techniques sont alors possibles en fonction du type d’analyse que l’on veut réaliser sur les co-immunoprécipitats (reprendre dans du tampon SDS/DTT et chauffer si on veut faire une séparation sur gel polyacrylamide dénaturant, élution par la force ionique si on ne veut pas récupérer notre protéine, élution par un peptide compétiteur, élution par choc acide, …) 4 – 4 : Identification des protéines du complexe : Une fois les complexes isolés, elles devront être identifiées. Cependant, une étape de séparation des
membres du complexe est généralement utilisée comme étape préliminaire.
Séparation des protéines
Gel 1D (dénaturant, de type SDS-PAGE)
C’est la façon la plus simple de procéder mais on peut être limité par la qualité de la résolution (bandes trop proches sur le gel pour pouvoir être séparées). Pour améliorer la résolution du gel, on peut jouer : Sur le % en acrylamide, éventuellement en gradient Sur le tampon de migration (tris tricine par exemple) Sur la taille du gel Une fois les protéines séparées, il faut les identifier. Identification des protéines
Western blot
Si on présume de la présence d’une protéine partenaire P déjà connue, on peut faire un western blot à l’aide d’un anticorps anti-P. Si les protéines ont été récupérées par co-immunoprécipitation, on peut, dans certains cas, rencontrer un autre problème : la protéine que l’on veut détecter migre à la même vitesse que les chaînes lourdes ou légères des anticorps qui nous ont servi à immunoprécipiter. Dans ce cas là, deux solutions sont possibles.

24/10/2011 196
Utiliser des anticorps secondaires qui détectent la forme native (avec les ponts disulfide) des
immunoglobulines, permettant ainsi de ne pas révéler les anticorps qui ont permis de réaliser
l’immunoprécipitation qui eux sont dénaturés (traités au SDS).
Une deuxième possibilité consiste à utiliser pour le western blot un anticorps réalisé dans une
espèce différente de celle qui a été utilisée pour fabriquer l’anticorps utilisé en immunoprécipitation.
Digestion trypsique / Spectroscopie de Masse Maldi-ToF
La première étape consiste à découper la/les bandes d’acrylamide qui contiennent des protéines. Pour cela, il faut colorer le gel. Le bleu de coomassie est le plus souvent utilisé pour cette étape. Une alternative à cela consiste à analyser l’ensemble du gel. Pour cela, la piste de migration sera découpée en bandes de tailles égales et chacune de ces bandes sera analysée. Les bandes découpées sont alors digérée à la trypsine (ou toute autre protéase spécifique). La masse précise de l’ensemble des peptides obtenus est alors déterminée par spectrométrie Maldi ToF (ou autre spectrophotomètre de masse présentant ce type d’ionisation). La comparaison avec des banques de données nous permet d’identifier la protéine recherchée. Si des ambiguïtés restent à lever, un séquençage par LC/MS/MS est possible, il nous permettra d’obtenir la séquence de 5 à 10 acides aminés. La digestion trypsique est alors toujours une digestion totale et la séparation des peptides est réalisée grâce à un système de chromatographie en phase liquide (LC) qui est couplé au Spectro de Masse (en tandem). Le premier étage de MS sert à sélectionner un ion, et le second analysera les ions issus de la fragmentation de celui-ci. La fragmentation se réalise par coupure au niveau des liaisons peptidiques principalement (et ce à partir des deux extrémités). Il est alors possible de déduire de l'ensemble des ions obtenus la séquence peptidique lue simultanément dans les deux sens. 4 – 5 : Avantages/inconvénients : Avantages de la co IP ::
Le complexe s’est formé in vivo avec de la protéine «endogène» .
L’affinité Ag/Ac est généralement très forte et spécifique.
Inconvénient de la coIP :
Il faut avoir un Ac immunoprécipitant c'est-à-dire un anticorps reconnaissant la protéine appât
au sein d’un complexe avec à la fois une forte affinité et une grande spécificité.
Un autre inconvénient est la quantité de protéine appât endogène dans les cellules. Si elle est
trop faible, la quantité de complexe isolée ne sera pas suffisante pour isoler les complexes.
Dans le cas de complexes de tailles très importantes, le/les épitope(s) peuvent être masqués,
on ne pourra pas alors utiliser cette méthode.

24/10/2011 197
Pour cette technique également, il faudra réaliser différents contrôles et en particulier extrait
+billes protéine A/G+Ac non spécifique.
4 – 6 : Alternatives à la Co Immunoprécipitation : - Chromatographie d’affinité, pull down.
Quand l’immunoprécipitation n’est pas possible (on n’a pas d’anticorps utilisable pour
l’immunoprécipitation ou la protéine appât est en concentration trop faible dans nos cellules pour
récupérer suffisamment de complexes pour leur analyse ultérieure), on utilisera des variantes à cette
techniques que sont les chromatographies d’affinité ou les « pull down » (chromatographies d’affinité
pour lesquelles la phase stationnaire n’est pas insérée dans une colonne mais est récupérée par
centrifugation.
Pour la purification d’un complexe protéique associé à une protéine X, on utilise un support solide constitué de billes d’agarose ou de sépharose sur lesquelles on a greffé un ligand présentant une forte affinité avec la protéine X.
Ces billes seront alors introduites dans une colonne. Ensuite on fait passer l’extrait de protéine contenant la protéine X. Après différents lavages, l’élution se fera le plus souvent à l’aide d’un compétiteur. Une autre possibilité est d’utiliser un gradient de force ionique. Dans ce dernier cas, sortiront les premières les protéines faiblement associées dans le complexe (souvent par des liaisons indirectes), puis celles plus fortement liées.
Si la protéine appât ne possède pas naturellement de ligand connu, on utilisera une protéine X taguée. C tag sera éloigné de notre protéine grâce à un espaceur qui permettra l’interaction quelque soit la taille et la géométrie du complexe. On a alors deux possibilités : soit on transfecte (transforme) nos cellules par un vecteur d’expression portant l’ADNc de la protéine appât fusionné avec une (ou plusieurs) étiquette(s), soit la protéine tagguée aura été au préalablement préparée. Dans ces cas là, c’est le ligand du tag qui sera fixé sur les billes. Si on a inséré un site reconnu par une protéase spécifique entre le tag et la protéine X, l’élution pourra se faire par digestion de ce site. L’inconvénient de ces deux variantes est qu’on ne travaille plus sur une protéine sauvage mais sur une protéine modifiée. La quantité de protéine appât est généralement plus importante que la protéine endogène ce qui peut engendrer des faux positifs.
♦ Pull down
Une variante de la chromatographie d’affinité est utilisée mais, au lieu de retenir les billes dans une colonne, on les sédimentera par centrifugation. Une autre variante consiste à utiliser des billes magnétiques. La récupération se fera alors par l’intermédiaire d’un aimant. Ex. GST pull down, billes magnétiques, complexe formé in vitro.
Support solide
Protéinecible
PartenaireTag

24/10/2011 198
GST : glutathione Sulfo Transférase – fusionné avec la protéine X – entre les deux, un site de digestion à la thrombine.
Si on ne veut pas marquer la protéine Y, on peut la détecter par western.
La même technique est utilisée pour aller « pêcher » des partenaires de la protéine X dans un
extrait protéique complexe.
! Penser aux témoins : billes + extraits, billes + GST + extraits car des interactions non spécifiques de
ce type peuvent être obtenues et ce, indépendamment de la protéine X.
Autres systèmes utilisés : steptavidine/biotine, Ni/6xHis, maltose/MBP, …
Avantage de cette technique : simple et rapide
Inconvénients :
Dans le cas des GST-pull-down, la taille importante de la GST peut entrainer un
encombrement stérique qui peut gêner la fixation de certains membres du complexe. On peut dans ce
cas là essayer de mettre le tag en C-terminal si il était en N (ou vice-versa). On peut aussi mettre un
« espaceur » entre le tag et notre protéine. Enfin, on peut aussi essayer avec un autre tag plus petit
(flag, 6xHis, …)
Souvent des « faux positifs », valider la validité de ces interactions par une autre technique, si
possible « in vivo ».
Quelle que soit la technique utilisée, les interactions doivent ensuite être vérifiées in vitro et même de préférence in vivo afin de
S’assurer de la validité de l’interaction
Vérifier si cette interaction est directe ou indirecte.
Autoradiographie Coloration coomassie
Protéine X, fusionnée à la GST Bille de glutathione sépharose Protéine Y, radioactive Protéines compétiteurs
Fixation et lavage
SDS-PAGE
On veut tester la liaison de la protéine
X avec la protéine Y. Pour cela, on va
:
1 - fusionner la protéine X avec la
GST et marquer la protéine Y.
2 - fixer la protéine X sur des billes de
glutathione sépharose
3 - ajouter la protéine Y en présence

24/10/2011 199
5 Etude/vérification des interactions protéine/protéine.
5.1 -> Vérification par une des techniques précédemment citées
Pour tester l’interaction entre une protéine X et une protéine Y ou vérifier la validité d’un partenaire
identifié par l’une des techniques précédemment citées, toutes les techniques permettant de
rechercher des partenaires protéiques peuvent être utilisées. Cependant, dans le cas de la vérification
d’une interaction, il faudra changer de type d’expérience mais aussi inverser la nature de l’appât et de
la proie (ex. : si la protéine Y a été identifiée comme partenaire de la protéine X en utilisant cette
dernière comme appât, la vérification se fera en prenant la protéine Y comme appât).
Pour vérifier si l’interaction est directe, on fera un pull down à partir des deux partenaires purifiés.
5.2 -> Vérification de l’interaction in vivo
♦ Colocalisation par immunofluorescence
Rappels sur l’immunofluorescence : L'immunofluorescence est une technique qui permet la
détection et la localisation d'une ou plusieurs protéines grâce à l'utilisation d'anticorps spécifiques. La
réalisation d'un marquage passe par différentes étapes : (i) prétraitement (fixation et perméabilisation)
du tissu ou des cellules, (ii) ajout de l'anticorps primaire puis lavages, (iii) ajout de l'anticorps
secondaire spécifique de l’anticorps primaire couplé à un fluorochrome puis lavages, (iv) révélation
du signal fluorescent à l'aide d'un microscope de fluorescence ou par microscopie confocale.
L'immunofluorescence est utilisée sur les
cellules en culture (on parlera alors
d'immunocytochimie) ou sur des coupes de
tissus (immunohistochimie).
I ii iii iv
Utilisation de l’immunofluorescence pour faire de la colocalisation de protéines :
L’immunofluorescence sera réalisée de la même façon que décrite précédemment mais on met les
deux anticorps primaires dirigés contre les deux protéines dont on veut tester la colocalisation puis les
deux anticorps secondaires dirigés contre les deux anticorps primaires et portant des fluorochromes
différents. Il est impératif que les deux anticorps primaires soient de types différents afin que l’on
puisse différencier les deux signaux et que les deux fluorophores aient des spectres d’émission

24/10/2011 200
suffisamment éloignés. L’acquisition des deux signaux se fait indépendamment puis on superpose les
deux images pour voir s’il y a colocalisation.
Dans l’exemple montré ci-contre, la première protéine est détectée avec
un anticorps préparé dans la chèvre et la deuxième avec un anticorps
préparé dans la souris. L’anticorps secondaire permettant de révéler la
première protéine est un anticorps anti anticorps de chèvre couplé à la
Cy3 (fluorescence rouge) et l’anticorps secondaire permettant de révéler
la deuxième protéine est un anticorps anti anticorps de souris couplé au
FITC (fluorescence verte). La superposition des deux images montre un
signal jaune, ce qui nous permet de valider l’hypothèse d’une
colocalisation de ces deux protéines.
Avantages de cette technique : la visualisation de la colocalisation se fait in vivo sur des protéines
endogènes.
Inconvénients :
Il faut avoir des anticorps spécifiques des deux protéines à tester, les anticorps doivent pouvoir
être différenciés (organismes à partir desquels ils ont été préparés différents ou isotypes différents
IgG pour un et IgM pour l’autre par exemple).
La concentration de chacune des protéines doit être suffisamment importante pour pouvoir
être révélée par cette technique qui n’est pas très sensible.
Parfois, on fait de l’immunofluorescence directe. Dans ce cas particulier c’est l’anticorps primaire qui
porte le fluorochrome, la révélation se fait alors sans passer par la troisième étape. Ceci permet d’une
part de diminuer le bruit de fond dû à des interactions non spécifiques des divers anticorps utilisés et
d’autre part permet de faire de la colocalisation alors que les deux anticorps dont nous disposons sont
du même type (IgG de type 1 de souris par exemple).
♦ Protéines de fusion
Dans le cas où des anticorps ne sont pas disponibles ou si la quantité de protéine à tester est
insuffisante pour la détection en immunofluorescence, une alternative est l’utilisation de protéine de
fusion entre la protéine d’intérêt et des protéines naturellement fluorescentes comme la GFP (Green
Fluorescent Protein) ou un de ses mutants (BFP – Blue Fluorescent Protein, CFP - Cyan Fluorescent
Protein ou encore YFP – Yellow Fluorescent Protein). Le gène de la GFP a été initialement identifié

24/10/2011 201
chez la méduse Aequorea Victoria. Les autres protéines (BFP, CFP, YFP, …) ont été produites par
biotechnologie (mutagénèse de la séquence sauvage).
Avantage de cette technique : la visualisation est directe, on n’a donc plus de problèmes de bruit de
fond lié à des interactions non spécifiques des anticorps (primaires et secondaires avec d’autres
constituants cellulaires). D’autre part, comme on contrôle la quantité de protéine produite, on n’est
donc plus limité par cette quantité.
Inconvénients : on ne travaille plus avec des protéines endogène ni à des concentrations
physiologiques. On n’est donc pas assuré d’avoir la localisation naturelle des protéines à tester.
Pour ces deux dernières techniques, l’interaction n’est pas prouvée, on montre juste que cette
interaction est possible de part la localisation des partenaires. Ces expériences ne sont donc en
aucun cas suffisantes pour démontrer une interaction.
♦ FRET (Föster Resonance Energy Transfer)
Le FRET est une technique qui permet de détecter la proximité de deux molécules. Chacune de ces molécules porte un groupement fluorescent et la technique repose sur le transfert, par résonance, de l’excitation de l’un de ces groupements (le donneur) à l’autre (l’accepteur) sans émission d’un photon. Pour cela, les deux partenaires de l’interaction (protéine A et B) doivent être conjugués avec un couple de fluorophores de couleurs différentes (donneur et accepteur). On excite ensuite le donneur A. Quand l’interaction entre A et B amène les colorants fluorescents à faible distance l’un de l’autre, un signal de fluorescence nouveau (issu de l’émission du receveur B) est développé. La présence de ce signal confirme la faible distance et donc l’interaction entre A et B. Ces interactions peuvent être suivies en temps réel grâce à une caméra jointe au microscope à fluorescence. Pour réaliser le transfert d’énergie de fluorescence, les spectres d’absorption de la première molécule
fluorescente (le donneur) et de la deuxième molécule fluorescente (l’accepteur) doivent se
superposer.
La famille des GFP (Green Fluorescent
Protein) offre différentes paires de mutants
utilisables pour des expériences de FRET.
Par exemple, le mutant appelé EGFP
(Enhanced GFP) et le mutant appellé BFP
(Blue Fluorescent Protein).

24/10/2011 202
Avantage de cette technique : La visualisation de l’interaction se fait dans les cellules vivantes
Inconvénients : on ne travaille pas sur des protéines endogènes.
♦ BRET
Une variation de cette technique est le BRET (Bioluminescence Resonance Energy Transfert).
Dans cette variante, le « donneur » est une molécule capable d’émettre des photons. Les photons émis
iront, si la distance entre le donneur et l’accepteur est suffisamment faible, exciter l’accepteur.
Généralement, le donneur est la luciférase de Renillla, qui sera fusionnée à une protéine d’intérêt
(protéine A). La GFP est fusionnée à l’autre protéine d’intérêt (protéine B), susceptible d’interagir
avec la protéine A. La luciférase est excitée à l’aide d’un substrat qui pénètre dans la cellule (la
cœlenterazine).
La luciférase de Renilla, initialement isolée à partir de l’organisme Renilla Reniformis (pensée de
mer) est une enzyme de 36 kDa qui catalyse l'oxydation de la coelentérazine en présence d’oxygène.
Cette réaction catalytique produit une lumière bleue avec un pic d'émission à 480 nm.
Si les deux protéines n’interagissent pas, on ne détecte que le signal émis par la luciférase. Si les deux
protéines interagissent, il y a transfert d’énergie entre la luciférase et la GFP et l’on peut mesurer un
signal supplémentaire émis par la GFP à 530 nm. Le transfert d’énergie reflète une interaction
physique étroite : pour qu’il y ait transfert d’énergie, la distance entre la luciférase et la GFP doit être
inférieure à 100 Å.

24/10/2011 203
Avantage de cette technique : Les mêmes que pour le FRET mais, en plus, le BRET est plus sensible
que le FRET et elle permet de s’affranchir d’une source excitatrice extérieure évitant ainsi les
problèmes de photoblanchiment des GFP, d’effet de filtre des milieux à la longueur d’onde
d’excitation du donneur ou encore de l’excitation croisée de l’accepteur.
Inconvénients : Les mêmes que pour le FRET
6 - Cross-linking (association par des agents pontants) On l’utilisera en complément d’une des techniques décrites ci-dessus chaque fois que l’on voudra
stabiliser une interaction.
Le principe consiste à associer de façon covalente la protéine à tester avec son partenaire avant
d’isoler le complexe. Cette technique nous permet de révéler des complexes présentant des
interactions de faible affinité. Elle peut aussi se réaliser in vivo, à condition que les agents cross-
linkant utilisés traversent les membranes de la cellule. Enfin, en utilisant un ensemble d’agents
pontants qui agissent à des distances variables, on pourra positionner les différents constituants au
sein d’un complexe multimoléculaire.
Les agents cross-linkant chimiques :
Il existe un très grand nombre d’agents cross-linkant chimiques. Ils se distinguent :
• Par la nature du résidu impliqué dans le cross-link (amine, sulfhydryls, carboxyls, hydroxyls,
…)

24/10/2011 204
• Par la distance entre les deux espèces à cross-linker (de 1,5 à plus de 30 A)
• Par la possibilité de réverser le cross-link (certains ne sont pas réversibles, d’autres le sont en
présence de thiols, d’un milieu basique, …)
• Le milieu dans lequel ils sont solubles (on choisira de préférence ceux qui le sont en milieu
aqueux pour un cross-link in vivo)
• Leur capacité à traverser les membranes de la cellule (indispensable pour un cross-link in
vivo)
• Le nombre de partenaires (deux ou plus) qu’ils peuvent assembler, …
La société Pierce en commercialise un très grand nombre, toutes leurs caractéristiques sont
répertoriées dans leur catalogue. Leur site sur le web – www.piercenet.com permet même un guide de
sélection de l’agent cross-linkant en fonction de l’expérience que l’on veut réaliser.
Cette technique ne permet pas d’affirmer qu’il y a interaction directe entre les deux partenaires, il
peut y avoir seulement une proximité dans l’espace qui peut être due à une interaction indirecte.
1. Caractérisation de l’interaction
♦ Quelle partie de la protéine X interagit avec la protéine Y
Beaucoup de protéines, en particulier des protéines eucaryotes, présentent une structure avec différents domaines structuraux indépendants. Dans ce cas, la protéine peut être « découpée » en différents morceaux capables de se structurer dans l’espace indépendamment les uns des autres. Dans ce cas là (et dans ce cas là uniquement), on peut rechercher le ou les domaines impliqués dans l’interaction protéine/protéine.
définition des domaines structuralement indépendants
il est nécessaire de connaître la structure de la protéine soit directement (RMN ou
cristallographie), soit parce qu’elle appartient à une famille dont la structure est connue,
soit parce qu’elle présente des domaines structuralement identifiés.
Ex : 1 séquence protéique pour X – bioinfo (voir TP bioinfo)-> 1 domaine POU (lui-même constitué de deux sous domaines POUh et POUs), 1 domaine GLU, 1 domaine ring. Pour Y, pas de domaines structuraux définis POU GLU RING

24/10/2011 205
Clonage de ces sous domaines
clonage dans un vecteur d’expression, production, éventuellement purification (pas obligé)
PCR
Etude de leur interaction (far western, coIP, pull down…) avec Y repérable (marquage par exemple).
♦ Kd, Kon, stœchiométrie de l’interaction, détermination des paramètres de tampon, …
Ultracentrifugation analytique
L'ultracentrifugation analytique (UCA) est une méthode de choix pour l'étude des interactions en solution. En effet, elle permet de déterminer les stœchiométries, les processus d'association, l'agencement spatial des sous-unités dans des complexes, l'influence des paramètres du solvant (pH, sel, température).
Il existe deux types principaux d’expériences de sédimentation :
vitesse de sédimentation
équilibre de sédimentation
La plus utilisée pour étudier les interactions protéine/protéine est la deuxième.
Pratiquement, on va mesurer précisément la masse des particules présentes dans des solutions
présentant des concentrations croissantes en protéine A, protéine B, et protéine A + protéine B. On
pourra ainsi en déduire
• si la protéine A est présente en solution sous forme d’un monomère ou d’un multimère
• idem pour la protéine B
• si les protéines A et B s’associent. Si cela est le cas, la stœchiométrie de l’interaction sera
déduite de la masse du complexe.
Des expériences de diffusion de lumière (DLS – Dynamic Light scattering) peuvent nous apporter en
plus une information sur la forme du complexe, information qui peut nous aider à proposer un
modèle d’interaction pour ce complexe.
Biacore

24/10/2011 206
La fonction de cet appareil est déjà suggérée par son appellation : "BIA" signifie Biospecific
Interaction Analysis et "core", au cœur de. En effet, le BIAcore est un automate qui permet de
mesurer en temps réel toute interaction biologique sans marquage de molécules. L'intérêt majeur de
cette mesure en temps réel est de pouvoir visualiser, sur l'écran, la cinétique de l'interaction et d'en
dégager ses caractéristiques propres. On peut ainsi mesurer les vitesses d'association et de
dissociation et en déduire la constante d'affinité et la constante de dissociation
Son principe de fonctionnement est d'enregistrer, en continu, à la surface d'une lamelle réactive
(appelée "sensor chip") la modification de résonance induite par toute interaction moléculaire
[résonance plasmonique de surface ( SPR ) ] . Cette modification est directement proportionnelle à la
masse de molécule liée et à sa quantité fixée sur le chip.
Le système de mesure est composé de trois éléments :
1. Le sensor chip
Il comprend une lamelle de verre sur laquelle est déposée une fine pellicule d'or elle-même
recouverte d'une couche de dextran carboxyméthylé sur lequel on couple de façon covalente la
première molécule impliquée dans la réaction que l'on veut étudier. Le couplage peut se faire par les
groupements amines, par les sucres ou par les groupements thiols selon des procédés de chimie
classique.
2. La micro-plaquette fluidique
Elle contrôle l'injection (de 5 à 300 microlitres) et le débit (de 1 à 100 microlitres/mn) des réactifs
à la surface du sensor chip.
3. L'unité optique
Une lumière polarisée est envoyée sur un prisme de verre, en contact direct avec la lamelle de
verre du sensor chip. L’appareil mesure et enregistre les changements de l’indice de réfraction
générés par un faisceau de lumière polarisée dirigée vers la face opposée de la feuille d’or lors du
processus d’association et de dissociation de A et B. Le faisceau réfléchi, qui est analysé en temps
réel par un ensemble de diodes, montre une extinction partielle pour un angle précis d’incidence, qui
dépend de l’indice de réfraction au voisinage de la feuille d’or. Les signaux obtenus, mesurés en
unités arbitraires (unités de résonance ou RU) sont proportionnels à la masse de B adsorbée à la

24/10/2011 207
surface (pour les protéines, 1 RU approximativement égal à 1 pg.mm-2). Les courbes obtenues sont
alors traitées par le logiciel BIAevaluation -> constante d’équilibre à l’interface K’d. L'interaction
peut être analysée rapidement en quelques minutes et nécessite de très petits volumes de réactifs (80
microlitres en moyenne).
En pratique, une protéine ligand (A) est immobilisée sur un support (chip).
On distingue alors trois phases :
1 - Phase d’adsorption : un tampon circule en flux continu à sa surface. Dans ce tampon se trouve la
protéine (B) qui interagit avec la protéine (A).
2 - Phase de désorption : un tampon n’incluant pas B est injecté, conduisant à une dissociation
progressive des complexes AB formés.
3 – Phase de régénération : la dissociation est brutalement accélérée, aboutissant à une surface vierge
de B, comme au début de l’expérience.
Microcalorimétrie [Microcalorimétrie isotherme à titration (ITC)]
1) Principe et méthode

24/10/2011 208
Afin de mesurer l’interaction entre deux molécules et les paramètres thermodynamiques qui en découlent, il est nécessaire de mesurer les échanges thermiques dus aux associations entre ces molécules. Le principe est le suivant : une macromolécule est placée dans la cellule de mesure d’un calorimètre isotherme puis est progressivement saturée par injection à l’aide d’une seringue de petits volumes de la deuxième molécule. L’interaction entre 2 molécules peut s’accompagner d’un échange de chaleur (absorption ou émission). Le suivi des interactions va se faire indirectement par suivi des échanges thermiques. Prenons l’exemple d’un ligand se fixant sur une protéine : En fonction de la quantité de complexe formée, on obtient des signaux thermiques proportionnels qui seront visualisables sous forme de pics grâce à un logiciel (Origin). La mesure du signal thermique permet donc de mesurer la quantité de complexe formé, et ainsi de mesurer les paramètres thermodynamiques de l’interaction.
Ainsi, la microcalorimétrie isotherme à titration permet de mettre en évidence et de quantifier les interactions entre diverses familles de protéines et leurs ligands potentiels et les modifications structurales qui en découlent.
♦ Interactions moléculaires – structure 3D
Une protéine se replie et acquiert une conformation particulière dans l'espace. C'est cette structure tridimensionnelle qui détermine la fonction que va jouer la protéine dans la cellule. Connaître cette structure est donc une étape-clé si l'on veut comprendre le détail des mécanismes de la cellule, par
Ligand + protéine = complexe + ΔH

24/10/2011 209
exemple pour concevoir des médicaments. Pour étudier la structure tridimensionnelle des protéines, mais aussi des complexes protéine/protéine, il y a plusieurs approches possibles :
La RMN (Resonnance Magnétique Nucléaire) On excite les atomes de la molécule étudiée. Chaque atome résonne à une fréquence qui lui est propre et qui dépend de son environnement magnétique. L'atome réémet un signal à cette fréquence. La superposition des signaux provenant de tous les atomes est enregistrée puis analysée. En jouant sur les différents types d'atomes et en utilisant des séquences d'excitation particulières, on peut faire apparaître de façon sélective certaines propriétés géométriques comme la proximité de deux atomes à travers l'espace ou leur proximité dans le squelette de la molécule. Contrairement à la cristallographie, l'analyse des spectres de RMN ne donne pas accès immédiatement à la structure tridimensionnelle. Il faut d'abord attribuer chaque fréquence de résonance à l'atome correspondant dans la molécule, puis utiliser les données géométriques observées pour calculer la structure tridimensionnelle globale. Aujourd'hui, la RMN est limitée par la résolution des spectres, qui dépend directement de la puissance des champs magnétiques utilisés pour exciter les atomes.
La RMN en phase liquide est la seule méthode amenant à la structure atomique de macromolécules en solution. Elle comporte cependant des contraintes significatives sur la taille des molécules étudiées (30 - 40 kDa au plus) comme sur leurs propriétés de solubilité. La RMN du solide ouvre quant à elle des possibilités intéressantes, mais encore peu exploitées, pour l’étude de macromolécules sous forme de poudre (donc non cristallisable) et pour les protéines membranaires.
Cristallographie et diffraction aux rayons X
En envoyant des rayons X sur un cristal de protéines, on observe un spectre de diffraction. Grâce à une transformation mathématique simple, il est possible d'accéder rapidement à la structure tridimensionnelle des protéines dans le cristal. L'étape limitante de cette méthode est la production d'un cristal pur (un monocristal). Elle peut être plus ou moins difficile, de façon plutôt imprévisible. Amélioration de la technique : le rayonnement synchrotron. Le rayonnement synchrotron permet la production d’un rayonnement X intense, stable, de longueur d’onde variable et de grande qualité optique. Ils offrent des données exploitables pour des cristaux de macromolécules que des méthodes classiques ne permettaient pas d’étudier : mailles plus grandes (et donc molécules ou complexes plus grands), cristaux plus petits voire imparfaits. D’autre part, le rayonnement synchrotron a considérablement accéléré la vitesse d’acquisition des données, réduisant celle-ci à quelques heures au lieu de quelques semaines. La très grande majorité des études cristallographiques contemporaines reposent sur l’utilisation de données collectées à partir d’un synchrotron.
In silico
Il est possible de prédire in silico la structure tridimensionnelle d'une protéine X par homologie par rapport à une autre protéine, de structure connue et présentant des homologies structurales avec la protéine X.

24/10/2011 210
La prédiction ab-initio du repliement, sans autre information que la seule séquence de la protéine, reste encore largement hors de portée, sauf cas exceptionnels. La méthode ab-initio nécessite des puissances de calcul importantes. Toute molécule dessinée doit être minimisée, c’est à dire que sa conformation doit être amenée à une position stable. Cette procédure appelée minimisation est un processus itératif dans lequel les coordonnées des atomes sont constamment ajustées jusqu'à amener la molécule à une énergie minimale. La conformation ayant la plus basse énergie est considérée comme la plus stable. Le docking est l'étude des interactions entre molécules. D’autre part, différents programmes ou serveurs permettent soit de prédire les régions protéiques permettant éventuellement à une protéine d’interagir avec une autre, soit de retrouver une interaction déjà mise en évidence dans le passé. Quelques exemples de ce type de sites :
banques d’interaction protéine/protéine
DIP : Database of Interacting Proteins : http://dip.doe-mbi.ucla.edu
Cette banque de donnée catalogue les interactions (déterminées expérimentalement) entre protéines.
Ce site présente un accès libre sur Internet et est supposé aider ceux qui étudient les interactions
protéine/protéine, les voix de signalisation les interactions multiples et les systèmes complexes.
prédiction de surfaces potentielles d’interaction
Protein – protein Interaction server : http://www.biochem.ucl.ac.uk/bsm/PP/server
Ce serveur nous fourni un moyen de calculer une série de paramètres descriptifs sur l’interface entre
deux chaînes polypeptidiques dans une structure protéique tridimensionnelle. En comparant ces
résultats avec des résultats obtenus avec des protéines connues pour interagir les unes avec les autres,
on peut estimer la validité de l’interface prédite entre nos deux protéines.
Inconvénient : ce programme ne fonctionne actuellement que sous Unix et nécessite une bonne
connaissance en biologie structurale et en bioinformatique.
Applications – conception de peptides inhibiteurs
Lorsqu'un composé actif de base (LEAD) a été identifié (expérimentalement ou in silico) et sa structure
chimique déterminée, on peut améliorer l'activité et/ou réduire les effets secondaires en modifiant sa
structure chimique de base.
Les peptides sont subséquemment optimisés par des modifications apportées aux chaînes latérales ou au
squelette de base. On peut ainsi concevoir des peptides avec des affinités supérieures à la cible (par

24/10/2011 211
rapport à la molécule de départ). Mais le plus grand défi qui reste à surmonter dans le cas des inhibiteurs
peptidomimétiques est de modifier ceux-ci pour qu'ils puissent être absorbés oralement et qu'ils ne se
décomposent pas rapidement dans l’organisme. Il faut alors trouver des inhibiteurs de type
nonpeptidique.

24/10/2011 212
Références
Adams M.D., Kelley J.M., Gocayne J.D., Dubnick M., Polymeropoulos M.H., Xiao H., Merril
C.R., Wu A., Olde B., Moreno R.F., Kerlavage A.R. McCombie W.R. et Venter J.C.
(1991) Science 252, 1651-1656.
Ahmad A., Akhtar M.S. et Bhakuni V. (2001) Biochemistry 40, 1945-55.
Ahern T.J. et Klibanov A.M. (1986) Dans Protein Structure, Folding and Design, 283-286, Alan
R. Liss ed.
Ahmadian A., Gharizadeh B., Gustafsson A.C., Sterky F., Nyrén P., Uhlén M. et Lundeberg J.
(2000) Anal. Biochem. 280, 103-110.
Andersson M.M., Breccia J.D. et Hatti-Kaul R. (2000) Biotechnol. Appl. Biochem. 32, 145-53.
Arakawa T. et Timasheff S. (1982) Biochemistry 21, 6545-6552.
Arakawa T., Bhat R. et Timasheff S. (1990) Biochemistry 29, 1914-1923.
Aronheim A., Zandi E., Hennemann H., Elledge S.J., Karim M. (1997) Mol. Cell. Biol. 17, 3094-
3102.
Bachman B., Luke W. et Hunsmann G. (1990) Nucleic Acid Res. 18, 1309.
Bachmsair A., Findley D. et Varshavsky A. (1986) Science 234, 179-186.
Baird G.S., Zacharias D.A. et Tsien R.Y. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 11984-11989.
Baldwin E. Xu J. Hajiseyedjavadi O. Baase W. A. Matthews B. W. (1996) J. Mol. Biol. 259, 542-
559.
Bambara R.A., Uyemura D. et Choi, T. (1978) J. Biol. Chem. 253, 413-423
Baskakov I. et Bolen D.W. (1998) Biophys J. 74, 2658-2665.
Barnard G.C., Henderson G.E., Srinivasan S. et Gerngross T.U. (2004) Protein Expression Purif.
(In press)
Barnes W.M. (1994) Proc. Natl. Acad. Sci. 91, 2216-2220.
Barr P.J. et al. (1986) Biotechniques 4, 428.
Bernard (1995) Gene 162, 159-160.
Bielefeldt-Ohmann et Fitzpatrick (1997) Biotechniques 23, 822-824.
Bevis B.J. et Click B.S. (2002) Nat. Biotechnol. 20, 83-87.
Blaber M., Baase W. A., Gassner N. et Matthews, B. W. (1995) J Mol Biol 246, 317-30

24/10/2011 213
Blais B.W., Turner G., Sooknanan R. et Malek L.T. (1997) Appl. Environ. Microbiol. 63, 310-
313.
Brenner M., Kisseberth W.C., Su, Y., Besnard F. et Messing A. (1994) J. Neurosci. 14, 1030-
1037.
Brunk C. F., Jones K.J. et James T.W. (1979) Anal. Biochem. 92, 497-500.
Burton K. (1956) Biochem. J. 62, 315-323.
Carninci P., Shiraki T., Mizuno Y., Muramatsu M., Hayashizaki Y. (2002) BioTechniques 32,
984-985.
Cha J. (1993) Gene 136, 369-370.
Chakrabarti G., Kim S., Gupta M.L. Jr, Barton J.S. et Himes R.H. (1999) Biochemistry 38, 3067-
3072.
Chakrabarti R. et Schutt C.E. (2002) BioTechniques 32, 866-874.
Chakrabartty A., Schellman J. A. et Baldwin R. L. (1991) Nature 351, 586-588.
Chakrabartty A. et Baldwin, R. L. (1995) Adv Protein Chem 46, 141-76.
Chamberlain J.S., Gibbs R.A., Ranier J.E. Nguyen P.N.et Casley C.T. (1988) Nucleis Acid Res.
16, 11141-11156.
Chatterjee D.K. et Esposito D. (2005) Protein Exp Purif sous presse
Chen B. et Przybyla A.E. (1994) Biotechniques 17, 657-659.
Cheng S., Fockler C., Barnes W.M. et Higuchi R. (1994) Proc. Natl. Acad. Sci. USA 91, 5695-
5699.
Cho R., Campbell M.J., Winzeler E.A., Steinmetz L., Conway A., Wodicka L., Wolfsberg T.G.,
Gabrielian A.E., Landsman D., Lockahart D.J. et Davis R.W. (1998) Mol. Cell. 2, 65-73.
Chong S., Shao Y., Paulus H., Benner J., Perler F.B. et Xu M-Q. (1996) J. Biol. Chem. 271,
22159-22168.
Chong S., Mersha F.B., Comb D.G., Scott M.E. Landry D., Vence L.M., Perler F.B., Benner J.,
Kucerra R.B., Hirnoven C.A., Pelletier J., Paulus H. et Xu M-Q. (1997) Gene 192, 271-
281.
Chong S., Montello G.E., Zhang A., Cantor E.J., Liao W., Xu M-Q. et Benner J. (1998) Nucl. Acid
Res. 26, 5109-5115.
Chong Y. et Chen H. (2000) BioTechniques 29, 1166-1167.

24/10/2011 214
Citti L., Rovero P., Colombo M.G., Mariani L., Poliseno L. et Rainaldi G. (2002) BioTechniques
32, 172-177.
Clark (1988) Nucleic Acid Res. 16, 9677-9686.
Compton J. (1991) Nature 350, 91-92.
Cormack B.P., Valdivia R.H., Falkow S. (1996) Gene 173, 33-38.
Cotton R.G.H., Rodrigues N.R. et Campbell R.D. (1988) Proc. Natl. Acad. Sci. U.S.A. 85, 4397-
4401.
Crameri A., Whitehorn E.A. Tate E., Stemmer W.P. (1996) Nat. Biotechnol. 14, 315-319.
Dao-pin, S., Sauer, U., Nicholson, H. et Matthews, B.W. (1991) Biochemistry 30, 7142-7153.
Davies M. J., Taylor J. I., Sachsinger N. et Bruce I.J. (1998) Anal. Biochem. 262, 92-94.
Daxelet G.A., Coene M.M., Hoet P.P. et Cocito C.G. (1989) Anal. Biochem. 179, 401-403.
De Boer H.A., Comstock L.J. et Vasser M. (1983) Proc. Natl. Acad. Sci. USA 80, 21-25.
DePaz R.A., Barnett C.C., Dale D.A., Carpenter J.F., Gaertner A.L. et Randolph TW. (2000)
Arch. Biochem. Biophys. 384,123-32.
Di Donato A., de Nigris M., Russo N., Di Biase S. et D’Alessio G. (1993) Anal. Biochem. 212,
291-293.
Ebel C., Faou P., Kernel B. et Zaccai G.(1999) Biochemistry 38, 9039-47
Felix C. F., Moreira C. C., Oliveira M. S., Sola-Penna M., Meyer-Fernandes J. R., Scofano H. M.
et Ferreira-Pereira A. (1999). Eur. J. Biochem. 266, 660-664.
Feng L., Chan W.W., Roderick S.L. et Cohen D.E. (2000) Biochemistry 39, 15399-15409.
Fisher C.L. et Pei (1997) Biotechniques 23, 570-574.
Finucane M. D. Woolfson D. N. (1999) Biochemistry 38, 11613-11623.
Flory D.J. (1956) J. Am. Chem. Soc. 78, 5222-5235.
Futami J., Tada H., Seno M., Ishikami S. et Yamada H. (2000) J Biochem (Tokyo). 128, 245-50.
Ganter C. et Pluckthun A. (1990) Biochemistry 29, 9395-9402.
Gossen M. et Bujard H. (1992) Proc. Natl. Acad. Sci. USA, 89, 5547-5551.
Gossen M., Freundlieb S., Bender G., Müller G., Hillen W. et Bujard H. (1995) Science 268,
1766-1769.
Gould S. J., Subramani S. et Scheffler I.E. (1989) Proc. Natl. Acad. Sci. USA 86, 1934-1938.
Graham Jr R.C. et Karnovsky M.J. (1966) J. Histochem. Cytochem. 14, 291-302.
Griffin B.A., Adams S.R. et Tsien R.Y. (1998) Science 281, 269-272.

24/10/2011 215
Gritson T.S., Mikhailov M.V., Roy P. et Gould E.A. (1977) Nucleic Acid Res. 25, 1864-1865.
Grutter M.G., Gray, T.M., Weaver, L.H., Wilson, T.A. et Matthews B.W. (1987). J Mol Biol. 197,
315-29.
Guan C. di, Li P., Riggs P.D. et Inouye H. (1988) Gene 67, 21-30.
Hanes O. et Plückthun A. (1997) Proc. Natl. Acad. Science USA 94, 4937-4942.
Hasan N et Szybalski W. (1995) Gene 163, 35-40.
Hawley D.K. et McClure W.R. (1983) Nucleic Acids Res. 11, 2237-2255.
Hecht M. H., Sturtevant J. M. et Sauer R. T. (1986) Proteins 1, 43-46.
Hill B.T. (1976) Anal. Biochem. 70, 635-638.
Hirschel B.J., Shen V. et Schlessinger D. (1980) J. Baceriol. 143, 1534-1537.
Ho A. et al. (2001) Cancer Res. 61, 474-477.
Hoff H., Random J. et Bricca G. (1999) Anal. Biochem. 268, 398-401.
Hom L.G. et Volkman L.E. (1998) Biotechniques 25, 20-22.
Hopp T et al., (1988) Biotechnology 6, 1204-1210.
Irun M.P., Maldonado S.et Sancho J. (2001) Protein Eng. 4, 173-81
Jiang X., Kowalski J. et Kelly J.W. (2001) Protein Sci.10, 1454-65.
Johnson R.E., Adams P. et Rupley J.A. (1978) Biochemistry 17, 1479-1484.
Johnsson N. et Varshavsky A. (1994) Proc. Natl. Acad. Sci. USA, 91, 10340-10344.
Kaplan, H.A., Welpy, J.K. et Lennarz, W.J. (1987) Biochim. Biophys. Acta 906, 161-173.
Karpeisky M.Y., Senchenko V.N., Dianova M.V. et Kanevsky V (1994) FEBS Lett 339, 209-212.
Keefe A.D., Wilson D.S., Seelig B. et Szostak J.W. (2001) Protein Expr Purif 2001, 440-446.
Kennedy D.G., Nelson J., van den Berg H.W. et Murphy R.F. (1987) Anal. Biochem. 167, 127-
127.
Kinney J., Leippe D., Lewis K., Lyke B., Mandrekar M. et Shultz J. Promega notes 73, 3-7.
Khanna R., Myers M.P., Laine M. et Papazian D.M. (2001) J Biol Chem. Jun 26 [epub ahead of
print])
Kholod N. et Mustelin T. (2001) Biotechniques, 31,322-328.
Kondoh N., Wakatsuki T., Ryo A., Tanaka K., Hada A., Shichita M. et Yamamoto M. (1999)
Anal. Biochem. 269, 427-430.
Krest I. et Keusgen M.(1999) Pharmazie 54, 289-93
Kunkel T.A. (1985) Proc. Natl. Acad. Sci. U.S.A. 82, 488-492.

24/10/2011 216
Kwon Y.K., Murray M.T. et Hecht N.B. (1993) Dev. Biol. 158, 90-100.
LaVallie E.R., DiBlasio E.A., Kovacic S., Grant K.L., Schendel P.F. et McCoy J.M. (1993)
Bioctechnology 11, 187-193.
Langemeier J.L., Cook F., Issel C.J.et Montelaro R.C. (1994) Biotechniques 17, 486-490.
Lelong JC, Prevost G, Lee K, Crepin M.(1989) Anal. Biochem. 179, 299-303
Le Peck, J.-B. et Paoletti , C. (1966) Anal. Biochem. 17, 100-107.
Letsinger, R.L. et al. (1989) Proc. Natl. Acad. Sci - USA 86, 6553.
Leush M.S., Lee S.C. et Ollins P. (1995) Gene 160, 191-194.
Lewis,R.V., Hinman, M., Kothakota, S., Fournier, M.J. (1996) Protein expression Purif. 7, 400-
406.
Liang P. et Pardee (1992) Science 257, 967-969.
Lin S.H., Konishi Y., Denton M.E. et Scheraga, H.A. (1984) Biochemistry 23, 5504-5512.
Lippert K. et Galinski E.A. (1992) Appl. Microbiol. Biotechnol. 37, 61-65.
Looman A.C., Bodlaender J., Comstock L.J., Eaton D., Jhurani P., de Boer H.A. et van
Knippenberg P.H. (1987) EMBO J. 6, 2489-2492.
Luckow V.A., Lee S.C., Barry G.F. et Olins P.O. (1993) J. Virol. 67, 4566-4579.
Lutz R. et Bujard H. (1997) Nucl. Acid. Res. 25, 1203-1210.
Majunder K. (1992) Gene 110, 89-94.
Makarova O., Kamberov E. et Margolis B. (2000) BioTechniques 29, 970-972.
Manstein D.J., Schuster H-P, Morandini P. et Hunt D.M. (1977) Gene 162, 129-134.
Margarit, I., Campagnoli, S., Frigerio, F., Grandi, G., De Filippis, V. et Fontana, A.(1992) Protein
Eng 5, 543-50.
Mariac C., Trouslot P., Poteaux C., Bezançon G. et Renno J.-F. Biotechniques 28, 110-113.
Maruyama, I.N., Rakow, T.L., Maruyama, H.I. (1995) Nucl. Acid. Res. 23, 3796-3797.
Matsumura M., Becktel W.J., Levitt M. et Matthews B.W. (1989) Proc. Natl. Acad. Sci. USA 86,
6562-6566
Meerman H.J. et Georgiou G. (1994) Biotechnology 12, 1107-1110.
Mitraki A. et King J. (1989) BioTechnology 7, 690-697.
Mok Y.K., Elisseeva E.L., Davidson A.R. et Forman-Kay J.D. (2001) J Mol Biol. 307, 913-28.
Monia B.P., Lesnik E.A., Gonzalez C., Lima W.F., McGee D., Guinosso C.J., Kawasaki A.M.,
Cook P.D. et Freier S.M. (1993) J. Biol. Chem., 268, 14514–14522.

24/10/2011 217
Morawski B., Quan, S. et Arnold F.H; (2001) Biotech. Bioengin. 76, 99-107.
Müller W., Weber H., Meyer F. et Weissmann C. (1978) J. Mol. Biol. 124, 343-358.
Munoz V. et Serrano L. (1994) Proteins 20, 301-311.
Myers R.M., Larin Z. et Maniatis T. (1985) Science 230, 303-305.
Myers R.M., Lumelsky N., Lerman L.S. et Maniatis T. (1985) Nature 313, 495-498.
Nakamaye K. et Eckstein F. (1986) Nucleic Acid Res. 14, 9679-9698.
Nilsson B. et Abrahmsen L. (1990) Methods Enzymol. 185, 144-161.
Noren C.J., Anthony-Cahill M.C., Griffith M.C., Schultz P.G. (1989) Science 244, 182-188.
O’Hare P. et Elliot G. (1997) Cell 88, 223-233.
O’Neil K.T. et DeGrado W.F. (1990) Science 250, 646-651.
Ochman H., Gerber A.S. et Hartl D.L. (1988) Genetics 120, 621-623.
Oefner P.J. et Underhill P.A. (1995) Am. J. Hum. Genet. 57 suppl, A266
Ohmura T., Ueda T., Ootsuka K., Saito M. et Imoto T (2001) Protein Sci. 10, 313-320.
Oleykowski C.A., Bronson C.R., Godwin A.K. et Yeung A.T. (1998) Nucleic Acid Res. 26, 4597-
4602.
Oliphant A.R et Struhl K. (1988) Nucleic Acid Res. 16, 7673-7683.
Oliphant A.R et Struhl K. (1989) Proc. Natl. Acad. Science USA 86, 9094-9098.
Olson P, Zhang Y., Olsen D., Owens A., Cohen P., Nguyen K., Ye J-J., Bass S. et Mascarenhas D.
(1998) Prot. Exp. Purif. 14, 160-166.
Olson C.A., Spek E.J., Shi Z., Vologodskii A. et Kallenbach N.R. (2001) Proteins 44,123-132.
Orita M. Iwahana H., Kanazawa K., Hayashi K et Sekiya T. (1989) Proc. Natl. Acad. Sci. 86,
2766-2770.
Ozawa T., Nogami S., Sato M., Ohya Y. et Umezawa Y. (2000) Anal. Chem. 72, 5151-5157.
Paabo S., Irwin D.M. et Wilson A.C. (1990) J. Biol. Chem. 265, 4718-4721.
Pace, C. N. et Scholtz, J. M. (1998) Biophys J. 75, 422-7.
Pace C.N., Horn G., Hebert E.J., Bechert J., Shaw K., Urbanikova L., Scholtz J.M. et Sevcik J.
(2001) J. Mol. Biol. 312, 393-404.
Pakula AA et Sauer RT (1990) Nature 344, 363-364.
Pan S-H. et Malcolm B.A. (2000) Biotechniques 29, 1234-1238.
Panjkovich A. et Melo F. (2005) Bioinformatics 21, 711-722.

24/10/2011 218
Pantoliano M.W., Ladner R.C., Bryan P.N., Rollence M.L., Wood J.F. et Poulos T.L. (1987)
Biochemistry 26, 2077-2082
Parker J.D., Rabinovitch P.S. et Burmer G.C. (1991) Nucl. Acid Res. 19, 3055-3060.
Parks D., Leuther K.K., Howard E., Johnston S.A. et Dougherty W.G. (1994) Anal. Biochem. 216,
416-417.
Paris P.L., Langenham J.M. et Kool E.T. (1998) Nucl. Acid Res. 26, 3789-3793.
Paulmurugan, R. et Gambhir, S.S. (2003) Anal. Chem. 75, 1584-1589.
Pelletier J. et Sonenberg N. (1988) Nature 334, 320-325.
Perl D. Mueller U. Heinemann U. et Schmid F. X. (2000) Nature Struct. Biol. 7, 380-383
Persson M., Bergstrand M.G., Bulow L.et Mosbach K. (1988) Anal. Biochem. 172, 330-337.
Perutz, M.F. and Raidt, H. (1975) Nature 255, 256-259.
Peterson, R.W., Nicholson, E.M., Thapar, R., Klevit, R.E. et Scholtz, J.M. (1999) J. Mol. Biol.
286, 1609-1619.
Porath J., Carlsson J., Olsson I. et Belfrage G. (1975) Nature 258, 598-599.
Powell J. (1998) Nucleic Acid Res. 26, 3445-3446.
Prasher D.C., Eckenrode V.K., Ward W.W., Prendergast F.G., Cormier M.J. (1992) Gene 111,
229-233.
Predki P. F., Nayak L. M., Gottlieb M. B. et Regan L. (1995) Cell 80, 41-50.
Raschthian A., Buchman G.W., Schuster D.M. et Berninger M.S. (1992) Anal. Biochem. 206, 91-
97.
Reddy A., Grimwood B.G., Plummer T.H. et Tarentino A.L. (1998) Glycobiology 8, 633-638.
Ringquist S., Shinedling S., Barrick L., Green L., Bindley J., Stormo G.D. et Gold L. (1992) Mol.
Microbiol. 6, 1219-1229.
Ronaghi M., Uhlén M. et Nyrén P. (1998) Science 281, 363-364
Rosenthal A. et Jones D.S.J. (1990) Nucl. Acid Res. 18, 3095-3096.
Ross-Macdonald P., et al. (1999) Nature 402, 413-417.
Rossi, F., Charlton C.A. et Blau, H.M. (1997) Proc. Natl. Acad. Sci. USA, 94, 8405-8410.
Russel M., Kidd S. et Kelly M.R. (1986) Gene 45, 333-338.
SantaLucia J.Jr., Allawi H.T. et Seneviratne P.A. (1996) Biochemistry 35, 3555-3562.
Sarkar G. et Sommer S.S. (1990) Biotechniques 8, 404-407.
Sarkar G., Yoon H.S. et Sommer S.S. (1992) Genomics 13, 441-443.

24/10/2011 219
Sassenfeld H.M. et Brewer S.J. (1984) Bio/Technology 2, 76-81
Sauge-Merle S., Falconet D. et Fontecave M. (1999) Eur. J. Biochem. 266, 62-69.
Schauder B et McCarthy J.E.G. (1989) Gene 78, 59-72.
Schaeffer B.C. (1995)
Schalling M., Hudson T.J, Buctow K.H.et Housman D.E. (1993) Nat. Genet. 4, 135-139.
Schatz P.J. (1993) Biotechnology 11, 1138-1143.
Schildkraut C et Lifton S. (1965) Biopolymers 3, 3555-3562.
Schmidt T.G.M. et Skerra, A. (1993) Protein Eng. 6, 109-122.
Scholz O., Thiel A., Hillen W. et Niederweis M., (2000) Eur. J. Biochem. 267, 1565-1570.
Scott M.R.D., Westphal K-H et Rigby P.W.J. (1983) Cell 34, 557-567.
Sektas M., Hasan N. et Szybalski W. (2001) Gene 267, 213-220.
Shakin-Eschleman S., Spitalnik S. L. et Kasturi L. (1996) J. Biol. Chem. 271, 6363-6366.
Shpigel E., Elias D., Cohen I.R. et Shoseyov O. (1998) Prot. Exp. Purif. 14, 185-191.
Short J.M., Fernandez J.M., Sorge J.A. et Huse W.D. (1988) Nucleic Acid Res. 16, 7583-7600.
Shortle D. et Nathans D. (1978) Proc. Natl. Acad. Sci. USA 75, 2170-2174.
Shuman S. (1994) J. Biol. Chem. 269, 32678-32684.
Siebert P.D., Chenchik A., Kellogg D.E., Lukyanov K.A. et Lukyanov S.A. (1995) Nucl. Acid .
Res. 23, 1087-1088.
Skerra A. (1994) Gene 151, 131-135.
Smith H.W. (1975) Nature 255, 500-502.
Smith D.B. et Johnson K.S. (1988) Gene 67, 31-40.
Sommer S.S., Groszbbach A.R. et Bottenma C.D.K. (1992) BioTechniques 12, 82-87.
Southern E.M. (1975) J. Mol. Biol. 98, 503-517.
Song S.B., Kim K.K.et Kim K.E. (2002) BioTechniques 32, 248-252.
Srinivasan S., Barnard G.C. et Gerngross T.U. (2002) Applied Environ. Microbiol. 68, 5912-5932.
Stemmer, W.P.C., Crameri, A., Ha, K.D., Brennan, T.M. et Heyneker, H.L. (1995) Gene 164, 49-
53.
Stofko-Hahn R.E., Carr D.W. et Scott J.D. (1992) FEBS Lett. 302, 274-278.
Studier F.W. (1991) J. Mol. Biol. 219, 37-44.
Studier F.W. et Moffatt B.A. (1986) J. Mol. Biol. 189, 113-130.
Tabor S. et Richardson C.C. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 6339-6343.

24/10/2011 220
Tams J.W. et Welinder K.G. (2001) Biochem. Biophys. Res. Comm. 286, 701-706.
Taniguchi K., Nomura K., Hata Y., Nishimura T., Asami Y., Kuroda A. (2007) Biotechnol.
Bioeng. 96, 1023–1029.
Thalhammer, S., Stark, R.W., Schütze, K et Heckl, W.M. (1997)
Thomas C.A.J. et Dancis B.M. (1973) J. Mol. Biol. 77, 43-55.
Ueda T., Yamada H., Hirata M. et Imoto T. (1985) Biochemistry 24, 6316-6322.
de Valdivia E.I.G et Isaksson L.A. (2005) FEBS J. 272, 5306-5316.
Van Dyke M.W., Sirito M. et Sawadogo M. (1992) Gene 111, 99-104.
Van Hest J.C.M., Kiick K.L., Tirrell D.A., (2000) J. Am. Chem. Soc. 122, 1282-
Vartamian J.P., Henry M., Wain-Hobson, S. (1996) Nucleic Acids Res. 24, 2627-2631.
Velculescu V.E., Zhang L., Volgelstein B. et Kinzler K.W. (1995) Science 270, 484-487.
Viazov S., Zibert A., Ramakrishnan K., Widell A., Cavicchini A., Schreier E. et Roggendorf M.
(1994) J Virol Methods 48, 81-91.
Waldburger C. D. Schildbach J. F. et Sauer R. T. (1995) Nature Struct. Biol. 2, 122-128.
Wang M., Doyle M.V. et Park D.F. (1989) Proc. Natl. Acad. Science USA 86, 9717-9721.
Wang D.G. et al. (1998) Science 280, 1077-1082.
Wang L., Brock A., Herberich B., Schultz P.G. (2001) Science 292, 498-500.
Watson D.E. et Bennett G.N. (1997) BioTechniques 23,858-862.
Weijers S. R. et van’t Riet K. (1992) Biotechnol. Adv. 10, 237–249.
West M. et Wilson V.G. (2002) BioTechniques 32, 44-46.
Wetzel R., Perry L.J., Baase W.A. et Becktel W.J. (1988) Proc. Natl. Acad. Sci. USA 85, 401-405.
Wieland I., Bolger G, Asouline G., Wigler M. (1990) Proc. Natl. Acad. Sci. 87, 2720-2724.
Zamecnik P.C. et Stephenson M.L. (1978). Proc. Natl. Acad. Sci. USA, 75, 280-284.
Zabner J., Fasbender A.J., Moninger T, Poellinger K.A. et Welsh M.J. (1997) J. Biol. Chem. 270,
18997-19007.
Zaccolo M., Williams D.M., Brown D.M., Gherardi E. (1996) J. Mol. Biol. 225, 589-603.
Zhen G. et Lyons G. (2002) BioTechniques 33, 32-36.
Zhou M. et Hatalet Z. (1995) Nucleic Acid Res. 23, 1089-1090.
24/10/2011 221
Fournisseurs
Clonteck : www.clonteck.com
In Vitrogene : www.invitrogen.com
Perbio : www.perbio.com
Gene Therapy Sustem Inc : www.genetherapysystems.com

24/10/2011 222
Index
A adenovirus, 285 ADNc, 58 ADN-T, 274 Adriamycine, 12 AFLP, 230 Agrobacterium tumefasciens, 274 alfavirus, 287 Aminoglycosides, 44 Aminopteridine, 46 ampicilline, 45 Amplification, 99 Amplification d’ARN, 105 AP-PCR, 234 ARN polymérase, 29
B BAC, 40 Bacillus, 263 Baculovirus, 276 banque, 58 banque de cDNA, 124 Banque de saut, 147 biotine, 68 bisbenzimide, 10 Blasticidine, 46 blot overlay, 348 Bovine Papilloma Virus, 284 Bridging PCR, 106 Bromodesoxyuridine, 47 bromure d'éthidium, 9
C chitine, 318 Chloramphenicol, 44 corps d’inclusion, 327 cosmide, 41 Cristal violet, 12
D DAF, 234 DAPI, 11 ddF, 224 DDPCR, 160 dénaturation, 78 DGGE, 224 Dictyostelium discoideum, 272 didéoxynucléotide, 77 differential display, 160
digoxigénine, 69 diphenylamine, 13 DNase I, 17 DNases, 16 double hybride, 150
E enterokinase, 319 Erythromycine, 43 exonucléases, 16
F facteur Xa, 319 Far-Western, 347
G G418, 47 GFP, 317 glancicovir, 47 glutathion S-transférase, 315 glycosidase, 32 Green fluorescent protein, 317 GST pull down, 347
H HcRed, 317 HIS-tag, 316 Histidinol, 47 Hoechst, 10 hot start, 101 Hybridation, 78 hybridation soustractive, 157 Hygromycine B, 48
I IMA, 234 Intein, 320
J Jumping PCR, 106
K kanamycine, 45
L lentivirus, 284

24/10/2011 223
ligase, 30
M M13, 38 MAAP, 234 Macrolides, 43 maltose binding protéine, 315 Maxam et Gilbert, 89 Methotrexate, 48 méthylase, 32 Microsatellites, 232 minisatellites, 232 mithramycine, 13 Mitomycine C, 48 Molecular beacons, 103 mung bean, 18 mutagenèse, 108 mutation, 213
N NASBA, 105 nick translation, 73 northern, 86 North-Western, 350 Nucléase S1, 17
O Oubaine, 48
P PAC, 40 PASA, 219 PCR, 99 phage, 36 phage display, 131 phagemide, 40 Phosphatase, 31 Pichia pastoris, 270 plasmide, 34 polymérase, 23 polymorphisme, 229 protéase, 318 Puromycine, 49 Pyrosequencing, 92
R Ramdom priming, 74 RAPD, 234 restriction, 18 rétrovirus, 286 reverse transcriptase, 27
RFLP, 222, 229 ribosome display, 122 Rifampicine, 46 RNases, 22
S Saccharomyces cerevisiae, 266 Sanger, 90 Schizosaccharomyces pombe, 269 scintillation proximity assay, 347 séquence, 89 sonde, 68 Southern, 83, 216 South-Western, 350 SPA, 347 SSCP, 223 streptavidine, 69, 315 SV40, 284
T Taq DNA pol, 26 télomérase, 28 Terminal transférase, 28 Tétracycline, 44 Tetracystéine, 318 TEV, 319 Thioredoxine, 316 thrombine, 319 Tm, 78 topoisomérase, 30 traduction, 236 Trichoderma reesei, 272 tsp, 180
V vaccine, 285 Vancomycine, 45 Viomycine, 45
X Xénope, 283 Xyl-A, b-D-Xylofuranosyl, 49
Y YAC, 40
� λ Zap, 41