Embed Size (px)
Citation preview

of July 14, 2018.This information is current as
Epithelial CellsInnate Immune Mechanisms in Respiratory FOXO Transcription Factors Regulate
Menger, Robert Bals and Christoph BeisswengerLanger, Hans-Joachim Schäfers, Frank Lammert, Michael D.Kamyschnikow, Christian Herr, Markus Bischoff, Frank Frederik Seiler, Jan Hellberg, Philipp M. Lepper, Andreas
ol.1200596http://www.jimmunol.org/content/early/2013/01/11/jimmun
published online 11 January 2013J Immunol
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2013 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on July 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on July 14, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
FOXO Transcription Factors Regulate Innate ImmuneMechanisms in Respiratory Epithelial Cells
Frederik Seiler,* Jan Hellberg,* Philipp M. Lepper,* Andreas Kamyschnikow,*
Christian Herr,* Markus Bischoff,† Frank Langer,‡ Hans-Joachim Schafers,‡
Frank Lammert,x Michael D. Menger,{ Robert Bals,* and Christoph Beisswenger*
Bacterial pathogens are a leading cause of lung infections and contribute to acute exacerbations in patients with chronic respiratory
diseases. The innate immune system of the respiratory tract controls and prevents colonization of the lung with bacterial pathogens.
Forkhead box transcription factor family O (FOXO) transcription factors are key regulators of cellular metabolism, proliferation, and
stress resistance. In this study, our aim was to investigate the role of FOXO transcription factors in innate immune functions of re-
spiratory epithelial cells. We show that bacterial pathogens potently activate FOXO transcription factors in cultured human respi-
ratory epithelial cells in vitro. Infection of mice with bacterial pathogens resulted in the activation of FOXO transcription factors in
alveolar and bronchial epithelial cells in vivo. Active FOXOwas also detectable in human bronchial tissue obtained from subjects with
different infection-related lung diseases. Small interfering RNA–mediated knockdown of FOXO in bronchial epithelial cells resulted
in reduced expression of factors of the innate immune system such as antimicrobial peptides and proinflammatory cytokines, both
under basal conditions and upon infection. FOXO deficiency further affected internalization of Haemophilus influenzae in bronchial
epithelial cells. Finally, we show that TLR3 activates innate immune responses in a FOXO-dependent manner. In conclusion, FOXO
transcription factors are involved in the cellular responses to bacterial stimuli and act as central regulators of innate immune
functions in respiratory epithelial cells. The Journal of Immunology, 2013, 190: 000–000.
Mucosal surfaces, as in the respiratory tract, are con-tinuously exposed to microbes. In the lung, residentmyeloid cells (e.g., alveolar macrophages) and respi-
ratory epithelial cells are principle components of the innate hostimmunity required to prevent bacterial colonization and infectionas well as to initiate adaptive immunity (1, 2). Innate immunefunctions of alveolar and airway epithelial cells include formationof mechanical barriers, production of mucus, mucociliary clearance,secretion of antimicrobial factors and peptides, and recruitment ofprofessional immune cells by cytokine release (1, 3, 4). Some ofthese functions are constitutively active. However, in case of in-fection, epithelial cells directly detect pathogen-associated molec-ular patterns (PAMPs) in a pattern recognition receptor–dependent
manner, resulting in enhanced innate immune responses (2, 3, 5).In addition, inflammatory cytokines (e.g., IL-1b and IL-17) releasedby professional immune cells induce innate immune functions ofairway epithelial cells (6, 7). Thus, epithelial cells initiate andamplify immune responses needed for clearance of pathogens inthe respiratory tract during infection.Numerous in vivo and in vitro studies have examined the sig-
naling events required for activation of respiratory epithelial cellsby microbes (3, 8–11). As in immune cells, the cellular responseof epithelial cells activated by TLRs can be MyD88-dependentor -independent (12). In respiratory epithelial cells, activation ofcell surface–expressed TLR2, TLR4, and TLR5 by their spe-cific ligands (lipoproteins, LPS, and flagellin) leads to the expres-sion of inflammatory mediators and antimicrobial peptides (AMPs)mediated by NF-kB and MAPK signaling cascades, whereas acti-vation of endosomal TLR3 and TLR4 results in the expression oftype 1 IFNs in a TRIF-dependent manner (12). TLR3 is commonlyknown to be important during viral infection recognizing dsRNA.However, recent studies have shown that TLR3 may also be as-sociated with bacterial infection. Sajjan et al. (13) have reportedthat TLR3 expression is upregulated in airway epithelial cellsduring infection with nontypeable Haemophilus influenzae (NTHi),resulting in enhanced rhinovirus-induced chemokine release. Inaddition, NTHi is capable of directly activating TLR3 (14).Previous studies have linked the forkhead box transcription factor
family O (FOXO) to innate immunity (15, 16). FOXO transcriptionfactors are highly conserved among distantly related species. Inhumans, four isoforms (FOXO 1–4) exist (17). FOXOs regulate awidespread array of cellular functions on a transcriptional levelsuch as energy metabolism (18), cell-cycle arrest (19), DNA repair(20), oxidative stress resistance (21, 22), and apoptosis (23, 24).Becker et al. (15) reported that the expression of AMPs in Dro-sophila melanogaster is induced by active FOXO independentfrom NF-kB. Furthermore, recent studies showed that FOXOregulates inflammation by enhancing TLR4-mediated signaling
*Department of Internal Medicine V–Pulmonology, Allergology and RespiratoryCritical Care Medicine, Saarland University Medical Center, 66421 Homburg/Saar,Germany; †Institute of Medical Microbiology and Hygiene, Saarland UniversityMedical Center, 66421 Homburg/Saar, Germany; ‡Department of Thoracic and Car-diovascular Surgery, Saarland University Medical Center, 66421 Homburg/Saar,Germany; xDepartment of Internal Medicine II–Gastroenterology, Hepatology andEndocrinology, Saarland University Medical Center, 66421 Homburg/Saar, Ger-many; and {Institute for Clinical and Experimental Surgery, Saarland UniversityMedical Center, 66421 Homburg/Saar, Germany
Received for publication February 17, 2012. Accepted for publication December 6,2012.
This work was supported by grants from Deutsche Forschungsgemeinschaft (BE4813/1-1 to C.B. and Ba 1641/12 to R.B.).
Address correspondence and reprint requests to Dr. Christoph Beisswenger, SaarlandUniversity Medical Center, Department of Internal Medicine V–Pulmonology, Aller-gology and Respiratory Critical Care Medicine, D-66421 Homburg/Saar, Germany.E-mail address: [email protected]
Abbreviations used in this article: AMP, antimicrobial peptide; ARDS, acute respi-ratory distress syndrome; COPD, chronic obstructive pulmonary disease; FOXO,forkhead box transcription factor family O; hBD-2, human b-defensin 2; HBEC,human bronchial epithelial cell; NTHi, nontypeable Haemophilus influenzae; PAMP,pathogen-associated molecular pattern; Poly-(I:C), polyinosinic-polycytidylic acid;qRT-PCR, quantitative RT-PCR; siRNA, small interfering RNA.
Copyright� 2013 by The American Association of Immunologists, Inc. 0022-1767/13/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1200596
Published January 11, 2013, doi:10.4049/jimmunol.1200596 by guest on July 14, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

(25) and IL-1b expression (16) in human macrophages, suggestingthat FOXO transcription factors also act as modulators of innateimmune functions in vertebrates. The transcriptional activity ofFOXO is regulated at different levels. Primarily, FOXOs havebeen described to be negatively regulated by growth factors. In thepresence of growth signals such as insulin or IGF-1, activation ofthe PI3K signaling pathway leads to phosphorylation of FOXOproteins by the kinase AKT (26, 27). Phosphorylated FOXO trans-locates from the nucleus to the cytosol, where it becomes ubiq-uitinated, leading to its degradation by the proteasome (28, 29). Inthe absence of external growth signals, the PI3K–AKT axis is inac-tive, and unphosphorylated FOXO binds to its DNA consensus se-quence to promote target gene transcription. Besides this regulatorymechanism, FOXO is activated by several stress pathways inducingnuclear translocation of FOXO. Oxidative stress and TNF, for in-stance, activate FOXO in a JNK- or STE20-like kinase 1 (MST1)–dependent manner (29). This activation overcomes AKT-dependentinhibition, resulting in active FOXO independent from the presenceof growth factors (30, 31). Furthermore, the histone deacetylase SIRT1has been shown to switch FOXO-dependent gene expression fromapoptosis to a repair and survival mode under oxidative stress (31).In this study, we aimed to investigate the role of FOXO tran-
scription factors in respiratory epithelial cells responding to infec-tious stimuli. We identify FOXO as a key player in innate immuneregulation. We show that FOXO is activated in epithelial cells byinfectious stressors and that expression and release of inflammatorymediators is FOXO dependent.We further demonstrate that FOXO isrequired for TLR3 signaling to promote innate immune responses.Finally, we reveal a regulatory network of FOXO and insulinthat modulates innate immune functions of respiratory cells.
Materials and MethodsCell culture
The bronchial epithelial cell line Calu-3, originally obtained from humanlung adenocarcinoma, was cultured in DMEM/F12 (Life Technologies)supplemented with 10% FCS, 100 IU/ml penicillin G, and 100 IU/mlstreptomycin sulfate. Prior to cell-culture experiments, Calu-3 cells wereserum-starved overnight. All cell-culture experiments were carried out inserum-free DMEM/F12. Primary human bronchial epithelial cells (HBEC)were isolated from large airways resected during surgery and cultivated assubmersed cultures as described previously (32). Primary cell-cultureexperiments presented in this study were performed with cells obtainedfrom six individual patients who underwent surgery because of lungcancer. Only cancer-free tissue was used for cell isolation. The protocolwas approved by the Institutional Review Board (ethics committee) of theLandesarztekammer des Saarlandes, and informed consent was obtainedfrom the patients. Cells were cultivated using Airway Epithelial CellGrowth Medium (PromoCell) supplemented with bovine pituitary extract(0.004 ml/ml), epidermal growth factor (recombinant human; 10 ng/ml),insulin (recombinant human; 5 mg/ml), hydrocortisone (0.5 mg/ml), epineph-rine (0.5 mg/ml), triiodo-L-thyronine (6.7 ng/ml), transferrin holo (human,10 mg/ml), and retinoic acid (0.1 ng/ml).
Bacterial strains and products
A patient isolate of nontypeableH. influenzae (NTHi) was grown overnight at37˚C and 5% CO2 on chocolate agar (Becton-Dickinson Biosciences).Bacteria were taken from the plate, resuspended, and washed in PBS. Forcell-culture experiments, the bacterial suspension was adjusted to anOD600 0.5 (∼5 3 108 CFU/ml). For experiments with inactivated NTHi,bacteria were treated with 100 IU/ml penicillin G and 100 IU/ml strep-tomycin sulfate for 1 h prior to cell-stimulation experiments and during thecell-stimulation itself. For experiments with viable NTHi, bacteria wereresuspended in DMEM/F12. For mouse experiments, bacteria were resus-pended in PBS and adjusted to ∼107 CFU/ml. Appropriate dilutions wereplated out to determine the NTHi titer. Pseudomonas aeruginosa strainPAO1 was grown overnight at 37˚C on Luria-Bertani agar plates. Bacterialcells were taken from the plate, resuspended in Luria-Bertani medium, andincubated for 2–4 h at 37˚C and 150 rpm. After washing with PBS, bacteriawere adjusted to OD600 1 and consequently heat-inactivated for 10 min at95˚C. Stocks were stored at 280˚C. For cell-culture experiments, the stock
solutions were diluted to OD600 = 0.01 in cell-culture medium. Polyinosinic-polycytidylic acid [Poly-(I:C)], Pam3CSK4, and LPS were purchased fromInvivogen.
FOXO DNA-binding activity assay
Calu-3 cells were serum-starved overnight and incubated with viable NTHifor 30 min. Nuclear extracts were prepared using the Nuclear extract kit(Active Motif) as instructed by the manufacturer. DNA-binding activity ofnuclear FOXO1 was assessed using the Trans-AM FKHR kit (Active Motif)as described by themanufacturer, using a TecanUltra 384 ELISA reader andthe software Magellan (Tecan).
Small interfering RNA experiments
Calu-3 cells or primary HBEC were transfected with small interfering RNA(siRNA) specific for FOXO1 and FOXO3 (each at 50 nM; Cell SignalingTechnology) using Lipofectamine RNAiMAX (Qiagen, Germany). Con-trols were transfected with unspecific control siRNA that does not lead tospecific degradation of any cellular message (at 100 nM; Cell SignalingTechnology). For experimental procedures, siRNA-transfected Calu-3 cellswere incubated overnight in DMEM/F12 medium without FBS andantibiotics and subsequently treated with bacterial stressors as indicated.Primary HBEC were incubated for 24 h with insulin (1 or 5 mg/ml) andsubsequently treated with bacterial stressors as indicated. Supernatantsand cell lysates were kept at 280˚C. Silencing of FOXO1 and FOXO3was assessed by quantitative RT-PCR (qRT-PCR) 24 h after transfectionor at the end of experimental procedures (data not shown).
RNA isolation and qRT-PCR
Total RNA from cells was isolated using the NucleoSpin RNA II Kit(Macherey-Nagel) according to the manufacturer’s manual. A total of 1 mgtotal RNA was reversely transcribed to cDNA using the RevertAid FirstStrand cDNA Synthesis Kit (Thermo Scientific) according to the manu-facturer’s manual. qRT-PCR was performed with a reaction mix includingthe SensiMix SYBR& Fluorescein Kit (Bioline) using the iCycler (Bio-Rad)with a two-step protocol (15 s at 95˚C and 45 s at 60˚C). Specificity ofamplification was controlled by melt-curve analysis and gel electropho-resis. RT-PCR results were analyzed with the DD threshold cycle method(33) using b-actin as an internal standard to normalize mRNA amounts.Primer sequences were used as follows: b-actin, 59-AGA GCT ACG AGCTGC CTG AC-39 (forward) and 59-AGC ACT GTG TTG GCG TAC AG-39 (reverse); human b-defensin 2 (hBD-2), 59-TCA GCT CCT GGT GAAGCT C-39 (forward) and 59-GGG CAAAAGACT GGATGACA-39 (reverse);IP-10, 59-TGA AAT TAT TCC TGC AAG CCA A-39 (forward) and 59-CAGACATCT CTT CTC ACC CTT CTT T-39 (reverse); IL-6, 59-CAC ACA GACAGC CAC TCA CC-39 (forward) and 59-TTT TCT GCC AGT GCC TCT TT-39 (reverse); TNF, 59-CCC AGG CAG TCAGAT CAT CTT C-39 (forward) and59-AGC TGC CCC TCA GCT TGA-39 (reverse); Bim, 59-CCG CCC TTATGA TGA AGT GT-39 (forward) and 59-AAA GCC TGG AGT CAG CAAAA-39 (reverse); FOXO1, 59-TGG ACA TGC TCA GCA GAC ATC-39 (for-ward) and 59-TTG GGT CAG GCG GTT CA-39 (reverse); and FOXO3, 59-AAA TGT TCG TCG CGG CGG AAC-39 (forward) and 59-GTC GCC CTTATC CTT GAAGTA-39 (reverse). All primers were purchased from Metabion.
Determination of cytokine concentrations
Concentrations of the inflammatory cytokines IL-6, IL-8, and IP-10 in cell-culture supernatants were assessed by ELISA using a Tecan Ultra 384ELISA reader and the software Magellan (Tecan). All ELISA kits werepurchased from R&D Systems and used as instructed by the manufacturer.
NTHi uptake assay
Calu-3monolayers were incubated overnight in DMEM/F12mediumwithoutFBS and antibiotics. Monolayers were infected with viable NTHi in DMEM/F12 for different periods with or without the PI3K inhibitor LY294002(50 mM; Cell Signaling Technology). Postinfection, cells were treated withDMEM/F12 containing 50 mg/ml gentamicin for 45–60 min to kill extra-cellular bacteria. Cells were washed three times with DMEM/F12 to removegentamicin and lysed with 1% saponin in PBS. Appropriate dilutions wereplated to detect intracellular bacterial titers. Efficacy of extracellular bacteriaremoval by gentamicin was verified in control experiments.
Immunofluorescence microscopy
Cells were cultured on LabTek Chamberslides (Thermo Scientific). HBECwere insulin depleted overnight. After 30 min preincubation with or withoutinsulin, HBEC were treated with Poly-(I:C) at 1 mg/ml for 60 min. Calu-3cells were serum-starved overnight and incubated for 60 min with viable
2 FOXO TRANSCRIPTION FACTORS REGULATE INNATE IMMUNE MECHANISMS
by guest on July 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
NTHi prepared as described above. Cells were fixed for 15 min with 4%paraformaldehyde in PBS and washed twice with ice-cold PBS. To per-meabilize the cells and block unspecific binding sites, slides were treatedwith 1% BSA and 0.1% Tween-20 in PBS for 1 h. Subsequently, cells wereincubated overnight at 4˚C with the primary Ab (anti-FOXO3A; Abcam)diluted 1:1000 in PBS containing 1% BSA and 0.1% Tween-20. Cells wereincubated for 1 h with the secondary Ab (polyclonal swine anti-rabbitTRITC; DakoCytomation) and phalloidin (Alexa Fluor-488 Phalloidin;Life Technologies). Between each two steps, cells were washed threetimes with PBS. Cells were mounted using Fluorescent Mounting Me-dium (DakoCytomation). To analyze images and merge channels, we usedImageJ software (National Institutes of Health).
Mouse experiments
Mice (C57BL/6N) were maintained under specific pathogen-free con-ditions. All animal experiments were approved by the Landesamt furSoziales, Gesundheit und Verbraucherschutz of the State of Saarland fol-lowing the national guidelines for animal treatment. Mice were slightlyanesthetized by i.p. injection of 2.6 mg ketaminhydrochloride (Ketanest;Pfizer) and 0.18 mg xylazinhydrochloride (Rompun; Bayer) per mouse andinfected intranasally with heat-inactivated P. aeruginosa (107 CFU/ml) inPBS, viable NTHi (107 CFU/ml) in PBS, or PBS alone as control. Threehours after bacterial infection, the animals were euthanized, and the lungwas removed for immunohistochemistry.
Immunohistochemistry
Formalin-fixed, paraffin-embedded human bronchial samples were obtainedfrom patients who underwent lung surgery for transplantation (cystic fi-brosis, chronic obstructive pulmonary disease [COPD], or acute respiratorydistress syndrome [ARDS] pneumonia) or cancer resection. The protocolwas approved by the Institutional Review Board (ethics committee) of theSaarland University Hospital, and informed consent was obtained from thepatients. Lungs of mice were embedded in paraffin, and immunohisto-chemistry was performed as described earlier (2). FOXO3 was detectedwith primary Abs (anti-FOXO3A; Abcam) diluted 1:200 in TBS (13 PBS[pH 7.2], 0.1% BSA). A biotinylated anti-rabbit secondary Ab was added(DakoCytomation), followed by avidin-HRP reagent (EnVision System;DakoCytomation). Imaging was performed using the software Cell SenseDimension (Olympus).
Statistical analysis
Values are displayed as mean 6 SD. Comparisons between groups wereanalyzed by t test (two-sided). Post hoc range tests were performed with
the t test (two-sided) with Bonferroni adjustment. Results were consideredstatistically significant for p , 0.05. All statistical tests were performedusing Prism software (GraphPad, San Diego, CA).
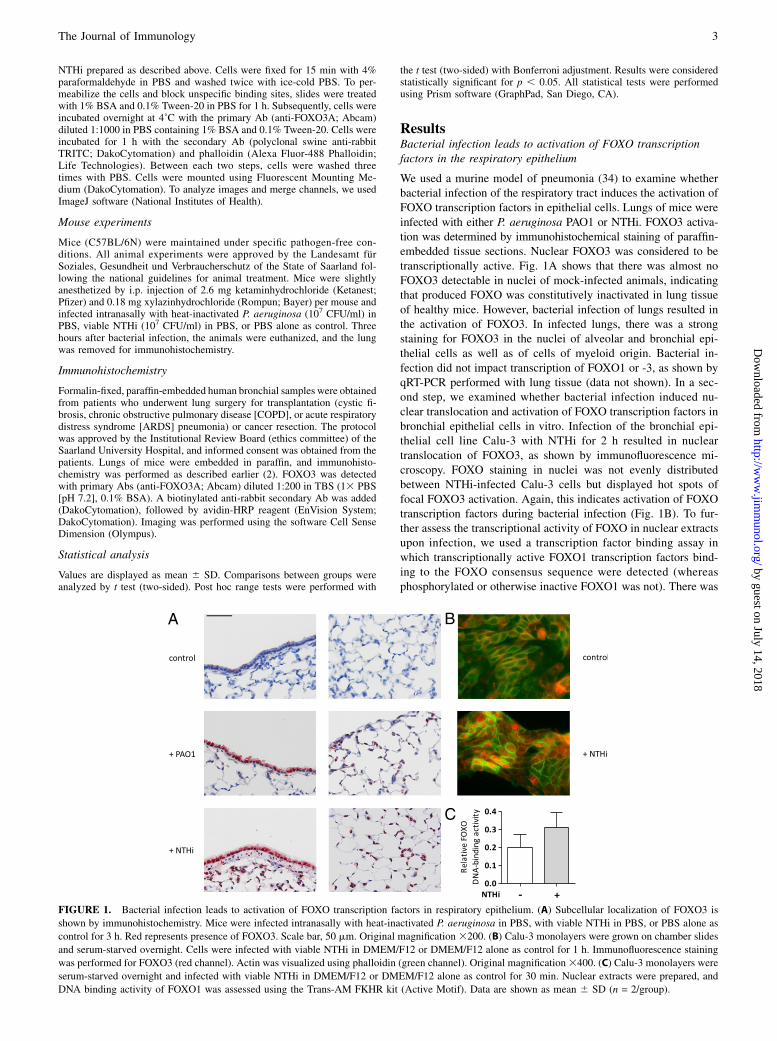
ResultsBacterial infection leads to activation of FOXO transcriptionfactors in the respiratory epithelium
We used a murine model of pneumonia (34) to examine whetherbacterial infection of the respiratory tract induces the activation ofFOXO transcription factors in epithelial cells. Lungs of mice wereinfected with either P. aeruginosa PAO1 or NTHi. FOXO3 activa-tion was determined by immunohistochemical staining of paraffin-embedded tissue sections. Nuclear FOXO3 was considered to betranscriptionally active. Fig. 1A shows that there was almost noFOXO3 detectable in nuclei of mock-infected animals, indicatingthat produced FOXO was constitutively inactivated in lung tissueof healthy mice. However, bacterial infection of lungs resulted inthe activation of FOXO3. In infected lungs, there was a strongstaining for FOXO3 in the nuclei of alveolar and bronchial epi-thelial cells as well as of cells of myeloid origin. Bacterial in-fection did not impact transcription of FOXO1 or -3, as shown byqRT-PCR performed with lung tissue (data not shown). In a sec-ond step, we examined whether bacterial infection induced nu-clear translocation and activation of FOXO transcription factors inbronchial epithelial cells in vitro. Infection of the bronchial epi-thelial cell line Calu-3 with NTHi for 2 h resulted in nucleartranslocation of FOXO3, as shown by immunofluorescence mi-croscopy. FOXO staining in nuclei was not evenly distributedbetween NTHi-infected Calu-3 cells but displayed hot spots offocal FOXO3 activation. Again, this indicates activation of FOXOtranscription factors during bacterial infection (Fig. 1B). To fur-ther assess the transcriptional activity of FOXO in nuclear extractsupon infection, we used a transcription factor binding assay inwhich transcriptionally active FOXO1 transcription factors bind-ing to the FOXO consensus sequence were detected (whereasphosphorylated or otherwise inactive FOXO1 was not). There was
FIGURE 1. Bacterial infection leads to activation of FOXO transcription factors in respiratory epithelium. (A) Subcellular localization of FOXO3 is
shown by immunohistochemistry. Mice were infected intranasally with heat-inactivated P. aeruginosa in PBS, with viable NTHi in PBS, or PBS alone as
control for 3 h. Red represents presence of FOXO3. Scale bar, 50 mm. Original magnification 3200. (B) Calu-3 monolayers were grown on chamber slides
and serum-starved overnight. Cells were infected with viable NTHi in DMEM/F12 or DMEM/F12 alone as control for 1 h. Immunofluorescence staining
was performed for FOXO3 (red channel). Actin was visualized using phalloidin (green channel). Original magnification3400. (C) Calu-3 monolayers were
serum-starved overnight and infected with viable NTHi in DMEM/F12 or DMEM/F12 alone as control for 30 min. Nuclear extracts were prepared, and
DNA binding activity of FOXO1 was assessed using the Trans-AM FKHR kit (Active Motif). Data are shown as mean 6 SD (n = 2/group).
The Journal of Immunology 3
by guest on July 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

increased DNA-binding activity of nuclear FOXO1 after 30 min ofincubation with viable NTHi (Fig. 1C). These results stronglysuggest that bacterial infection results in the activation of FOXOtranscription factors in respiratory epithelial cells.
FOXO regulates epithelial innate immune functions
As we have shown that FOXO is activated during the course of anairway infection and based on previous publications reporting thatFOXO promotes inflammation by regulating the expression ofTLR4 and IL-1b in macrophages (16, 25), we sought to investigatethe function of FOXO in innate immune functions of epithelialcells. Therefore, we generated FOXO-deficient respiratory epi-thelial cells via siRNA-mediated silencing of FOXO1 and FOXO3in the bronchial epithelial cell line Calu-3. Downregulation ofFOXO was confirmed by qRT-PCR (data not shown). Silencing ofFOXO resulted in a reduced expression of innate immune factorsboth under basal conditions and after bacterial stimulation (Figs.2, 3). In unstimulated cells, silencing of FOXO led to significantdecreases in the basal release of the proinflammatory cytokinesIL-6 (60%; Fig. 2A), IP-10 (84%; Fig. 3C), and IL-8 (55%; Fig.3D) and in the basal expression of the AMP hBD-2 (73%; Fig. 2C)as shown by ELISA and qRT-PCR. Furthermore, additionalchallenge of Calu-3 cells with bacteria resulted in a pathogen-specific response. Cells were stimulated for 4 h with either via-ble NTHi or heat-inactivated P. aeruginosa (Fig. 2). Stimulationwith viable NTHi for 4 h caused an increase in IL-6 release (Fig.2A) and expression (Fig. 2B) that was significantly suppressed inFOXO-deficient cells. However, silencing of FOXO did not affectIL-6 release and expression of cells stimulated with P. aeruginosafor 4 h (Fig. 2A, 2B), whereas the induction of expression of hBD-2 (Fig. 2C) and IP-10 (Fig. 2D) was diminished in FOXO-silencedcells stimulated with P. aeruginosa. As shown in Fig. 2C and 2D,stimulation with viable NTHi for 4 h was not sufficient to induceexpression of hBD-2 and IP-10. Therefore, cells were stimulatedwith NTHi for 24 h. To prevent bacterial outgrowth on cells, NTHiwere inactivated with antibiotics prior to their addition to cells,and antibiotics were given into the culture media. Under theseconditions, stimulation with inactivated NTHi for 24 h resulted inincreased expression or release of IL-6 (Fig. 3A), hBD-2 (Fig.3B), IP-10 (Fig. 3C), and IL-8 (Fig. 3D), whereas these effects
were significantly impaired in cells treated with siRNA specificfor FOXO1/3.NTHi has been shown to invade the alveolar epithelial cell line
A549 in a PI3K-dependent manner (35). To examine if PI3K andFOXO play a role in the internalization of NTHi into bronchialepithelial cells, we infected Calu-3 monolayers with viable NTHiin the presence of the PI3K inhibitor LY294002 (Fig. 4A). After2 h of bacterial incubation, extracellular bacteria were killed bygentamicin treatment, and intracellular bacteria were counted incell lysates. As shown before for A549 cells, LY294002-treatedcells contained significantly lower amounts of viable NTHi. Be-cause FOXO transcription factors are major downstream targets ofPI3K signaling (26), we determined whether FOXO is involved inbacterial uptake by epithelial cells. Calu-3 cells were treated withsiRNA specific for FOXO1/3 and control siRNA, respectively, andinfected as described above (Fig. 4B). siRNA-mediated silencingof FOXO1/3 resulted in significantly higher numbers of intra-cellular bacteria if compared with control cells, suggesting thatFOXO is involved in preventing bacterial internalization.These results show that FOXO transcription factors regulate
innate immune functions of bronchial epithelial cells duringbacterial infection.
FOXO silencing does not affect TLR5-related andIL-1b–induced innate immune responses
To identify the underlying mechanisms of FOXO-dependent innateimmune regulation, we tested several TLR agonists in their abilityto induce an innate immune response in Calu-3 cells after FOXOsilencing. The TLR agonists LPS (TLR4 ligand) and PAM3CSK4(TLR2 ligand) were not able to induce release of IL-6 in eithercontrol-siRNA– or FOXO1/3-siRNA–treated cells (data not shown).However, the TLR5 ligand flagellin caused a robust release of IL-6(Fig. 5A). In line with our findings with the flagellin-expressingP. aeruginosa strain PAO1, IL-6 release in response to stimulationwith flagellin was not affected by silencing FOXO (Fig. 5A). In-nate immune mechanisms of respiratory epithelial cells are notonly activated through pattern recognition receptors, but also bycytokines released from activated immune cells, such as alveolarmacrophages. To examine whether this indirectly triggered innateimmune response is also affected by FOXO knockdown, siRNA-
FIGURE 2. FOXO regulates expression and
release of innate immune factors in respiratory
epithelial cells. Calu-3 cells were transfected
with either unspecific control siRNA (RNA in-
terference [RNAi] 2) or siRNA specific for
FOXO1 and FOXO3 mRNA (RNAi +). At 24 h
after transfection, cells were serum-starved overnight.
After that, cells were treated with viable NTHi, heat-
inactivated P. aeruginosa PAO1, or medium alone as
control for 4 h. (A) IL-6 concentrations were
measured in supernatants by ELISA. mRNA levels
of IL-6 (B), hBD-2 (C), and IP-10 (D) were
assessed by qRT-PCR. Data are shown as mean 6SD (n $ 3/group). Presented data are representa-
tive for two independent experiments. *p , 0.05,
**p , 0.01, ***p , 0.001.
4 FOXO TRANSCRIPTION FACTORS REGULATE INNATE IMMUNE MECHANISMS
by guest on July 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

treated cells were stimulated with IL-1b for 24 h. Silencing ofFOXO did not affect the release of IL-6 in response to stimulationwith IL-1b (Fig. 5B).
FOXO mediates TLR-3 signaling
To examine whether FOXO mediates endosomal TLR signaling inepithelial cells, FOXO was silenced in Calu-3 cells using siRNAspecific for FOXO1/3, and cells were treated with Poly-(I:C),a synthetic ligand specific for TLR3. Because endosomal TLR3signals through TRIF and is easy to specifically address, it is anappropriate model for endosomal TRIF-dependent TLR signalingin general. Stimulation of epithelial cells with Poly-(I:C) at 1mg/mlresulted in a significantly increased secretion of IL-6 and IP-10 incontrol siRNA-treated cells but not in cells treated with siRNAspecific for FOXO1/3 (Fig. 6A, 6B). In the latter, secretion of IL-6and IP-10 was suppressed to basal levels. Induction of IL-8 byPoly-(I:C) was affected to a lesser extent by FOXO (Fig. 6C). Thedata on the protein level were confirmed by corresponding qRT-PCR results for IL-6 and IP-10 (data not shown). Analogous qRT-PCR results were also observed for hBD-2 and TNF (Fig. 6D, 6E),
with decreased mRNA levels in FOXO-deficient cells upon Poly-(I:C)stimulation. To exclude the possibility that these findings are asimple consequence of TLR3 downregulation through FOXOknockdown, we assessed TLR3 gene expression by qRT-PCR (Fig.6F). However, there was no difference in TLR3 transcription be-tween knockdown and control cells. In a former study, it has beenshown that Poly-(I:C) at 10 mg/ml induces TLR3-dependent celldeath in respiratory epithelial cells (36). Therefore we assessed ifcell death occurs in our particular setting. In a lactate dehydroge-nase cytotoxicity assay, we did not find any increase in cytotoxicityafter treatment with Poly-(I:C) at 1 mg/ml (data not shown). We alsoassessed the expression of Bim, a well-known proapoptotic FOXOtarget gene (24), to determine whether Poly-(I:C) induced a proap-optotic transcriptional program in our experiment (Fig. 6G). This wasnot the case.
Insulin interferes with FOXO-regulated innate immunefunctions of primary HBEC
As insulin negatively regulates the transcriptional activity of FOXOvia the PI3K–AKT axis, we hypothesized that insulin treatmentimpacts innate immune functions of respiratory epithelial cells. Toestablish more physiologic conditions and diminish cell line–specific effects, such as constitutively active insulin signaling,which is common in cancer cells, we used primary HBEC (ob-tained from nondiabetic subjects) cultured in defined airway epi-thelial growth media. To study the function of FOXO transcriptionfactors in primary HBEC, expression of FOXO1/3 was silencedwith siRNA. Downregulation of FOXO was confirmed by qRT-PCR (Fig. 7). In addition, HBEC were incubated with two dif-ferent concentrations of insulin (1 and 5 mg/ml) and stimulatedwith Poly-(I:C). Fig. 8A shows that downregulation of FOXOresulted in reduced expression of hBD-2 after stimulation withPoly-(I:C) for 24 h. This is in line with the results obtained withCalu-3 cells. In addition, hBD-2 expression was affected by insulin.In control-siRNA–treated cells, insulin had no effect on hBD-2expression. However, in HBEC treated with FOXO1/3-specificsiRNA and incubated with insulin at a high dose, the expression ofhBD-2 mRNA was significantly reduced compared with HBECtreated with FOXO-specific siRNA and incubated with a low doseof insulin. Furthermore, silencing of FOXO1/3 resulted in a re-duced release of IL-6 and IP-10 from HBEC under basal con-ditions (Fig. 8B, 8C) in agreement with our data obtained with thecell line Calu-3. FOXO knockdown reduced the release of IL-6
FIGURE 3. FOXO regulates expression and re-
lease of innate immune factors in respiratory epi-
thelial cells. Calu-3 cells were transfected with
either unspecific control siRNA (RNA interference
[RNAi] 2) or siRNA specific for FOXO1 and
FOXO3 mRNA (RNAi +). At 24 h after transfec-
tion, cells were serum-starved overnight. After that,
cells were treated with inactivated NTHi or me-
dium alone as control for 24 h. Concentrations of
IL-6 (A), IP-10 (C), and IL-8 (D) were measured in
supernatants by ELISA. hBD-2 mRNA levels (B)
were assessed by qRT-PCR. Data are shown as
mean 6 SD (n $ 3/group). Presented data are
representative for two independent experiments. *p
, 0.05, **p , 0.01.
FIGURE 4. FOXO affects NTHi internalization in respiratory epithelial
cells. (A) Calu-3 monolayers were serum-starved overnight and infected
with viable NTHi for 2 h with or without LY294002 at 50 mM. Intracel-
lular titers of NTHi were quantified using the NTHi uptake assay as de-
scribed in the Materials and Methods. Data are shown as mean 6 SD (n $
3/group). (B) Calu-3 cells were transfected with either unspecific control
siRNA (RNA interference [RNAi] 2) or siRNA specific for FOXO1 and
FOXO3 mRNA (RNAi +). At 24 h after transfection, cells were serum-
starved overnight and subsequently infected with viable NTHi for 2 h.
Intracellular titers of NTHi were quantified using the NTHi uptake assay.
Data are shown as mean 6 SD (n $ 6/group). Presented data are repre-
sentative for two independent experiments. ***p , 0.001.
The Journal of Immunology 5
by guest on July 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

and IP-10 after stimulation with Poly(I:C) in an insulin-dependentmanner. Although cells incubated with low-dose insulin were notconsiderably affected in IL-6 and IP-10 production by silencing ofFOXO, both cytokines were significantly downregulated underhigh-dose insulin conditions (Fig. 8B, 8C).Because FOXO activation induced by oxidative stress overcomes
growth factor and insulin-dependent inhibition of FOXO (37), weassumed a similar relation between infectious triggers and insulintreatment. Considering our FOXO-mediated TLR3 signaling data,
we stimulated HBEC with Poly-(I:C) in the presence or absence ofinsulin and evaluated FOXO activation with immunofluorescencemicroscopy. Fig. 9 shows that in insulin-treated cells FOXO3 wassequestered in the cytoplasm, whereas it was predominantly lo-calized in the nucleus in insulin-depleted cells. After Poly-(I:C)treatment, there was a strong nuclear FOXO3 signal not only ininsulin-depleted, but also in insulin-treated cells, indicating thatTLR3-induced FOXO activation partly overcomes PI3K-AKT–de-pendent FOXO inhibition through insulin. However, FOXO ac-
FIGURE 5. FOXO silencing does not affect TLR5-
related and IL-1b–induced innate immune responses.
Calu-3 cells were transfected with either unspecific
control siRNA (RNA interference [RNAi]2) or siRNA
specific for FOXO1 and FOXO3 mRNA (RNAi +). At
24 h after transfection, cells were serum-starved over-
night. Subsequently, cells were stimulated with flagel-
lin at 1 mg/ml (A) or IL-1b at 1 ng/ml (B) for 24 h. IL-
6 concentrations were measured in supernatants by
ELISA. Data are shown as mean 6 SD (n = 4/group).
Presented data are representative for two independent
experiments. **p , 0.01.
FIGURE 6. FOXO mediates TLR3 signaling.
Calu-3 cells were transfected with either unspe-
cific control siRNA (RNA interference [RNAi] 2)
or siRNA specific for FOXO1 and FOXO3 mRNA
(RNAi +). At 24 h after transfection, cells were
serum-starved overnight and subsequently treated
with Poly-(I:C) at 1 mg/ml for 24 h. IL-6 (A), IP-
10 (B), and IL-8 (C) concentrations were measured
in supernatants by ELISA, and mRNA levels of
hBD-2 (D), TNF (E), TLR3 (F), and Bim (G) were
assessed by qRT-PCR. Data are shown as mean 6SD (n $ 3/group). Presented data are representa-
tive for two independent experiments. *p , 0.05,
**p , 0.01, ***p , 0.001.
6 FOXO TRANSCRIPTION FACTORS REGULATE INNATE IMMUNE MECHANISMS
by guest on July 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

tivation seemed to be weaker in insulin exposed cells, indicatingan immune-inhibitory character of high insulin levels.We further examined the impact of insulin on epithelial uptake
of NTHi (Fig. 10). HBEC were insulin-depleted overnight andincubated with or without 5 mg/ml insulin and 50 mM LY294002for 30 min. After that, we performed the NTHi uptake assay asdescribed above. Consistent with our previous results, we founda clear dependency of NTHi uptake by HBEC on PI3K activity,with the highest intracellular bacterial counts in insulin-treatedcells (maximal FOXO inhibition) and lowest counts in insulin-unsupplemented and LY294002-treated cells (maximal FOXOactivation).Altogether, these results suggest that insulin modulates FOXO-
dependent innate immune functions of HBEC in an immune-suppressive manner.
FOXO is activated in inflammatory lung diseases
To assess the clinical significance of our results, we analyzed FOXO3expression in tissue samples from patients undergoing lung surgery(Fig. 11). Strong nuclear staining of FOXO3 was present in thelungs of patients with different infection-related lung diseases, in-cluding cystic fibrosis, COPD, and severe ARDS pneumonia. Nu-clear localized FOXO3 staining was weaker in nonafflicted lungtissue from a lung cancer patient (smoker) and virtually nonex-istent in lung tissue from a healthy organ donor. These observa-tions imply an important role of FOXO in acute and chronic lunginflammation and support our findings obtained from in vivo andin vitro experiments presented in this study.
DiscussionIn this study, we analyzed the function of FOXO transcription factorsin the innate immune response of epithelial surfaces of the respi-ratory tract. Our principle finding is that FOXO transcription factorsoperate as key modulators of innate immunity under basic conditionsand in the case of bacterial infection. We show that FOXO tran-scription factors are activated during bacterial infection in respiratoryepithelial cells and in human bronchial epithelium of patients withinfection-related lung diseases, whereas inactivation and degradationseems to be the default state in uninfected epithelial cells. To ourknowledge, this is the first time that FOXO activation upon infectionhas been detected in a vertebratemodel organism and in patients withrespiratory tract diseases. We further demonstrate that FOXO de-ficiency results in suppression of various epithelial innate immunefunctions, including secretion of AMPs and cytokines, and in anincrease of pathogen uptake. Finally, we reveal that TLR3-mediated
innate immune responses of bronchial epithelial cells depend onFOXO transcription factors and that FOXO and insulin forma regulatory network modulating the innate immune response.
FIGURE 7. Downregulation of FOXO1 and 3 by siRNA in primary
HBEC. Primary HBEC were transfected with either unspecific control
siRNA (RNA interference [RNAi] 2) or siRNA specific for FOXO1 and
FOXO3 mRNA (RNAi +). After 24 h, mRNA levels of FOXO1 and
FOXO3 were assessed by qRT-PCR. Data are shown as mean6 SD (n = 6/
group). Presented data are representative for two independent experiments.
*p , 0.05, ***p , 0.001.
FIGURE 8. Insulin interferes with FOXO-regulated innate immune
functions. Primary HBEC were transfected with either unspecific control
siRNA (RNA interference [RNAi] 2) or siRNA specific for FOXO1 and
FOXO3 mRNA (RNAi +). After 24 h, the medium were changed. Cells
were incubated with medium containing insulin at 1 or 5 mg/ml. After
24 h, the medium was changed again, and cells were stimulated with Poly-
(I:C) at 1 mg/ml for 24 h (insulin concentrations as before). mRNA levels
of hBD-2 were assessed by qRT-PCR (A). Concentrations of IL-6 (B) and
IP-10 (C) were measured in supernatants by ELISA. Data are shown as
mean 6 SD (n $ 3/group). Presented data are representative for two in-
dependent experiments. *p , 0.05, **p , 0.01, ***p , 0.001.
The Journal of Immunology 7
by guest on July 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

In recent years, several studies provided evidence that FOXOtranscription factors play a role in inflammation and infection.Becker et al. (15) demonstrated that FOXO regulates the expres-sion of several AMPs in D. melanogaster independent from Tollpathway activation. They showed that FOXO is a transcriptionfactor of AMPs in D. melanogaster by binding to regulatoryDNA regions of AMP genes and further demonstrated that insulin-dependent inhibition of FOXO resulted in downregulation ofb-defensin baseline expression in human tissues. Our data un-derline this role of FOXO transcription factors as regulators ofinnate immunity in the setting of the bronchial epithelium. In linewith Becker et al. (15), we show that the expression of AMPs suchas the b-defensin hBD-2 is regulated by FOXO under basalconditions. Another study presented by Fan et al. (25) showedthat FOXO1 promotes TLR-mediated inflammation by increasingTLR4 signaling in macrophages. This is accomplished by bindingof FOXO1 to enhancer-like elements within the TLR4 gene. In ourexperiments, however, we did not find any changes in TLR4 geneexpression in FOXO-deficient bronchial epithelial cells (data notshown), indicating that the impact of FOXO on innate immunity inepithelial cells is not only triggered by modulation of TLR4 ex-
pression. In addition to TLR4, Fan et al. (25) described manyother factors involved in TLR signaling cascades and in innateimmunity to be potential FOXO target genes. This candidate listincluded IL-6, IP-10, and TNF, which is consistent with ourpresent data. IL-8 is not predicted to be a direct FOXO targetgene; still, we found IL-8 expression to be FOXO dependent. Thismay be due to the ability of FOXO to regulate the expression ofseveral mediators within the TLR cascade like TRIF and MyD88.Therefore, FOXO might also indirectly affect the expression offactors downstream of TLRs, such as IL-8, even though they arenot FOXO targets in a strict sense.In this study, we show that silencing of FOXO expression in
bronchial epithelial cells resulted in a diminished expression andrelease of inflammatory mediators and AMPs during bacterialinfection with NTHi and P. aeruginosa. Yet, this was not the casefor IL-6 after P. aeruginosa stimulation. Because IL-6 is a targetgene of various pathways, we assumed that the effect of FOXOsilencing may be masked in this case due to the broad spectrum ofPAMPs expressed by P. aeruginosa. Indeed, stimulation of cellswith flagellin, a TLR5 agonist expressed by the PAO1 strain thatwe used in this study, was sufficient to induce an innate immuneresponse in FOXO-deficient cells, showing that silencing ofFOXO does not impair TLR5-dependent innate immunity. Wefurther show that FOXO is focally activated during NTHi infec-tion in vitro, suggesting that cells that internalized bacteria showFOXO activation. Moreover, NTHi only induced a robust immuneresponse after prior destruction with antibiotics, indicating thatviable NTHi are insufficient to activate epithelial innate immunity,and NTHi destruction is required to reach adequate immunestimulation. Degradation and discharge of microbial PAMPs canalso be associated with endosomal processing of internalizedbacteria. In fact, it has been shown that bacteria internalized byrespiratory epithelial cells end up in the endosomal compartmentof the cell (35). Considering these points, we assumed that FOXOis involved in an endosomal pathway that requires uptake anddegradation of the pathogen. TLR3 is one of the pattern recog-nition receptors localized in the endosomal compartment (12, 38)and can easily be targeted, unlike TLR4, which is additionallyexpressed on the cell membrane, thereby triggering distinct sig-naling mechanisms that are difficult to discriminate (39). There-fore, we decided to focus on TLR3 signaling. Even though TLR3is mainly known as a receptor for viral dsRNA, there is rising
FIGURE 9. TLR3-mediated FOXO activation over-
comes insulin-dependent FOXO inhibition. Subcellular
localization of FOXO3 in HBEC is shown by immu-
nofluorescence. HBEC were insulin-depleted over-
night and subsequently pretreated with (+) or without
(2) insulin at 5 mg/ml. After 30 min, cells were
stimulated with Poly-(I:C) at 1 mg/ml for 1 h.
FIGURE 10. Insulin modulates NTHi internalization in respiratory ep-
ithelial cells. HBEC were insulin-depleted overnight and subsequently
pretreated with (+) or without (2) insulin at 5 mg/ml and LY294002 at 50
mM. After 30 min, cells were infected with viable NTHi for 2 h. Intra-
cellular titers of NTHi were quantified using the NTHi uptake assay as
described in the Materials and Methods. Data are shown as mean 6 SD
(n = 3/group). Presented data are representative for two independent
experiments. *p , 0.05, **p , 0.01.
8 FOXO TRANSCRIPTION FACTORS REGULATE INNATE IMMUNE MECHANISMS
by guest on July 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

evidence that TLR3 is also involved in antibacterial responses (13,14). Beyond that, upon activation, TLR3 induces the same TRIF-dependent signaling events as endosomal TLR4, resulting in ac-tivation of the MyD88-independent pathway. This makes TLR3 aninteresting model to study both endosomal TLR3- and TLR4-triggered innate immune responses. To determine whether FOXOplays a role in endosomal TLR3 signaling, we used the syntheticagonist Poly-(I:C) to specifically activate TLR3 in FOXO knock-down cells. Although Poly-(I:C) induced a robust innate immuneresponse in control-siRNA–treated cells, expression of inflamma-tory markers was suppressed below baseline levels in stimulatedFOXO-silenced cells. These results show that FOXO transcriptionfactors are crucial factors for maintenance of the TLR3 signalingcascade.Another approach that we used to evaluate FOXO innate immune
functions was an NTHi uptake assay, a common method to detectintracellular bacteria (35, 40). In our experiments, we found thatactivation of the PI3K pathway, which causes FOXO inhibition(26), is capable of enhancing invasion of NTHi in airway epi-thelial cells as previously described for alveolar epithelial cells(35). Beyond that, we found that silencing of FOXO results inhigher intracellular bacterial counts. We therefore deduced thatinhibition of FOXO is a possible mechanism by which PI3K en-hances bacterial invasion. It can be argued that enhanced bacterialcounts in FOXO-deficient cells could have two different reasons.Both are consistent with our findings to some extent. One possi-bility is that NTHi can invade FOXO-deficient cells more easilygiven their reduced ability to produce antimicrobial factors likedefensins, which are likely to affect intracellular killing of bac-teria. Thus, internalization may be promoted by the pathogen toevade the immune response, persist intracellularly, and establishcolonization (35). The second possibility is that epithelial cellsinternalize bacteria to endosomally process them. Activation ofseveral pathways by PAMPs, including signaling cascades acti-vated by endosomal TLRs, requires uptake and consequent deg-radation of pathogens (41). This is consistent with the observationthat heat- or antibiotic-inactivated bacteria stimulate the immuneresponse more effectively than viable microbes. According to thishypothesis, pathogen internalization is an innate immune functionof the host cell for the purpose of sensing microbial PAMPs toactivate immune responses. In our experiments, FOXO-deficientcells had a significant impairment of innate immune functions. Itis possible that FOXO-deficient cells fail to kill bacteria once theyare internalized, which would explain higher intracellular bacterialcounts. This would also impede PAMP sensing and thus affectadditional effector mechanisms.
So far, the role of FOXO in innate immunity is incompletelyunderstood, with literature reporting inflammatory (15, 25) andanti-inflammatory effects (42). This juxtaposition is not neces-sarily a paradox, as it is comparable to former findings regardingFOXO in the context of oxidative cell damage, in which it hasbeen reported that FOXO is able to promote apoptosis as well ascell survival. The reason why FOXO can have seemingly contraryfunctions is the variety of possible posttranslational modifications.Thus, the transcriptional program is determined not only by nuclearFOXO activity, but also by the individual pattern of phosphoryla-tion, ubiquitination, and acetylation (17). In our experiments, wemonitored cell integrity by morphological examination and lactatedehydrogenase cytotoxicity assay (data not shown). We did notdetect any relevant levels of cell death [e.g., after treatment ofinsulin-depleted cells with 1 mg/ml Poly-(I:C)], although this ledto a strong nuclear FOXO activity. We also did not find any in-crease in the expression of the well-described FOXO target geneBim, a proapoptotic factor of the Bcl-2 family that is induced uponseveral stress stimuli (24). Numata et al. (36) reported that treatmentof insulin-depleted HBEC with 10 mg/ml Poly-(I:C) leads to apo-ptosis via TLR3. The authors did not investigate the role of FOXOin these findings, but, considering our data, FOXO is a potentialcoeffector.In the current understanding, basal FOXO activity is controlled
by the PI3K–AKT pathway. Activation of this pathway by insulinor growth factors results in sequestration of FOXO in the cytosol(26, 27) and polyubiquitination, which is followed by proteasomaldegradation. Inactivation of FOXO can be overcome under stressconditions by stress kinases like JNK, which activates FOXO di-rectly, or indirectly by inhibition of FOXO-binding proteins (17)and deacetylases (31, 37). We observed a very similar effect whenFOXO inactivation by insulin was overcome by Poly-(I:C) treat-ment in primary HBEC. Insulin treatment alone excluded FOXOfrom the nucleus, as previously described in many reports, whereasinsulin-depleted cells showed a predominantly nuclear localizationof FOXO. After treatment of HBEC with Poly-(I:C), we observeda strikingly strong FOXO activation in insulin-depleted cells, ex-ceeding that of only insulin-depleted cells. Interestingly, in insulin-treated cells, Poly-(I:C) was capable of activating FOXO also, thusovercoming the inhibitory effect of insulin. However, the nuclearFOXO signal was weaker than in insulin-depleted cells. This resultsuggests that insulin slightly impairs FOXO-dependent innate im-munity rather than completely suppressing it. This further under-lines the results of our siRNA studies in primary HBEC. In thisstudy, we found that even though minor tendencies could be ob-served, insulin does not significantly impair respiratory cell innate
FIGURE 11. FOXO is activated in infection-related lung diseases in vivo. Subcellular localization of FOXO3 is shown by immunohistochemistry.
FOXO3 was detected in formalin-fixed, paraffin-embedded human bronchial samples from a healthy donor (A) and from patients with lung cancer (smoker,
nonafflicted tissue) (B), cystic fibrosis (C), COPD (E), and severe ARDS pneumonia (D). Red represents presence of FOXO3. Scale bar, 50 mm.
The Journal of Immunology 9
by guest on July 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

immune functions. Furthermore, silencing of FOXO was not ca-pable of suppressing IL-6 and IP-10 release upon Poly-(I:C)treatment under low-insulin conditions. Because it is not possibleto entirely knock out FOXO with siRNA, we assumed that remainingFOXO protein is sufficient to maintain innate immune functionswhen it is in a highly activated state. Indeed, when remainingFOXO was inhibited by high-insulin conditions, IL-6 and IP-10release was significantly downregulated in FOXO-deficient cells.In the case of hBD-2, FOXO silencing resulted in a downregula-tion already under low-insulin conditions compared with control-siRNA–treated cells. Under treatment with the higher insulinconcentration, hBD-2 expression after Poly-(I:C) stimulation waseven suppressed to baseline levels in FOXO-deficient cells. Insummary, hyperinsulinism may impair innate immune functions inrespiratory epithelial cells by adverse modulation of FOXO.To evaluate the significance of our findings for respiratory tract
diseases in vivo, we assessed FOXO activity in human bronchialtissue from patients with different inflammatory or infectious lungdiseases. Analyzing samples from patients suffering from COPD,cystic fibrosis, and pneumonia/ARDS, we found that FOXO isactive during acute and chronic lung infection, which confirms ourin vitro data. In contrast to our observations, a recent study by Hwanget al. (42) reported that total FOXO is decreased in the lungs ofpatients with COPD. This discrepancy may be due to differences inpatient recruitment or state of disease.In our current report, we characterize FOXO transcription factors
as central factors within airway epithelial innate immunity. Weindicate that FOXO transcription factors are highly activated in thebronchial epithelium upon infectious stimuli, as shown in vitro withrespiratory epithelial cells, in a mouse model of pneumonia, andin human tissue samples. We further reveal a novel role of FOXOtranscription factors as regulators of innate immune functions ofrespiratory epithelial cells, introducing FOXO1 and -3 as criticalfactors for endosomal TLR3-dependent immune responses. Finally,we show that FOXO and insulin form a regulatory network, withinsulin acting in an immunosuppressive manner. Future studiesmay focus on the specific impact of FOXO on different respiratorytract diseases, on FOXO-dependent innate immune functions ofother epithelial surfaces, and on deepening the insights into theunderlying mechanistic details.
AcknowledgmentsWe thank Anja Honecker and Lena Arnold for excellent technical assistance
and Michelle Bill and Kerstin Kaendler for careful reading of the manu-
script.
DisclosuresThe authors have no financial conflicts of interest.
References1. Martin, T. R., and C. W. Frevert. 2005. Innate immunity in the lungs. Proc. Am.
Thorac. Soc. 2: 403–411.2. Hess, C., C. Herr, C. Beisswenger, T. Zakharkina, R. M. Schmid, and R. Bals.
2010. Myeloid RelA regulates pulmonary host defense networks. Eur. Respir. J.35: 343–352.
3. Beisswenger, C., E. S. Lysenko, and J. N. Weiser. 2009. Early bacterial colo-nization induces toll-like receptor-dependent transforming growth factor betasignaling in the epithelium. Infect. Immun. 77: 2212–2220.
4. Beisswenger, C., and R. Bals. 2005. Antimicrobial peptides in lung inflamma-tion. Chem. Immunol. Allergy 86: 55–71.
5. Ratner, A. J., J. L. Aguilar, M. Shchepetov, E. S. Lysenko, and J. N. Weiser.2007. Nod1 mediates cytoplasmic sensing of combinations of extracellularbacteria. Cell. Microbiol. 9: 1343–1351.
6. Kao, C. Y., Y. Chen, P. Thai, S. Wachi, F. Huang, C. Kim, R. W. Harper, andR. Wu. 2004. IL-17 markedly up-regulates beta-defensin-2 expression in humanairway epithelium via JAK and NF-kappaB signaling pathways. J. Immunol. 173:3482–3491.
7. Huang, F., C. Y. Kao, S. Wachi, P. Thai, J. Ryu, and R. Wu. 2007. Requirementfor both JAK-mediated PI3K signaling and ACT1/TRAF6/TAK1-dependent NF-kappaB activation by IL-17A in enhancing cytokine expression in human airwayepithelial cells. J. Immunol. 179: 6504–6513.
8. Chen, R., J. H. Lim, H. Jono, X. X. Gu, Y. S. Kim, C. B. Basbaum, T. F. Murphy,and J. D. Li. 2004. Nontypeable Haemophilus influenzae lipoprotein P6 inducesMUC5AC mucin transcription via TLR2-TAK1-dependent p38 MAPK-AP1 andIKKbeta-IkappaBalpha-NF-kappaB signaling pathways. Biochem. Biophys. Res.Commun. 324: 1087–1094.
9. Shuto, T., H. Xu, B. Wang, J. Han, H. Kai, X. X. Gu, T. F. Murphy, D. J. Lim,and J. D. Li. 2001. Activation of NF-kappa B by nontypeable Hemophilusinfluenzae is mediated by toll-like receptor 2-TAK1-dependent NIK-IKK alpha/beta-I kappa B alpha and MKK3/6-p38 MAP kinase signaling pathways inepithelial cells. Proc. Natl. Acad. Sci. USA 98: 8774–8779.
10. Schmeck, B., S. Huber, K. Moog, J. Zahlten, A. C. Hocke, B. Opitz,S. Hammerschmidt, T. J. Mitchell, M. Kracht, S. Rosseau, et al. 2006. Pneu-mococci induced TLR- and Rac1-dependent NF-kappaB-recruitment to the IL-8promoter in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 290:L730–L737.
11. Beisswenger, C., C. Hess, and R. Bals. 2012. Aspergillus fumigatus conidiainduce interferon-b signalling in respiratory epithelial cells1. Eur. Respir. J. 39:411–418.
12. Parker, D., and A. Prince. 2011. Innate immunity in the respiratory epithelium.Am. J. Respir. Cell Mol. Biol. 45: 189–201.
13. Sajjan, U. S., Y. Jia, D. C. Newcomb, J. K. Bentley, N. W. Lukacs, J. J. LiPuma,and M. B. Hershenson. 2006. H. influenzae potentiates airway epithelial cellresponses to rhinovirus by increasing ICAM-1 and TLR3 expression. FASEB J.20: 2121–2123.
14. Teng, F., V. Slavik, K. E. Duffy, L. San Mateo, and R. Goldschmidt. 2010. Toll-like receptor 3 is involved in airway epithelial cell response to nontypeableHaemophilus influenzae. Cell. Immunol. 260: 98–104.
15. Becker, T., G. Loch, M. Beyer, I. Zinke, A. C. Aschenbrenner, P. Carrera,T. Inhester, J. L. Schultze, and M. Hoch. 2010. FOXO-dependent regulation ofinnate immune homeostasis. Nature 463: 369–373.
16. Su, D., G. M. Coudriet, D. Hyun Kim, Y. Lu, G. Perdomo, S. Qu, S. Slusher,H. M. Tse, J. Piganelli, N. Giannoukakis, et al. 2009. FoxO1 links insulin re-sistance to proinflammatory cytokine IL-1beta production in macrophages. Di-abetes 58: 2624–2633.
17. Greer, E. L., and A. Brunet. 2005. FOXO transcription factors at the interfacebetween longevity and tumor suppression. Oncogene 24: 7410–7425.
18. Haeusler, R. A., K. H. Kaestner, and D. Accili. 2010. FoxOs function syner-gistically to promote glucose production. J. Biol. Chem. 285: 35245–35248.
19. Tran, H., A. Brunet, J. M. Grenier, S. R. Datta, A. J. Fornace, Jr., P. S. DiStefano,L. W. Chiang, and M. E. Greenberg. 2002. DNA repair pathway stimulated bythe forkhead transcription factor FOXO3a through the Gadd45 protein. Science296: 530–534.
20. Furukawa-Hibi, Y., K. Yoshida-Araki, T. Ohta, K. Ikeda, and N. Motoyama.2002. FOXO forkhead transcription factors induce G(2)-M checkpoint in re-sponse to oxidative stress. J. Biol. Chem. 277: 26729–26732.
21. Nemoto, S., and T. Finkel. 2002. Redox regulation of forkhead proteins througha p66shc-dependent signaling pathway. Science 295: 2450–2452.
22. Kops, G. J., T. B. Dansen, P. E. Polderman, I. Saarloos, K. W. Wirtz, P. J. Coffer,T. T. Huang, J. L. Bos, R. H. Medema, and B. M. Burgering. 2002. Forkheadtranscription factor FOXO3a protects quiescent cells from oxidative stress.Nature 419: 316–321.
23. Modur, V., R. Nagarajan, B. M. Evers, and J. Milbrandt. 2002. FOXO proteinsregulate tumor necrosis factor-related apoptosis inducing ligand expression.Implications for PTEN mutation in prostate cancer. J. Biol. Chem. 277: 47928–47937.
24. Dijkers, P. F., R. H. Medema, J. W. Lammers, L. Koenderman, and P. J. Coffer.2000. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated bythe forkhead transcription factor FKHR-L1. Curr. Biol. 10: 1201–1204.
25. Fan, W., H. Morinaga, J. J. Kim, E. Bae, N. J. Spann, S. Heinz, C. K. Glass, andJ. M. Olefsky. 2010. FoxO1 regulates Tlr4 inflammatory pathway signalling inmacrophages. EMBO J. 29: 4223–4236.
26. Brunet, A., A. Bonni, M. J. Zigmond, M. Z. Lin, P. Juo, L. S. Hu,M. J. Anderson, K. C. Arden, J. Blenis, and M. E. Greenberg. 1999. Akt pro-motes cell survival by phosphorylating and inhibiting a Forkhead transcriptionfactor. Cell 96: 857–868.
27. Kops, G. J., N. D. de Ruiter, A. M. De Vries-Smits, D. R. Powell, J. L. Bos, andB. M. Burgering. 1999. Direct control of the Forkhead transcription factor AFXby protein kinase B. Nature 398: 630–634.
28. Huang, H., and D. J. Tindall. 2007. Dynamic FoxO transcription factors. J. CellSci. 120: 2479–2487.
29. Sunayama, J., F. Tsuruta, N. Masuyama, and Y. Gotoh. 2005. JNK antagonizes Akt-mediated survival signals by phosphorylating 14-3-3. J. Cell Biol. 170: 295–304.
30. Essers, M. A., S. Weijzen, A. M. de Vries-Smits, I. Saarloos, N. D. de Ruiter,J. L. Bos, and B. M. Burgering. 2004. FOXO transcription factor activation byoxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 23: 4802–4812.
31. Brunet, A., L. B. Sweeney, J. F. Sturgill, K. F. Chua, P. L. Greer, Y. Lin, H. Tran,S. E. Ross, R. Mostoslavsky, H. Y. Cohen, et al. 2004. Stress-dependent regu-lation of FOXO transcription factors by the SIRT1 deacetylase. Science 303:2011–2015.
32. Bals, R., C. Beisswenger, S. Blouquit, and T. Chinet. 2004. Isolation and air-liquid interface culture of human large airway and bronchiolar epithelial cells. J.Cyst. Fibros. 3(Suppl 2): 49–51.
10 FOXO TRANSCRIPTION FACTORS REGULATE INNATE IMMUNE MECHANISMS
by guest on July 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

33. Pfaffl, M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29: e45.
34. Beisswenger, C., K. Kandler, C. Hess, H. Garn, K. Felgentreff, M. Wegmann,H. Renz, C. Vogelmeier, and R. Bals. 2006. Allergic airway inflammationinhibits pulmonary antibacterial host defense. J. Immunol. 177: 1833–1837.
35. Morey, P., V. Cano, P. Martı-Lliteras, A. Lopez-Gomez, V. Regueiro, C. Saus,J. A. Bengoechea, and J. Garmendia. 2011. Evidence for a non-replicative in-tracellular stage of nontypable Haemophilus influenzae in epithelial cells. Mi-crobiology 157: 234–250.
36. Numata, T., J. Araya, S. Fujii, H. Hara, N. Takasaka, J. Kojima, S. Minagawa,Y. Yumino, M. Kawaishi, J. Hirano, et al. 2011. Insulin-dependent phosphati-dylinositol 3-kinase/Akt and ERK signaling pathways inhibit TLR3-mediatedhuman bronchial epithelial cell apoptosis. J. Immunol. 187: 510–519.
37. Frescas, D., L. Valenti, and D. Accili. 2005. Nuclear trapping of the forkheadtranscription factor FoxO1 via Sirt-dependent deacetylation promotes expressionof glucogenetic genes. J. Biol. Chem. 280: 20589–20595.
38. Kawai, T., and S. Akira. 2011. Toll-like receptors and their crosstalk with otherinnate receptors in infection and immunity. Immunity 34: 637–650.
39. Zanoni, I., R. Ostuni, L. R. Marek, S. Barresi, R. Barbalat, G. M. Barton,F. Granucci, and J. C. Kagan. 2011. CD14 controls the LPS-induced endocytosisof Toll-like receptor 4. Cell 147: 868–880.
40. Swords, W. E., B. A. Buscher, K. Ver Steeg Ii, A. Preston, W. A. Nichols,J. N. Weiser, B. W. Gibson, and M. A. Apicella. 2000. Non-typeable Haemophilusinfluenzae adhere to and invade human bronchial epithelial cells via an interactionof lipooligosaccharide with the PAF receptor. Mol. Microbiol. 37: 13–27.
41. Campodonico, V. L., M. Gadjeva, C. Paradis-Bleau, A. Uluer, and G. B. Pier.2008. Airway epithelial control of Pseudomonas aeruginosa infection in cysticfibrosis. Trends Mol. Med. 14: 120–133.
42. Hwang, J. W., S. Rajendrasozhan, H. Yao, S. Chung, I. K. Sundar, H. L. Huyck,G. S. Pryhuber, V. L. Kinnula, and I. Rahman. 2011. FOXO3 deficiency leads toincreased susceptibility to cigarette smoke-induced inflammation, airspace en-largement, and chronic obstructive pulmonary disease. J. Immunol. 187: 987–998.
The Journal of Immunology 11
by guest on July 14, 2018http://w
ww
.jimm
unol.org/D
ownloaded from





























