Embed Size (px)
Citation preview

PEDIATRIC PHARMACOLOGY AND THERAPEUTICS
Enhanced hepatic drug clearance patients with cystic fibrosis
in
Gregory L. Kearns, PharmD, G e o r g e B. Mallory, Jr., MD, William R, Crom, PharmD, and William E. Evans, PharmD
From the Department of Pediatrics, University of Arkansas for Medical Sciences, and the Divi- sions of Pediatric Clinical Pharmacology and Pulmonary Medicine, Arkansas Children's Hospi- tal, Little Rock, Arkansas; the Departments of Clinical Pharmacy and Pediatrics, University of Tennessee, Memphis; and the Center for Pediatric Pharmacokinetics and Therapeutics and the Pharmaceutical Division, St, Jude Children's Research Hospital, Memphis
To examine whether hepat ic drug metabol ism is a l tered in patients with cystic fibrosis (CF), we eva luated the pharmacokinet ics of three model pharmaco- logic substrates (antipyrine, a marker of hepat ic ox idat ive metabolism; Iorazepam, a marker of hepat ic glucuronosyltransferase activity; and indocy- anine green [ICG], a marker of hepat ic b lood f low and bil iary secret ion) in 14 patients with CF (14.6 to 29.2 years of age) and in 12 chi ldren and adolescents with cancer (7.2 to 19.4 years of age), which was t reated with only surgery and radiation. Each study subject rece ived a single intravenous dose of the combined model substrates (0.03 mg/kg Iorazepam, 10 mg/kg antipyrine, and 0.5 mg/kg ICG) for 5 minutes, fo l lowed by repeated b lood sampling (n = 10) during a 24-hour postinfusion period. Patients with CF had a signif icantly greater plasma c learance of Iorazepam (56.5 • 5.2 vs 25.9 __+ 1.9 ml /min/m 2) and ICG (892.5 • 176.4 vs 256.5 • 41.7 ml /min/m 2) but not of antipyrine (27.2 • 3.8 vs 20.7 • 2.0 ml /min/m 2) in comparsion with control subjects. The apparent steady- state volume of distribution for Iorazepam, ICG, and antipyrine was signif icantly higher in the patients with CF (2.0-, 3.1-, and 1.4-fold, respectively) than in con- trol subjects. C learance of the model substrates d id not correlate with standard b iochemica l markers of hepat ic function. Similarly, no signif icant relationships were observed between the c learance or steady-state volume of distribution of the compounds and the National Institutes of Health prognost ic scores for the patients with CF. These data demonstrate that the plasma c learance of Iorazepam and ICG is increased in patients with CF and suggest that hepat ic glucuronosyltransferase act ivi ty and bil iary secretory capac i ty are enhanced in this disease. (J PEDIATR 1990;117:972-9)
Supported in part by a grant (No. A028 0-1) from the Cystic Fi- brosis Foundation, a National Institutes of Health grant (No. R37CA36401), a Center of Excellence Grant from the State of Tennessee, and American Lebanese and Syrian Associated Char- ities.
Submitted for publication March 19, 1990; accepted July 18, 1990.
Reprint requests: Gregory L. Kearns, PharmD, Associate Profes- sor of Pediatrics, Division of Pediatric Clinical Pharmacology, Ar- kansas Children's Hospital, 800 Marshall St., Little Rock, AR 72202.
9/25/23850
During the past l 5 years, controlled studies in patients with
cystic fibrosis have shown lower serum concentrations, in-
creased total plasma clearance, and an increase in the ap-
parent steady-state volume of distribution for a number of drugs, including gentamicin, 1 tobramycin, 2 dicloxacillin, 3
cloxacillin, 4 theophylline, 5 ceftazidime, 6 methicillin, 7 and
cefoperazonel 8 Recently, enhanced metabolism and nonre-
nal clearance of theophylline 9 and furosemide, 1~ two drugs
that undergo extensive hepatic biotransformation, also have
been demonstrated in patients with CF. A single mechanism
9 7 2

Volume 117 Enhanced hepatic drug clearance in CF 9 7 3 Number 6
ALT Alanine aminotransferase AST Aspartate aminotransferase BMI Body mass index BSA Body gurface area CF Cystic fibrosis CLcr Creatinine clearance GGT ~/-Gluta myltranspeptidase ICG Indocyanine green VDss Apparent steady-state volume of distribution
that accounts for and adequately describes these observed increases in renal 11 or hepatic 12 drug clearance in CF has not been determined. Indeed, the attendant renal 13 and hepatiO 4-17 pathophysiologic findings in many patients with the disease might be expected to impair rather than enhance
drug clearance. In a recent investigation we (W.R.C., W.E.E.) coadmin-
istered three pharmacologic "model substrates" to children: antipyrine, a marker of hepatic oxidative metabolism; lorazepam, a marker of hepatic glucuronosyltransferase activity; and indocyanine green, an indicator of hepatic blood flow and extraction and biliary secretory function) 8 The objectives of this study were to evaluate the pharma- cokinetics of these drugs to examine the effects of leukemia therapy on the function of three different important path- ways (Figure) for hepatic biotransformation and drug clearance, is In our study we used this pharmacologic tech- nique to determine whether specific alterations in these major pathways of hepatic drug clearance have increased
activity in patients with CF.
METHO DS
Patients. Fourteen patients with CF (14.6 to 29.2 years; seven females) and nine patients without CF (7.2 to 19.4 years; six females) were enrolled in the study after informed parental consent and patient assent were obtained. The di- agnosis of CF was based on clinical history, symptoms, and diagnostic sweat chloride results (chloride >60 mEq/L). All patients with CF had pancreatic insufficiency, which required exogenous enzyme replacement and vitamin sup- plementation. Additionally, each patient with CF received individualized doses of tobramycin and either ceftazidime (n = 9) or ticarcillin-clavulanic acid (n = 5) for treatment of a pulmonary exacerbation. The National Institutes of Health (NIH) prognostic scores t9 were determined for these patients at the conclusion of their hospitalization and ranged between 22 and 71, reflecting a range of moderate to severe disease. Patients with CF were studied on the final 2 days of a 14- to 21-day hospital course of treatment, at which time pulmonary function and clinical condition were stable and improved. The control population comprised nine children and adolescents with cancer who were disease free
after successful therapy for either Hodgkin disease (n = 6) or brain tumor (n = 3). These subjects were studied 3 to 6 months after hospital discharge for treatment of their ma- lignancies, which had included surgical excision and, for some, regional irradiation that did not involve exposure of the liver or kidneys. None of these patients had received any antineoplastic drugs at any time. Additionally, they were afebrile, clinically stable, and not receiving concomitant medications at the time of study. The experimental proto- col was approved by the human research advisory commit- tee of the University of Arkansas for Medical Sciences and by the clinical trials committee of St. Jude Children's Re- search Hospital.
Experimental procedures. Before administration of the model substrates, prothrombin time, complete blood cell count, serum urea nitrogen, serum creatinine, cholesterol, alkaline phosphatase, 7-glutamyltranspeptidase, ALT, AST, lactate dehydrogenase, total protein and albumin, and prothrombin time were determined for all patients (Table I). Creatinine clearance was estimated in all subjects by the following equation: CLcr = (0.55 X Height in centimeters)/ Serum creatinine. In six subjects with CF, CLcr was also determined from a quantitative urine collection. A physical examination was performed on each subject, and drug and dietary histories were obtained. For 72 hours before study, all subjects were instructed to avoid certain foods (e.g., al- cohol, chocolate, crueiferous vegetables, charcoal-broiled meats) and specific drugs (e.g., caffeine, cimetidine, pheny- toin, phenobarbital, carbamazepine, ketoconazole, ri- fampin) that might affect drug metabolism. Adherence to dietary and pharmacologic exclusions was verified by history and examination of patient diet and medication logs. Strenuous exercise was not permitted for 48 hours before and 24 hours after model substrate administration. None of the subjects had clinical evidence of an acute viral respiratory infection or a history of smoking at the time of study.
Drug administration and sampling. For 4 to 6 hours be- fore and 2 to 4 hours after administration of the model sub- strates, the subjects fasted and continued bed rest. In the morning on the day of study, each subject received a single intravenous injection containing lorazepam (Ativan), 0.03 mg/kg; ICG (Cardio-Green), 0.5 mg/kg; and antipyrine (Sigma Chemical Co., St. Louis, Mo., formulated for injection by the Department of Pharmaceuties, University of Tennessee, Memphis, as lnvestigational New Drug No. 23-409), 10 mg/kg for 5 minutes by constant rate infusion. During drug infusion and for 2 hours afterward, patients were kept recumbent, with the head of the bed elevated at 30 degrees. With the exception of transient sedation, which presumably was due to lorazepam, all subjects tolerated the model substrates without adverse effect.

9 7 4 Kearns et al. The Journal of Pediatrics December 1990
LORAZEPAM o
C I ~ OH
uridine diphosphate ~.~1 glucuronyt glucuronic " ~ transferase acid
GLUCUNONIDATION
INDOCu GREEN
CHs ella C. , " ] ~ CH'CH'(C'CH)z~CH " ~ - - CH,
Taken up by the hepatocytes rate-limiting factor is liver blood flow
LIVER BLOOD FLOW
0 OHA
ANTIPYRINE
�9 / I ~ CytOchrOme P450
�9 ymes
�9 �9 ~ C H ~
HMA NORA
OXIDATIVE METABOLISM
Figure. Summary of hepatic biotransformation routes for lorazepam, ICG, and antipyrine.
A venous blood sample (3.0 ml) was collected from a contralateral extremity into a heparinized tube immedi- ately before infusion of the model substrates and at 1, 3, 7, 10, and 15 minutes and 1, 1.25, 6, 23.75, and 24 hours after the end of the infusion. These sampling times were selected with the DESIGN computer program 2~ to obtain the most information about parameters to be estimated with the use of weighted nonlinear least-squares regression for classic compartmental modeling of serum concentration versus time data. Samples were immediately refrigerated and then centrifuged at 2500 rpm and 4 ~ C for 10 minutes, and the plasma was removed and frozen at - 70 ~ C until analysis.
Laboratory, pharmacokinetic, and statistical analysis. Plasma ICG concentrations were measured by a spectro- photometric technique within 2 weeks of collection. 21 The minimal detection limit for this assay was 0.2 mg/L; with- in- and between-analysis coefficients of variation were con- sistently <6%. Lorazepam and antipyrine were measured simultaneously with the use of high-performance liquid chromatography with ultraviolet light detection. 22 The be- tween-analysis coefficients of variation for antipyrine were 9.4% and 6.4% at concentrations of 60 and 5.5 rag/L, re- spectively, and for lorazepam were 9.1% and 9.9% at 110 and 10.5 ng/ml, respectively. 18 The mean concentration of duplicate samples for each analyte was used in the pharma-
cokinetic analysis. Initial polyexponential parameter estimates were ob-
tained from curve fits of plasma concentration versus time data for each subject by means of a peeling algorithm with correction for infusion time. 23 Final estimates were ob- tained by weighted nonlinear least-squares regression, 24 with weighting according to the reciprocal of observations. Selection of a two-compartment model to describe the pharmacokinetics of antipyrine and lorazepam was made
after comparison of respective individual curve fits with the Akaike information criterion. 25 All concentration versus time data for ICG were fitted to a monoexponential func- tion with a weighting scheme of the reciprocal of the square of the observed concentration. Pharmacokinetic parameters for each model substrate were calculated by standard equations applicable to classic compartmental modeling of the data. 26
Demographic data are presented as mean _+ SD, and the pharmacokinetic parameters for lorazepam, ICG, and an- tipyrine are presented as mean and range about the mean. Comparison of a given pharmacokinetic parameter between subject groups was performed with the Wilk-Shapiro test for normality of distribution, the F test for equality of vari- ance, and the appropriate version of the two-tailed Student t test for either pooled or unpooled variance. Relationships between demographic factors and pharmacokinetic param- eters were examined with the use of least-squares linear re- gression, which included an analysis of variance for linear- ity. All statistical analyses used previously described meth- Ods and were performed with the RS/1 statistical analysis system (BBN Software Products Co., Cambridge, Mass.). The level of significance accepted for all statistical evalua-
tions was a = 0.05.
R E S U L T S
Demographic and laboratory data for both the study and control groups are summarized in Table I. The NIH scores f9 in the subjects with CF ranged between 22 and 71, with a mean _+ SD and median value of 47.9 _+ 14.9 and 46.5, respectively. There was a significant age difference (p = 0.001) between the control and study groups, although weight, height, and body surface area were not statistically different. To afford a gross comparison of body composition

Volume 117 Enhanced hepatic drug clearance in CF 9 7 5 Number 6
between the subject groups, we calculated the body mass
indexes for each subject according to the following equa-
tion: BMI = (Weigh t /He igh t 2) • 100, where weight and
height are expressed as kilograms and centimeters, respec-
tively. The BMI for those subjects with CF (16.5 +_ 2.5;
range 12.5 to 21.3 k g / m 2) was not significantly different
(p = 0.07) from that observed in the control group
(19.7 +- 4.6; range 14.7 to 27.4 kg/m2).
Abnormalities in results of routine liver function tests
were found in 21% to 50% of the patients with CF but not
in any of the subjects in the control group (Table I). Addi-
tionally, none of the subjects in either group had a serum
creatinine, serum urea nitrogen, total serum protein, pro-
thrombin time, platelet count, hemoglobin, or hematocrit
value that was outside the expected normal range for age.
Four patients with CF had leukocyte counts that were
greater than the upper limit of normal for age, probably
because of pulmonary exacerbations. Four patients with CF
had calculated CLer values that were greater than the up-
per limit of normal for age, although these subjects'
estimated values were not significantly different from those
of the control group (Table I). One subject in the control
group had a serum cholesterol concentration that exceeded
the normal range, and one subject with CF had a value that
was less than the lower limit of the normal range for age.
The disposition profile for antipyrine and lorazepam in
serum was best described by a two-compartment open
model, and for ICG by a one-compartment model. The re-
sultant pharmacokinetic parameters for each of the model
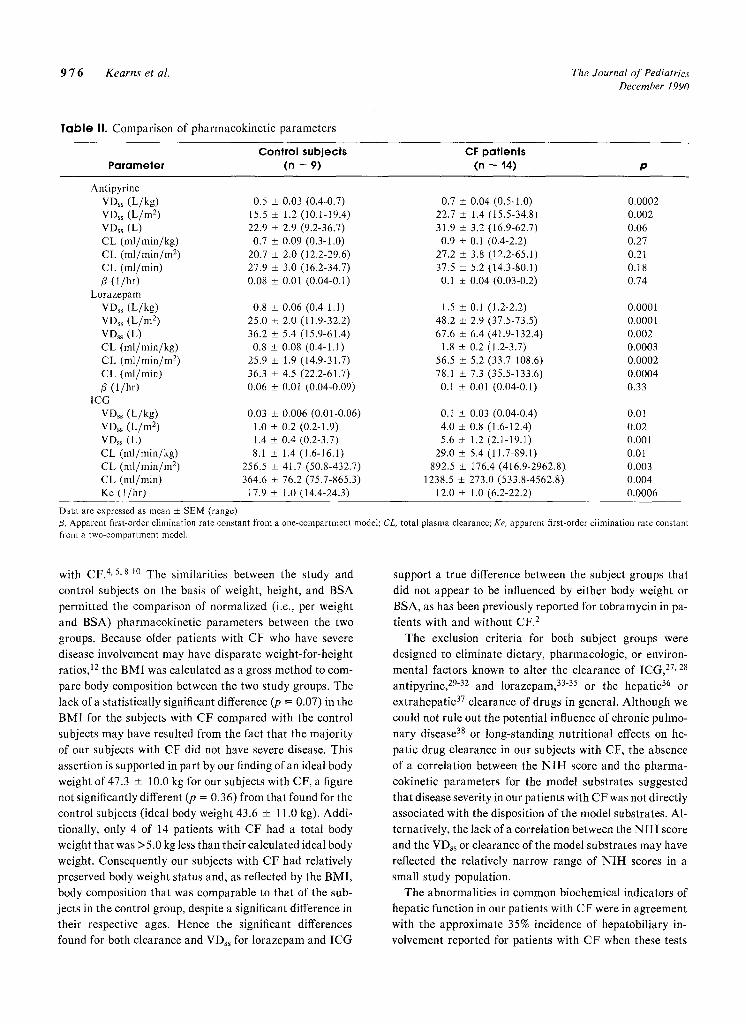
substrates are summarized in Table II. The mean clearance
of both lorazepam and I C G was significantly (p <0.01)
higher in the patients with CF (i.e., 2.2-fold for lorazepam
and 2.7-fold for ICG) than in control subjects. The mean
clearance of antipyrine in the patients with CF (0.9 + 0. l
m l /min /kg ) was not statistically different (p = 0.21) from
that in the control subjects (0.7 _+ 0.1 ml /min /kg ) . The
VDss values for lorazepam, ICG, and antipyrine were sig-
nificantly higher in the patients with CF (1.9-, 3.3-, and
1.4-fold, respectively, when normalized to body weight)
than in the Control subjects. The significant differences re-
mained when the values for VDss and clearance were nor-
malized on the basis of BSA and, with the exception of the
VDss for antipyrine, when they were not normalized for
bo(ty weight or BSA (i.e., VD~s expressed in liters and
clearance in milliliters per minute) (Table II). Significant
correlations were not observed between the clearance of any
of the model substrates and the biochemical markers of liver
function (GGT, lactate dehydrogenase, AST, ALT, alka-
line phosphatase, albumin) in the study and control popu-
lations. Additionally, no significant linear relationships
were found between disease severity (as assessed by the
N I H score) in the patients with CF and the VDss or clear-
ance for any of the model substrates.
T a b l e I. Study population data summary
Control subjects CF Patients (n = 9) (n = '14)
Demographic data Age (yr) 12.7 +__ 4,2 20.9 _+ 4.6* Weight (kg) 47.2 + 20.4 43.6 • 11.0 Height (cm) 150.7 ___ 22.4 161.3 _+ 9.7 Surface area (m 2) 1.4 • 0.1 1.4 • 0.2 Hematologic tests Hematoerit (%) 38.3 _+ 2.4 42.6 _+ 3.2*
(L) (0.38 + 0.02) (0.43 _+ 0.03) Hemoglobin (gm/dl) 13.2 + 1,0 13.9 + 1.2
(gin/L) (132 + 10) (139 _+ 12) Leukocyte count
(• 3) 7.7 • 3.5 13.5 _+ 5.1"[4]~ (109/g) (7.7 _+ 3.5) (13.5 _+ 5.1)
Platelets (Xl03/mm 3) 322.2 • 164.8 450.6 +_ 154.3" (109/L) (322 +_ 165) (451 _+ 154)
Prothrombin time (see) 10.3 _+ 0.4 12.5 + 1.0" Renal function tests Serum creatinine (mg/dl) 0.7 • 0,2 0.6 • 0.3
(#mol/L) (61.9 • 17.7) (53.0 • 26.5) BUN (mg/dl) 12.0 _ 3.7 11.4 • 5.2
(mmol/L urea) (4.3 • 1.3) (4.1 _+ 1.8) CLef (ml/min/1.73 m 2) 124.5 • 23.9 176.2 • 91.2 [4]
(ml/sec/l.73 m 2) (2.08 • 0.40) (2.94 • 1.52) Hepatic function tests ALT (IU/L) 23.1 • 4.4 48.0 _+ 58.9 [5]
(~kat/L) (0.38 • 0.07) (0.80 • 0.98) AST (IU/L) 25.7 • 13.9 53.2 + 61.2 [5]
(#kat/L) (0.43 • 0.23) (0.89 • 1.02) LDH (IU/L) 242.6 • 218.1 549.1 • 207,8* [7]
(ukat/L) (4.04 _+ 3.63) (9.15 _+ 3.46) GGT (IU/L) ND 44.9 _+ 50.7 [3]
(ukat/L) ND (0.75 _+ 0.84) ALP (IU/L) 343.4 • 261.2 174.4 • 163.5 [1]
0zkat/L) (5.7 • 4.3) (2.9 • 2.7) Other tests Total protein (gm/dl) 6.5 • 0,3 7.4 • 0.8*
(gin/L) (65 • 3) (74 • 8) Albumin (gm/dl) 4.1 • 0.2 3.7 _+ 0.3* [15]
(gin/L) (41 • 2) (37 • 3) Cholesterol (mg/dl) 166.6 • 45.9 [1] 121.8 _+ 43.1" [1:~]
(mmol/L) (4.31 _+ 1.18) (3.15 • 1.11)
Data are expressed as mean +- SD. SI values are indicated in parentheses below the metric values. ALP, Alkaline phosphatase; BUN, serum urea nitrogen; LDH, lactate dehy- drogenase; ND, not determined. *Significant (p <0.05) difference between study groups. "~Brackets denote number of subjects with value greater than upper limit of normal range for age. :~Number of subjects with value less than lower limit of normal range for age.
D I S C U S S I O N
The pharmacokinetie data from our investigation support
the findings of previous investigators, who have suggested
that both the clearance and VD~s of drugs with major he-
patic routes of biotransformation are increased in patients
9 7 6 Kearns et al. The Journal of Pediatrics December 1990
Table II. Comparison of pharmacokinetic parameters
Control subjects CF pat ients P a r a m e t e r (n = 9) (n = 14) p
Antipyrine VDss (L/kg) 0.5 _+ 0.03 (0.4-0.7) VD~s (L/m 2) 15.5 • 1.2 (10.1-19.4) VDss (L) 22.9 + 2.9 (9.2-36.7) CL (ml/min/kg) 0.7 • 0.09 (0.3-1.0) CL (rnl/min/m 2) 20.7 _+ 2.0 (12.2-29.6) CL (ml/min) 27.9 • 3.0 (16.2-34.7) [3 (1/hr) 0.08 _+ 0.01 (0.04-0.1)
Lorazepam VDss (L/kg) 0.8 + 0.06 (0.4-1.1) VD~s (L/m 2) 25.0 • 2.0 (11.9-32.2) VDss (L) 36.2 • 5.4 (15.9-61.4) CL (ml/min/kg) 0.8 + 0.08 (0.4-1.l) CL (ml/min/m 2) 25.9 • 1.9 (14.9-31.7) CL (ml/min) 36.3 _+ 4.5 (22.2-61.7) /3 (1/hr) 0.06 • 0.01 (0.04-0.09)
1CG VDss (L/kg) 0.03 • 0.006 (0.01-0.06) VDss (L/m 2) 1.0 • 0.2 (0.2-1.9) VDss (L) 1.4 • 0.4 (0.2-3.7) CL (lnl/min/kg) 8.1 + 1.4 (1.6-16.1) CL (ml/min/m 2) 256.5 • 41.7 (50.8-432.7) CL (ml/min) 364.6 + 76.2 (75.7-865.3) Ke (1/hr) 17.9 • 1.0 (14.4-24.3)
0.7 _+ 0.04 (0.5-1.0) 0.0002 22.7 • 1.4 (t5.5-34.8) 0.002 31.9 • 3.2 (16.9-62.7) 0.06 0.9 • 0.1 (0.4-2.2) 0.27
27.2 • 3.8 (12.2-65.1) 0.21 37.5 • 5.2 (14.3-80.1) 0.18 0.1 • 0.04 (0.03-0.2) 0.74
1.5 • 0.1 (l.2-2.2) 0.0001 48.2 + 2.9 (37.5-73.5) 0.0001 67.6 • 6.4 (41.9-132.4) 0.002
1.8 _+ 0.2 il.2-3.7) 0.0003 56.5 • 5.2 (33.7-108.6) 0.0002 78.1 • 7.3 (35.5-133.6) 0.0004 0.1 • 0.01 (0.04-0.1) 0.33
0.1 + 0.03 (0.04-0.4) 0.01 4.0 • 0.8 (1.6-12.4) 0.02 5.6 • 1.2 (2.1-19.I) 0.001
29.0 _+ 5.4 (11.7-89./) 0.01 892.5 +_ 176.4 (416.9-2962.8) 0.003
1238.5 • 273.0 (533.8-4562.8) 0.004 12.0 + 1.0 (6.2-22.2) 0.0006
Data are expressed as mean + SEM (range) ~, Apparent first-order elimination rate constant from a one-compartment model; CL, total plasma clearance; Ke, apparent first-order elimination rate constant from a two-compartment model.
with CF. 4, 5,8-10 The similarities between the study and
control subjects on the basis of weight, height, and BSA
permitted the comparison of normalized (i.e., per weight
and BSA) pharmacokinetic parameters between the two
groups. Because older patients with CF who have severe
disease involvement may have disparate weight-for-height
ratios] 2 the BMI was calculated as a gross method to com-
pare body composition between the two study groups. The
lack of a statistically significant difference (p = 0.07) in the
BMI for the subjects with CF compared with the control
subjects may have resulted from the fact that the majority
of our subjects with CF did not have severe disease. This
assertion is supported in part by our finding of an ideal body
weight of 47.3 _+ 10.0 kg for our subjects with CF, a figure
not significantly different (.p = 0.36) from that found for the
control subjects (ideal body weight 43.6 _+ 11.0 kg). Addi-
tionally, only 4 of 14 patients with CF had a total body
weight that was >5.0 kg less than their calculated ideal body
weight. Consequently our subjects with CF had relatively
preserved body weight status and, as reflected by the BMI,
body composition that was comparable to that of the sub-
jects in the control group, despite a significant difference in
their respective ages. Hence the significant differences
found for both clearance and VDss for lorazepam and ICG
support a true difference between the subject groups that
did not appear to be influenced by either body weight or
BSA, as has been previously reported for tobramycin in pa-
tients with and without CF. 2
The exclusion criteria for both subject groups were
designed to eliminate dietary, pharmacologic, or environ- mental factors known to alter the clearance of ICG, 27, 28 antipyrine, 29-32 and lorazepam, 3335 or the hepatic 36 or
extrahepatic 37 clearance of drugs in general. Although we
could not rule out the potential influence of chronic pulmo-
nary disease 38 or long-standing nutritional effects on he-
patic drug clearance in our subjects with CF, the absence
of a correlation between the N I H score and the pbarma-
cokinetic parameters for the model substrates suggested
that disease severity in our patients with CF was not directly
associated with the disposition of the model substrates. Al-
ternatively, the lack of a correlation between the N I H score
and the VDss or clearance of the model substrates may have
reflected the relatively narrow range of N I H scores in a
small study population.
The abnormalities in common biochemical indicators of
hepatic function in our patients with CF were in agreement
with the approximate 35% incidence of hepatobiliary in-
volvement reported for patients with CF when these tests

Volume 117 Enhanced hepatic drug clearance in CF 9 7 7 Number 6
were used. 16' 17 Although none of our patients with CF had laboratory or clinical evidence of cirrhosis, we could not rule out the presence of subclinical biliary tract abnormalities 15 or qualitative or quantitative changes in bile that are often found in patients with CF39-41; such conditions may result
in decreased activity of certain phase I and phase II hepatic biotransformation reactions. 4z' 43
The three model substrates used in this study were cho- sen for specific reasons. First, coadministration of lorazepam, ICG, and antipyrine to adults 44 and children 18 has been shown to be safe and to permit accurate assess- ments of their respective plasma clearances during a 24-hour period. Second, each substrate is eliminated by in- dependent hepatic pathways (Figure) that constitute major routes of xenobiotic biotransformation: microsomal oxida- tion (antipyrine), a phase I reaction; glucuronidation (lorazepam), a phase II reaction; and hepatic blood flow and biliary secretion (ICG). Third, characterization of the clearance of antipyrine, which reflects N-demethylation and hydroxylation, 45,46 and of lorazepam, a compound that primarily undergoes glucuronidation at the 3-hydroxy position, 47 may yield predictive information for the metab- olism of similar substrates that share the same cytochrome P-450 enzymes. 48' 49 Last, clearance of the three model
substrates correlates more closely with functional hepatic metabolic capacity TM 5o than ALP or GGT values, 44 which
in our population did not correlate with the clearance of any of the model substrates.
The increase in VDss found for each of the model substrates in our study population agrees with previous re- ports of expanded distribution volumes for drugs commonly used in the treatment of patients with CF. 17, 9 Although the VDss for any of the model substrates did not correlate with the NIH score in the subjects with CF, the apparent increase in this parameter may have a physiologic explana- tion that is related to disease severity. In patients with CF, chronic reduction in systemic arterial oxygen saturation, which is observed with increasing severity of pulmonary disease, is often associated with increases in both erythro- cyte count and plasma volume.12 These alterations result in an increase in the body water/body mass ratio, effectively increasing the available distribution space for drugs that partition to both intravascular and extravascular fluid spaces.Z, 11 This assertion is also supported by our demon- stration of a significantly larger non-normalized VDss for the model substrates for the patients with C F .
The approximate twofold increase in lorazepam clear- ance in our study population corroborates a recent report of patients with CF I~ in whom there was increased nonrenal clearance of furosemide, a compound that, like lorazepam, is extensively metabolized via hepatic glucuronidation. 51 As shown for furosemide, 1~ one possible mechanism of in-
creased clearance of lorazepam in subjects with CF might be a decrease in plasma protein binding. Although we did not evaluate the protein binding of lorazepam, the absence of significant hypoalbuminemia and the demonstration by previous investigations of similar clearance of free drug in adult and pediatric patients 52 suggest that alterations in protein binding were not the cause of our pharmacokinetic findings. This contention is further supported by a recent study of 50 children with cancer and 10 healthy adults, which demonstrated no significant differences in the lorazepam free fraction or clearance of free drug between the study populations (Evans WE: personal communica-
tion, June 1990). A more likely explanation for increased lorazepam clear-
ance in patients with CF may relate to the recent demon- stration of increased glucuronidation activity in experimen- tally induced cholestasis, 42' 53 which would be expected to increase the clearance of lorazepam as well as the nonrenal clearance of furosemide. However, because of the presence of multiple isoenzymes of glucuronosyltransferase 49 and the possible enterohepatic recirculation and extrahepatic me- tabolism of lorazepam, 54 its increased clearance in patients with CF may not be directly extrapolated to the clearance of other drugs that undergo hepatic glucuronidation.
Potential explanations for the 1.8-fold to twofold increase in the clearance of ICG in our patients with CF arc less clear. ICG is taken up by hepatocytes through the sinuso- idal cell membrane and is stored on binding sites before its predominant excretion into bile. 5~ 55 In view of the biliary tract abnormalities 15-17 and the absence of data supporting an increase in hepatic blood flow 41 in patients with CF, the increased clearance of ICG in our subjects with CF was un- expected. Characterization of the cellular mechanisms that govern ICG transport and their evaluation in hepatocytes from patients with CF will be necessary to address the un- derlying mechanism for increased ICG clearance in these patients.
Increased nonrenal and plasma clearance of theophylline consequent to enhanced N-demethylation and 8-hydroxy- lation has recently been reported in patients with CF. 9 Like theophylline, antipyrine is a polyfunctional substrate that undergoes N-demethylation and hydroxylation via separate cytochrome P-450 enzymes. 48 Given the possibility of genetic cosegregation for enzyme systems that regulate the biotransformation of ~mtipyrine and theophylline, 48' 56 one might expect enhanced clearance for both these agents in patients with CF. However, this was not found for antipy- rine in our study; instead, the clearance in subjects with CF was not significantly greater than that observed in the con- trol subjects. The lack of a statistical difference for antipy- rine clearance between our study groups may have resulted from the inherent wide variability in clearance previously

9 7 8 Kearns et al. The Journal o f Pediatrics December 1990
reported for this model substrate, 18, 29, 30 a factor com-
pounded by the relatively small size of our study groups.
This consideration, together with the multiplicity of sub-
types of hepatic microsomal enzymes, 57 their substrate
specificity, s8 and the inherent variability in the activity of
selected enzymes, 59 raises important questions regarding
the comparison of our data with those previously reported
for theophylline in patients with CF. 9 Additionally, these
factors focus important limitations on using a single sub-
strate to make broad extrapolations to other drugs that un-
dergo hepatic oxidative metabolism.
The mechanisms underlying the apparent increased he-
patic monooxygenase activity in patients with CF are not
known. Braganza 6~ hypothesized that the basic biochemical
defect in CF may be the result of genetic or environmental
induction of cytochrome P-450 activity. Indeed, the mor-
phologic findings in liver tissue from patients with CF
reported by Hultcrantz et al. 61 are compatible with the
known effects of induction on hepatic cellular architecture.
As recently reviewed by Shapiro, 62 investigations of sub-
jects with CF have demonstrated increased mitochondrial
calcium uptake, increased oxygen consumption in multiple
cell types, increased energy expenditure, and altered affin-
ity of mitochondrial N A D H dehydrogenase for its substrate
in cells from persons who were homozygous or heterozygous
for the CF gene. Therefore it appears that mitochondrial
dysfunction may be a fundamental expression of the CF
gene product, 62 which in turn may also influence the
biotransformation of various xenobiotics. Detailed investi-
gation will be required to evaluate the complex associations
between CF and drug metabolism.
We conclude that the use of antipyrine, lorazepam, and
ICG as model substrates demonstrates an increased VDs~ in
patients with CF and that the clearance of lorazepam and
ICG is markedly increased in those patients with the
disease. At present, exogenous factors or a common mech-
anism to explain increased hepatic drug clearance in CF
have not been demonstrated. It may therefore be plausible
to consider the apparent disease-specific alterations in drug
disposition to be the consequence of both the primary (i.e.,
genetic) and secondary (i.e., pathophysiologic) aspects of
the disease.
We gratefully acknowledge the clinical and scientific contribu- tions of Drs. Robert H. Warren, Robert H. Fiser, Jr., and Helen B. Casteel, and the technical contributions of Paula G. Stracener, RNP, and Ms. Cindy Stewart. The editorial assistance of Drs. Stephen F. Kemp, John T. Wilson, and Thomas G. Wells, and of Mrs. Sharon Kemp, is also sincerely appreciated.
R E F E R E N C E S
1. Kearns GL, Hilman BC, Wilson JT. Dosing implications of altered gentamicin disposition in patients with cystic fibrosis. J PEDIATR 1982;100:312-8.
2. Levy J, Smith AL, Koup JR, et al. Disposition of tobramycin in patients with cystic fibrosis--a prospective, controlled study. J PEDIATR 1984;105:117-24.
3. Jusko W J, Mosovich LL, Gerbracht LM, et al. Enhanced re- nal excretion of dicloxacillin in patients with cystic fibrosis. Pediatrics 1975;56:1038-44.
4. Spino M, Chai RP, Isles AF, et al. Cloxacillin absorption and disposition in cystic fibrosis. J PEDIATR 1984;105:829-35.
5. Isles AF, Spino M, Tabachnik E, et al. Theophylline disposi- tion in cystic fibrosis. Am Rev Resp Dis 1983;127:417-21.
6. Leeder JS, Spino M, Isles AF, et al. Ceftazidime disposition in acute and stable cystic fibrosis. Clin Pharmacol Ther 1984;36:355-62.
7. Yaffe S J, Gerbracht LM, Mosovich LL, et al. Pharmacoki- netics of methicillin in patients with cystic fibrosis. J Infect Dis 1977;135:828-31.
.8. Schacter SH, Spino M, Isles AF, et al. Pharmacokinetics of eefoperazone (CFP) in patients with cystic fibrosis and healthy volunteers. In: Proceedings of the Thirteenth International Congress of Chemotherapy, 1983:7-12.
9. Knoppert DC, Spino M, Beck R, et al. Cystic fibrosis: enhanced theophylline metabolism may be linked to the disease. Clin Pharmacol Ther 1988;44:254-64.
10. Alvan G, Beermann B, Hjelte L, et al. Increased nonrenal clearance and increased diuretic efficiency of furosemide in cystic fibrosis. Clin Pharmacol Ther 1988;44:436-41.
11. Kearns GL, Trang JM. Introduction to pharmacokinetics: aminoglycosides in cystic fibrosis as a prototype. J PEDIATR 1986;108(suppl):847-53.
12. Prandota J. Drug disposition in cystic fibrosis: progress in un- derstanding pathophysiology and pharmacokinetics. Pediatr Infect Dis J 1987;6:t 111-26.
13. Abramowsky CR, Swinehart GL. The nephropathy of cystic fibrosis: a human model of chronic nephrotoxicity. Hum Pathol 1982;13:934-9.
14. Goodchild MC, Banks A J, Drolc Z, Anderson CM. Liver scans in cystic fibrosis. Arch Dis Child 1975;50:813-5.
15. Gaskin K J, Waters DL, Howman-Giles R, et al. Liver disease and common-bile duct stenosis in cystic fibrosis. N Engl J Med 1988;318:340-6.
16. Kattwinkel J, Taussig LM, Statland BE, Vertes JE. The effect of age on alkaline phosphatase and other serologic liver func- tion tests in normal subjects and patients with cystic fibrosis. J PED1ATR 1973;82:234-42.
17. Boat TF, Doerchuk CF, Stern R, Matthews LS. Serum alka- line phosphatase in cystic fibrosis. Clin Pediatr 1974;13:505- 12.
18. Relling MV, Crom WR, Pieper JA, et al. Hepatic drug clear- ance in children with leukemia: changes in clearance of model substrates during remission-induction therapy. Clin Pharma- col Ther 1987;41:651-60.
19. Taussig LM, Kattwinkel J, Friedewald WT, et al. A new prognostic score and clinical evaluation system for cystic fibrosis. J PEDIATR 1973;58:740-3.
20. Metzler CM, Elfing GC, McEwen AJ. A package of computer programs for pharmacokinetic modeling [Letter]. Biometrics 1974; 30:3.
21. Caesar J, Shaldon S, Chiandussi L, et al. The use of indocy- anine green in the measurement of hepatic blood flow and as a test of hepatic function. Clin Sci 196I;21:43-57.
22. Riley CA, Evans WE. Simultaneous analysis of antipyrine and lorazepam by high performance liquid chromatography. J Chromatogr 1986;382:199-205.

Volume 117 Enhanced hepatic drug clearance in CF 9 7 9 Number 6
23. Gomeni R, Gomeni C. AUTOMOD: a polyalgorithm for an integrated analysis of linear pharmacokinetic models. Comput Biol Med 1979;9:38-48.
24. Marquardt DW..Algorithm for least-squares estimation of non- linear parameters. J Soe Indust Appl Math 1963;11:43t-41.
25. Yamaoka K, Nakagawa T, Uno T. Application of Akaike's information criterion (AIC) in the evaluation of linear phar- macokinetic equations. J Pharmacokinet Biopharm 1978; 6:165-75.
26. Wagner JG. Linear pharmacokinetics equations allowing direct calculation of needed pharmacokinetic parameters from the coefficients and exponents of polyexponential equations which have been fitted to the data. J Pharmacokinet Biopharm 1976;4:443-67.
27. Nies AS, Shand DG, Wilkinson GR. Altered hepatic blood flow and drug disposition. Clin Pharmacokinet 1976;1:135-55.
28. McDevitt DG, Nies AS, Wilkinson GR. Influence of phe- nobarbital on factors responsible for hepatic clearance of indocyanine green in the rat: contributions of induction and altered liver blood flow. Biochem Pharmacol 1977;26:1247-50.
29. Vesell ES. The antipyrine test in clinical pharmacology: con- ceptions and misconceptions. Clin Pharmacol Ther 1979; 26:275-86.
30. Stevenson IH. Factors influencing antipyrine elimination. Br J Clin Pharmacol 1977;4:261-5.
31. Kramer P, McClain CH. Depression of aminopyrine metabo- lism by influenza vaccination. N Engl J Med 1981;305: 1262-4.
32. Hepner GW, Vesell ES, Lopton A, et al. Disposition of ami- nopyrine, antipyrine, diazepam and indoeyanine green in pa- tients with liver disease or an anticonvulsant drug therapy: di- azepam breath test and correlations in drug elimination. J Lab Clin Med 1977;90:440-56.
33. Greenblatt D J, Shader RI, Franke K, et al. Pharmacokinetics and bioavailability of intravenous, intramuscular and oral lorazepam in humans. J Pharm Sci 1979;68:57-63.
34. Desmond PV, Roberts RK, Wood A J J, et al. Effect of heparin administration on plasma binding of benzodiazepines. Br J Clin Pharmacol 1980;9:171-5.
35. Kraus JW, Desmond PV, Marshall JP, et al. Effects of aging and liver disease on the disposition of lorazepam. Clin Phar- macol Tiler 1978;24:4t 1-9.
36. Bass NM, William RL. Guide to drug dosage in hepatic dis- ease. Clin Pharmacokinet 1988;15:396-420.
37. Farrell GC. Drug metabolism in extrahepatic diseases. Phar- macol Ther 1987;35:375-404.
38. Du Souich P, McLean A J, Lalka D, et al. Pulmonary disease and drug kinetics. Clin Pharmacokinet 1978;3:257-66.
39. Oppenheimer EH, Esterly JR. Pathology of cystic fibrosis: re- view of the literature and comparison with 146 autopsied cases. Perspect Pediatr Pathol 1975;2:241-78.
40. Craig JM, Haddad H, Shwachman H. The pathological changes in the liver in cystic fibrosis of the pancreas. Am J Dis Child 1957;93:357-69.
41. Oppenheimer EJ, Esterly JR. Hepatic changes in young infants with cystic fibrosis: possible relation to 'focal biliary cirrhosis. J PEDIATR 1975;86:683-9.
42. Galinsky RE, Chalasinska B. Effect of taurolithocholate on in vivo sulfation and glucuronidation of acetaminophen in rats. Pharm Res 1988;5:61-4.
43. Olomu AB, Vickers CR, Waring RH, et al. High incidence of poo r sulfoxidation in patients with primary biliary cirrhosis. N Engl J Med 1988;318:1089-92.
44. Crom WR, Webster SL, Bobo L, et al. Simultaneous admin- istration of multiple model substrates to assess hepatic drug clearance. Clin Pharmacol Ther 1987;41:645-50.
45. Penno MB, Vesell ES. Monogenic control of variations in an- tipyrine metabolite formation: new polymorphism of hepatic drug oxidation. J Clin Invest 1983;71:1698-709.
46. Penno MB, Dvorchik BH, Vesell ES. Genetic variation in rates of antipyrine metabolite formation: a study in uninduced twins. Proc Natl Acad Sei USA 1981;78:5193-6.
47. Kyriakopoulos AA, Greenblatt D J, Shader RI. Clinical phar- macokinetics of lorazepam: a review. J Clin Psychiatry 1978;39:16-23.
48. Miller CA, Slusher LB, Vesell ES. Polymorphism of theo- phylline metabolism in man. J Clin Invest 1985;75:1415-25.
49. Leakey JEA, Hume R, Burchell B. Development of multiple activities of UDP-glucuronyltransferase in human liver. Bio- them J 1987;243:859-61.
50. Kawasaki S, Sugiyama Y, Iga T, et al. Hepatic clearances of antipyrine, indocyanine green and galactose in normal subjects and in patients with chronic liver diseases. Clin Pharmacol Ther 1988;44:217-24.
51. Cutler RE, Blair AD. Clinical pharmacokinetics of furosem- ide. Clin Pharmacokinet 1979;4:279-96.
52. Relling MV, Mulhern RK, Dodge RK, et al. Lorazepam pharmacodynamies and pharmacokinetics in children. J PED1- ATR 1989;114:641-6.
53. Vermeulen JP, Ziurys JC, Gollan JL. Hepatic cytochrome P- 450 and UDP-glueuronyltransferase activities are rapidly modulated by bile salts [Abstract]. Hepatology 1986;6:1178.
54. Herman R J, Duc Van Pham J, Szakaes CBN. Disposition of lorazepam in human beings: enterohepatic recirculation and first-pass effect. Clin Pharmacol Ther 1989;46:18-25.
55. Paumgartner G. The handling of indocyanine green by the liver. Sehweiz Med Wochenschr 1975;105(suppl):5-30.
56. Nebert DW, Jensen NM. The Ah locus: genetic regulation of the metabolism of carcinogens, drugs and other environmen- tal chemicals by cytochrome P-450-mediated monooxygena- ses. CRC Crit Rev Biochem 1979;6:401-37.
57. Nebert DW, Adesnik M, Coon M J, et al. The P-450 gene su- perfamily: recommended nomenclature. DNA Cell Biol 1987;6:1-11.
58. Boobis AR, Murray S, Clare-Kahn G, et al. Substrate speci- ficity of the form of cytochrome P-450 catalyzing the 4- hydroxylztion of debrisoquine in man. Mol Pharmaeol 1983; 23:474-81.
59. Meier P J, Mueller HK, Dick B, Meyer UA. Hepatic monoox- ygenase activities in subjects with a genetic defect in drug ox- idation. Gastroenterology 1983;85:682-92.
60. Braganza J. Cystic fibrosis: a casualty of "detoxification?" Med Hypotheses 1986;20:233-43.
61. Hultcrantz R, Mengarelli S, Strandvik B. Morphological findings in the liver of children with cystic fibrosis: a light and electron microscopical study. Hepatology 1986;6:881- 9.
62. Shapiro BL. Evidence for a mitochondrial lesion in cystic fi- brosis. Life Sei 1989;44:1327-34.






