Embed Size (px)
Citation preview

Dent Mater 9:300-305, September, 1993
Enhanced evaporation of mercury from amalgams in non-oxidizing environments
J.L. Ferracane ~, H. Nakajima 2, T. Okabe 2
1 Department of Dental Materials Science, Oregon Health Sciences University, Portland, OR, USA 2 Department of Biomaterials Science, Baylor College of Dentistry, Dallas, TX, USA
Abstract. The release of mercury from two freshly triturated amalgams exposed to a reducing atmosphere, hydrogen, was quantitated at three different temperatures. A low-copper and a high-copper amalgam were placed into a flowing hydrogen gas atmosphere for 60 min after trituration, and then the hydrogen was replaced by compressed air. The results were compared to those obtained in a previous study in which air and argon atmospheres were used under identical conditions. At 37°C, the rate of evaporation of mercury from the amalgams was similar when they were exposed to hydrogen before being exposed to air. During exposure to hydrogen, the evaporation rate appeared to exceed the limit of the gold film mercury analyzer (284 pg/mm2/s), but was rapidly reduced upon expo- sure to air. The results were identical to those from argon exposure. When the same experiment was performed at 80°C, the evaporation rate after hydrogen exposure was greater than that after exposure to argon, and far greater than that during exposure to air alone. Similar results were achieved at 110°C, but there was less difference between hydrogen and argon exposure. The results support previous studies which show that the evaporation of mercury from amalgam is mainly limited by the formation of an oxide film.
INTRODUCTION The thermal analysis of amalgams in air and argon gas environ- ments has produced different results with respect to the release of mercury. Tsutsumi et al. (1988) found no weight change for amalgams heated to 130°C in air, signifying no loss of mercury. In contrast, Hanawa et al. (1986) showed that a considerable amount of mercury was liberated from amalgams heated in an argon environment. These studies suggest that the evaporation of mercury is retarded by an oxide film formed on the surface of the amalgam during aging in air.
Hanawa et al. (1987) previously showed that within 20 min after trituration, tin and/or zinc had preferentially diffused to the surface of amalgam and combined with oxygen to form a surface film, thereby diluting the mercury concentration on the surface. Although it is probable that mercury release from amalgam decreases with time simply because unreacted mercury is de- pleted as the setting reaction progresses, it is likely that the oxide-containing film formed during setting further suppresses
mercury release. In a recent study, Ferracane et al. (1992) showed the rate of mercury release from amalgams to be significantly greater in an inert argon environment than it was in air. They further showed that the mercury release declined dramatically once the amalgam was exposed to air and oxidation was allowed to occur. A similar result has been reported by Mahler et al. (1993) for abraded amalgams. Further evidence for the role of oxygen in minimizing the evaporation of mercury from amalgams would be provided by studying the release rate in a reducing gas, such as hydrogen. Hydrogen should inhibit the formation of the oxide film, and also has the potential to eliminate existing oxides.
The objective of this study was to compare the rate of mercury evaporation from setting amalgam exposed to a reducing environ- ment, such as hydrogen, to that occurring after exposure to air or argon. The aim was to provide further support for the hypothesis that the formation of an oxide film is the most important factor limiting the release of mercury vapor from dental amalgam.
MATERIALS AND METHODS Amalgam specimens were prepared from two types of alloy: Tytin (Kerr, Romulus, MI, USA), a high-copper, single-composition, spherical alloy, and Velvalloy (Kerr), a non-zinc containing, low- copper, lathe-cut alloy (Table 1).
The amalgams were triturated according to the manufactur- ers' recommendations (Table 1) and condensed into cylindrical specimens (4.0 mm diameter x 5.0 mm height; surface area = 88 mm 2) in a steel mold under 14 MPa of pressure by following the procedure outlined in ADA Specification No. 1. The mass% of residual Hg in each amalgam was determined by collecting and weighing the mercury that was expressed during condensation and subtracting this amount from that originally used in the mix.
Five min after trituration, one specimen was placed within a 5 cm long glass tube connected to Tygon tubing (4.75 mm I.D., 8.0 mm O.D.). The glass tube was then suspended in a furnace held at either 37°C, 80°C, or ll0°C. Mercury release was exam- ined at the non-physiologic temperatures of 80°C and ll0°C because the vapor pressure of mercury is raised at elevated temperatures, and because a previous study revealed that low-copper and high-copper amalgams form a liquid phase, presumably via the peritectic reaction of ~'1 + ~ .... > ~1 + L (containing Hg), at approximately 80°C and 100°C, respectively
3{)0 Ferracane et aL/Mercury release in hydrogen

TABLE 1: CHEMICAL COMPOSITION OF THE AMALGAM ALLOYS AND TRITURATION CONDITIONS USED
Alloy %Ag %Sn %Cu %Hg* Trit time Trit # Batch No. (mass) (mass) (mass) (mass) (s) speed
Tytin 59.3 27.1 13.3 41.4 16 M2 3604-20004
Velvalloy 71.1 26.0 2.93 44.4 18 M2 52187-71111
* mass % of residual Hg in the amalgam (coefficient of variation = 0.4- 0.9%) # The triturator was a Varimix II (L.D. Caulk, Milford, DE, USA) The composition of the alloys was taken from De Freitas (1979).
Tsutsumi et al., 1988). The formation of a liquid phase might influence the mercury vaporization behavior.
Hydrogen gas (99.99% pure) was continuously blown through the tubes at a flow rate of 750 mL/min for the first 60 rain to maintain the amalgam in a reducing atmosphere. The flow rate of 750 mL/min was chosen to match the flow rate of the gold film mercury analyzer. No attempt was made to evaluate mercury evaporation during the time that the amalgam was exposed to hydrogen gas because the mercury vapor analyzer could not be reliably operated under the conditions of a reducing environ- ment. After 60 rain, the hydrogen gas flow was stopped and compressed air was continuously blown through the tubes at a flow rate of 750 mL/min. The relative humidity of the air in the oven at 37°C was examined several times throughout the course of the experiment and found to be 22.7 _+ 1.0%. The value for saturated aqueous vapor in air at 37°C is 43.96 g of water/m '~ (Weast and Astle, 1979-80). Therefore, the air in the experiment at 37°C contained approximately 10.0 g of water vapor/m ~.
The flow rate was measured with a flowmeter (J-3218-17, Cole-Palmer Instrument Company, Chicago, IL, USA) placed at the inlet of the Tygon tube. A frequent check of the flow rate at the outlet was made using a second flowmeter to verify that no leakage occurred at the connections. A filter consisting of a glass tube (4.0 mm I.D.; 6 mm O.D.) filled with activated charcoal nuggets (J.T. Baker Chemical Co., Phillipsburg, NJ, USA) was connected to the outlet in order to prevent mercury from being exhausted to the air.
Mercury release was monitored with a gold film mercury vapor analyzer (Model 411, Arizona Instrument Corp., Tempe, AZ, USA). Once the hydrogen was stopped and the air was introduced, the tubing connection to the charcoal filter was detached and immediately connected to the intake of the mercury analyzer. After the air was sampled, the tubingwas re-connected to the exhaust filter. Measurements were made at 10 min intervals during the first 60 min, at 30 rain intervals for the next 120 min, and at 60 min intervals thereafter. When very high levels of mercury were released, the time intervals differed to avoid saturation of the gold film in the analyzer. The air was sampled by the analyzer for 10 s at a flow rate of 750 mL/min, providing direct readings of mercury vapor concentration in mg/m :~. The minimum detectable concentration was i pg/m 3 per sampling. The calibration of the gold film analyzer was routinely checked during the experimental period by following the manufacturer's procedure.
Each experiment was repeated three times. All mercury readings (mg/m '~) were converted to units of mass per sampling time per unit surface area of the specimen (pg/mm2/s). The values
at each period were plotted against time. Because identical procedures were per- formed, the results from this experi- ment in hydrogen were compared to those obtained previously in air and argon environments (Ferracane et al., 1992).
RESULTS
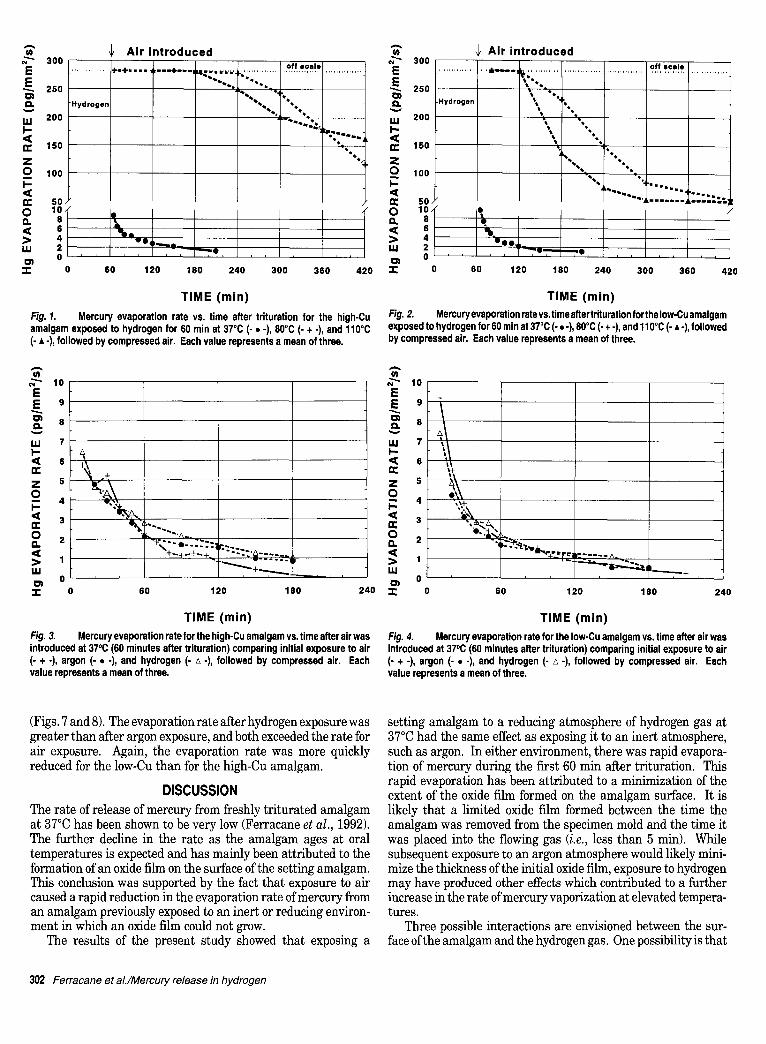
The rate of evaporation of mercury at each time period for the three tempera- tures was averaged for the three trials for both the high-Cu amalgam (Fig. 1)
and the low-Cu amalgam (Fig. 2). The coefficient of variation for the evaporation rate at each time period ranged from a low of 5% to a high of 78%. The average coefficient of variation at the different temperatures for the high-Cu amalgam was: 20% at 37°C, 12% at 80°C, and 42% at 110°C. The average variation for the low-Cu amalgam was: 39% at 37°C, 26% at 80°C, and 50% at 110°C.
During the initial 60 min in hydrogen gas, the evaporation rate at all three temperatures was assumed to exceed the maxi- mum capacity of the gold film analyzer (i.e., 284 pg/mm%) because the first reading taken after air was introduced was offscale. The rate of mercury release decreased with time for both types of amalgam after the introduction of air. At 37°C, the vapor level declined below the detectable limit within 180 to 240 min after trituration (Figs. 1 and 2), and there were no differences between the behaviors of the two amalgams.
When the experiment was performed at either 80°C or 110°C, the levels ofmercurywere significantly higher than at 37°C (Figs. 1 and 2). The rate of vaporization of mercury from both amalgams was similar. The evaporation rate was reduced for both amal- gams after the introduction of air, but to a lesser extent than at 37°C. The reduction in the evaporation rate as a result of air exposure was more slowly achieved for the high-Cu than for the low-Cu amalgam. For example, at the higher temperatures, the evaporation rate remained above the maximum level of the analyzer for 180 - 240 min after trituration for the high-Cu amalgam, but was below the maximum value before 120 min after trituration for the low-Cu amalgam.
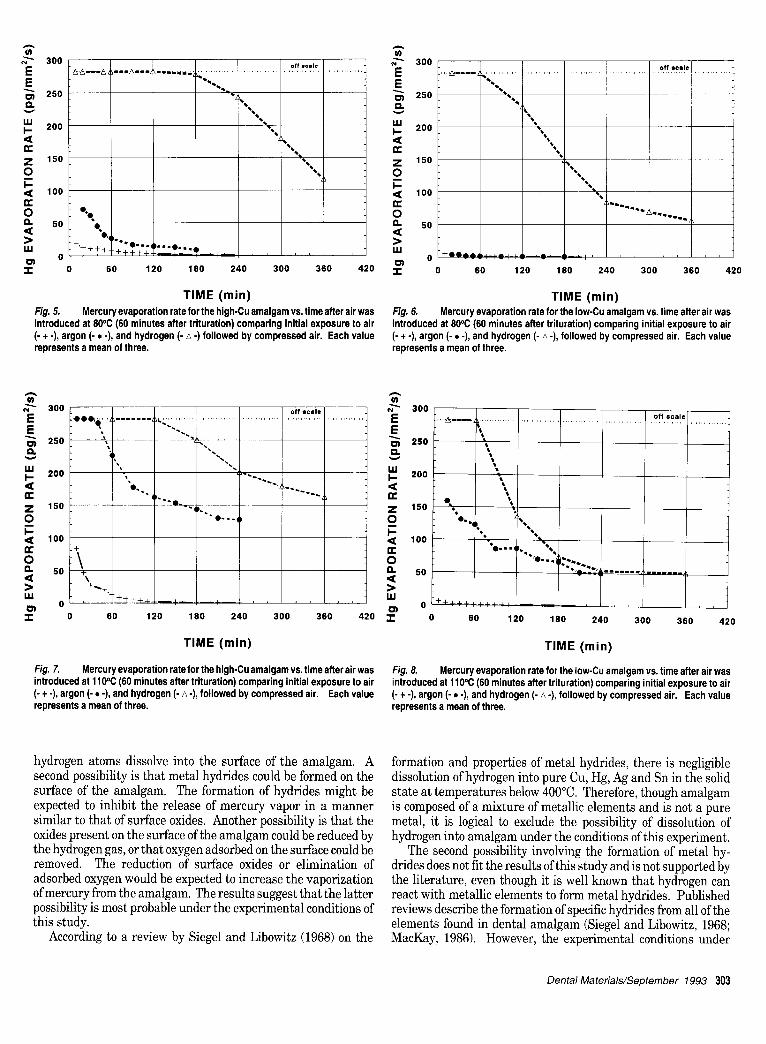
The results from this study were similar to those from previous work by the present authors in that once air was introduced, the evaporation rate after hydrogen or argon exposure was greater than after exposure to air alone, but only at the elevated tempera- tures (Ferracane et al., 1992). Figs. 3-8 show the data for expo- sures to hydrogen (from this study) and to argon (Ferracane et al., 1992). The data for the argon and hydrogen exposures were plotted by shifting the ordinates by 60 rain to the left so all three curves could be directly compared with respect to the time at which exposure to air occurred (i.e., immediately for the air exposed samples and after 60 min for the hydrogen- or argon- exposed samples). The graphs show that the mercury evapora- tion rate at 37°C was independent of previous environmental exposure for either amalgam (Figs. 3 and 4). For both amalgams at 80°C, the evaporation rate after hydrogen exposure was signifi- cantly greater than after exposure to argon or air (Figs. 5 and 6). The reduction in evaporation rate was achieved sooner for the low-Cu than for the high-Cu amalgam, as previously stated. The results for 110°C were similar to those obtained at 80°C
Dental Materials~September 1993 31)1
300 E
250 C~ Q.
IJ.l 200
n- 15o
Z O 100 I-- ,< rr SO o
~ 2 o
~ o
J, A i r i n t r o d u c e d
I... ~"+ . . . . : " ' ' + ' ' ' ~ "=*""~::i''" ' '
-Hydrogen
%. , , ~ 7 e ,
60 120 180
::-.. off scale
t t t t
L--t~ t t
. : - . . . .
240 300 360 420
T I M E ( m i n )
Fig. 1. Mercury evaporation rate vs. time after trituration for the high-Cu amalgam exposed to hydrogen for 60 rain at 37°C (- • -), 80°C (- + .), and lt0°C (- • -), followed by compressed air. Each value represents a mean of three.
10 E E 9
8
IJJ 7 I-- < S n" Z 5 o - - 4 I-- <: 3 n- O 2 n < > 1 UJ
0 01 'I- 0
+ ~ + ~ + ~ + ~ .
60 120 180
300 E ~- 250 O) Q.
LIJ 200 I--
150
Z O 100 k- < n" 50 O 180~
m 2 ~ 0 " r 0
A i r i n t r o d u c e d
, . . . . . . . . . . . . . I , , , t ~ I . . . . . . . . . . . . . .
• e , ,_
60 120 180
4b~
off scale
240 300 360 420
T I M E ( r a i n )
Fig. 2. Mercury evaporation rate vs.time after trituration for the Iow-Cu amalgam exposed to hydrogen for 60 min at 37°C (- s -), 80°C (- + -), and 110°C (- •-), followed by compressed air. Each value represents a mean of three.
9 +
f_. ,
e i i
24O
T I M E ( m i n )
Fig. 3. Mercury evaporation rate for the high-Cu amalgam vs. time after air was introduced at 37°C (60 minutes after trituration) comparing initial exposure to air (- + -), argon (- • -), and hydrogen (- ~ -), followed by compressed air. Each value represents a mean of three.
. ' . ' ' - - - A-
. - - - - - . _ . +
"I" 0 60 120 180 240
T I M E ( m i n )
Fig. 4. Mercury evaporation rate for the Iow-Cu amalgam vs. time after air was introduced at 37°C (60 minutes after trituration) comparing initial exposure to air (- + -), argon (- = -), and hydrogen (- A -), followed by compressed air. Each value represents a mean of three.
(Figs. 7 and 8). The evaporation rate after hydrogen exposure was greater than after argon exposure, and both exceeded the rate for air exposure. Again, the evaporation rate was more quickly reduced for the low-Cu than for the high-Cu amalgam.
DISCUSSION
The rate of release of mercury from freshly triturated amalgam at 37°C has been shown to be very low (Ferracane et al., 1992). The further decline in the rate as the amalgam ages at oral temperatures is expected and has mainly been attributed to the formation of an oxide film on the surface of the setting amalgam. This conclusion was supported by the fact that exposure to air caused a rapid reduction in the evaporation rate of mercury from an amalgam previously exposed to an inert or reducing environ- ment in which an oxide film could not grow.
The results of the present study showed that exposing a
setting amalgam to a reducing atmosphere of hydrogen gas at 37°C had the same effect as exposing it to an inert atmosphere, such as argon. In either environment, there was rapid evapora- tion of mercury during the first 60 min after trituration. This rapid evaporation has been attributed to a minimization of the extent of the oxide film formed on the amalgam surface. It is likely that a limited oxide film formed between the time the amalgam was removed from the specimen mold and the time it was placed into the flowing gas (i.e., less than 5 min). While subsequent exposure to an argon atmosphere would likely mini- mize the thickness of the initial oxide film, exposure to hydrogen may have produced other effects which contributed to a further increase in the rate of mercury vaporization at elevated tempera- tures.
Three possible interactions are envisioned between the sur- face of the amalgam and the hydrogen gas. One possibility is that
302 Ferracane et al./Mercury release in hydrogen
E E
Q.
200 ,< n" Z 150 O
.~ 100 n- O O. 50 .< > LU
0 01 "I"
300
250 ~'z
%4
'q ) . • +'--+~ +. : : " • ' " ' .... •
- + + + + + -
0 60 120 180 240 300 360 420
T I M E ( m i n )
Fig. 5. Mercury evaporation rate for the high-Cu amalgam vs. time after air was introduced at 80°C (60 minutes after trituration) comparing initial exposure to air (- + -), argon (- • -), and hydrogen (- A -) followed by compressed air. Each value represents a mean of three.
E E
D.
f_ < IZ
Z O
n- O a. < > ILl O1
300
250
200
150
100
50
\ %
o f f s c a l e
0 + J i l l . . . .
0 60 120 180 240 300 360 420
T I M E ( m i n )
Fig. 6. Mercury evaporation rate for the Iow-Cu amalgam vs. time after air was introduced at 80°C (60 minutes after trituration) comparing initial exposure to air (- + -), argon (- • -), and hydrogen (- A -), followed by compressed air. Each value represents a mean of three.
E E
Q.
200
=< 150
100 rr o Iz. 5o ,<
U.l oi o "I-
300 .ee• :~
i
250 ",
+
\ t
" + ~ + + ~ _
0 60 120 180
`% %% %,%
'%%%%
off s c a l e
240 300 360 420
TIME (min)
Fig. 7. Mercury evaporation rate for the high-Cu amalgam vs. time after air was introduced at 110°C (60 minutes after trituration) comparing initial exposure to air (- + -), argon (- • -), and hydrogen (- A -), followed by compressed air. Each value represents a mean of three.
300
i ,,.~ ..... i i i~ I 25O
200
150 ~o,
1 O0 - ~ °o.--,,... ",,
so I;2 ..... , > uJ O1 0 i + + 4 - J - - F ÷ ± ! ! ! , ~ ~ ,
'1" 0 60 120 180 240
r t r o f f s c a l e j
300 360 420
TIME (min)
Fig. 8. Mercury evaporation rate for the Iow-Cu amalgam vs. time after air was introduced at 110°C (60 minutes after trituration) comparing initial exposure to air (- + -), argon (- • -), and hydrogen (- A -), followed by compressed air. Each value represents a mean of three.
hydrogen atoms dissolve into the surface of the amalgam. A second possibility is that metal hydrides could be formed on the surface of the amalgam. The formation of hydrides might be expected to inhibit the release of mercury vapor in a manner similar to that of surface oxides. Another possibility is that the oxides present on the surface of the amalgam could be reduced by the hydrogen gas, or that oxygen adsorbed on the surface could be removed. The reduction of surface oxides or elimination of adsorbed oxygen would be expected to increase the vaporization of mercury from the amalgam. The results suggest that the latter possibility is most probable under the experimental conditions of this study.
According to a review by Siegel and Libowitz (1968) on the
formation and properties of metal hydrides, there is negligible dissolution of hydrogen into pure Cu, Hg, Ag and Sn in the solid state at temperatures below 400°C. Therefore, though amalgam is composed of a mixture of metallic elements and is not a pure metal, it is logical to exclude the possibility of dissolution of hydrogen into amalgam under the conditions of this experiment.
The second possibility involving the formation of metal hy- drides does not fit the results of this study and is not supported by the literature, even though it is well known that hydrogen can react with metallic elements to form metal hydrides. Published reviews describe the formation of specific hydrides from all of the elements found in dental amalgam (Siegel and Libowitz, 1968; MacKay, 1986). However, the experimental conditions under
Dental Mater ia ls~September 1993 303

which the hydrides are reported to be formed are vastly differ- ent from those described in this study. In addition, most of the hydrides are very unstable and would decompose immediately at or above oral temperatures. Therefore, though metal hy- drides can be formed from the elements in dental amalgam, they are not stable in the tem- perature range employed in this study, and this interaction can also be disregarded.
The final possibility for the interaction between hydrogen and the amalgam involves the reduction of surface oxides or elimination of adsorbed oxygen. Previous studies using x-ray photoelectron spectroscopy have revealed that the surface of amalgam is rapidly oxidized
TABLE 2: CALCULATED VALUES FOR THE STANDARD FREE ENERGY OF FORMATION
AND REDUCTION OF OXIDES(G, kcal/mol)
Temperature
37°C 80°C 110°C
Oxides Formation Reduction Formation Reduction Formation Reduction
Ag20 + 2.5 - 57.0 + 3.8 - 57.9 + 4.8 - 58.5
Ag202 + 7.1 -116.1 + 8.9 -117.0 +10.2 -117.6
Hg20 - 12.5 -42.0 - 11.3 -42.8 - 10.4 -43.3
HgO - 13.7 -40.8 - 12.6 -41.5 - 11.8 -41.9
Cu20 - 34.8 - 19.7 - 34.0 - 20.0 - 33.5 - 20.2
CuO - 30.7 - 23.8 - 29.7 - 24.4 - 29.0 - 24.7
SnO - 61.1 + 7.1 - 60.1 + 6.1 - 59.4 + 5.7
SnO 2 -123.7 +14.7 -121.6 +12.7 -120.1 +12.7
H20 (gas) - 54.5 . . . . 54.1 . . . . 53.7 ---
after exposure to air, even under partial vacuum where the concentration of oxygen is very low (Hanawa et al., 1987; Okabe et al., 1989). Potential oxides for amalgams are listed in Table 2, along with values for the standard free energy of formation, AG, calculated from available data (Weast and Astle, 1979) for the three temperatures used in the study. In addition, the AG value for the formation of H20 (gas) from oxygen and hydrogen gas is also included for the three temperatures. The table demonstrates that based upon the very low free energy of formation, which makes the reaction favorable, the major constituents of the surface film that forms on amalgams are likely to be oxides of tin, such as SnO 2 and/or SnO. This is consistent with experimental data showing tin oxides as the predominant oxides formed on zinc-free dental amalgams (Hanawa et al, 1987; Okabe et al., 1989). In addition, conditions are highly favorable for the reac- tion between adsorbed oxygen and hydrogen to form water vapor.
Using the values of standard free energy of formation for each oxide and H20 (gas), the values for the standard free energy of reduction for each oxide can also be calculated (Table 2), assum- ing the following reaction with hydrogen:
MmO n (solid) + n H 2 (gas) ---> m M (solid) + n H20 (gas) (m,n>_l) (1)
This redox reaction is obtained by combining the following oxidation and reduction reactions:
n H 2 (gas) + n 1/~ O2 (gas) ---> n H20 (gas) (2) MmO n (solid) ---> m M (solid) + n ½ O2 (gas) (3)
If G 2 and G 3 are the standard free energies for Eq. 2 and Eq. 3, respectively, then the free energy of the reduction of oxides by hydrogen (G 1) can be given by equation 4:
G 1 = G 2 + G 3 (4)
Table 2 shows that there is only a slight decrease in standard free energy of reduction of 1 - 2 kcal/mole for each oxide as the
temperature is increased. Thus, the tendency for a reduction in the oxide film by hydrogen is essentially constant at the tempera- tures employed in the experiment. The G values for each oxide are negative, except for the tin oxides. This implies that under the conditions of the experiment, SnO and SnO 2 are less likely to be reduced by hydrogen than are the other oxides. It should be noted that this thermodynamic explanation is only valid under equilib- rium conditions, and therefore can only be used as an approxima- tion. However, this observation is consistent with the published reviews of Mellor (1922; 1923; 1927), which report on studies showing the reduction of oxides by a flow of hydrogen to begin at 0°C for AgO, at 64-76°C for HgO, at 90°C for CuO and at 170- 180°C for SnO2. Therefore, all oxides except SnO 2 would be expected to be reduced by hydrogen at the maximum tempera- ture employed in this study, i.e. ll0°C.
Table 2 shows that the formation of water vapor under the experimental conditions is favorable due to a negative AG for this reaction. This suggests that any oxygen atoms which had adsorbed on the surface of the amalgam without forming oxides would react with hydrogen to form water vapor and would subsequently be carried away by the stream of hydrogen gas. Therefore, though the removal of tin oxides through reduction by hydrogen does not appear to be favorable, the elimination of adsorbed oxygen would be expected to contribute to the increased vaporization of mercury from the amalgams in hydrogen gas at elevated temperatures.
Another possibility also exists to explain the elevation in mercury vaporization from amalgam in hydrogen. The two high temperatures were chosen to increase the mercury vaporization rate. The vapor pressure of pure mercury is elevated by nearly one order of magnitude for each of the temperature increases used in the study. In addition, 80 - 110 °C is in the temperature range where a peritectic reaction for 7, (Ag2Hg3) occurs. This reaction results in the formation of a liquid phase containing mercury (Tsutsumiet al., 1988). The generation of liquid mercury might be expected to contribute to an increase in the rate of mercury release if the liberated mercury does not become oxi- dized by oxygen in the surface film. The AG values in Table 2
304 Ferracane et aL/Mercury release in hydrogen

suggest that in a hydrogen atmosphere, the reduction of mercury oxides is more favorable than is the formation of mercury oxides. Thus, at the higher temperatures where liquid mercury may be formed due to the peritectic reaction, no inhibiting film is formed to minimize the rate of vaporization.
The results of the previous study showed the rate of mercury evaporation in air to be essentially independent of temperature, supporting the hypothesis that the oxidation of the surface was the primary cause for the observed decline in mercury release with time (Ferracane et al., 1992). However, the results in argon and hydrogen demonstrate a clear dependence of the mercury evaporation rate on temperature when amalgams are exposed to non-oxidizing environments which inhibit oxide film formation. Though the vaporization of mercury is elevated during hydrogen or argon exposure at 37°C, subsequent exposure to air allows the formation of an oxide film, which reduces the rate of evaporation (Figs. 3 and 4). Note that although the amalgams subjected to the inert environments had been aged for 60 min before being exposed to air, the reduction in evaporation rate was identical to that of the amalgam immediately exposed to air. This supports the hypothesis that the oxide film is the primary factor limiting mercury evaporation during the setting of amalgam. In contrast, though an oxide film begins to form once air is introduced to the specimens previously exposed to the non-oxidizing environments, the mercury vaporization rate at the higher temperatures re- mains elevated for a substantial period of time (Figs. 5-8). Appar- ently a much thicker film must be needed to effectively inhibit mercury vaporization due to the higher vapor pressure at the high temperatures. The effect is most obvious after the hydrogen exposure. Although it is likely that some of the original oxide film and/or adsorbed oxygen was eliminated during the hydrogen exposure, it is unclear why there is such a long delay before inhibition of vapor release occurs once the amalgams are exposed to air.
The difference in the rate of decline of the evaporation rates for the amalgams after exposure to air at the elevated temperatures suggests that the high-Cu amalgam is less thermally stable than the low-Cu amalgam. This difference may be due to variations in the tin content in the silver-mercury matrix phase (71), being greater in the low-Cu than in the high-Cu amalgam (Mahler, 1993). Another possibility is that the differences in the alloy particle shape and composition of the amalgams influences the manner in which mercury liberated during heating is further reacted by the alloy, thus altering the vaporization rate (Ferracane et al., 1992).
The results of this study lend further support to the hypothesis that the formation of an oxide film is the primary factor limiting mercury evaporation from amalgams. The rapid formation of this film minimizes the extent of mercury release, even when the amalgam is exposed to elevated temperatures where the vapor pressure of mercury is significantly increased over that at oral temperatures. Elimination of the surface oxide and/or adsorbed oxygen, either by physical abrasion or by chemical reduction, as achieved in this study, can contribute to an increase in the rate of evaporation of mercury from amalgam. However, the rate of evaporation is rapidly reduced once the amalgam is exposed to oxygen, except at temperatures far in excess of normal oral conditions. These data support studies showing a minimal exposure to mercury for individuals with amalgam restorations.
ACKNOWLEDGMENTS We thank Mr. Thien Nguyen and Mr. Robert Spears for their assistance with this study. This investigation was supported in part by USPHS Research Grant DE 07644-06 from the National Institute of Dental Research, National Institutes of Health, Bethesda, MD 20892.
Received June 21, 1993/Accepted August 25, 1993
Address correspondence and reprint requests to: J.L. Ferracane Department of Dental Materials Science Oregon Health Sciences University 611 S.W. Campus Drive Portland, OR 97201 USA
REFERENCES De Freitas JF (1979). A survey of the elemental composition of
alloy for dental amalgam. Aust Dent J 24:17-25. Ferracane JL, Hanawa T, Okabe T (1992). Effectiveness of oxide
films in reducing mercury release from amalgams. JDent Res 71:1151-1155.
Hanawa T, Matsuda K, Ota M, Shimokobe H, Ferracane JL, Nelson P, Okabe T (1986). Thermal behavior of amalgams under argon atmosphere. JDent Res 65:192, Abst. No. 208.
Hanawa T, Takahashi T, Ota M, Pinizzotto RF, Ferracane JL, Okabe T (1987). Surface characterization of amalgams using x-ray photoelectron spectroscopy. J Dent Res 66:1470-1478.
MacKay KM (1966). Chapter 3: Transition metal hydrides; and Chapter 4: Covalent hydrides. In: Hydrogen compounds of the metallic elements. London: E & F N Spon Ltd, 45-137.
Mahler DB, Adey JD, Fleming-Slayden MA (1993). Hg emission from amalgam related to Sn in Ag-Hg phase. J Dent Res 72:112, Abst. No. 71.
Mellor JW, editor (1922). Chapter VII: Hydrogen. 8. The chemical properties of hydrogen. In: A comprehensive treatise on inor- ganic and theoretical chemistry. Vol. I. London: Longmans, Green and Co., 325-338.
Mellor JW, editor (1923). Chapter XXXI: Mercury. 6. Mercurous oxides; and 7. Mercuric oxides. In: A comprehensive treatise on inorganic and theoretical chemistry. Vol. IV. London: Longmans, Green and Co., 768-784.
Mellor JW, editor (1927). Chapter XLVI: Tin. 11. Stannous oxide and hydroxide; and 12. Stannic oxides. In: A comprehensive treatise on inorganic and theoretical chemistry. Vol. VII. London: Longmans, Green and Co., 386-404.
Okabe T, Gnade BE, Nakajima H, Hashimoto H, Ferracane JL, Hanawa T, Nguyen T (1989). Rapid formation of surface oxides on amalgams. JDent Res 68:210, Abst. No. 227.
Siegel B, Libowitz FF (1968). The covalent hydrides and hydrides of the group V to VIII transition metals. In: Metal Hydrides. Mueller WM, Blackledge JP, Libowitz GG, eds. New York: Academic Press, 545-674.
Tsutsumi S, Nakamura M, Schiller TL, Ferracane JL, Okabe T (1988). Thermal analysis of amalgams. Dent Mater 4:307-311.
Weast RC, Astle MJ (1979-80): CRC Handbook of Chemistry and Physics, 60th edition, Boca Raton, Florida: CRC Press, D45- Dh0; E41.
Dental Materials~September 1993 305





























