Embed Size (px)
Citation preview

Enantioselective Hydroarylation of Unactivated Ketones with Aryl Pinacolboronic Esters
and
A Hydrindanone-Based Approach Toward Natural Products
by
Gary Michael Gallego
A dissertation submitted in partial satisfaction of the requirements
for the degree of
Doctor of Philosophy
in
Chemistry
in the
GRADUATE DIVISION
of the
UNIVERSITY OF CALIFORNIA, BERKELEY
Committee in charge:
Professor Richmond Sarpong (Chair) Professor Thomas Maimone Professor Mary Wildermuth
Fall 2013 !

!

! "!
Abstract
Enantioselective Hydroarylation of Unactivated Ketones with Aryl Pinacolboronic Esters and
A Hydrindanone-Based Approach Toward Natural Products
By
Gary Michael Gallego
Doctor of Philosophy in Chemistry
University of California, Berkeley
Professor Richmond Sarpong, Chair
The first section of this dissertation describes our efforts to further develop the rhodium-catalyzed 1,2-addition of pinacolboronic ester nucleophiles into unactivated ketones. This reaction represents a mild and efficient alternative for performing this carbon-carbon forming transformation, which traditionally employs harsh conditions, and is especially useful in the context of total synthesis. Our observation that bisphosphine ligands are a beneficial additive in this transformation led to its elaboration into an asymmetric reaction manifold. The second section of this dissertation centers on the use of a key hydrindanone intermediate as a valuable synthetic precursor to the diterpenoid alkaloids and the phragmalin-type limonoids. Chapter 2 begins with a discussion of the isolation and classification of the diterpenoid alkaloids. In addition, major synthetic contributions made by others toward the synthesis of these natural products are also discussed. Our own synthetic approach to these compounds is then detailed, including the construction of the versatile denudatine-type core. Central to the success of this strategy was a highly diastereoselective Diels-Alder cycloaddition to furnish a functionalized hydrindanone, the stereochemical information of which was relayed throughout the rest of the synthesis. Chapter 3 focuses on the phragmalin-type limonoids. Previous landmark syntheses of limonoid natural products are first summarized following a brief overview of their isolation and classification. Our strategy for accessing the bridging framework of these natural products is then disclosed. Key to the success of this approach was the concept of network analysis allowing for disconnection of the bridging framework into a fused [6,5] system, thus simplifying its construction. !

! "!
Table of Contents
Acknowledgments ii Chapter One: Enantioselective Hydroarylation of Unactivated Ketones with Aryl Pinacolboronic Esters 1.1 Introduction 01 1.2 1,2-Additions of Organoboron Reagents 02 1.3 Reaction Optimization 04 1.4 Extension to an Asymmetric Manifold 09 1.5 Conclusion 11 1.6 Experimental Methods 11 1.7 References and Notes 29 Appendix One: Spectra Relevant to Chapter One 32 Chapter Two: Diterpenoid Alkaloids 2.1 Introduction 46 2.2 Isolation and Classification of the Diterpenoid Alkaloids 47 2.3 Syntheses and Approaches 49 2.4 Construction of the Tricyclic Core of the Denudatine-Type Diterpenoid Alkaloids 51 2.5 Construction of the Hexacyclic Core of the Denudatine-Type Diterpenoid Alkaloids 62 2.6 Elaboration of the Hexacyclic Core of the Denudatine-Type Diterpenoid Alkaloids 66 2.7 Conclusion 72 2.8 Experimental Methods 73 2.9 References and Notes 84 Appendix Two: Spectra and Crystallographic Data Relevant to Chapter Two 87 Chapter Three: Phragmalin-Type Limonoids 3.1 Introduction 149 3.2 Isolation and Classification of Limonoids 150 3.3 Syntheses and Approaches 152 3.4 Construction of the Bridging Framework of the Phragmalin-Type Limonoids 156 3.5 Conclusion 161 3.6 Experimental Methods 161 3.7 References and Notes 168 Appendix Three: Spectra and Crystallographic Data Relevant to Chapter Three 170

! ""!
Acknowledgments
I am truly grateful to my advisor, Richmond Sarpong, for giving me the opportunity to work in his lab. His dedication to the education and development of his students was a most valuable asset in my graduate career. Richmond has always been supportive and continuously pushed me to better myself. He respected my ideas while simultaneously offering his own. As I developed as a chemist through graduate school, I came to realize what a rarity it was to have an advisor with such a vested interest in the success of each of his students. Richmond encourages his students to work hard by working hard for them in turn.
An important lesson I have learned here is how valuable it is to be surrounded by supportive colleagues and friends. I am forever indebted to my best friend, Terry Lebold. Terry joined the Sarpong lab during my second year of school and he took a genuine interest in my progress and that of all of our labmates. I was fortunate to be on the diterpenoid and phragmalin projects with him; while working together, he taught me more about synthetic techniques, lab efficiency, and problem solving than I could have ever learned otherwise. I will also always appreciate Terry’s positive attitude and encouraging nature and the Berkeley family that he and his wonderful wife Jen have been to me. Our trips to the dog park and the aquarium or to crossfit followed by cooking dinner together and watching reality TV were some of my favorite times in grad school.
There have been a few other people who helped make my time at Berkeley unforgettable. Gabriela de Almeida’s friendship has been invaluable, and our short, daily visits to each other’s labs would brighten my day. She has been a great source of advice for science and life in general.
Jack Lee, who joined our group in my last year, has been a major contributor to the diterpenoid project and a fantastic coworker. He is a great problem solver and a team player when it comes to moving the project forward. I am grateful to know that the diterpenoid project will remain in good hands. It has also been wonderful to work with Helene (“LN”) Viart on a mechanistic and computational project pertaining to the yohimbine alkaloids; she quickly became a friend and source of support.
I was fortunate to join the group with four other wonderful students. Erica Schultz was my partner in crime during many outings to the movies with a bottle of wine. Rebecca Murphy’s warm and caring personality always shone through, and I owe all of my cardiovascular exercise completed at Berkeley to her. Jessica Kisunzu has had to sit next to me since our first year of school, and I especially appreciate that she was always an ear to talk to and never judged me for constantly talking to myself. Amy Hamlin was never afraid to tell it to me straight, and has been great fun throughout the years.
Jim Newton has been an amazing labmate and roommate for the last two years of school, and, most importantly, a close friend throughout. Between working and whiskey and Halo nights, I am not quite sure how we ever got groceries. We were recently joined by a French-Canadian semi-roommate, Vincent Lindsay. Vince has taught me about chemistry and French, mostly while drinking the whiskey he brought to the cottage; it was certainly more fun that way.

! """!
Senior students and post-docs helped me through my first few years in lab. Laura Miller, my first year mentor, was an endless source of advice, and her eye-rolls were an uncannily accurate meter for how silly my questions were. Alison Hardin Narayan had an unrivaled love for Justin Bieber and Justin Derulo and was a great source of cake and a cozy guest bed after a long night at “The Graduate.” Jessica Wood is an awesome friend and an even better softball teammate. Stephen Heller was always patient and a fantastic chemistry teacher; he also throws one heck of a St. Patrick’s Day party. I will always be grateful to Ethan Fisher for bringing Oriental Barbecue Chicken Town into my life.
Chris Canlas has kept the NMR facilities in top shape and was always receptive to questions. Antonio DiPasquale has been amazingly quick at providing crystal structures. The Toste, Bertozzi, Hartwig and Maimone groups have been very helpful both for chemical discussion and sharing of equipment and chemicals.
Finally, I would like to thank my family, who have been unconditionally supportive through all of the trials and tribulations of graduate school. Their encouragement and love reminded me that I always had somebody there for me.

! "!
Chapter I Enantioselective Hydroarylation of Unactivated Ketones with Aryl Pinacolboronic Esters A portion of the work described below has been previously published: Gallego, G. M.; Sarpong, R. Chem. Sci. 2012, 3, 1338-1342. 1.1 Introduction
Within the context of natural product synthesis, there exists the necessity for the development of mild and efficient methods for performing particular transformations. Among them, construction of carbon-carbon bonds remains at the forefront as a central challenge that continues to produce an active area of research. A premier method for the generation of these bonds is through the 1,2-addition of organometallic reagents into carbonyl electrophiles. Traditional organometallic reagents (e.g., organolithium, Grignard reagents) are limited by their relatively high basicity, and low functional group tolerance and selectivity. Additionally, their high air and moisture sensitivity make them less than ideal for complex molecule synthesis.
In 2009, the Sarpong group reported the total synthesis of Galbulimima akaloid G.B. 13; a pentacyclic alkaloid isolated from the tree Galbulimima.1 A key strategic maneuver during this synthesis was transforming pyridine 1.1 (R = Br, Mg, Li, B(OH)2, Sm) into 1,2-adduct 1.2 (Scheme 1.1). A number of traditional tactics were investigated (e.g., Rieke magnesiation,2 metal-halogen exchange with tBuLi or Knochel conditions3) all of which failed to deliver the desired product. Counter to intuition, this transformation presented a heightened challenge as an intramolecular reaction, due to the fact that many methods for the generation of reactive nucleophiles from the halide are incompatible with ketonic groups. Ultimately, the 1,2-addition was accomplished by conversion of the halide to a pinacolboronic ester (1.1, R = BPin), and exposure to catalytic [Rh(cod)(MeCN)2]BF4 and triethylamine.
Scheme 1.1 Initial discovery of Rh-catalyzed hydroarylation.
Pinacolboronic esters offer a range of advantages as latent nucleophiles. These groups can be introduced onto organic frameworks under a variety of conditions. Standard methods to install pinacolboronic esters involve generation of a reactive organometallic species and quenching with an electrophilic source of the pinacolboron
HOO
HN
R
OMe
HOHO
H NOMe
cat. [Rh(cod)(MeCN)2]+BF4-
Et3N (2 equiv)
PhMe, 80 ºC, 77% yield
Variousmethods
(R = BPin)
(R = Br, Mg, Li, B(OH)2, Sm)
1.1
1.2

! #!
subunit (e.g., iPrOBPin, HBPin).4 Milder (those more suited for complex molecule synthesis) include palladium-catalyzed cross-coupling by the method of Miyaura5 and Ir(I)- and Rh(I)-catalyzed C-H functionalization.6 In addition to their ease of installation, pinacolboronic esters offer the advantage of air and moisture stability, making them especially easy to handle and purify in a laboratory setting.7 Furthermore, their robust nature allows them to be carried through multiple synthetic manipulations before being unveiled as a nucleophile, making them ideal for total synthesis, where intramolecular applications are more common. The many advantages these functionalities have to offer as latent nucleophiles prompted us to further investigate their use in carbonyl 1,2-addition reactions. 1.2 1,2-Additions of Organoboron Reagents Given the challenges associated with more traditional organometallic nucleophiles, much work has been described involving the use of organoboron reagents as nucleophilic partners. Several reports describe the use of boronic acids and esters in the metal-catalyzed addition into electron-deficient alkene and alkyne groups. These reactions can now be applied to generate tertiary and quaternary carbon centers in a racemic and non-racemic sense.8-10 The scope of 1,2-additions, however, remains rather limited. 1,2-Addition into more electrophilic carbonyl groups remains the most common implementation of organoboron reagents as nucleophiles. For example, additions into aldehydes has received great attention and been described in a racemic and an enantioenriched sense with a variety of metal catalysts, including Cu,11 Fe,12 Ir,13 Ni,14-17 Pd,18-20 Pt,21 and Rh13,22,23. Analogously, aldimines24-26 and ketimines27 (typically bearing an electron withdrawing group) have also been shown to produce non-racemic, chiral amines utilizing boronic acid or tetrarylboronate nucleophiles. Ketone groups, on the other hand, have been seldom used as electrophiles. These reactions suffer from the challenge of decreased reactivity and increased steric encumbrance associated with the ketone functional group. As a result, these reactions are largely limited to boronic acid 1,2-additions into activated ketone groups. Strained ring systems have been exploited extensively as a method for increasing the reactivity of carbonyl groups. The distortion of the orbitals within the cyclic ketone group of strained cycloalkanones in combination with the driving force behind rehybridization from sp2 to sp3 give them enhanced reactivity to nucleophiles and organometallic complexes. 1,2-Addition of boronic acids into cyclobutanones, for example, has been described. These processes also result in ! – carbon scission to produce acyclic ketones, via rhodium alkoxide 1.4 (Scheme 1.2).28,29 The intermediacy of alkoxide 1.4 has allowed for interception by rhodation of tertiary cyclobutanols to undergo similar ring opening-type reactions.30-32

! $!
Scheme 1.2 1,2-addition and ring opening reaction catalyzed by [Rh].
In a similar fashion to cyclobutanones, 1,2-dicarbonyl groups are also reported to undergo 1,2-addition by organoboron reagents in a racemic and non-racemic fashion. Isatins are a valuable substrate in this regard, producing enantio-enriched hydroxy-oxindoles (Scheme 1.3), which are prevalent in natural products and in biologically active compounds.33 The scope of these reactions have been expanded to include more general 1,2-dicarbonyls such as 1,2-diketones and 1,2-ketoester to produce !-hydroxy-ketones.26,34
Scheme 1.3 Rh-catalyzed 1,2-addition into isatins.
Reports of unactivated ketone groups undergoing 1,2-addition with arylboron species are much more scarce and typically require specialized substrates. Lu has shown that, under palladium catalysis, boronic acids will effectively add into unactivated ketones in an enantioselective fashion.35,36 This chemistry takes advantage of the enhanced electrophilicity and vacant coordination site of a Pd(II)-catalyst system, allowing for the circumvention of redox chemistry associated with more traditional Pd(0)-catalyzed additions of aryl halides. While this chemistry is elegant, for the majority of substrates demonstrated by Lu, an oxygen atom in the linker was required to act as a directing and activating component (Scheme 1.4). Scheme 1.4 Lu’s asymmetric 1,2-addition.
O
R
Rh(acac)(C2H4)2,P(tBu)3, ArB(OH)2
CsCO3, dioxane, 100 °CR Me
Ar
O
ArO[Rh]
R
1.3 1.5
1.4
NH
O
OR
[(C2H4)2Rh(acac)] P(OPh)3 or L1.8
ArB(OH)2, solvent NH
OR
HO Ar
OO
P N
L1.81.6 1.7
O
B(OH)2
R
O PdPP
HOOH
PdPP* *
PP* = (R)-BINAP
·2TfOPd complex 1.11
Pd complex 1.11
O
HO R*
1.9 1.10

! %!
As an alternative to boronic acid additions, two other arylboron species have been employed. Tetraarylboronates have effectively added into nitriles, ketones and imines in the presence of a rhodium catalyst.37 The necessity for a tetraarylboronate, however, is not ideal, especially for intramolecular applications. The second aryl organoboron nucleophile is a boronic ester. Itami demonstrated the first application of this chemistry where, in the presence of a nickel catalyst and an N-heterocyclic carbene ligand, intermolecular additions of glycolatoboronic esters into ketones can take place (Scheme 1.5).16 This chemistry serves as a nice addition to the aforementioned boronic acid chemistry. It is noteworthy that, shortly after publication of the chemistry described in the following section, an additional report of an asymmetric organoboronic ester addition into ketone groups was described by Lam and coworkers utilizing chiral, non-racemic sulfinamide ligands.38 Scheme 1.5 Itami’s Ni-catalyzed, intermolecular 1,2-addition.
1.3 Reaction Optimization Although the Rh(I)-catalyzed 1,2-addition of pyridine-derived pinacolboronic ester 1.1 in to a ketone group was successful in the synthesis of G.B. 13, the reaction conditions proved to be capricious at best for other substrates. For example, the tertiary alcohol that arises from when simple ketone 1.15 is used as a substrate is isolated in 32% yield following the completion of the reaction (Scheme 1.6). In an effort to develop a truly generally method, we embarked on an optimization campaign. Scheme 1.6 Initial result in extension of Rh-catalyzed 1,2-addition.
At the forefront of our thinking was an analysis of this reaction from a mechanistic perspective. As such, we first considered the proposed catalytic cycle by which hydroarylation proceeds (Figure 1.1). We propose that an initial transmetallation from boron to rhodium, likely facilitated by the amine base, delivers aryl-rhodium intermediate 1.18. The role of the amine base in this step may be to coordinate to the boron center to generate a more nucleophilic boronate complex, thus promoting nucleophilic attack onto the cationic rhodium metal center. Following coordination of the ketone group to the metal center, migratory insertion furnishes rhodium alkoxide 1.20. A second, key transmetallation with another molecule of the starting material regenerates 1.18 and boron alkoxide 1.21 (which liberates the alcohol upon aqueous workup). This second transmetallation may also benefit from the amine base additive by displacing the alkoxide ligand from intermediate 1.20 to generate an even more electrophilic, cationic
O
R2R1Ar B
O
O
Me
Me Ni(cod)2, IPr·HCl
CsF, Toluene, 80 °CR1
Ar R2OH
+
1.12 1.13 1.14
BPin HO nPr
[Rh(cod)(MeCN)2]BF4 (10 mol%)
Et3N, PhMe, 120 °C, 3 dnPr
O
32%1.15 1.16

! &!
metal center. Corey has proposed a similar amine-base assisted transmetallation in the context of 1,4-additions of organoboron reagents.39
Figure 1.1 Proposed catalytic cycle for Rh-catalyzed 1,2-addition. Given the large role the amine base may play in the catalytic cycle, we first investigated a range of amine bases, more specifically, those which vary in nucleophilicity.40 As can be seen in Table 1.1, the highly nucleophilic base DABCO, proved to be most beneficial in promoting reactivity (compare entries 1 and 2). Complete consumption of the starting material could be observed by decreasing the reaction temperature from 120 °C to 80 °C, albeit over extended reaction times. Finally, a remarkable increase in reactivity was observed in the change from toluene to benzene as a solvent. While, it is not entirely clear why such a dramatic effect was observed between these solvents, it maybe attributed to subtleties in solubility of the rhodium complexes.
[Rh]+BF4-R3N
[PinB-NR3]BF4
Transmetallation
Coordination
Migratory Insertion
Transmetallation
R
O[Rh]
O
R
BO
O
O
R
[Rh]
R O[Rh]
O
R
BO
O
R OBO
O
1.17
1.18
1.19
1.20
1.17
1.21

! '!
Table 1.1 First optimization campaign.
With these newly established conditions in hand, we explored the substrate scope of the 1,2-addition. As can be seen in Table 1.2, a range of substrates undergo hydroarylation. Methoxypyridine pinacolboronic esters produce the bicyclic, tertiary alcohols in excellent yields (entry 1). Less activated pinacolboronic esters are also smoothly converted to the desired 1,2-adducts in good yields with aryl ketones being the exception (entry 2c). Notably, sterically encumbered ketone substrates readily participate in this reaction (entries 2f and 3c). Oxygen atoms in the tether are also well tolerated, even in the presence of a strongly deactivating group on the nucleophilic, arene component (entry 3c).
BPin HO nPr
[Rh(cod)(MeCN)2]BF4 (10 mol%)
conditions
Baseb Solvent Temperature (ºC) Time (h) Pdt : SMc
Et3N PhMe 120 26 1.2 : 1 (32%)DABCO PhMe 120 26 14 : 1
DABCO PhH 80 22 1.0 : 0 (81%)DABCO PhMe 80 50 1.0 : 0
aVaiues in parenthesis are isolated yields b 2 equiv c Ratios determined by 1H NMR
nPr
O
Entry
12
43
1.15 1.16

! (!
Table 1.2 Substrate scope of hydroarylation of alkyl ketones.
The low conversion and isolated yields associated with aryl ketone substrates (e.g., entry 2c, Table 1.2), prompted us to undertake a second optimization campaign focusing on these particularly challenging systems. The increase in reaction efficiency noted upon addition of DABCO, and its possible role in facilitating transmetallation led us to hypothesize that a hydroxide ligand on the catalyst could also prove beneficial. The role of alkoxide bases as ligands in promoting transmetallation from boron to palladium and rhodium has been discussed in the context of Suzuki couplings41-43 and
entry substrate product yield (%)atime (h)
NMeO
BPin
O
n
OH
NMeO
n
1b
2c
BPin HO RR
O
n n
a) n = 1, R = nPr
b) n = 1, R = nBu
c) n = 1, R = p-F-C6H4
d) n = 2, R = Me
20
20
58
24
81
58
23d
78
e) n = 2, R = iBu 24 83f) n = 2, R = iPr 24 94
3c
BPin
OR
O
O
HO R
a) R = iBu, R' = H 24 62b) R = iPr, R'= H 6724
R'R'
c) R = iPr, R' = F 8024
a) n = 1 24 96b) n = 2 24 90c) n = 3 24 87
1.22 1.23
1.24 1.25
1.26 1.27
aIsolated Yields. b Reaction conditions: 2 equivalents of DABCO, 10 mol% [Rh(cod)(MeCN)2]BF4, 0.10 M in PhMe, 100 °C. c Reaction conditions: 2 equivalents of DABCO, 10 mol%[Rh(cod)(MeCN)2]BF4, 0.10 M in PhH, 80 °C.d15 mol% catalyst loading.

! )!
conjugate additions.44 In replacing the [Rh(cod)(MeCN)2]BF4 pre-catalyst with [Rh(cod)(OH)]2, a modest improvement in reactivity was observed (compare entries 1 and 3, Table 1.3). Further, we anticipated a more electron rich metal center would more easily promote migratory insertion (see Figure 1.1) and, as such, began investigating a range of bisphosphine ligands. After screening several bisphoshines, dppe emerged as a superior additive and led to complete conversion of 1.28 to tertiary alcohol 1.29 (entry 7). This result is in contrast to that observed with metal-catalyzed organoboron additions to aldehydes, where larger bite angle ligands are quite beneficial.45 The contrast in these results can likely be attributed to increased steric demands of both pinacolboronic esters over acids as well as ketones over aldehydes. Table 1.3 Optimization campain of aryl ketones.
Having found conditions that led to complete conversion of aryl ketones to the corresponding tertiary alcohol, we were delighted to find that isolated yields were high for these substrates (Table 1.4). Additionally, we found that the conditions for aryl ketones were comparable to those found with alkyl ketones (compare entry 1d, Table 1.4, with entry 2f, Table 1.2). Table 1.4 Substrate scope for aryl ketone addition.
catalyst (mol %) pdt : SMa
[Rh(cod)(MeCN)2]BF4 (10)
[Rh(cod)(OH)]2 (5)
ligand
DABCO (2 equiv)
-[Rh(cod)(OH)]2 (5) DABCO (2 equiv)
Ph
OBPin
[Rh], Ligand
C6D6, 80 °C, 24 hHO Ph
[Rh(cod)(OH)]2 (5)
[Rh(cod)(OH)]2 (5)
dppe (0.1 equiv)
dppf (0.1 equiv)
[Rh(cod)(OH)]2 (5) BINAP (0.1 equiv)
2.0 : 1.03.7 : 1.02.6 : 1.0
> 20 : 17.9 : 1.0
0.9 : 1.0
entry
1234
67
[Rh(cod)(OH)]2 (5) dppm (0.1 equiv) 0.2 : 1.08
[Rh(cod)(OH)]2 (5) dppb (0.1 equiv) 0.7 : 1.05
1.28 1.29
entry substrate product yield (%)btime (h)
1
BPin HO RR
O
n n
a) n = 1, R = Ph 24 85%b) n = 1, R = p-F-C6H4 24 74%c) n = 2, R = Ph 24 72%
1.30 1.31
d) n = 2, R = iPr 24 96%
aReaction conditions: 5 mol% [Rh(cod)(OH)]2 , 10 mol% dppe, 0.10 M in PhH, 80 °C. bIsolated Yields.

! *!
1.4 Extension to an Asymmetric Manifold Having established a trend between bisphosphine additives and catalytic activity for the 1,2-addition of pinacolboronic esters into ketones, we recognized that employing a chiral, non-racemic bisphophine could potentially translate to an asymmetric synthesis of tertiary alcohols. Thus, we undertook a study investigating the effect of various bisphosphines and reaction conditions (Table 1.5). We first elected to employ ligands which bear the same two carbon backbone as dppe (entries 1-3). While these did not promote high levels of enantioselectivity, they did provide proof of principle that these transformations could be rendered enantioselective. Table 1.5 Asymmetric hydroarylation optimization.
Hayashi and others have developed numerous conditions for the asymmetric addition of organoboron compounds into Michael acceptors. Among the most successful ligands for these transformations have been chiral, non-racemic cyclohexa- and cyclooctadienes46,47 as well as axially chiral bisphosphines such as BINAP.8
(R,R) - Chiraphos (10)2 > 20 : 1.0 11C6D6
Ph
BPin
O
Pdt : SMLigand (mol %)Entry
[Rh(cod)(OH)]2 (5 mol%)
Ligand, Solvent, Temperature HO Ph
% eeSolvent
(R,R) - 1-Napthyl DIPAMP (10) > 20 :1. 01 5C6D6
(R,R) - CatASium D (10)3 0.6 : 1.0 12C6D6
(R) - BINAP (10) > 20 : 1.04 49C6D6
(R,S) - Josiphos SL-J003-1 (10)5 8.1 : 1.0 59C6D6
(R,S) - Josiphos SL-J002-1 (11)6 1.5 : 1.0 85C6D6
Temperature (°C)
(R,S) - Josiphos SL-J002-1 (11)7 > 20 : 1.0 94PhMe
80808080808085
PPh2
PPh2
(R) - BINAP
Fe(Cy)2PP(Cy)2
(R,S) - Josiphos SL-J003-1
FePh2PP(t-Bu)2
(R,S) - Josiphos SL-J002-1
Ph2PPPh2
Me
Me
(R,R) - Chiraphos
PP
(R,R) - 1-Napthyl DIPAMP
NBn
Ph2P PPh2
(R,R) - CatASium D
1.32 1.33

! "+!
Having found that bisphosphines were more productive ligands then cyclooctadiene in our racemic studies, we elected to investigate axially chiral bisphosphines. These ligands proved to be quite effective with BINAP emerging as the best among the class, providing the tertiary alcohol as a scalemic mixture (entry 4). Further enhancements in enantioselectivity with these types of ligands was not observed under various conditions.
Josiphos ligands have arisen as powerful bisphoshine ligands in a wide range of contexts, including from C–N bond formation48 to asymmetric hydrogenation.49 We were intrigued by a report by Cramer in which aryl-rhodium intermediates were generated via metallation of cyclobutanols, "-carbon scission and hydrogen atom transfer.30 These intermediates effectively added into newly generated ketones in a diastereo- and enantioselective fashion. With the understanding that we too were generating an aryl-rhodium intermediate, we screened a range of Josiphos ligands, and found Josiphos SL-J002-1 produces the highest enantiomeric excess (entry 6). Recognizing the strong effect solvent choice had in our previous studies, we elected to revisit toluene as a solvent, and after minor optimization, found that complete conversion of the aryl ketones into tertiary alcohols could be observed and, importantly, maintain high levels of enantiomeric excess (entry 7).
Having established conditions for a highly enantioselective 1,2-addition, we explored the substrate scope of this transformation (Table 1.6). A wide range of aryl ketones undergo highly enantioselective hydroarylation including electron-neutral (entry 1a), electron-rich (entry 1b), relatively sterically encumbered (entry 1c), and electron-poor (entry 1d). An alkyl ketone was also shown to be a productive substrate in this transformation (entry 1f). Extension of this transformation to larger ring forming systems, proved to be challenging in promoting high levels enantiocontrol (entry 1g). Table 1.6 Substrate scope for asymmetric hydroarylation.
entry substrate product % eecyield (%)b
1
BPin HO RR
O
a) n = 1, R = Ph 78 94b) n = 1, R = m-MeO-C6H4 85 95c) n = 1, R = 2-Nap 50 93
1.34 1.35
d) n = 1, R = p-F-C6H4 54 92
aReaction conditions: 5 mol% [Rh(cod)(OH)]2 , 11 mol% Josiphos SL-J002-1, 0.10 M in PhMe, 85 °C. bIsolated Yields. cDetermined by chiral HPLC.
e) n = 1, R = p-Tol 90 94f) n = 1, R = Me 90 95
n n
g) n = 2, R = iPr 86 46

! ""!
1.5 Conclusion
We have demonstrated the Rh-catalyzed hydroarylation of unactivated ketones by pinacolboronic esters, which are readily accessible, air- and moisture-stable boronic acid derivatives. Historically, these pro-nucleophiles have been deemed inert in reactions with unactivated ketones. The results presented here represent an efficient and high yielding process to furnish tertiary alcohol products. This chemistry serves as a compliment to more traditional organometallic reagents, such as Grignard reagents and organolithiums (which are most beneficial in intermolecular additions), as well as the limited in scope additions of boronic acid and glycolotoboronic ester into ketones. Furthermore, through judicious choice of ligand and reaction conditions, the aforementioned hydroarylation can be performed with high levels of enantiocontrol, to produce enantioenriched tertiary alcohols. We anticipate that this methodology will be particularly beneficial in the context of total synthesis, where mild reaction conditions are likely required.
1.6 Experimental Methods General: Unless otherwise stated, reactions were performed in flame-dried glassware or dried in an oven overnight. All reaction vessels were fitted with rubber septa or Teflon screw caps and kept under an atmosphere of nitrogen. Liquid reagents and solvents were transferred via syringe under nitrogen using standard Schlenk techniques. Tetrahydrofuran, toluene, and benzene were sparged with argon and passed through an alumina column. Dichloromethane was distilled over calcium hydride. All other solvents were used as received unless otherwise noted. Reaction temperatures above 23 °C refer to oil bath temperature which was controlled by an IKA® temperature modulator. Reactions were monitored by thin layer chromatography using SiliCycle silica gel 60 F254 precoated plates (0.25 mm), which were visualized using UV irradiation, p-anisaldehyde stain or KMnO4 stain. Sorbent Technologies silica gel (particle size 40-63 µm) was used for column chromatography. 1H and 13C NMR were recorded on Bruker AVB-400, AVQ-400, DRX-500 or AV-600 spectrometers with 13C operating frequencies at 100, 125 and 150 MHz, respectively, in deuterated chloroform, benzene or p-xylene at 23 °C. Chemical shifts are reported relative to residual solvent signal (! = 7.26 for 1H NMR and 77.00 for 13C NMR in chloroform, 7.16 for 1H NMR in benzene, 6.91 for 1H NMR in p-xylene). Data for 1H NMR are reported as follows: chemical shift (multiplicity, coupling constant, number of hydrogens). Multiplicity is abbreviated as follows: s (singlet), br s (broad singlet), d (doublet), dd (doublet of doublets), t (triplet), tt (triplet of triplets), q (quartet), aq (apparent quartet), ap (apparent pentent), hept (heptet), m (multiplet). Signals marked by an asterisk (*) denote the minor rotamer. IR spectra were recorded on a Nicolet MAGNA-IR 850 spectrometer and are reported in frequency of absorption (cm-1). Only selected IR absorbencies are reported. Enantiomeric excess (ee) was determined by HPLC analysis on a Waters chromatography system (1525 binary pump, 717+ autosampler, 2487 dual wavelength detector) using a Chiralcel OD-H (0.46 cm x 25 cm)(from Daicel Chemical Ind., Ltd.) stationary phase and 97:3 hexanes/isopropanol mobile phase (1 mL/min) at 220 nm. Mass spectra were recorded on an LTQ Orbitrap XL (ThermoFisher Scientific) for ESI

! "#!
and AutoSpec Premier (Waters) for EI through the mass spectral facility at the University of California, Berkeley. General synthetic route for the preparation of substrates in Table 1.2, entry 1:
Representative experimental procedure for picoline alkylation (1.36):
LDA was generated over 1 h in a 250-mL, flame-dried round-bottom flask by the slow addition of n-BuLi (2.5 M, 3.47 mL, 8.69 mmol, 2.3 equiv) to diisopropylamine (1.25 mL, 8.87 mmol, 2.35 equiv) in 20 mL of THF at -78 °C. 3-Bromo-6-methoxy-2-methylpyridine (763 mg, 3.78 mmol, 1.0 equiv) in 10 mL of THF was then added to the LDA and the resulting mixture was allowed to stir for 1 h at -78 °C. In a separate, flame-dried 50-mL pear-shaped flask, cyclopentenone (372 mg, 4.53 mmol, 1.2 equiv) in 8 mL of THF was cooled to -78 °C and transferred via cannula into the reaction mixture, which was stirred for 1 h at that temperature. The reaction mixture was quenched with 2 mL saturated NH4Cl solution and extracted with Et2O (2 x 50 mL). The combined organic phases were dried over MgSO4, filtered and concentrated. The crude product was purified via silica gel column chromatography (10:1 hexanes/EtOAc) to deliver tertiary alcohol 1.36 (545 mg, 1.92 mmol, 51% yield). Rf 0.40 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 7.69 (d, J = 8.7 Hz, 1H), 6.56 (d, J = 8.7 Hz, 1H), 5.90 (d, J = 2.5 Hz, 1H), 5.72 (br s, 2H), 3.88 (s, 3H), 3.17 (s, 2H), 2.53 (br s, 1H), 2.38 – 2.30 (m, 1H), 2.04 – 1.92 (m, 2H); 13C NMR (125 MHz, CDCl3) # 162.2, 155.1, 143.3, 136.0,
NMeO Me
Br
LDA
O
n
NMeO
Br
OH PCCNMeO
Br
O
Rh/C, H2
NMeO
Br
O
PdCl2(dppf)·CH2Cl2KOAc, B2Pin2
NMeO
BPin
O
n n
nn
NMeO
Br
OH
1.36

! "$!
133.5, 112.8, 110.9, 85.5, 54.1, 44.7, 38.2, 31.2; IR (film) "max 3412, 1579, 1462, 1057 cm-1; HRMS (EI) calc’d for [C12H14NO2Br]+ m/z 283.0208, found 283.0206. Representative experimental procedure for oxidative allylic transposition (1.37):
An oven-dried 25 mL round-bottom flask equipped with a stir bar was purged with nitrogen and charged with 1-((3-bromo-6-methoxypyridin-2-yl)methyl)cyclopent-2-enol (531 mg, 1.87 mmol, 1.0 equiv) followed by dichloromethane (19 mL), celite (805 mg) and PCC (806 mg, 3.74 mmol, 2.0 equiv) to yield a dark colored solution. The reaction flask was then sealed and allowed to stir at ambient temperature for 3.5 h, after which time it was passed through a plug of silica, which was washed with dichloromethane (5 mL), and concentrated under reduced pressure. The crude product was purified via silica gel column chromatography (gradient of 10:1 hexanes/EtOAc ! 6:1 hexanes/EtOAc ! 4:1 hexanes/EtOAc) to deliver 1.37 (125 mg, 441 $mol 24% yield). Rf 0.31 (2:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) # 7.66 (d, J = 8.7 Hz, 1H), 6.55 (d, J = 8.7, 1H), 5.91 (t, J = 1.4 Hz, 1H), 3.97 (s, 2H), 3.86 (s, 3H), 2.70 – 2.67 (m, 2H), 2.44 – 2.41 (m, 2H); 13C NMR (125 MHz, CDCl3) # 209.9, 178.4, 162.9, 152.8, 142.9, 131.4, 112.1, 111.3, 53.9, 41.4, 35.7, 31.8; IR (film) "max 1708, 1617, 1462, 1016 cm-1; HRMS (EI) calc’d for [C12H12NO2Br]+ m/z 281.0051, found 281.0043. Representative experimental procedure for enone reduction (1.38):
3-((3-Bromo-6-methoxypyridin-2-yl)methyl)cyclopent-2-enone (124 mg, 440 $mol, 1.0 equiv) was added to a 25-mL round-bottom flask equipped with a stir bar and was diluted with 7.30 mL of THF. Rh/C (5% Rh by weight, 109 mg, 52.7 µmol, 0.12 equiv) was then added and the reaction vessel was purged with hydrogen gas for 10 min. The reaction mixture was allowed to stir under an atmosphere of hydrogen gas for 7 h, after which it was filtered through Celite and concentrated under reduced pressure. The crude residue was purified by column chromatography (gradient of 10:1 hexanes/EtOAc ! 5:1 hexanes/EtOAc ! 4:1 hexanes/EtOAc) to furnish 1.38 (89.0 mg, 313 $mol, 71% yield). Rf 0.43 (3:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 7.63 (d, J = 8.6 Hz, 1H), 6.49 (d, J = 8.6 Hz, 1H), 3.88 (s, 3H), 3.02 (dd, J = 14.2, 6.8 Hz, 1H), 2.94 (dd, J = 14.2, 7.7 Hz, 1H), 2.83 – 2.74 (m, 1H), 2.42 (dd, J = 18.4, 7.4 Hz, 1H), 2.37 – 2.30 (m, 1H), 2.22 – 2.17 (m, 2H), 2.03 (dd, J = 18.4, 9.9 Hz, 1H), 1.77 – 1.67 (m, 1H); 13C NMR
NMeO
Br1.37
O
NMeO
Br1.38
O

! "%!
(125 MHz, CDCl3) # 219.8, 162.6, 155.6, 142.6, 112.0, 110.3, 53.8, 45.1, 42.0, 38.5, 36.2, 29.4; IR (film) "max 1740, 1460, 1298 cm-1; HRMS (EI) calc’d for [C12H14NO2Br]+ m/z 283.0208, found 283.0202. Representative experimental procedure for borylation (1.39):
Potassium acetate (154 mg, 1.57 mmol, 5.0 equiv) was added to a 25-mL Schlenk tube equipped with a stir bar. The reaction vessel was then placed under vacuum and flame-dried. Once cool, [1,1-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (dichloromethane adduct, 25.6 mg, 31.3 $mol, 0.10 equiv) was added followed by bis(pinacolato)diboron (398 mg, 1.56 mmol, 5.0 equiv). 3-((3-Bromo-6-methoxypyridin-2-yl)methyl)cyclopentanone (89.0 mg, 313 µmol, 1.0 equiv) in 3.2 mL of DMF was then transferred from a screw cap vial to the Schlenk tube via cannula. The reaction mixture was sparged with nitrogen for 10 min, sealed with a Teflon stopper and heated at 80 °C for 22 h. After this time, the reaction mixture was diluted with 3 mL of Et2O and washed with 3 mL of water. The organic layer was isolated and the aqueous layer was extracted with Et2O (2 mL). The combined organic phase was washed with water (2 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was first purified via column chromatography (13:1 hexanes/EtOAc ! 6:1 hexanes/EtOAc) to remove bulk impurities followed by a second purification via column chromatography using slower elution conditions (13:1 hexanes/EtOAc ! 10:1 hexanes/EtOAc) to yield 1.39 (44.8 mg, 135 µmol 43% yield). Rf 0.54 (2:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) # 7.92 (d, J = 8.3 Hz, 1H), 6.54 (d, J = 8.3 Hz, 1H), 3.92 (s, 3H), 3.12 (d, J = 7.2 Hz, 2H), 2.73 – 2.64 (m, 1H), 2.38 – 2.25 (m, 2H), 2.18 – 1.99 (m, 3H), 1.78 – 1.66 (m, 1H), 1.31 (s, 12H); 13C NMR (125 MHz, CDCl3) # 220.4, 165.5, 164.9, 146.5, 107.2, 83.6, 53.2, 44.8, 42.0, 38.3, 37.9, 29.0, 24.9, 24.8. The boron-bound carbon was not detected likely due to quadropolar relaxation; IR (film) "max 1742, 1589, 1344, 1305, 1145, 1026 cm-1; HRMS calc’d for [C18H27O4NB]+: m/z 332.2028, found 332.2031.
1.40 was prepared using the representative synthetic route. Bulb-to-bulb distillation (120 °C, 0.1 torr, 1 h) was necessary to purify final product. Rf 0.35 (4:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) # 7.91 (d, J = 8.3 Hz, 1H), 6.53 (d, J = 8.3 Hz, 1H), 3.91 (s, 3H), 3.07 - 3.02 (m, 1H), 3.01 - 2.97 (m, 1H), 2.40 - 2.31 (m, 2H), 2.30 - 2.22 (m, 2H), 2.19 - 2.13 (m, 1H), 2.08 - 2.02 (m, 1H), 1.88 (d, J = 11.5 Hz, 1H), 1.66 - 1.54 (m, 1H), 1.50 -
NMeO
BPin1.39
O
NMeO
BPin1.40
O
! "&!
1.39 (m, 1H), 1.32 (s, 12H); 13C NMR (150 MHz, CDCl3) # 212.2, 165.3, 164.9, 146.5, 107.1, 83.6, 53.2, 48.0, 43.8, 41.5, 40.5, 31.2, 25.3, 24.89, 24.86. The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max 2977, 1710, 1588, 1345, 1308, 1145, 1023 cm-1; HRMS (ESI) calc’d for [C19H29O4NB]+: m/z 346.2184, found 346.2184.
1.41 was prepared using the representative synthetic route. Bulb-to-bulb distillation (120 °C, 0.1 torr, 1 h) was necessary to purify final product. Rf 0.37 (4:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) # 7.91 (d, J = 8.3 Hz, 1H), 6.53 (d, J = 8.3 Hz, 1H), 3.91 (s, 3H), 3.01 - 2.91 (m, 2H), 2.55 - 2.50 (m, 1H), 2.49 - 2.44 (m, 3H), 2.31 - 2.23 (m, 1H), 1.95 - 1.81 (m, 3H), 1.65 - 1.55 (m, 1H), 1.44 - 1.27 (m, 14H); 13C NMR (150 MHz, CDCl3) # 214.6, 165.6, 164.8, 146.5, 107.1, 83.6, 53.2, 49.8, 44.2, 44.0, 37.1, 36.4, 28.8, 24.9, 24.6. The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max 1700, 1344, 1267, 1145 cm-1; HRMS (ESI) calc’d for [C20H31NO4B]+: m/z 360.2341, found 360.2341. General synthetic route for the preparation of substrates in Table 1.2, entries 2a-c and Table 1.6, entries 1a-f:
Representative experimental procedure for Wittig homologation (1.42):
In a 20 mL screw cap vial, 1-(triphenylphosphoranylidene)pentan-2-one50 (430 mg, 1.2 mmol, 1.3 equiv) was diluted with 2-formylphenylboronic acid pinacol ester51 (220 mg, 0.96 mmol, 1.0 equiv) in toluene (7 mL). The vial was then purged with nitrogen, sealed with a Teflon cap and heated to 90 °C for 20 h. The reaction mixture was then allowed to cool to room temperature and concentrated under reduced pressure. The resulting solids were suspended in Et2O and filtered through celite to remove excess triphenylphosphine oxide and concentrated. The crude product was purified via column chromatography (4:1 hexanes/EtOAc) to deliver 1.42 (170 mg, 0.56 mmol, 59%). Rf 0.41 (4:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) # 8.52 (d, J = 16.5 Hz, 1H), 7.87 (d, J = 7.4 Hz, 1H), 7.68 (d, J = 7.9 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 7.35 (t, J = 7.4 Hz,
NMeO
BPin1.41
O
H
O
BPin
PPh3
R
O
BPin
R
O
BPin
R
OPd/C, H2
BPin
nPr
O
1.42

! "'!
1H), 6.58 (d, J = 16.5 Hz, 1H), 2.71 (t, J = 7.5 Hz, 2H), 1.74 (h, J = 7.5 Hz, 2H), 1.37 (s, 12H), 1.00 (t, J = 7.4 Hz, 3H); 13C NMR (150 MHz, CDCl3) # 201.6, 144.2, 140.6, 136.4, 131.2, 129.0, 128.0, 125.4, 84.0, 41.2, 24.8, 18.2, 13.9. The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max 1668, 1612, 1347 cm-1; HRMS (ESI) calc’d for [C18H25O3BNa]+: m/z 323.1789, found 323.1788. Representative experimental procedure for the reduction of enones (1.15):
1.15 (160 mg, 0.54 mmol, 1.0 equiv) was diluted with EtOAc (5.4 mL) in a 20 mL screw cap vial equipped with a magnetic stir bar. Pd/C (10%, 58 mg, 54 $mol, 0.1 equiv) was then added and the vial was fitted with a rubber septum and purged with hydrogen. The reaction mixture was stirred under a balloon of hydrogen at ambient temperature for 2 h at which time it was filtered through celite and concentrated. The crude product was purified via column chromatography (4:1 hexanes/EtOAc) to deliver 1.15 (130 mg, 0.44 mmol, 81%). Rf 0.50 (4:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) # 7.79 (d, J = 7.4 Hz, 1H), 7.34 (td, J = 7.5, 1.6 Hz, 1H), 7.21 - 7.16 (m, 2H), 3.16 – 3.09 (m, 2H), 2.71 - 2.65 (m, 2H), 2.39 (t, J = 7.3 Hz, 2H), 1.61 (h, J = 7.4 Hz, 2H), 1.34 (s, 12H), 0.91 (t, J = 7.4 Hz, 3H); 13C NMR (150 MHz, CDCl3) # 210.7, 148.2, 136.3, 131.1, 129.3, 125.4, 83.5, 46.1, 44.6, 30.5, 24.8, 17.3, 13.8. The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max 2976, 1713, 1348, 1315, 1145 cm-1; HRMS (ESI) calc’d for [C18H27O3BNa]+: m/z 325.1945, found 325.1944.
1.43 was prepared using the representative synthetic route. Rf 0.60 (2:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) # 7.80 (dd, J = 7.2, 1.4 Hz, 1H), 7.35 (td, J = 7.5, 1.6 Hz, 1H), 7.19 (ddd, J = 12.8, 7.1, 1.1 Hz, 2H), 3.28 - 3.01 (m, 2H), 2.80 - 2.56 (m, 2H), 2.15 (s, 3H), 1.34 (s, 12H). 13C NMR (125 MHz, CDCl3) # 208.7, 148.0, 136.4, 131.2, 129.3, 125.5, 83.5, 47.1, 30.5, 29.8, 24.9. The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max 2978, 1712, 1348 cm-1; HRMS (ESI) calc’d for [C16H23O3BNa]+: m/z 297.1632, found 297.1632.
1.44 was prepared using the representative synthetic route. Rf 0.50 (4:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) # 7.79 (dd, J = 7.4, 1.5 Hz, 1H), 7.34 (td, J = 7.5, 1.5 Hz, 1H), 7.21 - 7.16 (m, 2H), 3.15 – 3.09 (m, 2H), 2.73 - 2.63 (m, 2H), 2.40 (t, J = 7.5 Hz, 2H), 1.56 (dt, J = 15.1, 7.5 Hz, 2H), 1.34 (s, 12H), 1.34 - 1.26 (m, 2H), 0.90 (t, J = 7.4 Hz, 3H); 13C NMR (150 MHz, CDCl3) # 210.9, 148.2, 136.3, 131.1, 129.3,
BPin
nPr
O
1.15
BPin
Me
O
1.43
BPin
nBu
O
1.44

! "(!
125.4, 83.5, 46.1, 42.4, 30.5, 26.0, 24.9, 22.4, 13.8. The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max1711, 1348 cm-1; HRMS (ESI) calc’d for [C19H29O3BNa]+: m/z 339.2102, found 339.2100.
1.32 was prepared using the representative synthetic route but employing PtO2 (10 mol%) in the presence of Na2CO3 (5.0 equiv) at 0 °C for the reduction of the enone. Rf 0.50 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 8.00 (dd, J = 8.3, 1.3 Hz, 2H), 7.83 (dd, J = 7.4, 1.5 Hz, 1H), 7.58 - 7.52 (m, 1H), 7.49 - 7.42 (m, 2H), 7.38 (td, J = 7.5, 1.6 Hz, 1H), 7.27 (d, J = 6.1 Hz, 1H), 7.23 (td, J = 7.4, 1.2 Hz, 1H), 3.35 - 3.23 (m, 4H), 1.31 (s, 12H); 13C NMR (125 MHz, CDCl3) # 199.8, 148.4, 137.1, 136.3, 132.8, 131.18, 129.4, 128.5, 128.1, 125.5, 83.5, 42.2, 30.8, 24.8. The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max1686, 1348, 743 cm-1; HRMS (ESI) calc’d for [C21H25O3BNa]+: m/z 359.1789, found 359.1787.
1.45 was prepared using the representative synthetic route but employing PtO2 (10 mol%) in the presence of Na2CO3 (5.0 equiv) at 0 °C for the reduction of the enone. Rf 0.59 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 8.01 (dd, J = 8.6, 5.6 Hz, 2H), 7.82 (dd, J = 7.4, 1.6 Hz, 1H), 7.40 - 7.34 (m, 1H), 7.28 - 7.19 (m, 2H), 7.11 (t, J = 8.6 Hz, 2H), 3.60 - 3.02 (m, 4H), 1.30 (s, 12H); 13C NMR (125 MHz, CDCl3) # 198.2, 165.6 (d, J = 254.2 Hz), 148.2, 136.4, 133.5 (d, J = 3.0 Hz), 131.2, 130.7 (d, J = 9.2 Hz), 129.4, 125.5, 115.5 (d, J = 21.8 Hz), 83.6, 42.1, 30.8, 24.8. The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max 2978, 1687, 1348, 1145 cm-1; HRMS (ESI) calc’d for [C21H25O3BF]+: m/z 355.1875, found 355.1880.
1.46 was prepared using the representative synthetic route but employing PtO2 (10 mol%) in the presence of Na2CO3 (5.0 equiv) at 0 °C for the reduction of the enone. Rf 0.71 (4:1 hexanes/EtOAc; buffered with 1% triethylamine; eluted twice); 1H NMR (600 MHz, CDCl3) # 7.82 (dd, J = 7.4, 1.5 Hz, 1H), 7.57 (d, J = 7.7 Hz, 1H), 7.51 (dd, J = 2.5, 1.6 Hz, 1H), 7.39 – 7.32 (m, 2H), 7.29 - 7.17 (m, 2H), 7.14 - 7.06 (m, 1H), 3.85 (s, 3H), 3.44 - 3.14 (m, 4H), 1.30 (s, 12H). 13C NMR (150 MHz, CDCl3) # 199.4, 159.7, 148.3, 138.4, 136.3, 131.1, 129.4, 125.4, 120.7, 119.3, 112.1, 83.5, 55.3, 42.2, 30.7, 24.7 (one sp2 carbon was not observed, likely due to signal overlap). The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max 2997, 1687, 1348 cm-1; HRMS (ESI) calc’d for [C22H27O4BNa]+: m/z 389.1895, found 389.1896.
BPin
Ph
O
1.32
BPin
O
1.45F
BPin
O
1.46
OMe

! ")!
1.47 was prepared using the representative synthetic route but employing PtO2 (10 mol%) in the presence of Na2CO3 (5.0 equiv) at 0 °C for the reduction of the enone. Rf 0.59 (4:1 hexanes/EtOAc; buffered with 1% triethylamine); 1H NMR (600 MHz, CDCl3) # 7.92 (d, J = 7.9 Hz, 2H), 7.85 (d, J = 7.4 Hz, 1H), 7.39 (t, J = 7.5 Hz, 1H), 7.31 - 7.19 (m, 4H), 3.35 - 3.30 (m, 2H), 3.29 - 3.24 (m, 2H), 2.43 (s, 3H), 1.33 (s, 12H). 13C NMR (125 MHz, CDCl3) # 199.4, 148.4, 143.4, 136.3, 134.6, 131.1, 129.4, 129.1, 128.2, 125.4, 83.5, 42.1, 30.9, 24.8, 21.5. The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max 2978, 1684, 1348 cm-1; HRMS (ESI) calc’d for [C22H27O3BNa]+: m/z 373.1945, found 373.1947.
1.48 was prepared using the representative synthetic route but employing PtO2 (10 mol%) in the presence of Na2CO3 (5.0 equiv) at 0 °C for the reduction of the enone. Rf 0.52 (4:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) # 8.46 (s, 1H), 8.07 (d, J = 8.6 Hz, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.88 (t, J = 9.3 Hz, 2H), 7.83 (d, J = 7.3 Hz, 1H), 7.62 - 7.57 (m, 1H), 7.56 - 7.51 (m, 1H), 7.39 (t, J = 7.5 Hz, 1H), 7.29 (d, J = 7.6 Hz, 1H), 7.24 (t, J = 7.3 Hz, 1H), 3.45 - 3.39 (m, 2H), 3.39 - 3.34 (m, 2H), 1.29 (s, 12H). 13C NMR (150 MHz, CDCl3) # 199.6, 148.5, 136.3, 135.5, 134.4, 132.6, 131.2, 129.6, 129.5, 128.31, 128.26, 127.7, 126.7, 125.5, 124.0, 83.6, 42.2, 30.7, 24.8. The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max 1680, 1348 cm-1; HRMS (ESI) calc’d for [C25H27O3BNa]+: m/z 409.1945, found 409.1945. General synthetic route for the preparation of substrates of Table 1.2, entries 2d-f and Table 1.4, entry c:
Representative experimental procedure for the synthesis of 4-(2-Bromophenyl)butanal:
A 250 mL round-bottom flask equipped with a magnetic stir bar was charged with (2-bromophenyl)butan-1-ol52 (2.3 g, 9.9 mmol, 1.0 equiv) in wet EtOAc (99 mL) followed by IBX (5.5 g, 20.0 mmol, 2.0 equiv). The reaction vessel was then fitted with a condenser,
BPin
O
1.47Me
BPin
O
1.48
Br
OH 1. PdCl2(dppf)·CH2Cl2 KOAc, B2Pin2
BPin
R
O1. IBX
2. RMgBr Br
R
OH 2. IBX
BrO
H

! "*!
purged with nitrogen, and heated to reflux temperature with vigorous stirring for 3 h at which time the mixture was allowed to cool to ambient temperature, filtered through a pad of silica and concentrated under reduced pressure. The crude product was purified by column chromatography (4:1 hexanes/EtOAc) to deliver 4-(2-bromophenyl)butanal (1.9 g, 8.4 mmol, 83%), which was found to be sensitive to oxidation upon standing. Rf 0.47 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 9.78 (s, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.27 - 7.19 (m, 2H), 7.07 (td, J = 7.6, 2.0 Hz, 1H). 2.78 (t, J = 7.6 Hz, 2H), 2.50 (td, J = 7.3, 1.6 Hz, 2H), 2.06 (ap, J = 15.0, 7.4 Hz, 2H); 13C NMR (150 MHz, CDCl3) # 202.0, 140.5, 132.9, 130.4, 127.8, 127.5, 124.4, 43.0, 35.2, 22.2; IR (film) "max 3427, 3059, 2918, 1725, 1020 cm-1; HRMS (EI) calc’d for [C10H11OBr]+: m/z 225.9993, found 225.9991. Representative experimental procedure for Grignard addition (1.49):
4-(2-Bromophenyl)butanal (670 mg, 3.0 mmol, 1.0 equiv) was diluted with dry THF (30 mL) in a 100 mL, flame-dried round bottom flask equipped with a magnetic stir bar and rubber septum. The reaction vessel was then submerged in an ice water bath and isobutylmagnesium chloride (2.0 M in Et2O, 1.9 mL, 3.8 mmol 1.3 equiv) was added dropwise via syringe. The reaction mixture was stirred at this temperature for 45 min before being quenched with saturated NH4Cl solution (2 mL), poured into water (15 mL) and extracted with EtOAc (3 x 15 mL). The combined organic phases were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (4:1 hexanes/EtOAc) to deliver 1.49 (400 mg, 1.4 mmol, 48%). Rf 0.37 (4:1 hexanes/EtOAc); 1H NMR (125 MHz, CDCl3) # 7.52 (d, J = 7.5 Hz, 1H), 7.24 - 7.20 (m, 2H), 7.08 - 7.02 (m, 1H), 3.75 - 3.69 (m, 1H), 2.81 - 2.70 (m, 2H), 1.81 - 1.71 (m, 2H), 1.72 - 1.61 (m, 1H), 1.58 - 1.47 (m, 2H), 1.46 – 1.42 (m, 1H) 1.41 -1.34 (m, 1H), 1.28 - 1.21 (m, 1H), 0.991 (t, J = 7.0 Hz, 6H); 13C NMR (125 MHz, CDCl3) # 141.6, 132.7, 130.3, 127.5, 127.4, 124.4, 69.7, 46.7, 37.6, 36.1, 26.0, 24.6, 23.5, 22.0; IR (film) "max 3347, 2953, 1470, 1022, 749 cm-1; HRMS (EI) calc’d for [C14H21OBr]+: m/z 284.0776, found 284.0779. Representative experimental procedure for borylation and oxidation of haloarenes (1.50):
A 25 mL Schlenk flask equipped with a magnetic stir bar and charged with potassium acetate (344 mg, 3.51 mmol, 5.0 equiv) was flame-dried under vacuum and cooled under a nitrogen atmosphere. Bis(pinacolato)diboron (355 mg, 1.40 mmol, 2.0 equiv) and [1,1%-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) (dichloromethane adduct, 57.2 mg, 0.70 mmol, 0.10 equiv) were added and the flask was evacuated/backfilled (3X) with nitrogen. 1-(2-bromophenyl)-6-methylheptan-4-ol (200
Br
iBu
OH
1.49
BPin
iBu
O
1.50

! #+!
mg, 0.70 mmol, 1.0 equiv) was then added in DMF (7 mL) and the resulting mixture was sparged with nitrogen for 5 minutes before being sealed and heated to 80 °C for 12 h. After this time, the mixture was cooled to room temperature, poured into water (10 mL) and extracted with Et2O (2 x 10 mL). The combined organic phases were washed with brine, dried over Na2SO4, filtered and concentrated. The crude residue was passed through a short column of silica (4:1 hexanes/EtOAc) and concentrated under reduced pressure. The resulting material was then diluted with wet EtOAc (10 mL) in a 20 mL screw cap vial equipped with a stir bar. IBX (566 mg, 2.02 mmol, 2.9 equiv) was added and the vial was purged with nitrogen, sealed with a Teflon cap and heated to 80 °C for 2 h while stirring rapidly. The heterogeneous mixture was then cooled to room temperature, filtered through a plug of silica and concentrated under reduced pressure. The crude product was purified by column chromatography (10:1 hexanes/EtOAc) to deliver 1.50 (148 mg, 0.45 mmol, 64%). Rf 0.37 (10:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 7.79 (d, J = 7.4 Hz, 1H), 7.34 (td, J = 7.5, 1.6 Hz, 1H), 7.20 - 7.15 (m, 2H), 2.89 (t, J = 7.6 Hz, 2H), 2.40 (t, J = 7.5 Hz, 2H), 2.26 (d, J = 7.0 Hz, 2H), 2.16 – 2.09 (m, 1H), 1.85 (pent, J = 7.7 Hz, 2H), 1.34 (s, 12H), 0.90 (d, J = 6.6 Hz, 6H); 13C NMR (150 MHz, CDCl3) # 210.9, 148.8, 136.2, 130.9, 129.2, 125.2, 83.4, 51.7, 43.0, 35.01, 27.1, 24.8, 24.5, 22.6. The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max 1712, 1348 cm-1; HRMS (ESI) calc’d for [C20H31O3BNa]+: m/z 353.2258, found 353.2256.
1.51 was prepared using the representative synthetic route. Rf 0.48 (4:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) # 7.78 (dd, J = 7.4, 1.5 Hz, 1H), 7.35 (td, J = 7.5, 1.5 Hz, 1H), 7.21 - 7.14 (m, 2H), 2.89 (t, J = 7.6 Hz, 2H), 2.44 (t, J = 7.5 Hz, 2H), 2.12 (s, 3H), 1.86 (dt, J = 15.1, 7.7 Hz, 2H), 1.34 (s, 12H); 13C NMR (125 MHz, CDCl3) # 209.21, 148.66, 136.20, 130.91, 129.25, 125.22, 83.44, 43.42, 34.85, 29.79, 27.14, 24.85 The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max 2977, 1712, 1348, 1145 cm-1; HRMS (ESI) calc’d for [C17H25O3BNa] : m/z 311.1789, found 311.1789.
1.52 was prepared using the representative synthetic route. Rf 0.38 (4:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) # 7.78 (d, J = 7.5 Hz, 1H), 7.34 (td, J = 7.5, 1.5 Hz, 1H), 7.21 - 7.14 (m, 2H), 2.89 (t, J = 7.6 Hz, 2H), 2.58 (hept, J = 6.9 Hz, 1H), 2.47 (t, J = 7.5 Hz, 2H), 1.85 (ap, J = 7.7 Hz, 2H), 1.34 (s, 12H), 1.08 (d, J = 7.0 Hz, 6H); 13C NMR (150 MHz, CDCl3) # 214.7, 148.8, 136.2, 130.9, 129.2, 125.2, 83.4, 40.7, 40.0, 35.0, 27.1, 24.9, 18.2. The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max 2975, 1712, 1348, 1145 cm-1; HRMS (ESI) calc’d for [C19H29O3BNa]+: m/z 339.2102, found 339.2100.
BPin
Me
O
1.51
BPin
iPr
O
1.52

! #"!
1.28 was prepared using the representative synthetic route. Rf 0.48 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 7.93 (d, J = 7.0 Hz, 2H), 7.80 (d, J = 6.8 Hz, 1H), 7.54 (t, J = 7.3 Hz, 1H), 7.44 (t, J = 7.7 Hz, 1H), 7.36 (td, J = 7.5, 1.6 Hz, 1H), 7.21 (d, J = 7.4 Hz, 2H), 3.00 (q, J = 7.4 Hz, 4H), 2.04 (dt, J = 15.0, 7.5 Hz, 2H), 1.33 (s, 12H); 13C NMR (125 MHz, CDCl3) # 200.4, 148.8, 137.0, 136.2, 132.8, 130.9, 129.3, 128.5, 128.0, 125.2, 83.4, 38.2, 35.0, 27.6, 24.8 The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max 2977, 1688, 1348 cm-1; HRMS (ESI) calc’d for [C22H27O3BNa]+: m/z 373.1945, found 373.1944. General synthetic route for the preparation of substrates of Table 1.2, entry 3:
Representative experimental procedure for the alkylation of benzyl alcohols (1.53):
In a 100 mL flame-dried round-bottom flask, NaH (60% dispersion in mineral oil, 1.07g, 26.7 mmol, 5.0 equiv) was suspended in dry DMF (24 mL). The reaction vessel, which was under a nitrogen atmosphere, was then submerged into an ice water bath and 2-bromobenzyl alcohol (1.00 g, 5.35 mmol, 1.0 equiv) was added in dry DMF (24 mL) via cannula. After 10 min at that temperature, the reaction mixture was allowed to warm to ambient temperature and stir for 1 h before being cooled back down to 0 °C. Bromoacetaldehyde dimethyl acetal (4.52 g, 3.16 mL, 26.7 mmol, 5.0 equiv.) was added dropwise via syringe and the resulting peach-colored solution was warmed to ambient temperature for 7 h at which time it was poured into ice water and extracted with Et2O (3 x 30 mL). The combined organic phases were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude mixture was evacuated for 12 h then dissolved in THF (50 mL) and aqueous HCl (1.0 M, 50 mL). The combined mixture was heated to 50 °C for 4 h. The mixture was then cooled, poured into water (50 mL) and extracted with EtOAc (3 x 50 mL). The combined organic phases were washed with saturated NaHCO3 solution (50 mL), water (50 mL) dried over MgSO4,
BPin
Ph
O
1.28
ROH
Br
1. NaH, bromoacetaldehyde dimethyl acetal, DMF2. 1.0 M HCl:THF
RO
Br
H
O
1. R'MgX2. PdCl2(dppf)·CH2Cl2 KOAc, B2Pin2
RO
BPin
R'
OHIBX
RO
BPin
R'
O
O
Br
H
O
1.53

! ##!
filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (4:1 hexanes/EtOAc) to furnish 1.53 (920 mg, 4.02 mmol, 75%). Rf 0.18 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 9.79 (s, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 1.8 Hz, 1H), 7.34 (t, J = 7.6 Hz, 1H), 7.19 (t, J = 7.6 Hz, 1H), 4.71 (s, 2H), 4.20 (s, 2H); 13C NMR (150 MHz, CDCl3) # 200.2, 136.4, 132.7, 129.5, 129.4, 127.6, 123.0, 75.82, 72.9; IR (film) "max 2870, 1738, 752 cm-1; HRMS (ESI) calc’d for [C9H9O2BrNa]+: m/z 250.9678, found 250.9678.
1.54 was prepared following the representative synthetic route and procedure for Grignard addition and borylation/oxidation sequence described above. Rf 0.50 (4:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) # 7.80 (d, J = 1.2 Hz, 1H), 7.51 - 7.42 (m, 2H), 7.30 (td, J = 7.2, 1.5 Hz, 1H), 4.84 (s, 2H), 4.06 (s, 2H), 2.35 (d, J = 6.9 Hz, 2H), 2.22 - 2.11 (m, 1H), 1.34 (s, 12H), 0.92 (d, J = 6.6 Hz, 6H); 13C NMR (150 MHz, CDCl3) # 209.1, 143.6, 135.8, 131.0, 127.8, 127.0 83.7, 75.8, 72.5, 47.8, 24.9, 24.2, 22.6. The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max
2977, 1718, 1349, 1146 cm-1; HRMS (ESI) calc’d for [C19H29O4BNa]+: m/z 355.2051, found 355.2049.
1.55 was prepared following the representative synthetic route and procedure for Grignard addition and borylation/oxidation sequence described above. Rf 0.41 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 7.81 (d, J = 5.9 Hz, 1H), 7.48 (d, J = 7.2 Hz, 1H), 7.44 (td, J = 7.4, 1.4 Hz, 1H), 7.29 (t, J = 7.3 Hz, 1H), 4.85 (s, 2H), 4.16 (s, 2H), 2.84 (hept, J = 6.9 Hz, 1H), 1.34 (m, 12H), 1.09 (d, J = 6.9 Hz, 6H); 13C NMR (150 MHz, CDCl3) # 212.5, 143.7, 135.8, 131.0, 127.9, 126.9, 83.7, 73.9, 72.5, 36.9, 24.9, 17.9. The boron-bound carbon was not detected likely due to quadropular relaxation; IR (film) "max 2976, 1712, 1349, 1146 cm-1; HRMS (ESI) calc’d for [C18H27O4BNa]+: m/z 341.1895, found 341.1894.
1.56 was prepared following the representative synthetic route and procedure for alkylation of benzyl alcohols, Grignard addition and borylation/oxidation sequence above. Rf 0.50 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 7.80 (dd, J = 8.3, 6.5 Hz, 1H), 7.28 - 7.20 (m, 1H), 6.96 (td, J = 8.4, 2.5 Hz, 1H), 4.84 (s, 2H), 4.20 (s, 2H), 2.84 (hept, J = 6.8 Hz, 1H), 1.32 (s, 12H), 1.11 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) # 212.2, 165.0 (d, J = 250.6 Hz), 147.4 (d, J = 7.5 Hz), 138.2 (d, J = 8.3 Hz), 114.3 (d, J = 21.4 Hz), 113.7 (d, J = 20.1 Hz), 83.7, 74.1, 71.8 (d, J = 1.6 Hz), 37.0, 24.8, 17.9. The boron-bound carbon was not detected likely due to quadrupolar relaxation; IR (film) "max 2977, 1716, 1348, 1145 cm-1; HRMS (EI) calc’d for [C18H27O4FB]+: m/z 336.2023, found 336.2011.
O
BPin
iBu
O
1.54
O
BPin
iPr
O
1.55
O
BPin
iPr
O
1.56
F

! #$!
Representative experimental procedure for hydroarylation of ketones in Table 1.2, entry 1 (1.57):
Inside a glove box, 1.41 (20.0 mg, 55 $mol, 1.0 equiv) was weighed into a 4 mL screw cap vial, equipped with a magnetic stir bar, and diluted with toluene (0.56 mL). DABCO (12.6 mg, 112 $mol, 2.0 equiv) and [Rh(cod)(MeCN)2]BF4 (2.1 mg, 5.6 $mol, 10 mol%) were then added and the vial was sealed with a Teflon cap and heated at 80 °C for 24 h at which time it was cooled to room temperature. The reaction mixture was then poured into water (1 mL) and extracted with EtOAc (3 x 1 mL) and the combined organic phases were dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (2:1 hexanes/EtOAc) to provide 1.57 (11.3 mg, 48.4 $mol, 87% yield). Rf 0.34 (2:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 7.71 (d, J = 8.5 Hz, 1H), 6.61 (d, J = 8.5 Hz, 1H), 3.92 (s, 3H), 2.99 (dd, J = 16.9, 5.2 Hz, 1H), 2.60 (d, J = 17.4 Hz, 1H), 2.47 – 2.39 (m, 1H), 2.34 (dt, J = 13.6, 2.7 Hz, 1H), 2.18 - 2.07 (m, 2H), 1.94 (dt, J = 13.5, 3.2 Hz, 1H), 1.76 (dd, J = 12.5, 4.9 Hz, 1H), 1.73 - 1.63 (m, 2H), 1.55 - 1.46 (m, 1H), 1.43 - 1.33 (m, 1H), 1.09 - 0.99 (m, 1H), 0.84 - 0.72 (m, 1H); 13C NMR (125 MHz, CDCl3) # 162.5, 154.1, 137.1, 129.6, 108.6, 73.1, 53.1, 44.2, 42.3, 40.8, 33.5, 28.3, 25.6, 24.1; IR (film) "max 3363, 2923, 1598, 1478, 1032, 830 cm-1; HRMS (ESI) calc’d for [C14H20NO2]+: m/z 234.1489, found 234.1487.
1.58 was prepared following the representative experimental procedure to yield the product (96%) after column chromatography (4:1 hexanes/EtOAc). Rf 0.13 (5:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 7.75 (d, J = 8.5 Hz, 1H), 6.56 (d, J = 8.5 Hz, 1H), 3.89 (s, 3H), 3.08 (dd, J = 17.6, 4.1 Hz, 1H), 2.69 (d, J = 17.7 Hz, 1H), 2.63 (d, J = 5.6 Hz, 1H), 2.22 – 2.13 (m, 1H), 2.02 (dd, J = 10.7, 6.1 Hz, 1H), 1.98 – 1.92 (m, 2H), 1.84 (t, J = 13.5 Hz, 2H), 1.53 – 1.46 (m, 1H); 13C NMR (125 MHz, CDCl3) # 162.9, 152.1, 134.0, 133.4, 107.6, 78.7, 53.4, 43.3, 42.8, 42.6, 32.6, 28.9; IR (film) "max 3384, 1593, 1304, 1246 cm-1; HRMS (ESI) calc’d for [C12H16O2N]+: m/z 206.1176, found 206.1181.
OH
NMeO
1.57
OH
NMeO
1.58

! #%!
1.59 was prepared following representative experimental procedure to yield the product (90%) after column chromatography (4:1 hexanes/EtOAc). Rf 0.19 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 7.73 (d, J = 8.4 Hz, 1H), 6.56 (d, J = 8.5 Hz, 1H), 3.90 (s, 3H), 3.11 (dd, J = 18.6, 7.2 Hz, 1H), 2.63 (d, J = 18.5 Hz, 1H), 2.49 (br s, 1H), 1.91 (d, J = 12.5 Hz, 1H), 1.82 (dd, J = 11.5, 2.1 Hz, 1H), 1.78 - 1.49 (m, 6H), 1.14 - 1.01 (m, 1H); 13C NMR (125 MHz, CDCl3) # 162.6, 154.7, 135.1, 130.9, 107.9, 70.9, 53.3, 41.0, 40.8, 38.0, 32.4, 29.4, 20.8; IR (film) "max 3385, 2931, 1580, 1478, 1310, 1023, 824 cm-1; HRMS (ESI) calc’d for [C13H18O2N]+: m/z 220.1332, found 220.1331. Representative experimental procedure for the hydroarylation of ketones in Table 1.2, entries 2 and 3 (1.60):
Inside a glove box, 1.50 (30 mg, 91 $mol, 1.0 equiv) was weighed into a 4 mL screw cap vial, equipped with a magnetic stir bar, and diluted with benzene (0.91 mL). DABCO (20 mg, 0.18 mmol, 2.0 equiv) and [Rh(cod)(MeCN)2]BF4 (3.5 mg, 9.1 $mol, 10 mol%) were then added and the vial was sealed with a Teflon cap and heated at 80 °C for 24 h at which time it was cooled to room temperature. The reaction mixture was then poured into water (1 mL) and extracted with EtOAc (3 x 1 mL) and the combined organic phases were dried over sodium sulfate, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (4:1 hexanes/EtOAc; buffered with 1% triethylamine) to deliver 1.6 (15.4 mg, 75 $mol, 83% yield). Rf 0.48 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 7.54 (d, J = 7.6 Hz, 1H), 7.21 (t, J = 7.1 Hz, 1H), 7.15 (td, J = 7.4, 1.5 Hz, 1H), 7.06 (d, J = 6.2 Hz, 1H), 2.89 - 2.66 (m, 2H), 2.17 - 2.00 (m, 1H), 1.98 - 1.70 (m, 6H), 1.68 (s, 1H), 1.08 (d, J = 6.1 Hz, 3H), 0.81 (d, J = 6.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) # 143.3, 136.4, 128.9, 127.0, 126.4, 126.1, 73.1, 50.8, 36.2, 29.9, 24.7, 24.5, 24.5, 19.9; IR (film) "max 3416, 2951, 1450, 757; HRMS (EI) calc’d for [C14H20O]+: m/z 204.1514, found 204.1512.
1.16 was prepared following the representative experimental procedure to yield the product (81%) after column chromatography (10:1 hexanes/EtOAc). Rf 0.33 (4:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) # 7.34 - 7.31 (m, 1H), 7.26 - 7.21 (m, 3H), 3.00 (ddd, J = 16.0, 8.6, 4.8 Hz, 1H), 2.86 - 2.79 (m, 1H), 2.36 - 2.27 (m, 1H), 2.12 - 2.06 (m, 1H), 1.89 (td, J = 13.0, 4.6 Hz, 1H), 1.76 - 1.68 (m, 2H), 1.52 - 1.43 (m, 1H),
OH
NMeO
1.59
HO iBu
1.60
nPr OH
1.16

! #&!
1.40 - 1.30 (m, 1H), 0.93 (t, J = 7.3 Hz, 3H); 13C NMR (150 MHz, CDCl3) # 147.7, 143.1, 128.2, 126.6, 124.9, 122.8, 83.8, 42.8, 40.0, 29.5, 17.6, 14.6; IR (film) "max 3362, 2957, 1172 cm-1; HRMS (EI) calc’d for dehydrated compound [C12H14]+: m/z 158.1096, found 158.1098.
1.61 was prepared following the representative experimental procedure to yield the product (58%) after column chromatography (10:1 hexanes/EtOAc). Rf 0.34 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 7.35 - 7.30 (m, 1H), 7.26 - 7.20 (m, 3H), 3.01 3(ddd, J = 16.1, 8.5, 4.9 Hz, 1H), 2.83 (ap, J = 7.9 Hz, 1H), 2.31 (ddd, J = 13.2, 8.3, 4.9 Hz, 1H), 2.09 (ddd, J = 13.2, 8.6, 6.4 Hz, 1H), 1.95 -1.87 (m, 1H), 1.76 (dd, J = 11.9, 4.1 Hz, 1H), 1.77 -1.67 (m, 1H), 1.46 - 1.38 (m, 1H), 1.37 - 1.27 (m, 3H), 0.90 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) # 147.6, 143.1, 128.2, 126.6, 124.9, 122.8, 83.8, 40.2, 40.0, 29.5, 26.4, 23.2, 14.1; IR (film) "max 3364, 2956, 1169 cm-1; HRMS (EI) calc’d for [C13H18O]+: m/z 190.1358, found 190.1354.
1.62 was prepared following the representative experimental procedure to yield the product (78%) after column chromatography (10:1 hexanes/EtOAc). 1H NMR spectral data are fully consistent with previously reported values.53
1.63 was prepared following the representative experimental procedure to yield the product (94%) after column chromatography (4:1 hexanes/EtOAc; buffered with 1% triethylamine). 1H NMR spectral data are fully consistent with previously reported values.53
1.64 was prepared following the representative experimental procedure to yield the product (62%) after column chromatography (4:1 hexanes/EtOAc; buffered with 1% triethylamine). Rf 0.33 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 7.57 (d, J = 7.7 Hz, 1H), 7.32 - 7.26 (m, 1H), 7.23 (td, J = 7.3, 1.2 Hz, 1H), 6.99 (d, J = 7.4 Hz, 1H), 4.79 (d, J = 14.9 Hz, 1H), 4.74 (d, J = 15.0 Hz, 1H), 3.92 (d, J = 14.6 Hz, 1H), 3.83 (d, J = 11.5 Hz, 1H), 2.16 (s, 1H), 1.86 - 1.84 (m, 2H), 1.81 - 1.77 (m, 1H), 1.57 (br s, 1H), 0.99 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 6.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) # 139.6, 133.8, 127.4, 127.1, 126.0, 123.9, 74.4, 69.9, 68.7, 47.1, 24.59, 24.56, 24.0; IR (film)
nBu OH
1.61
HO Me
1.62
HO iPr
1.63
O
HO iBu
1.64

! #'!
"max 3435, 2954, 1100, 946 cm-1; HRMS (EI) calc’d for [C13H18O2]+: m/z 206.1307, found 206.1302.
1.65 was prepared by following the representative experimental procedure to yield the product (67%) after column chromatography (4:1 hexanes/EtOAc; buffered with 1% triethylamine). Rf 0.28 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 7.55 (d, J = 7.7 Hz, 1H), 7.29 (t, J = 7.5 Hz, 1H), 7.23 (td, J = 7.4, 1.2 Hz, 1H), 6.99 (d, J = 7.5 Hz, 1H), 4.68 (aq, J = 14.8 Hz, 2H), 4.94 (d, J = 11.3 Hz, 1H), 3.85 (d, J = 11.3 Hz, 1H), 2.43 (hept, J = 7.1 Hz, 1H), 2.27 (s, 1H), 1.11 (d, J = 6.9 Hz, 3H), 0.79 (d, J = 7.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) # 138.7, 134.7, 127.2, 125.9, 124.0, 71.5, 71.3, 68.6, 35.1, 18.3, 16.3 (one sp2 carbon was not observed, likely due to signal overlap); IR (film) "max 3435, 2962, 1450, 1383, 1102, 760 cm-1; HRMS (EI) calc’d for [C12H16O2]+: m/z 192.1150, found 192.1144.
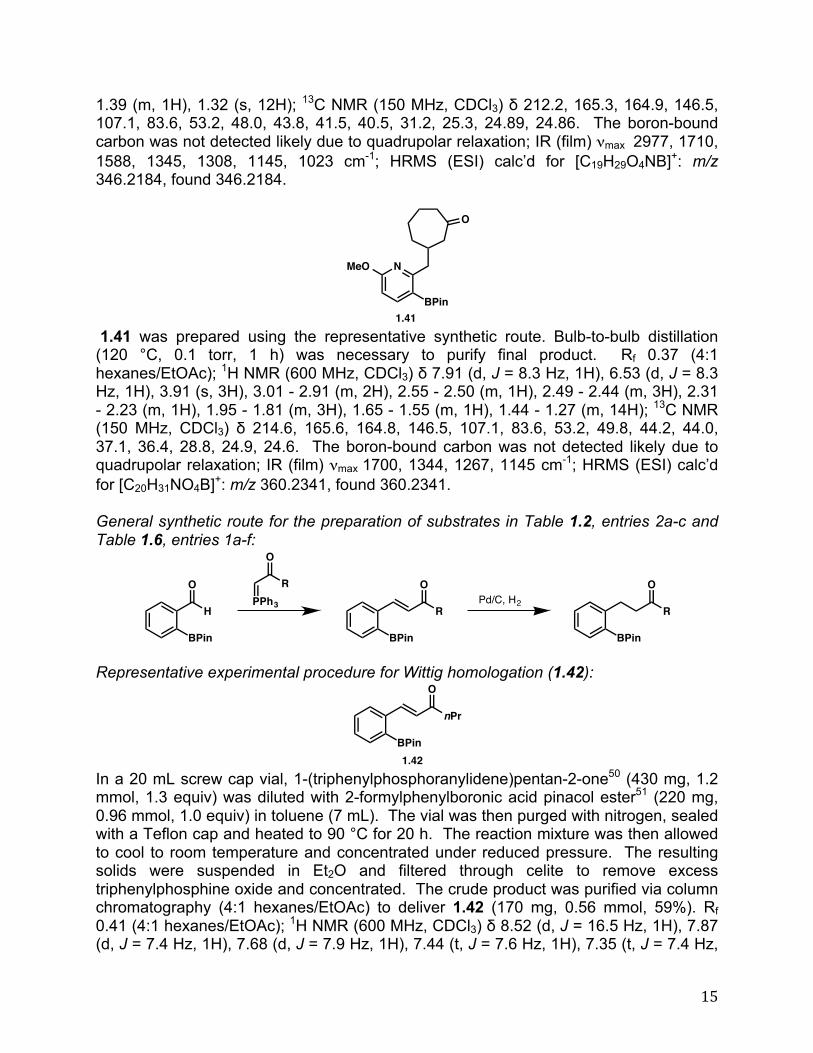
1.66 was prepared by following the representative experimental procedure to yield the product (80%) after column chromatography (4:1 hexanes/EtOAc; buffered with 1% triethylamine). Rf 0.29 (4:1 hexanes/EtOAc, 1% triethylamine); Rf 0.25 (4:1 hexanes/EtOAc, 1% triethylamine); 1H NMR (500 MHz, CDCl3) # 7.52 (dd, J = 8.7, 5.5 Hz, 1H), 6.98 (td, J = 8.6, 2.7 Hz, 1H), 6.69 (dd, J = 9.1, 2.4 Hz, 1H), 4.69 (aq, J = 15.1 Hz, 2H), 3.91 (d, J = 11.8 Hz, 1H), 3.83 (d, J = 11.8 Hz, 1H), 2.4 (hept, J = 7.0 Hz, 1H), 2.26 (br s, 1H), 1.09 (d, J = 6.9 Hz, 3H), 0.78 (d, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) # 161.6 (d, J = 246.8 Hz), 136.9 (d, J = 7.0 Hz), 134.5 (d, J = 3.1 Hz), 128.2 (d, J = 8.2 Hz), 114.5 (d, J = 21.5 Hz), 110.4 (d, J = 21.5 Hz), 71.5, 71.1 , 68.4 (d, J = 2.1 Hz), 35.2, 18.2, 16.3; IR (film) "max 3434, 2964, 1250, 1110 cm-1; HRMS (EI) calc’d for [C12H15O2F]: m/z 210.1056, found 210.1051. Representative experimental procedure for hydroarylation of ketones in Table 1.4, entries 1a-c (1.29):
Inside a glove box, 1.29 (30 mg, 86 $mol, 1.0 equiv) was weighed into a 4 mL screw cap vial, equipped with a magnetic stir bar, and diluted with benzene (0.86 mL). 1,2-Bis(diphenylphosphino)ethane (3.4 mg, 8.6 $mol, 10 mol%) and [Rh(cod)(OH)]2 (2.0 mg, 4.3 $mol, 5.0 mol%) were then added and the vial was sealed with a Teflon cap and heated at 80 °C for 24 h at which time it was cooled to room temperature. The reaction mixture was then poured into water (1 mL) and extracted with EtOAc (3 x 1 mL) and the combined organic phases were dried over sodium sulfate, filtered and
O
HO iPr
1.65
O
HO iPr
1.66
F
HO Ph
1.29

! #(!
concentrated under reduced pressure. The crude product was purified by column chromatography (10:1 hexanes/EtOAc; buffered with 1% triethylamine) to furnish 1.29 (13.8 mg, 61.5 $mol, 72% yield). 1H NMR spectral data agree are fully consistent with previously reported values.53
1.33 was prepared by following representative experimental procedure to yield the product (85%) after column chromatography (10:1 hexanes/EtOAc; buffered with 1% triethylamine). 1H NMR spectral data are fully consistent with previously reported values.54
1.67 was prepared by following the representative experimental procedure to yield the product (74%) after column chromatography (10:1 hexanes/EtOAc; buffered with 1% triethylamine). Rf 0.36 (4:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 7.42 - 7.28 (m, 4H), 7.23 (td, J = 7.5, 1.6 Hz, 1H), 7.09 (d, J = 7.6 Hz, 1H), 7.04 - 6.96 (m, 2H), 3.17 (dt, J = 16.2, 7.3 Hz, 1H), 2.94 (dt, J = 16.0, 6.4 Hz, 1H), 2.47 (dd, J = 7.4, 6.3 Hz, 2H), 2.07 (br s, 1H); 13C NMR (125 MHz, CDCl3) # 161.8 (d, J = 245.1 Hz), 147.7, 144.0, 142.1 (d, J = 3.1 Hz), 128.6, 127.4 (d, J = 8.0 Hz), 127.1, 125.0, 123.9, 114.7 (d, J = 21.2 Hz), 85.2, 44.9, 29.8; IR (film) "max 3395, 1223, 1158 cm-1; HRMS (EI) calc’d for [C15H13OF]+: m/z 228.0950, found 228.0949. Representative experimental procedure for asymmetric hydroarylation of ketones in Table 1.6 (1.68):
On the bench, 1.47 (50 mg, 0.14 mmol, 1.0 equiv), Josiphos SL-J002-1 (8.6 mg, 16 $mol, 11 mol%), and [Rh(cod)(OH)]2 (3.3 mg, 7.2 $mol, 5 mol%) were weighed into a 5 mL conical vial, equipped with a magnetic stir bar,. The vial was sealed with a Teflon septum, diluted with toluene (1.4 mL) and stirred at rt for 1 h. It was then heated to 85 °C and held at this temperature for 22 h at which time it was cooled to room temperature. The reaction mixture was then poured into water (2 mL) and extracted with EtOAc (3 x 2 mL). The combined organic phases were dried over sodium sulfate, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (4:1 hexanes/EtOAc; buffered with 1% triethylamine) to furnish 1.68 (29 mg, 13 $mol, 90% yield). Rf 0.41 (4:1 hexanes/EtOAc); 1H NMR (500 MHz,
Ph OH
1.33
HO
1.67
F
HO
1.68
Me

! #)!
CDCl3) # 7.37 - 7.27 (m, 4H), 7.23 (t, J = 7.3 Hz, 1H), 7.15 (d, J = 8.0 Hz, 2H), 7.11 (d, J = 7.5 Hz, 1H), 3.17 (dt, J = 15.1, 7.2 Hz, 1H), 3.94 (ddd, J = 15.9, 7.8, 5.1 Hz, 1H), 2.62 - 2.42 (m, 2H), 2.36 (s, 3H), 2.10 (br s, 1H).13C NMR (125 MHz, CDCl3) # 148.0, 144.1, 143.4, 136.5, 128.7, 128.4, 127.0, 125.6, 124.9, 123.9, 85.4, 44.8, 29.9, 21.0; IR (film) "max 3406, 817, 761 cm-1; HRMS (EI) calc’d for [C16H16O]+: m/z 224.1201, found 224.1206. The enantiomeric excess was determined to be 94% by chiral HPLC. The retention times for the enatiomers were 8.93 and 13.81 min.
1.33 was prepared in enantioenriched form using the representative experimental procedure over 72 h to yield the product (78%) after column chromatography (4:1 hexanes/EtOAc; buffered with 1% triethylamine). The enantiomeric excess was determined to be 94% by chiral HPLC. The retention times for the enatiomers were 10.24 and 15.50 min.
1.69 was prepared in enantioenriched form using representative experimental procedure over 48 h to yield the product (85%) after column chromatography (10:1 ! 4:1 hexanes/EtOAc; buffered with 1% triethylamine). Rf 0.30 (4:1 hexanes/EtOAc, 1% triethylamine). 1H NMR (500 MHz, CDCl3) # 7.36 - 7.27 (m, 2H), 7.27 - 7.18 (m, 2H), 7.11 (d, J = 7.5 Hz, 1H), 7.02 (t, J = 1.2 Hz, 1H), 6.91 (d, J = 7.7 Hz, 1H), 6.81 (dd, J = 8.1, 2.8 Hz, 1H), 3.80 (s, 3H), 3.17 (dt, J = 15.2, 7.2 Hz, 1H), 3.96 (ddd, J = 15.9, 7.9, 5.2 Hz, 1H), 2.63 - 2.36 (m, 2H), 2.08 (br s, 1H). 13C NMR (125 MHz, CDCl3) # 159.4, 148.1, 147.8, 144.1, 129.1, 128.5, 127.1, 125.0, 124.0, 118.2, 112.1, 111.6, 85.5, 55.2, 44.7, 29.9; IR (film) "max 3431, 1600, 1045 cm-1; HRMS (EI) calc’d for [C16H15O2]+: m/z 240.1150, found 240.1154. The enantiomeric excess was determined to be 95% by chiral HPLC. The retention times for the enatiomers were 15.24 and 25.22 min.
1.70 was prepared in enantioenriched form using representative experimental procedure over 98 h to yield the product (50%) after column chromatography (10:1 ! 4:1 hexanes/EtOAc; buffered with 1% triethylamine). Rf 0.39 (4:1 hexanes/EtOAc, 1% triethylamine). 1H NMR (400 MHz, CDCl3) # 7.91 (s, 1H), 7.84 - 7.79 (m, 3H), 7.48 - 7.43 (m, 3H), 7.35 (dt, J = 14.6, 7.5 Hz, 2H), 7.25 (m, 1H), 7.11 (d, J = 7.5 Hz, 1H), 3.24 (dt, J = 15.3, 7.4 Hz, 1H), 3.02 (ddd, J = 16.0, 8.1, 4.5 Hz, 1H), 2.66 - 2.58 (m, 1H), 2.58
Ph OH
1.33
HO
1.69
OMe
HO
1.70

! #*!
- 2.50 (m, 1H), 2.20 (s, 1H). 13C NMR (100 MHz, CDCl3) # 147.9, 144.2, 143.6, 132.9, 132.4, 128.6, 128.2, 127.8, 127.5, 127.1, 126.1, 125.8, 125.0, 124.6, 124.1, 124.0, 85.7, 44.6, 30.0; IR (film) "max 3394, 2941 cm-1; HRMS (EI) calc’d for [C19H16O]+: m/z 260.1201, found 260.1205. The enantiomeric excess was determined to be 93% by chiral HPLC. The retention times for the enatiomers were 17.62 and 24.73 min.
1.67 was prepared in enantioenriched form using general experimental procedure over 70 h to yield the product (54%) after column chromatography (10:1 hexanes/EtOAc; buffered with 1% triethylamine). The enantiomeric excess was determined to be 92% by chiral HPLC. The retention times for the enatiomers were 8.87 and 14.11 min.
1.71 was prepared in enantioenriched form using representative experimental procedure over 17 h to yield the product (90%) after column chromatography (10:1 ! 4:1 hexanes/EtOAc; buffered with 1% triethylamine). Rf 0.43 (2:1 hexanes/EtOAc,) 1H NMR spectral data were fully consistent with previously reported values.55 The enantiomeric excess was determined to be 95% by chiral HPLC. The retention times for the enatiomers were 8.92 and 10.59 min. 1.7 References and Notes (1) Larson, K. K.; Sarpong, R. J. Am. Chem. Soc. 2009, 131, 13244-13245. (2) Lee, J.-S.; Verlarde-Ortiz, R.; Guijarra, A.; Rieke, R. D. J. Org. Chem. 2000, 65, 5428-5430. (3) For recent discussions of metal-halogen exchange, see: (a) Blümke, T.; Chen, Y.-H.; Peng, Z.; Knochel, P. Nat. Chem. 2010, 2, 313-318. (b) Fleming, F. F.; Zhang, Z.; Knochel, P. Org. Lett. 2004, 6, 501-503. (c) Knochel, P.; Dohle, W.; Gommermann, N.; Kniesel, F. F.; Kopp, F.; Korn, T.; Sapountzis, I.; Vu, V. A. Angew. Chem., Int. Ed. 2003, 42, 4302-4320. (d) Chinchilla, R.; Najera, C.; Yus, M.; Chem. Rev. 2004, 104, 2667-2722. (4) Clary, J. W.; Rettenmaier, T. J.; Snelling, R.; Bryks, W.; Banwell, J.; Wipke, T.; Singaram, B. J. Org. Chem. 2011, 76, 9602-9610. (5) Tatsuo, I.; Murata, M.; Miyaura, N. J. Org. Chem. 1995, 60, 7508-7510. (6) For a discussion on C-H activation for the construction of C-B bonds, see: Mkhalid, I. A. I.; Barnard, J. H.; Marder, T. B.; Murphy, J. M.; Hartwig, J. F. Chem. Rev. 2009, 110, 890-931. (7) For stability studies on boronic esters, see: (a) Roy, C. D.; Brown, H. C. J. Organomet. Chem. 2007, 692, 784-790. (b) Roy, C. D.; Brown, H. C. Monatsh. Chem. 2007, 138, 879-887.
HO
1.67
F
Me OH
1.71

! $+!
(8) Hayashi, T. Pure Appl. Chem. 2004, 76, 465-475. (9) Hayashi, T.; Yamasaki, K.; Chem. Rev. 2003, 103, 2829-2844. (10) Nishimura, T.; Makino, H.; Nagaosa, M.; Hayashi, T. J. Am. Chem. Soc. 2010, 132, 12865-12867. (11) Hanmei, Z.; Zhang, Q.; Chen, J,; Liu, M.; Cheng, S.; Ding, J.; Wu, H.; Su, W. J. Org. Chem. 2008, 74, 943-945. (12) Zou, T.; Pi, S.-S.; Li, J.-H. Org. Lett. 2009, 11, 453-456. (13) Imlinger, N.; Mayr, M.; Want, D.; Wurst, K.; Buchmeiser, M. R. Adv. Synth. Catal. 2004, 346, 13-15. (14) Zhou, L.; Du, X.; He, R.; Ci, Z.; Bao, M. Tetrahedron Lett. 2009, 50, 406-408. (15) Arao, T.; Kondo, K.; Aoyama, T. Tetrahedron Lett. 2007, 48, 4115-4117. (16) Bouffard, J.; Itami, K. Org. Lett. 2009, 11, 4410-4413. (17) Takahashi, G.; Shirakawa, E.; Tsuchimoto, T.; Kawakami, Y. Chem. Commun. 2005, 11, 1459-1461. (18) Yamamoto, T.; Ohta, T.; Ito, Y. Org. Lett. 2005, 7, 4153-4155. (19) Yamamoto, T.; Lizuka, M.; Takenaka, H.; Ohta, T.; Ito, Y. J. Organomet. Chem. 2009, 694, 1325-1332. (20) Kuriyama, M.; Ishiyama, N.; Shimazawa, R.; Shirai, R.; Onomura, O. J. Org. Chem. 2009, 74, 9210-9213. (21) Liao, Y.-X.; Xing, C.-H.; He, P.; Hu, Q.-S. Org. Lett. 2008, 10, 2509-2512. (22) Sakai, M.; Ueda, M.; Miyaura, N. Angew. Chem., Int. Ed. 1998, 37, 3279-3281. (23) Duan, H.-F.; Xie, J.-H.; Shi, W.-J.; Zhang, Q.; Zhou, Q.-L. Org. Lett. 2006, 8, 1479-1481. (24) Tokunaga, N.; Otomaru, Y.; Okamoto, K.; Ueyama, K.; Shintani, R.; Hayashi, T. J. Am. Chem. Soc. 2004, 126, 13584-13585. (25) Weix, D. J.; Shi, Y.; Ellman, J. A. J. Am. Chem. Soc. 2005, 127, 1092-1093. (26) Dai, H.; Lu, X. Org. Lett. 2007, 9, 3077-3080. (27) Shintani, R.; Takeda, M.; Tsuji, T.; Hayashi, T.; J. Am. Chem. Soc. 2010, 132, 13168-13169. (28) Matsuda, T.; Makino, M.; Murakami, M. Bull. Chem. Soc. Jpn. 2005, 78, 1528-1533. (29) Matsuda, T.; Makino, M.; Murakami, M. Org. Lett. 2004, 6, 1257-1259. (30) Seiser, T.; Roth, O. A.; Cramer, N. Angew. Chem., Int. Ed. 2009, 48, 6320-6323. (31) Ishida, N.; Sawano, S.; Masuda, Y.; Murakami, M. J. Am. Chem. Soc. 2012, 134, 17502-17504. (32) Shigeno, M.; Yamamoto, T.; Murakami, M. Chem. Eur. J. 2009, 15, 12929-12931. (33) Toullec, P. Y.; Jagt, R. B. C.; de Vries, J. G.; Feringa, B. L.; Minnaard, A. J. Org. Lett. 2006, 8, 2715-2718. (34) Ganci, G. R.; Chisolm, J. D.; Tetrahedron Lett. 2007, 48, 8266-8269. (35) Liu, G.; Lu, X. J. Am. Chem. Soc. 2006, 128, 16504-16505. (36) Liu, G.; Lu, X. Tetrahedron. 2008, 64, 7324-7330. (37) Ueura, K.; Miyamura, S.; Satoh, T.; Miura, M. J. Organomet. Chem. 2006, 691, 2821-2826. (38) Low, D. W.; Pattison. G.; Wieczysty, M. D.; Churchill, G. H.; Lam, H. W. Org. Lett. 2012, 14, 2548-2551. (39) Lalic, G.; Corey, E. J. Tetrahedron Lett. 2008, 49, 4894-4896.

! $"!
(40) For nucleophilicity/basicity of amines, see: (a) Baidya, M.; Kobayashi, S.; Brotzel, F.; Schmidhammer, U.; Riedle, E.; Mayr, H. Angew. Chem., Int. Ed. 2007, 46, 6176-6179. (b) Ammer, J.; Baidya, M.; Kobayashi, S.; Mayr, H. J. Phys. Org. Chem. 2010, 23, 1029-1035. (41) Miyaura, N. J. Organomet. Chem. 2002, 653, 54-57. (42) Amatore, C.; Jutand, A.; Le Duc, G. Chem. Eur. J. 2011, 17, 2492-2503. (43) Carrow, B. P.; Hartwig, J. F. J. Am. Chem. Soc. 2011, 133, 2116-2119. (44) Itooka, R.; Iguchi, Y.; Miyaura, N. J. Org. Chem. 2003, 68, 6000-6004. (45) Sakai, M.; Ueda, M.; Miyaura, N. Angew. Chem., Int. Ed. 1998, 37, 3279-3281. (46) Okamoto, K.; Hayashi, T.; Rawal, V. H. Org. Lett. 2008, 10, 4387-4389. (47) Shintani, R.; Ichikawa, Y.; Takatsu, K.; Chen, F-X.; Hayashi, T. J. Org. Chem. 2009, 74, 869-873. (48) Vo, G. D.; Hartwig, J. F. J. Am. Chem. Soc. 2009, 131, 11049-11061. (49) For a discussion on the applications of Josiphos-type ligands, see: Blaser, H. U.; Brieden, W.; Pugin, B.; Spindler, F.; Studer, M.; Togni, A. Top. Catal. 2002, 19, 3-16. (50) Phosphoranes were prepared according to known methods, see: Ruijter, E.; Schultingkemper, H.; Wessjohan, L. A. J. Org. Chem. 2005, 70, 2820-2823. (51) Prepared according to known methods, see: Lautens, M.; Mancuso, S. J. Org. Chem. 2004, 69, 3478-3487. (52) Torraca, K. E.; Kuwabe, S.-I.; Buchwald, S. L. J. Am. Chem. Soc. 2000, 122, 12907-12908. (53) Hatano, M.; Ito, O.; Suzuki, S.; Ishihara, K. J. Org. Chem. 2010, 75, 5008-5016. (54) Hatano, M.; Miyamoto, T.; Ishihara, K. Org. Lett. 2007, 9, 4545-8603. (55) Bietti, M.; Lanzalunga, O.; Salamone, M. J. Org. Chem. 2005, 70, 1417-1422.

! $#!
APPENDIX ONE
Spectra Relevant to Chapter One:
Enantioselective Hydroarylation of Unacitvated Ketones With Aryl Pinacolboronic Esters
!!!!!!!!!!!!!!!!!!!!

! $$!
!
!Figure A1.1 Proton and carbon NMR spectra for compound 1.58
OH
NMeO

! $%!
Figure A1.2 Proton and carbon NMR spectra for compound 1.59
OH
NMeO

! $&!
Figure A1.3 Proton and carbon spectra for compound 1.57
OH
NMeO

! $'!
Figure A1.4 Proton and carbon NMR spectra for compound 1.16
nPr OH

! $(!
Figure A1.5 Proton and carbon NMR spectra for compound 1.61
nBu OH

! $)!
Figure A1.6 Proton and carbon NMR spectra for compound 1.60
HO iBu

! $*!
Figure A1.7 Proton and carbon NMR spectra for compound 1.64
O
HO iBu

! %+!
Figure A1.8 Proton and carbon NMR spectra for compound 1.63
O
HO iPr
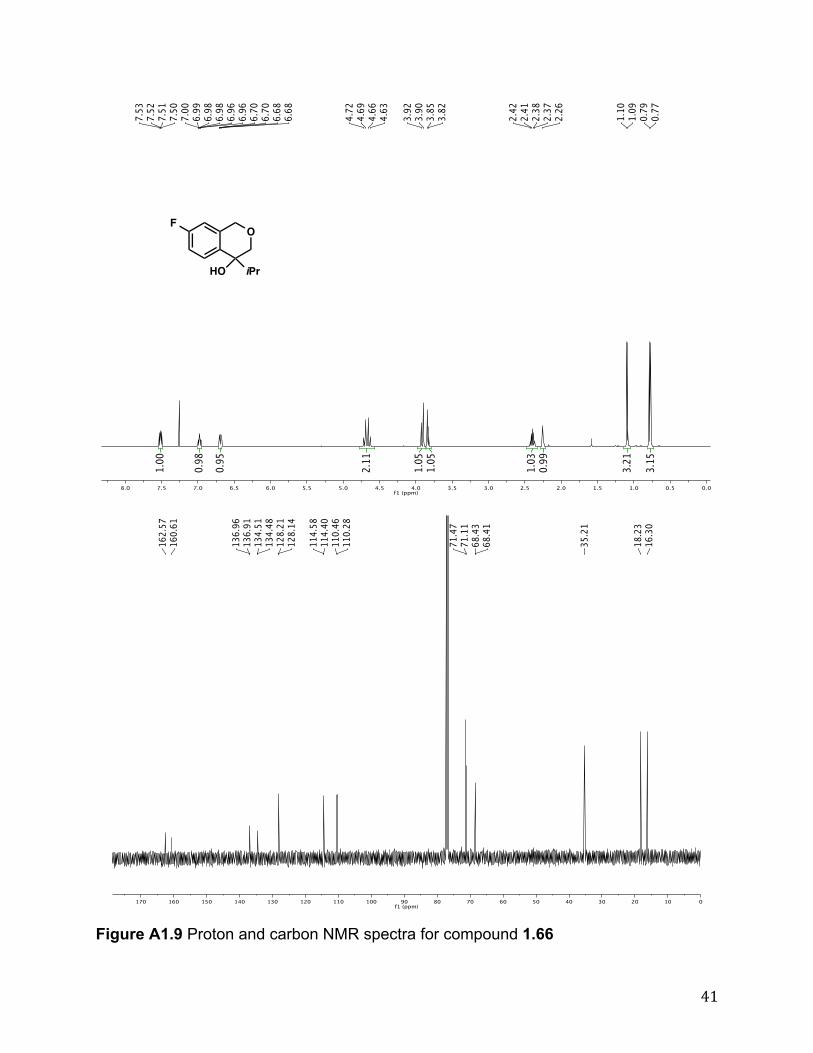
! %"!
Figure A1.9 Proton and carbon NMR spectra for compound 1.66
O
HO iPr
F
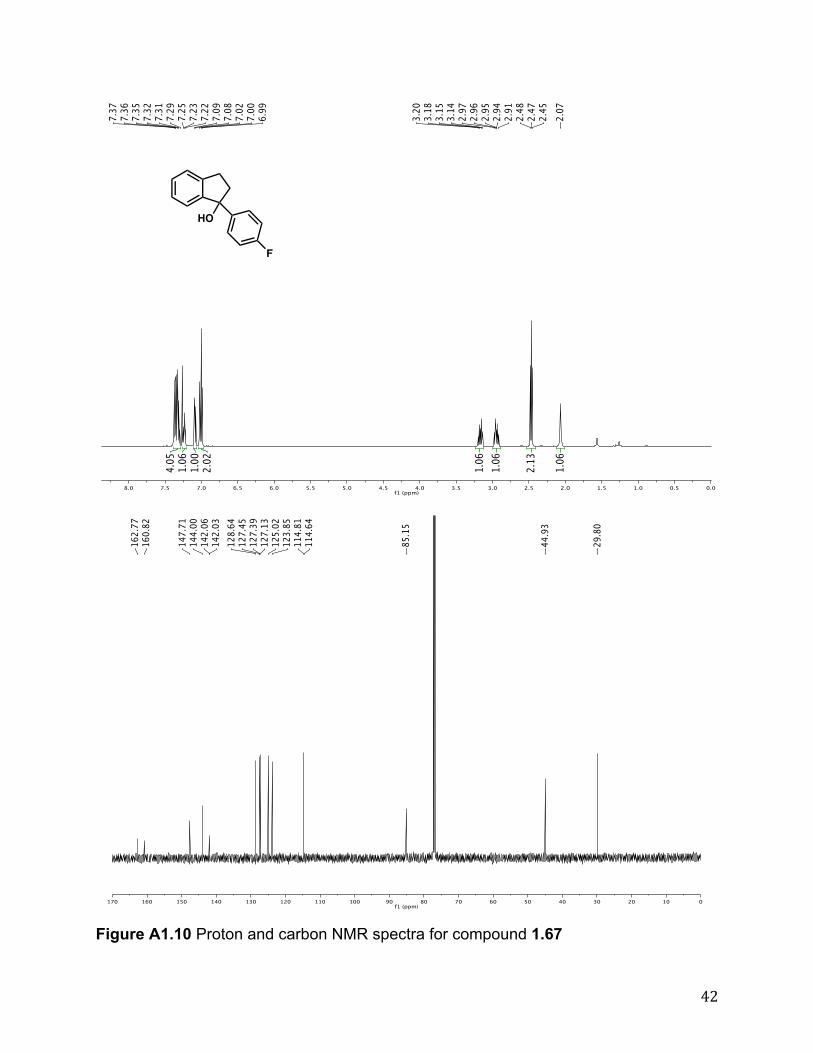
! %#!
Figure A1.10 Proton and carbon NMR spectra for compound 1.67
HO
F
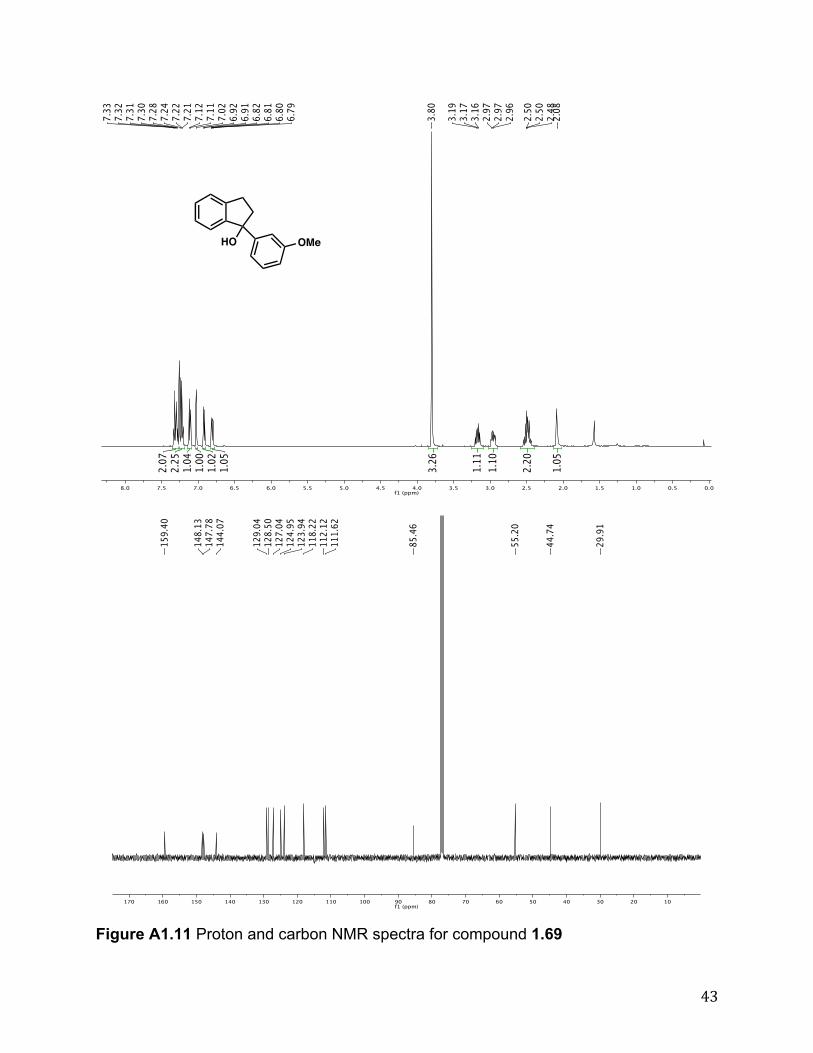
! %$!
Figure A1.11 Proton and carbon NMR spectra for compound 1.69
HO OMe

! %%!
Figure A1.12 Proton and carbon NMR spectra for compound 1.68
HO
Me

! %&!
Figure A1.13 Proton and carbon NMR spectra for compound 1.70
HO

! %'!
II. A Hydrindanone-Based approach to Natural Products Chapter 2 !Diterpenoid Alkaloids 2.1 Introduction ! The ability to access varied natural product frameworks from a common intermediate allows for exceptional synthetic efficiency and is a goal for which chemist strive. With this mantra in mind, we have begun to develop a research program centered on a hydrindanone precursor (2.1, Figure 2.1), which may provide access to a range of natural products and associated derivatives.
!Figure 2.1 A hydrindanone-based approach to natural products. ! There are multiple advantages to using precursors such as 2.1 in total synthesis. First, 2.1 contains a variety of functional groups, which, in large part, can be manipulated independently of one another. Second, the hydrindanone [6.5] system contains four, contiguous stereocenters, including a quaternary carbon center, which are set in a highly diastereoselective manner via a Diels-Alder cycloaddition from readily available starting materials (Scheme 2.1). Third, the keto-ester grouping of dienophile 2.7 bolsters the prospect for two-point binding of chiral, non-racemic metal complexes, which may render these cycladditions enantioselective. Finally, the dual electronics of the dienophile may allow for the reversal of the diastereoselectivity-controlling functional group.
Me
O
AcO CO2MeMe
HOHO
O Me
OH
O
O
Me
AcO
AcO
Xyloccensin O (2.2)(Phragmalin-Type Limonoid)
Alstovenine (2.4)(Yohimbinoid Alkaloid)
NHO
H
H
HNOMe
HO OMe
Gomandonine (2.3)(Diterpenoid Alkaloid)
N
Me OH
Me
H
H
OH
O
OHH
ROCO2Me
H
O
OTBS
2.1

! %(!
Scheme 2.1 Diels-Alder cycloaddition provides key hydrindanone.
This chapter describes the implementation of this strategy to the synthesis of the core framework of the denudatine-type diterpenoid alkaloids, as well as efforts toward its elaboration to the skeleton of the lappaconotine-type diterpenoid alkaloids. A brief discussion of the isolation and classification of this subset of diterpenoid alkaloids as well as other synthetic approaches to these molecules is also included. An extension of this strategy to the synthesis of the phragmalin-type limonoids is detailed in Chapter 3. 2.2 Isolation and Classification of The Diterpenoid Alkaloids The diterpenoid alkaloids boast an impressive diversity of compounds, both in terms of structure and biological activity. Diterpenoid alkaloids are defined1 as “bases that are derived from tetrayclic or pentacyclic diterepenes in which the nitrogen atom of methylamine, ethylamine, or "-aminoethanol is linked to the C-17 and C-19 in the C19-diterpenoid alkaloids, and to C-20 and C-19 in the C20-diterpenoid alkaloids, to form a substituted piperidine ring.” The history of these compounds (isolated from the Aconitum, Delphinium and Consolida genera of plants) begins with their use in traditional Chinese and Japanese folk medicine.2 The biological activity of these molecules is largely attributed to their interactions with Na+ and K+ voltage-gated ion channels.3 The most famous of these compounds is aconitine, a potent lipid soluble poison, which is known to interact with Na+ ion channels and block their inactivation.4 While the therapeutic window for these molecules is narrow, positive effects attributed to these compounds include anticancer5 analgesic6 and antibacterial7 properties. Their potency, in concert with the ease with which these molecules cross the blood brain barrier,8 offers an enticing opportunity to arrive at potentially highly effective therapeutics which may be realized through structure/activity studies. In this regard, access to novel derivatives of these molecules through synthesis is crucial. The diterpenoid alkaloids have been divided into many classes and types based on the number of carbons within the skeleton and the relative connectivity within the core as described by Wang9 (Figure 2.2). More specifically, the diterpenoids are separated into three main classes depending of the presence or absence of a C19 and C20 carbon. Molecules within each type vary widely in oxidation level.
ROCO2Me
H
O
OTBSOTBS
OR
MeO2C
O1.PhMe, 100 °C
2.Pd/C, H2, EtOAc
R = Me (2.5)R = Bn (2.6)
2.7 R = Me (78% yield, 2.8)R = Bn (85% yield, 2.9)
+
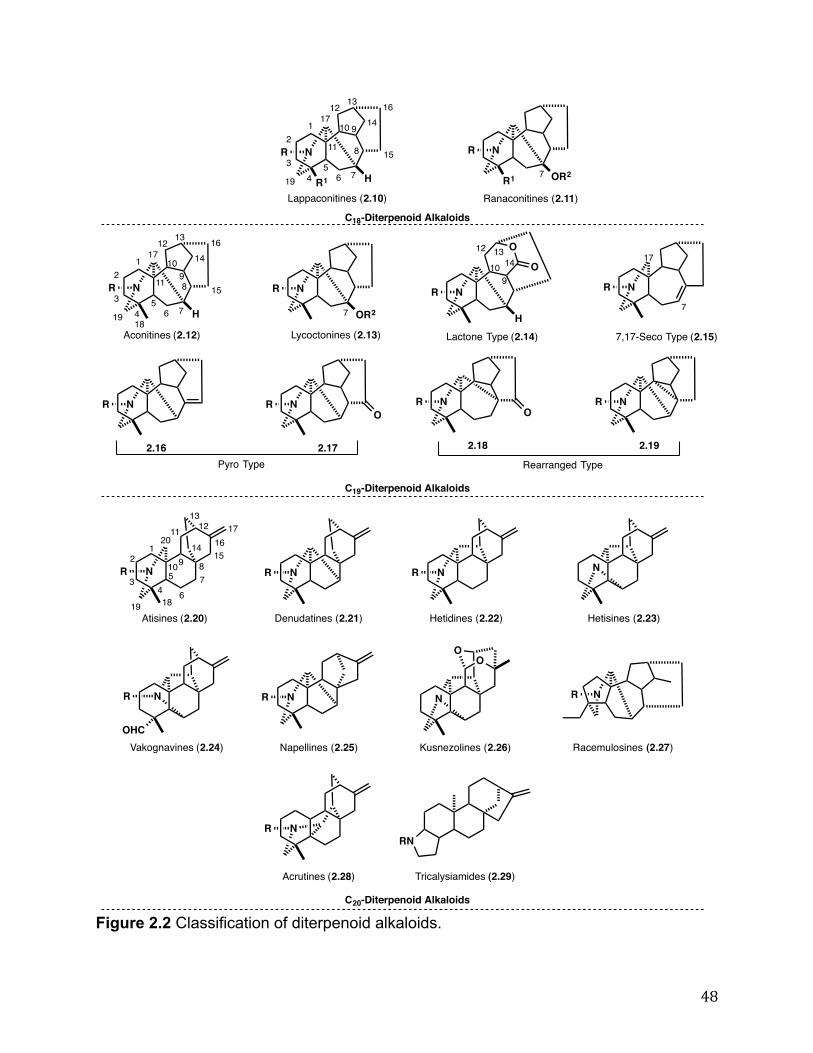
! %)!
Figure 2.2 Classification of diterpenoid alkaloids.
R1
NR
H
Lappaconitines (2.10)
12
34
56 7
8
910
11
12 13
14
15
1617
18
19 R1
NR
OR2
Ranaconitines (2.11)
C18-Diterpenoid Alkaloids
NR
H
Aconitines (2.12)
12
34
56 7
89
10
11
12 13
14
15
1617
19
NR
OR2
Lycoctonines (2.13)
NR
HLactone Type (2.14)
910
12 1314O
O
NR
7,17-Seco Type (2.15)
17
77
7
NR
Pyro Type
NRO
2.16 2.17
NRO
2.18
NR
2.19
Rearranged Type
NR
Atisines (2.20)
NR
Denudatines (2.21)
C19-Diterpenoid Alkaloids
NR
Hetidines (2.22)
N
Hetisines (2.23)
OHC
N
Vakognavines (2.24)
R NR
Napellines (2.25)
O
N
Kusnezolines (2.26)
O
NR
Racemulosines (2.27)
NR
Acrutines (2.28)
RN
Tricalysiamides (2.29)
C20-Diterpenoid Alkaloids
18
12
34
5
678910
11 1213
141516
17
19
20

! %*!
The main focus of the work presented here centers on the construction of the skeleton of the denudatine-type diterpenoids. The denudatines have been mostly isolated from the genera Aconitum although isolation from the genera Consolida has been noted.10 These natural products have yet to succumb to total synthesis, although the denudatine-type alkaloids have been elegantly employed as intermediates en route to other diterpenoid alkaloids (vide infra). 2.3 Syntheses and Approaches To date, there have been no reported syntheses of the denudatine-type diterpenoid alkaloids. However, the closely related atisine-type alkaloids have been synthesized. Specifically, the natural product atisine itself, has been the focus of multiple synthetic studies.11-17 The first studies, by Pelletier,11 centered on the aminal fragment installation from a known degradation product, in order to prove the assigned structure (Scheme 2.2). Secondary base 2.30 was exposed to ethylene chlorohydrin in the presence of base to furnish 2.31, which forms isoatisine upon oxidation with osmium tetroxide. Edwards and Wiesner have already demonstrated that 2.32 can be isomerized to atisine.18,19 This relatively short sequence has proved to be the crux of every synthesis of atisine, as all synthetic efforts toward atisine have intercepted the Pelletier intermediate. The first total synthesis of atisine was reported by Nagata and coworkers in 1963.12
Scheme 2.2 Pelletier’s partial synthesis of isoatisine.
Despite the fact that some members of the diterpenoid family were isolated almost a century ago, the number of synthesis of these natural products has been quite low. This can largely be attributed to the highly complex nature of these molecules. In addition to structural elucidation and degradation studies, Karel Wiesner made enormous contributions to the total synthesis of the diterpenoid alkaloids. One key insight that contributed to his success centered on the conversion of the atisine skeleton to the aconitine skeleton via a 1,2-alkyl shift (Figure 2.3).
Figure 2.3 Wiesner’s proposed rearrangement. Wiesner went on to apply this bioinspired transformation in the total synthesis of talatisamine20 (2.46, Scheme 2.3). First, tricycle 2.37 was accessed from vinyl nitrile
NH
MeH OH Cl
OH
Na2CO3, MeOH N
MeH OH
OsO4
Et2OHO
N
MeH OH
O
Pelletier Intermediate (2.30) 2.31 Isoatisine (2.32)
NR
LG
Atisine Core (2.33)
NR
NR
Aconitine Core (2.12)2.34

! &+!
2.35 and diene 2.36. The trimesylate was exposed to sodium hydride to furnish piperidine 2.38. After a few synthetic manipulations, Wiesner accessed enone 2.39, which could then undergo a [2+2] cycloaddition with ketene, to form 2.40. Elaboration of this system to cyclobutanol 2.41 set the stage for an acid-catalyzed retro-aldol/aldol sequence to furnish the atisine-type core. Further transformation to tosylate 2.43 set the stage for the key rearrangement from the [2.2.2] bicycle to the [3.2.1] bicycle present in talatisamine. Heating 2.43 in a 1:1 mixture of base and DMSO, [3.2.1] bicycle 2.44 was formed (only the productive olefin regioisomer shown). Following protective group removal and oxidation level adjustment, talatisamine (2.46) was accessed from 2.45 by oxidation to an iminium ion, aza-prins reaction and attack of water on the newly formed tertiary carbocation. Scheme 2.3 Wiesner’s synthesis of talatisamine.
Having established the rearrangement of the atisine core as a feasible route to talatisamine, an important extension of this chemistry came in the synthesis of 13-
CN
OAc
OAc
MeO
+
OMeMeO MsHN
HMsO
MsO
OMeMeO
HMsO
N Ms
NMs
MsO
OMe
OMeNAc
MeO
OMe
ONAc
MeO
OMe
O
NAc
MeO
OMe
O
O
OHNAc
OMe
O
OH
MeONAc
OMe
OMe
MeOO
O
OTs
NAc
OMeMeO
O
O
OMeN
OMeMeO
OMeN
OMe
MeO
OH
OH
Me
HOHOMe
MeTalatisamine (2.46)
NaHTHF
Ketene h!
THF
HClTHF
DMSO:Tetramethyl-
guanidine180 °C
Hg(OAc)2
H2O
2.45 2.44
2.432.422.41
2.40 2.392.38B
2.38A2.372.362.35

! &"!
desoxydelphonine and chasmanine.21 More specifically, Wiesner demonstrated that a similar rearrangement of the atisine core could be extended to the denudatine skeleton. A brief summary of this rearrangement in the context of the total synthesis of 13-desoxydelphonine (2.50) is shown in Scheme 2.4. Elaboration of methoxyindanone 2.47 to bromo-ketal 2.48 enabled smooth conversion to the rearranged product 2.49. Importantly, with the C17-C7 bond already intact (as our approach mirrors), the rearrangement and attendant elimination occurs with selectivity to yield 2.49. 2.49 was then taken on to 13-desoxydelphonine (2.50). Scheme 2.4 Wiesner rearrangement toward 13-desoxydelphonine.
In addition to the syntheses noted above, inspired approaches to and syntheses of these natural products by a variety of groups have been reported.23 Worth noting is the elegant syntheses recently disclosed by Gin of nominine24 and neofinaconitine.25 These reports are representative of a resurgence of interest in these complex natural products. We now disclose our approach toward these challenging molecules.
2.4 Construction of the Tricyclic Core of the Denudatine-Type Diterpenoid Alkaloids Our strategy to achieve a unified synthesis of the diterpenoid alkaloids is delineated in Figure 2.4. The natural products aconosine (2.53), gomandonine (2.3) and cossonidine (2.57) were chosen as initial targets to highlight the generality of our approach as these compounds differ significantly in their core frameworks. However, we recognized that they could all arise from a common hydrindanone precursor (2.8). It is important to note that this approach is not limited to these particular natural products, as they are representative of a much larger group of natural products bearing similar frameworks. As noted above, the various functional handles of hydrindanone 2.8 allows for further functionalization and access to more decorated and oxygenated cores.
MeO
ONMe OMe
O
O
Br
OMe
OMeO
MeO
N
OMe
O
O
OMeMe
MeO
MeO
DBNDMSO:o-xylene
180 °C
NMe OH
OHOMe
13-desoxydelphonine (2.50)
MeO
MeO
OMe
2.492.482.47
7
17 17
7

! &#!
Figure 2.4 Synthetic strategy toward diterpenoid alkaloids. As one example of our synthetic approach to these natural products, hydrindanone 2.8 has been elaborated into [6.7.6] tricycle via benzyne insertion chemistry.26 Sequential C–N bond forming reactions would furnish caged, tertiary amine 2.56, which upon Diels-Alder cycloaddition would deliver molecules such as cossonidine (2.57) as is being pursued by Jessica Kisunzu in our group.
The work described herein is centered on accessing molecules such as gomandonine and aconosine from 2.8. We anticipated this goal could be accomplished from 2.8 via !-arylation and installation of a nitrogen atom in place of the siloxy group to arrive at 2.51. A key C–N bond-forming event would deliver versatile tricycle 2.52. Depending upon the substitution pattern on the arene unit of 2.52, two molecular complexity-building pathways could be imagined. The first possibility is a &-arene photocycloaddition27 to produce a [3.2.1] bicycle and ultimately lead to molecules such as aconosine (2.53). Alternatively, a Diels-Alder reaction could furnish the [2.2.2] bicycle present in intermediate 2.54. A strategic choice of reaction pathways could then allow access to either set of natural products. A Wagner-Meerwein shift of the [2.2.2] bicycle as originally employed by Wiesner21 would again allow access to a [3.2.1] bicycle and natural products such as aconosine (2.53). In addition, maintenance of the [2.2.2] bicycle and manipulation of the oxygenation pattern on the periphery of the [2.2.2] bicycle would lead to natural products such as gomandonine (2.3).
Central to the success of this strategy are two key intermediates. The first is tricycle 2.52 which would serve as a branching point between the two natural product
benzyneinsertion
MeO
HPGHNMe
O
OR
CO2RN
OH
Me
Cossonidine (2.57)
MeO
HPGHN
O
R'
R''
!-arylation
C–N bondformation
π-arenephotocycloaddition
Diels–Aldercycloaddition
Aconosine (2.53)
N
H O
Me
H Me
H
OH
H
HO
H
OMeN
R' O
R
H Me
R''
N
HOMe
OHH H
H
H
MeOCO2Me
H
O
OTBS
N
R' O
R
H
OMe
OMe
OR
Gomandonine (2.3)
N
Me OH
Me
H
H
OH
O
OHH
O
Me
Wagner-Meerweinshift
C–N bondformation
Diels–Aldercycloaddition
2.51
2.8
2.55 2.56
2.54
2.52

! &$!
classes. Importantly, the installation of the aryl component must be versatile so that either pathway could be accessed. The second key intermediate is [2.2.2] bicycle 2.54. This hexacycle serves as a second branching point in our strategy, and will be key if attempts at a photocycloaddition prove to be challenging. This section details our synthetic approach to tricycle 2.52.
While great advances have been made in the area of ketone !-arylation chemistry, it still remains a challenging and unpredictable transformation, especially in the context of creating tertiary centers in sterically congested environments. Our initial strategy to install the requisite aryl group in intermediates such as 2.51 through the use of palladium catalysis failed to deliver any appreciable amounts of the !-arylated material. While these attempts were not exhaustive, we ultimately elected to employ the method of Pinhey28 to achieve arylation. In this regard, formation of a "-keto-benzylester from 2.8 (Scheme 2.5) followed by treatment with aryl-lead reagent 2.58 leads to the desired arylation. Importantly, the organo-lead compounds required for arylation can be synthesized via a transmetallation of an aryl boronic acid or stannane. Thus, the wide-ranging availability of these organometallics translate to a wide range of lead-arylating compounds. A debenzylation/decarboxylation sequence then generates arylated hydrindanone 2.59. Given the cupped nature of the [6.5] system, protonation occurs from the convex "-face to deliver the undesired diastereomer at the aryl bearing carbon (see 2.59) and efforts to epimerize this stereocenter were unsuccessful. Ablation of the stereocenter was effected by a reduction of the ketone group followed by a triflation and elimination sequence. Installation of the nitrogen atom via Mitsunobu reaction generates azide 2.62. With this compound in hand, many attempts were made to form tricycle 2.63. Methods such as nitrene insertions and generation of the tin-bound radical29 failed to furnish the desired product. Scheme 2.5 Initial synthetic efforts toward key tricycle 2.63.
1. KHMDS, CNCO2Bn THF, –78 °C
65% (3 steps)1% HCl/MeOH
94% (2 steps)
3. Raney Ni, H2, THF
2. pyridine, CDCl3, 60 °C
MeOCO2Me
H
O
OTBS
OMe
THF
MeOCO2Me
HOH
OMeMeOCO2Me
HN3
OMe
NH
OH Me
CO2Me
OMe
(AcO)3Pb OMe
MeOCO2Me
H
O
OTBS
1. NaBH4 MeOH:CH2Cl22. Tf2O, pyridine CH2Cl23. DBU CH2Cl2
MeOCO2Me
HOTBS
OMe
42% (3 steps)
PPh3, DIAD, DPPA
2.8 2.59 2.60
2.63 2.62 2.61
2.58

! &%!
Many derivatives of 2.62 were prepared in an attempt to form the C–N bond found within the tricycle, a brief summary of which is presented in Figure 2.5. As mentioned above, azide 2.62 could not be coaxed into a C–N formation. We further explored conjugate addition chemistry using 2.64 with the expectation that the ester functionality could be replaced at a later stage with an aryl group. Bridgehead imine forming reactions are known to proceed and can be trapped with an alcohol or hydride source.30,31 However, with 2.65, we were unable to carry out this chemistry. Finally, amino-alcohol 2.66 could not be employed in activation and displacement chemistry with agents such as thionyl chloride.32
Figure 2.5 Representative subset of hydrindanone derivatives for C–N bond formation. We hypothesized that perhaps the conformations of hydrindanone 2.8 did not favor C–N bond formation. A preliminary investigation into alleviating conformational restrictions focused on oxidative cleavage of the hydrindanone to furnish a highly substituted cyclohexanone (Scheme 2.6). To this end, azide 2.67 was accessed by deprotection of 2.8 and Mitsunobu reaction. !-Hydroxylation33 of the ketone group provides 2.68, which is a substrate that undergoes oxidative cleavage mediated by lead tetraacetate in methanol to yield an aldehyde. Reduction of the newly formed aldehyde with sodium borohydride, delivers cyclohexane 2.69. The primary alcohol was protected as the MOM ether, and the C–N bond could be formed by an aza-Wittig reaction. While this route did represent the first formation of the C–N bond, we were cognizant of the associated low yields, as well as the potential hardship of reforming the cleaved C–C bond at a later stage.
MeOCO2Me
HN3
OMe MeOCO2Me
H
O
N3
OMe MeOCO2Me
H
OH
NH2
OMeMeOCO2Me
HNHR
CO2Me
2.62 2.64 2.65 2.66
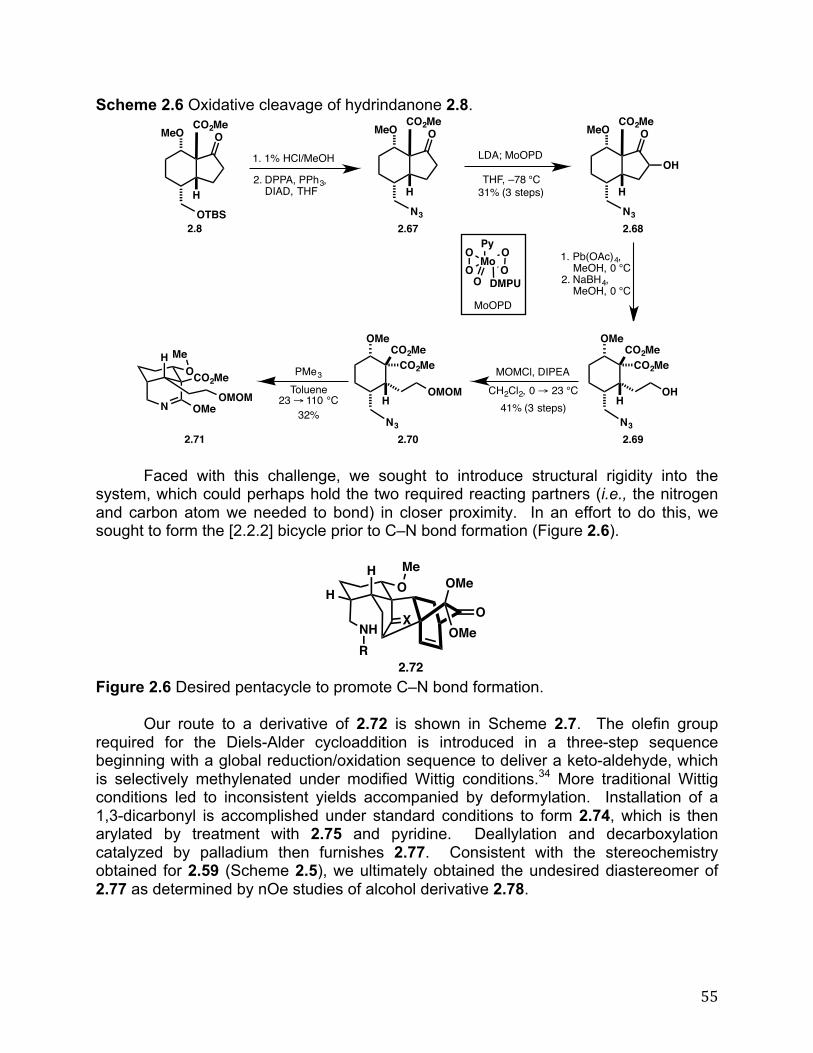
! &&!
Scheme 2.6 Oxidative cleavage of hydrindanone 2.8.
Faced with this challenge, we sought to introduce structural rigidity into the system, which could perhaps hold the two required reacting partners (i.e., the nitrogen and carbon atom we needed to bond) in closer proximity. In an effort to do this, we sought to form the [2.2.2] bicycle prior to C–N bond formation (Figure 2.6).
Figure 2.6 Desired pentacycle to promote C–N bond formation. Our route to a derivative of 2.72 is shown in Scheme 2.7. The olefin group required for the Diels-Alder cycloaddition is introduced in a three-step sequence beginning with a global reduction/oxidation sequence to deliver a keto-aldehyde, which is selectively methylenated under modified Wittig conditions.34 More traditional Wittig conditions led to inconsistent yields accompanied by deformylation. Installation of a 1,3-dicarbonyl is accomplished under standard conditions to form 2.74, which is then arylated by treatment with 2.75 and pyridine. Deallylation and decarboxylation catalyzed by palladium then furnishes 2.77. Consistent with the stereochemistry obtained for 2.59 (Scheme 2.5), we ultimately obtained the undesired diastereomer of 2.77 as determined by nOe studies of alcohol derivative 2.78.
1. 1% HCl/MeOH LDA; MoOPD
OO
MoOO
Py
O DMPU
31% (3 steps)
1. Pb(OAc)4, MeOH, 0 °C2. NaBH4, MeOH, 0 °C
MOMCl, DIPEA
41% (3 steps)
OMeCO2Me
CO2Me
HOH
N3
PMe3
Toluene23 ! 110 °C
32%
MeOCO2Me
H
O
OTBS
MeOCO2Me
H
O
N3
2. DPPA, PPh3, DIAD, THF
MeOCO2Me
H
O
N3
OH
CH2Cl2, 0 ! 23 °C
OMeCO2Me
CO2Me
HOMOM
N3
N
OH Me
CO2Me
OMOMOMe
THF, –78 °C
MoOPD
2.8 2.67 2.68
2.692.702.71
NH
H O
R
H
OMe
OMeMe
OX
2.72

! &'!
Scheme 2.7 Initial attempt to introduce [2.2.2] bicycle.
As a way to work around the challenging stereochemical outcome which results from the need to remove the ester functionality of 2.76, we moved to perform the ring closure prior to removal of the ester so that the aryl group is maintained on the "-face as in 2.79 (Scheme 2.8). To this end, cleavage of the methoxymethyl ether and Kita oxidation35,36 unveiled ortho-quinone dimethyl ketal 2.80. Exposure of this compound to elevated temperatures failed to yield the desired [2.2.2] bicycle by a [4+2] cycloaddition. Scheme 2.8 Initially attempted Diels-Alder cycloaddition.
It was hypothesized that the presence of the sp2-hybridized carbon within the five-membered ring of 2.80 was deleterious to productive overlap of the two reactive partners for the Diels-Alder cycloaddition. With this hypothesis in mind, we performed a global reduction of both the ester and ketone groups to furnish 2.83 (following deprotection of the phenol, Scheme 2.9). Oxidation of the arene to the ortho-quionone dimethyl ketal delivered 2.84, the key precursor for cycloaddition. Upon heating 2.84 in p-xylene to 150 °C, a Diel-Alder reaction proceeds in a modest 23% yield to furnish bicycle 2.85. Unfortunately, the diastereoselectivity of cycloaddition is undesired (as
1. LAH, THF 0 ! 23 °C2. DMP, NaHCO3, CH2Cl2, 0 °C
3. TMSCHN2, RhCl(PPh3)3, PPh3, i-PrOH, THF
55% (3 steps)
KHMDS, CNCO2Allyl
45%THF, –78 °C
MeO
HOTBS
O
CO2Allyl
MeOOMOM
pyridine, CDCl3, 60 °C65%
Pd(PPh3)4pyrrolidine, MeCN
78%
MeO
H
OTBS
O
MeO OMOM
MeOCO2Me
H
O
OTBS
MeO
H
O
OTBS
MeO
H
O
OTBS
CO2Allyl
(AcO)3PbOMe
OMOM
MeO
H
OHH
HH
OMeOMOM
OTBS
Key nOe signals
2.8 2.73 2.74
2.762.772.78
2.75
MeO
HOTBS
O
CO2Bn
MeOOMOM
MeO
HOH
O
CO2Bn
OMeOMeO
1. 2N HCl, i-PrOH
2. PIDA, MeOH, 0 °C76% (2 steps)
p-xylene
130-160 °C
OH
OMe
OMeMe
OOHO
BnO2C
2.79 2.80 2.81

! &(!
confirmed by X-ray crystallography of benzoate derivative 2.86), but efforts to influence this stereoselectivity (e.g., varying solvents) were futile. Scheme 2.9 First Diels-Alder cycloaddition to form a [2.2.2] bicycle.
The boat conformation of the Diels-Alder adduct (2.86) indicated that the six-membered ring would need to be constrained into a chair-conformation if the diastereoselectivity were to be significantly influenced. It seemed clear that we would need to revisit the formation of the piperidine ring from a [6.5] adduct as this would constrain the six-membered ring in the chair conformer (see 2.52, Figure 2.4). The insurmountable challenges associated with our original route (see Scheme 2.5 and Figure 2.5) required us to rethink our strategy.
A key result in our investigations into the phragmalin-type limonoids (see Chapter 3) was the intramolecular alkylation reaction of benzenesulfonate 2.87 (Scheme 2.10). In this transformation, it seemed clear that the alkylation must be proceeding through an
MeO
HOTBS
OH
OMeOMeO
OH
p-Xylene, 150 °CTBSO
OMe
HOH
HO OMeMeOO 23%
MeO
HOTBS
O
CO2Bn
MeOOAllyl
1. LAH, THF, 0 ! 23 °C
2. Pd(PPh3)4, K2CO3, MeOH
MeO
HOTBS
OHMeO
OH
OH43% (2 steps)
PIDAMeOH, 0 °C
95%
TBSO
OMe
HOH
O OMeMeOO
O
Br
2.82 2.83
2.85 2.84
2.86

! &)!
enolate with a conformation which resembles 2.88, in which the two reactive components are in a close enough proximity to undergo a C–C bond forming event. It occurred to us that if, by analogy, a nitrogen-centered anion could be generated (i.e., 2.91) a productive C–N bond formation could be achieved.
Scheme 2.10 Conformation insights into C–N bond formation.
Moving forward, we required an intermediate containing the particular stereotetrad within the northern portion of the [6.5] system of 2.90. A more formal depiction of our approach to this arrangement is shown in Figure 2.7. With the stereochemical insights we derived from manipulation of the hydrindanone described above, we were cognizant of the fact that incoming nucleophiles and electrophiles approach from the convex face of the molecule, and thus we could arrive at 2.90 by a conjugate addition of an aryl nucleophile into unsaturated system 2.93 to furnish the aryl group on the "-face. The C–N bond stereochemistry would also be set by protonation from the convex face.
BnOCO2Me
H
O
OBs
OH Bn
CO2Me
BsO O
K
OH Bn
CO2Me
O
OH Me
CO2Me
LG RN
K
KHMDS, TBAI
THF:Et3N, –78 ! 23 °C74%
Ar
MeOCO2Me
H
NHR
LG
ArO
H Me
CO2Me
NR Ar
Via2.87 2.89
2.90 2.91 2.92
2.88

! &*!
Figure 2.7 Approach toward piperidine forming precursor 2.90.
With 2.93 as our initial target, we investigated many ways to introduce unsaturation into the five-membered ring, which was bound to a nitrogen-centered electron-withdrawing group. A subset of our initial approaches to this structural motif is shown in Scheme 2.11. Condensation of hydroxylamine onto the ketone group of 2.8 followed by silylation and radical halogenation37 furnished 2.94. Traditional tactics for functionalization of these types of systems involve generation of a vinyl nitroso intermediate, which is then trapped by an organometallic nucleophile.38 All attempts to perform the transformation of 2.94 to 2.96 were unsuccessful with various fluoride sources and organometallics. Evidence for the formation of the vinyl nitroso was gathered by treatment of 2.94 with 1%HCl/MeOH to generate the !-methoxylated compound (2.97). It seemed the nitroso intermediate was not electrophilic enough to undergo Michael addition with our desired nucleophile. As a result we turned our attention to installation of a nitro group. Regioselective nitration of olefins remains a challenge. Corey has described the use of vinyl stannanes as way to control the regioselectivity of these reactions.39 In our hands, however, these efforts were ultimately unproductive in transforming 2.98 into 2.99.
MeOCO2Me
H
NOX
OTBS
MeOCO2Me
H
NHR
LG
Ar
2.90
MeOCO2Me
H
O
OTBS2.82.93

! '+!
Scheme 2.11 Initial attempts to form a Michael acceptor hydrindanone 2.8.
Faced with the hardship of introducing a nitrogen atom directly, we sought an alternative electron-withdrawing group that would serve as a nitrogen atom surrogate. Importantly, we needed a route in which the stereocenter introduced from protonation after conjugate addition, could be maintained. Particularly attractive was a Hofmann rearrangement40 of amide 2.100, which is known to proceed with stereoretention (Figure 2.8). Amide 2.100 could, in turn, arise from vinyl nitrile 2.101 using a conjugate addition/hydration sequence.
MeOCO2Me
H
O1. H2NOH·HCl, NaOAc, MeOH
MeOCO2Me
H
N
OTBS
OTBS
2. TBSOTf, Et3N, CH2Cl2 0 ! 23 °C3. NBS, (PhCO2)2 CCl4, 77 °C
56% (3 steps)
OTBS
F–
Ar[M]
MeOCO2Me
H
N
OTBS
OH
Ar
MeOCO2Me
H
N
OTBS
O
MeOCO2Me
H
SnMe3
OTBS
MeOCO2Me
H
O
OTBS
1. LiHMDS, PhNTf2 THF, –78 ! 23 °C
22% (2 steps)
Nitration
MeOCO2Me
H
NO2
OTBS
Br
2. (SnMe3)2, CuCl Pd(PPh3)4, LiCl DMSO, 70 °C
2.8 2.94 2.96
2.95
2.8 2.98 2.99
MeOCO2Me
H
N
OTBS
OTBS
Br1% HCl/MeOH
MeOCO2Me
H
N
OH
OH
OMe85%
2.972.94

! '"!
Figure 2.8 Second generation approach to alkylation intermediate.
The execution of this approach is demonstrated in Scheme 2.12. Hydrindanone 2.8 is readily converted into vinyl nitrile 2.101 by formation of vinyl triflate 2.102 followed by a palladium-catalyzed coupling with sodium cyanide. In a model study, we elected to introduce a phenyl group as our aryl component, which could be accomplished by treatment of 2.101 with phenylmagnesium bromide in the presence of a Cu(I) salt. The reaction delivers a mixture of diastereomers, which are epimerizable at the carbon bearing the cyano group, in favor of the desired diastereomer. Epimerization of the undesired diastereomer with basic methanol produces the desired diastereomer as the sole product (not shown). Hydration of the alkyl nitrile group can be effected with the platinum complex shown, developed by Ghaffar and Parkins41 without over hydration to the carboxylic acid and furnishes amide 2.104. Gratifyingly, Hofmann rearrangement proceeds without event upon reaction of carboxamide 2.104 with PIFA in methanol. The trifluoroacetic acid liberated from the oxidant, partially cleaves the TBS protecting group, and the resulting alcohol can be unveiled completely by addition of trace acid in methanol after complete consumption of the amide. The free primary alcohol was then activated as a leaving group by transformation into methanesulfonate 2.105.
Scheme 2.12 Synthesis of key alkylation intermediate 2.105.
MeOCO2Me
H
NHR
LG
Ar
MeOCO2Me
H
CONH2
OTBS
Ar
MeOCO2Me
H
CN
OTBS2.90 2.100 2.101
MeOCO2Me
H
O
OTBS
LiHMDS, PhNTf2
74% (2 steps)
MeOCO2Me
H
OTf
OTBS
MeOCO2Me
H
CN
OTBS
Ph
Me2 Me2Pt
PP
HP
O
OH OH
Me2
MeOCO2Me
H
CN
OTBS
NaCNCuI, Pd(PPh3)4 MeCN, 82 °C
THF, –78 ! 23 °C
PhMgBr, CuClTHF
2:1 dr86%
EtOH:H2O, 70 °C> 99%
MeOCO2Me
H
CONH2
OTBS
Ph1. PIFA, MeOH; 1% HCl/MeOH
MeOCO2Me
H
NHCO2Me
OMs
Ph
48% (2 steps)
2. MsCl CH2Cl2:Et3N
2.8 2.102 2.101
2.1032.1042.105

! '#!
With alkylation substrate 2.105 in hand, we then focused on achieving ring closure to access the first key intermediate. Gratifyingly, treatment of carbamate 2.105 with potassium tert-butoxide in THF smoothly converts mesylate 2.105 into tricycle 2.106 in good yield. This reaction represented the first proof of principle that this strategy to form the piperidine was feasible.
Scheme 2.13 First example of tricycle formation via C–N bond formation.
2.5 Construction of the Hexacyclic Core of the Denudatine-Type Diterpenoid Alkaloids With a route established to the tricycle present within the denudatine-type diterpenoid alkaloids, we then focused on accessing our second key intermediate (Figure 2.9). More specifically, we required hexacycle 2.107, which would require the synthesis of tricycle 2.108, which contains a functionalized arene and olefin group (bolded in Figure 2.9).
Figure 2.9 Approach toward second key intermediate. En route to a more functionalized tricycle, we first needed to effect a conjugate addition with an oxygenated aryl nucleophile (Scheme 2.14). While a Grignard reagent in the presence of a copper salt was successful in promoting conjugate addition of a simple phenyl group (2.101 to 2.103, Scheme 2.12), these conditions proved to be unproductive for a more substituted arene. We eventually discovered that Rh(I)-catalysis was essential in furnishing the addition product in good yields. This addition proceeds to give a mixture of diastereomers epimeric at the cyano bearing carbon. However the undesired diastereomer can be epimerized by the action of basic methanol at elevated temperatures, to deliver 2.110, which contains six contiguous stereocenters as a single diastereomer in multi-gram quantities. Given the array of functional groups
MeOCO2Me
H
NHCO2Me
OMs
PhKOtBu
THF, –78 ! 23 °C N
H O
CO2Me
H
CO2Me
Ph
Me
68%
2.105 2.106
OH Me
H
N
O OMeOMOMMeO
N
H OH
OMe
OMe
O
OMeO
Me
2.107 2.108

! '$!
present on 2.110 that would be sensitive to standard hydrolysis conditions (i.e., silyl ether, ester group), a method for mild hydration was necessary. This goal was accomplished by treatment of 2.110 with Wilkinson’s catalyst in the presence of acetaldehyde oxime to provide amide 2.111 following the method of Chang.42 Gratifyingly, conditions employed in our synthesis of the model tricycle (2.106) extended to this system. Thus Hofmann rearrangement and alcohol activation provided methanesulfonate 2.113, which could then be transformed into tricycle 2.114 upon exposure to base. Scheme 2.14 Synthesis of a tricycle containing a substituted arene.
With 2.114 in hand, all that remained was installation of the olefin group present in 2.116 (Scheme 2.15), which represents a tricycle possessing both reaction partners for the Diels-Alder cycloaddition. To that end, global reduction of tricycle 2.114 furnishes a primary alcohol and a tertiary amine, which results from reduction of the carbamate. Oxidation of the alcohol group is effected by IBX in refluxing ethyl acetate, which delivers aldehyde 2.115. Several methylenation reactions were investigated in order to transform 2.115 into 2.116. More traditional tactics, including Wittig, Julia, Tebbe and Peterson olefinations were unsuccessful in producing any appreciable amounts of the homologated product. Additionally, while methylmagnesium bromide was successful in a 1,2-addition reaction into the aldehyde to produce a secondary alcohol (not shown), dehydration of this alcohol to give the vinyl group proved to be challenging. We attribute the lack of reactivity toward methylenation of this system to steric encumbrance around the aldehyde functionality. The rigid framework of 2.115 flanks the aldehyde with the methoxy group and aryl component. Support for this hypothesis is found in the fact that upon treatment of 2.115 with traditional Wittig ylides (e.g., CH2PPh3) no reaction takes place, however, small amounts of product (2.116)
MeOCO2Me
H
CN
OTBS
MeOCO2Me
HOTBS
CN
MeO OMOM(HO)2BOMe
OMOM
1. [Rh(cod)(OH)]2, EtOH, 78 °C
72% (2 Steps)PIFA, MeOH;
1% HCl/MeOH
41% (3 steps)
OH
CO2Me
Me
H
N
O OMeOMOM
MeOCO2Me
HOTBS
CONH2
OMOMMeORhCl(PPh3)3PhMe, 110 °C
NOH
HMe
70%
2. K2CO3, MeOH, 65 °C
MeO
THF, 0 ! 23 °C
MeOCO2Me
HOH
NHCO2Me
OMOMMeO
MeOCO2Me
HOMs
NHCO2Me
OMOMMeO
MsCl
Et3N:CH2Cl2 0 ! 23 °C
KOtBu
2.101 2.110 2.111
2.1122.1132.114
2.109

! '%!
were observed in the crude reaction mixture when smaller ylides (e.g., CH2PMe3) were employed. Scheme 2.15 Initial route toward olefin group installation.
Faced with a challenging olefination reaction, it became clear that this group must be introduced earlier in our route. We had already established that hydrindanone 2.8 could be olefinated selectively under Lebel’s modified Wittig conditions (see Scheme 2.7). As such, we elected to first carry this olefin group through our established route (Scheme 2.16). While conversion to vinyl nitrile 2.117 proceeds smoothly under conditions described above, arylation proved to be an insurmountable challenge. Both rhodium catalysis and cuprate chemistry failed to furnish the desired product (2.118). We ascribe the lack of reactivity to the lesser electron withdrawing nature of the olefin group as opposed to the ester group as well as the ability of the vinyl group to act as a deactivating ligand on the metal center. Scheme 2.16 Second-generation approach toward olefin group installation.
We realized that since olefination was unsuccessful in the tricyclic system (Scheme 2.15), and also prior to conjugate addition (Scheme 2.16), it was important to carry out homologation at some point between those two steps. The challenge presented by this task was in choosing an appropriate point at which the selectivity issues were reasonable. Ultimately, we anticipated that differentiation of the ester and nitrile functional groups in 2.110 should be possible in a chemoselective reduction (Scheme 2.17). While more traditional tactics such as lithium borohydride failed and lithium aluminum hydride gave inconsistent reductions, Red-Al® was a competent reductant and delivered a primary alcohol but left the nitrile group intact (not shown). Oxidation of the alcohol to the aldehyde with Dess-Martin periodinane occurs without event. Gratifyingly, exposure of aldehyde 2.119 to Wittig conditions results in smooth formation of the olefinated compound (2.118) in good yield over the three steps.
1. LAH, THF 0 ! 66 °C
2. IBX, EtOAc, 77 °C
OH Me
H
NMe
OMOMMeO
Olefination
56% (2 steps)
OH
CO2Me
Me
H
N
O OMeOMOMMeO
2.114
OH
CHO
Me
H
NMe
OMOMMeO2.115 2.116
MeO
H
O
OTBS
MeO
H
CN
OTBS
2. NaCN, CuI, Pd(PPh3)4 MeCN, 82 °C
1. LiHMDS, PhNTf2 THF, –78 ! 23 °C
67% (2 steps)
Arylation
MeO
H
CN
OTBSMeO OMOM
2.73 2.117 2.118

! '&!
Scheme 2.17 Final olefination route.
Having achieved olefination with a substituted arene in place, we next focused on accessing key intermediate 2.108 (Figure 2.9), which would set the stage for a Diels-Alder cycloaddition. We found that 2.108 could be obtained as described in Scheme 2.18. Following established precedent from our previous tricycle studies on the synthesis of the tricycle (see Scheme 2.13), hydration of the nitrile occurs in good yield to deliver amide 2.120. The key tricycle was then synthesized by Hofmann rearrangement and hydrolysis of the TBS ether, activation of the alcohol group and ring closure upon exposure to base.
Scheme 2.18 Accessing key tricycle with Diels-Alder components intact.
With an efficient route to 2.108 in hand, we then focused on completing the synthesis of the core of the denudatine natural products via a Diels-Alder cycloaddition (Scheme 2.19). Cleavage of the MOM ether under acidic conditions delivered a phenol (not shown), which was then oxidized to the ortho-quinone dimethyl ketal (2.121) in excellent yield over the two steps. Upon heating to 150 °C, 2.121 smoothly converts to hexacycle 2.107 as a single diastereomer. The connectivity and relative stereochemistry were unambiguously confirmed by X-ray crystallography of p-nitrobenzoate derivative 2.122.
MeOCO2Me
H
CN
OTBSMeO OMOM
MeOCHO
H
CN
OTBSMeO OMOM
1. Red-Al®, CH2Cl2 0 ! 23 °C
2. DMP, CH2Cl2, 0 °C
Ph3PMeBr,LiHMDS
THF 0 ! 23 °C
MeO
H
CN
OTBSMeO OMOM66% (3 steps)
2.110 2.119 2.118
1. PIFA, MeOH; 1% HCl/MeOH
36% (3 steps)
RhCl(PPh3)3PhMe, 110 °C
NOH
HMe
MeO
HOTBS
CONH2
OMOMMeO80%
MeO
H
CN
OTBSMeO OMOM
OH Me
H
N
O OMeOMOMMeO
2. MsCl, Et3N:CH2Cl2 0 ! 23 °C 3. KOtBu, THF 0 ! 23 °C2.118 2.120 2.108

! ''!
Scheme 2.19 Accessing the core framework of the denudatine diterpenoids
2.6 Elaboration of the Hexacyclic Core of the Denudatine-Type Diterpenoid Alkaloids
Having established a route to the hexacyclic core of the denudatine-type diterpenoids, the current focus of our studies is to complete the total synthesis of gomandonine and aconosine.
In analyzing the route to completing the synthesis of gomandonine, one major obstacle becomes apparent. A clear discrepancy between the core framework accessed above and the core framework of the natural product is the presence of an additional quaternary carbon center (C4; Figure 2.10).
Figure 2.10 Comparison of structure of gomandonine (2.3) and 2.107.
Many tactics were investigated for the installation of the methyl group to deliver the quaternary center. Our initial strategy centered on the use of a trisubstituted diene in our initial Diels-Alder cycloaddition to construct a key hydrindinanone bearing a quaternary center (Figure 2.11).
1. 2N HCl/iPrOH
OH Me
H
N
O OMeOMeO OMe
96% (2 steps)
2. PIDA MeOH, 0 °C N
H OH
OMe
OMe
O
OMeO
Me
p-Xylene, 150 °C
55%
OH Me
H
N
O OMeOMOMMeO
2.108 2.121 2.107
N
H OH
OMe
MeO
OMeO
Me
O
O
NO2
H
2.122
N
H OH
OMe
OMe
O
OMeO
Me
2.107Gomandonine (2.3)
N
Me OH
Me
H
H
OH
O
OHH
44

! '(!
Figure 2.11 Initial approach to quaternary carbon center.
Several challenges are inherent in this approach. First, trisubstituted dienes are notoriously challenging in Diels-Alder reactions. Among the requirements for a successful cycloaddition of this type is that the diene component adopt an s-cis conformation. Given the increased steric interaction (as depicted in Figure 2.12) the majority of the diene sits in the s-trans conformer. Furthermore, when the diene is in the s-cis conformation, the temperatures required for cycloaddition (intermolecular) often match those required for an undesired 1,5-hydride shift (intramolecular) furnishing diene 2.126 from 2.125.
Figure 2.12 Challenges of trisubstituted dienes in Diels-Alder reactions. As such, we focused our attention on Lewis acid catalysis as well as high-pressure reactions to circumvent these challenges. Beginning with diene 2.127, we investigated a range of conditions for cycloaddition (Scheme 2.20). Unsurprisingly, traditional heating (up to 110 °C) led to no appreciable product formation. Lewis acid catalysis (e.g., with ytterbium triflate) also failed to furnish the substituted hydrindanone due to sensitivity of the diene to Lewis acid. High-pressure reactions have been shown to be beneficial in promoting or accelerating both inter- and intramolecular Diels-Alder reactions.43 Hoping to extend this precedent to our system, we pressurized 2.127 and 2.7 up to 7000 psi, but still observed no reaction. Scheme 2.20 Initial attempts at Diels-Alder cycloadditions with a trisubstituted ester diene.
Me
R
OMeO
MeO2CN
Me OH
OMe
OMe
O
OMeO
Me
2.123
4
MeO CO2Me
HR
O
2.124
Me
+4
4
2.125 2.7
Me
R
OMe
2.125B
OMe
2.125AR
Me
R
OMe
2.125
H !
R
OMe
2.126
HH
Me
CO2Me
OMe
2.127
O
MeO2C
2.7
MeO CO2Me
HCO2Me
O
2.128
Me
Conditions+

! ')!
The electron-withdrawing nature of the ester group on diene 2.127 was postulated to deactivate the diene toward cycloaddition, and therefore, we explored an alternative diene, 2.129, where the ester group has been reduced (Scheme 2.21). Unfortunately under Lewis acid catalysis, high pressure and elevated temperature, no productive reaction was observed. Scheme 2.21 Diels-Alder cycloaddition attempts with trisubstituted alcohol diene 2.129.
Faced with a difficult Diels-Alder reaction, we explored other points along the synthetic pathway where the quaternary center could be introduced. One promising route is detailed here: Immediately following Diels-Alder cycloaddition and hydrogenation, cleavage of the TBS ether and oxidation furnishes aldehyde 2.131, which was found to be unstable (Scheme 2.22). Therefore it was immediately exposed to various methylation conditions including basic methylation (e.g., with potassium tert-butoxide in the presence of methyl iodide) and Stork’s enamine conditions.44 All of these conditions failed to deliver the product and largely led to decomposition. Possibly contributing to the failed reaction was the presence of the 1,3-carbonyl, which would enable a retro-Michael reaction and other undesired reactions. Scheme 2.22 Attempts to methylate hydrindanone 2.8.
Our first successful methylation sequence is shown in Scheme 2.23. From vinyl nitrile 2.101, oxidation of the alcohol to the aldehyde following alcohol deprotection, provides 2.133. Enolization in the presence of methyl iodide provides the desired quaternary carbon center, and subsequent reduction, yields 2.134.
Me
OMe
2.129
O
MeO2C
2.7
MeO CO2Me
H
O
2.130
Me
Conditions+
OH OH
MeO CO2Me
H
O
OTBS
1. 1% HCl/MeOH
2. DMP, NaHCO3 CH2Cl2, 0 °C
MeO CO2Me
H
O
CHO
Conditions
MeO CO2Me
H
O
CHOMe
2.8 2.131 2.132

! '*!
Scheme 2.23 First successful methylation.
Methylated hydrindanones 2.134 and 2.135 were then explored as electrophiles in the previously elucidated conjugate addition chemistry (Scheme 2.24). We were disappointed to find that, under various conditions for conjugate addition, only minimal conversions were observed. The subtleties of the effects of the methyl group on the conformation of these compounds are not well understood at this time. However, it is clear that the conjugate addition chemistry was relatively sensitive to substitution about the hydrindanone, and thus, we elected to explore methylation following conjugate addition. Scheme 2.24 Attempted arylation of vinyl nitrile containing quaternary carbon-containing substrate 2.134 and 2.135.
In the forward sense, cleavage of the TBS ether of 2.118 liberates the free primary alcohol, which was oxidized to the corresponding aldehyde (Scheme 2.25). Selective methylation of the aldehyde (attributed to differences in pKa and steric encumbrance of the proton ! to the nitrile) generates 2.139 following reduction. Hydration and Hofmann rearrangement provides carbamate 2.140. Mesylation of the primary alcohol furnishes 2.141, which undergoes alkylation under basic conditions.
MeOCO2Me
H
CN
OTBS2.101
MeOCO2Me
HCHO
CN
2.133
1. 1% HCl/MeOH2. DMP, NaHCO3 CH2Cl2
84% (2 steps)
MeOCO2Me
H
CN
2.134
1. KOtBu, MeI CH2Cl2, 0 °C2. NaBH4 MeOH
OHMe41% (2 steps)
MeOCO2Me
H
CN
R = H (2.134)
ORMe
R = TBS (2.135)
Arylation
MeOCO2Me
H
CN
ORMe MeO OMOM
R = H (2.136)R = TBS (2.137)
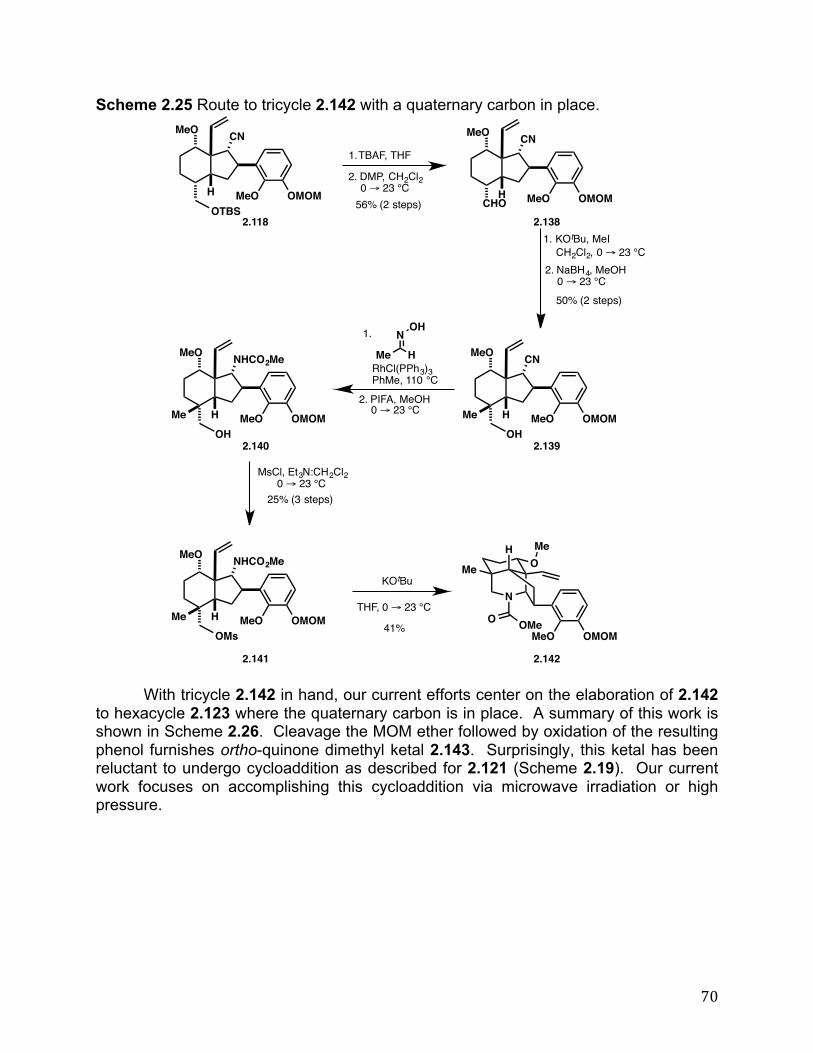
! (+!
Scheme 2.25 Route to tricycle 2.142 with a quaternary carbon in place.
With tricycle 2.142 in hand, our current efforts center on the elaboration of 2.142 to hexacycle 2.123 where the quaternary carbon is in place. A summary of this work is shown in Scheme 2.26. Cleavage the MOM ether followed by oxidation of the resulting phenol furnishes ortho-quinone dimethyl ketal 2.143. Surprisingly, this ketal has been reluctant to undergo cycloaddition as described for 2.121 (Scheme 2.19). Our current work focuses on accomplishing this cycloaddition via microwave irradiation or high pressure.
MeO
H
CN
OTBSMeO OMOM
1.TBAF, THF
2. DMP, CH2Cl2 0 ! 23 °C
MeO
HCHO
CN
MeO OMOM
1. KOtBu, MeI CH2Cl2, 0 ! 23 °C2. NaBH4, MeOH 0 ! 23 °C
1.
RhCl(PPh3)3 PhMe, 110 °C
NOH
HMe
2. PIFA, MeOH 0 ! 23 °C
MeO
H
NHCO2Me
OHMeO OMOMMe
25% (3 steps)
THF, 0 ! 23 °C
56% (2 steps)
50% (2 steps)
41%
MeO
H
CN
OHMeO OMOMMe
MsCl, Et3N:CH2Cl20 ! 23 °C
MeO
H
NHCO2Me
OMsMeO OMOMMe
OH Me
Me
N
O OMeOMOMMeO
KOtBu
2.142
2.118 2.138
2.1392.140
2.141

! ("!
Scheme 2.26 forward progress toward gomandonine.
Lastly, with regard to accessing the aconosine [3.2.1] bicycle (see Figure 2.4), we have begun a preliminary investigation into an appropriate rearrangement substrate. As shown in Figure 2.13, the substrate required for the Wagner-Meerwein shift contains an appropriately disposed leaving group on the [2.2.2] bicycle, and the chemistry discussed below describes our efforts toward its installation.
Figure 2.13 Representation of substrate for Wiesner rearrangement. Transformation of 2.107 into a substrate such as 2.144 first necessitated a change in oxidation level ! to the carbonyl. We sought to reduce the dimethyl ketal to the methylene by reaction with samarium diiodide in the presence of a proton source45 (Scheme 2.27). This reaction delivers 2.146 in modest yield along with significant formation of 2.147 for which the stereochemistry is not yet known. Scheme 2.27 Samarium diiodide mediated reduction of dimethyl ketal-containg substrate 2.107.
A proposed mechanism for the formation of 2.146 is shown in Figure 2.14. Ketyl radical 2.148 is likely formed from 2.107, which may be protonated to generate radical 2.149. Addition of a second electron to 2.149 from another molecule of samarium diiodide generates 2.150. Elimination of a molecule of methoxide then furnishes an enol which upon tautomerization forms ketone 2.151. Repeating this process to lose the second molecule of methoxide delivers the product (2.146). By analogy to the other
OH Me
Me
N
O OMeOMOMMeO
1. 2N HCl/iPrOH
2. PIDA, NaHCO3, MeOH, 0 °C
OH Me
Me
N
O OMeOMeO OMe
N
Me OH
OMe
OMe
O
OMeO
Me
!
45% (2 steps)
2.142 2.143 2.123
N
H O
R
H
LGX
Me
2.144
N
H O
R
H Me
H
X
H
2.145Aconosine (2.53)
N
H O
Me
H Me
H
OH
H
HO
H
OMe
N
H OH
OMe
OMe
O
OMeO
Me
2.107
SmI2MeOH:THF N
H OH
O
OMeO
Me
2.146 (31%)
N
H OH
OH
OMeO
Me
2.147 (19%)
OMe+

! (#!
single-electron reduction,46 side product 2.147 is likely formed from protonation of the ketyl anion (2.150) intermediate before elimination of the second molecule of methoxide.
Figure 2.14 Proposed mechanism for dimethyl ketal reduction. With ketone 2.146 in hand, our latest efforts are shown in Scheme 2.28. Reduction of the olefin group with palladium on carbon under a hydrogen atmosphere furnishes the saturated [2.2.2] bicycle. After some investigation, it was found that mono-bromination proceeds in good yield by reaction of 2.152 with elemental bromine in chloroform. The stereochemistry has not been unambiguously confirmed for this oxidation, however it is believed to be that which is represented by analogy to the results obtained by Wiesner (see 2.48, Scheme 2.4).21 Scheme 2.28 Latest efforts toward aconosine.
2.7 Conclusion We have described a concise approach to the tricyclic core of the denudatine- and lappaconitine-type diterpenoid alkaloids. We have further elaborated this structure to access the core framework of the denudatine-type diterpenoid alkaloids, as well as set the stage for accessing the lappaconitine-type diterpenoid alkaloids using a
N
H OH
OMe
OMe
O
OMeO
Me
2.107
SmI2
N
H OH
OMe
OMe
O
OMeO
Me
2.148
SmI2
H
N
H OH
OMe
OMe
OH
OMeO
Me
2.149
SmI2
N
H OH
OMe
OMe
OH
OMeO
Me
2.150
SmI2
N
H OH
OMe
OMeO
Me
2.151
TautomerizationO
Repeat 1x2.146
N
H OH
O
OMeO
Me
2.146
1. Pd/C, H2, EtOAc
2. Br2, CDCl3 N
H OH
O
OMeO
Me
2.152
Br>99% (2 steps)
N
H O
R
H Me
H
X
H
2.145

! ($!
rearrangement first described by Wiesner. Central to the success of this approach has been elaboration of a highly functionalized hydrindanone core, which is accessed via a highly diastereoselective Diels-Alder reaction. We relied heavily on conformational insights from the phragmalin-type limonoid chemistry (addressed in Chapter 3), which was critical to the success of the C–N bond-forming event to build the requisite tricycle. An oxidative dearomatization/Diels-Alder sequence led to the rapid construction of molecular complexity and completed the synthesis of the core of gomandonine. 2.8 Experimental Methods !General: Unless otherwise stated, reactions were performed in flame-dried glassware or dried in an oven overnight. All reaction vessels were fitted with rubber septa or Teflon screw caps and kept under an atmosphere of nitrogen. Liquid reagents and solvents were transferred via syringe under nitrogen using standard Schlenk techniques. Tetrahydrofuran, toluene, and benzene were sparged with argon and passed through an alumina column. Dichloromethane was distilled over calcium hydride. All other solvents were used as received unless otherwise noted. Reaction temperatures above 23 °C refer to oil bath temperature which was controlled by an IKA® temperature modulator. Reactions were monitored by thin layer chromatography using SiliCycle silica gel 60 F254 precoated plates (0.25 mm), which were visualized using UV irradiation, p-anisaldehyde stain or KMnO4 stain. Sorbent Technologies silica gel (particle size 40-63 µm) was used for column chromatography. 1H and 13C NMR were recorded on Bruker AVB-400, AVQ-400, DRX-500 or AV-600 spectrometers with 13C operating frequencies at 100, 125 and 150 MHz, respectively, in deuterated chloroform, benzene or p-xylene at 23 °C. Chemical shifts are reported relative to residual solvent signal (! = 7.26 for 1H NMR and 77.00 for 13C NMR in chloroform, 7.16 for 1H NMR in benzene, 6.91 for 1H NMR in p-xylene). Data for 1H NMR are reported as follows: chemical shift (multiplicity, coupling constant, number of hydrogens). Multiplicity is abbreviated as follows: s (singlet), br s (broad singlet), d (doublet), dd (doublet of doublets), t (triplet), tt (triplet of triplets), q (quartet), aq (apparent quartet), ap (apparent pentent), hept (heptet), m (multiplet). Signals marked by an asterisk (*) denote the minor rotamer. IR spectra were recorded on a Nicolet MAGNA-IR 850 spectrometer and are reported in frequency of absorption (cm-1). Only selected IR absorbencies are reported. Enantiomeric excess (ee) was determined by HPLC analysis on a Waters chromatography system (1525 binary pump, 717+ autosampler, 2487 dual wavelength detector) using a Chiralcel OD-H (0.46 cm x 25 cm)(from Daicel Chemical Ind., Ltd.) stationary phase and 97:3 hexanes/isopropanol mobile phase (1 mL/min) at 220 nm. Mass spectra were recorded on an LTQ Orbitrap XL (ThermoFisher Scientific) for ESI and AutoSpec Premier (Waters) for EI through the mass spectral facility at the University of California, Berkeley.

! (%!
Selenide 2.153: Bromine (4.50 g, 1.45 mL, 28.3 mmol) was added to diphenyl diselenide (9.05 g, 29 mmol) in CH2Cl2 (200 mL) at room temperature. After 25 min, the dark purple mixture was cooled to 0 °C and pyridine (5.70 g, 5.80 mL, 72.0 mmol) was added followed by methyl 2-oxocyclopentanecarboxylate (6.80 g, 5.96 mL, 48.0 mmol). 1 h after dicarbonyl addition, the reaction was deemed complete by TLC analysis and poured into water (200 mL) and extracted with CH2Cl2 (3 X 150 mL). The combined organic phases were dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (2:1 hexanes/EtOAc) to furnish selenide 2.153 (13.4 g, 45.1 mmol, 94% yield). The spectral data was identical to that which was previously reported.47
Enone 2.7: Selenide 2.153 (11.70 g, 39.38 mmol) was dissolved in CH2Cl2 (250 mL) and cooled to 0 °C. With vigorous stirring, H2O2 (8.0 mL, 30 wt. % solution in water) was slowly added in portions over 25 minutes. Upon complete consumption of the starting material by TLC analysis the reaction was poured into water (200 mL). The organic layer was separated and washed again with water. The combined aqueous layers were then back extracted with CH2Cl2. The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure furnish enone 2.7 (5.44 g, 38.83 mmol, 99% yield) as an orange oil. The spectral data was identical to that which was previously reported.47
Hydrindenone 2.154 (Major, endo product): In a 100 mL round bottom flask equipped with a magnetic stir bar and a reflux condenser, a mixture of freshly prepared 2.7 (6.70 g, 47.8 mmol, 1.0 equiv) and diene 2.5 (25.1 g, 110 mmol, 2.3 equiv) in toluene (20 mL) were heated to 100 °C. After 12 h, nmr analysis indicated complete consumption of the starting material and the solvent was removed under reduced pressure. The crude mixture was purified by column chromatography on silica gel (19:1 hexanes/EtOAc) to deliver hydrindenone 2.154 as a yellow oil (13.8 g, 78% yield). Rf = 0.43 (9:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3): # 5.99 (dt, J = 11, 3.6 Hz, 1H), 5.71 (d, J = 10 Hz, 1H), 4.35 (s, 1H), 3.73 (s, 3H), 3.63 (dt, J = 20, 9.6 Hz, 2H), 3.31 (s, 3H), 2.99 (q, J = 6.6 Hz, 1H ), 2.47 (d, J = 6.6 Hz, 1H), 2.38 (dd, J = 19, 8.4 Hz, 1H), 2.24 (q, J = 9.0 Hz, 1H), 1.96 – 1.87 (m, 2H), 0.89 (s, 9H), 0.06 (s, 6H); 13C NMR (150 MHz, CDCl3):
O
MeO2C
(PhSe)2, Br2Pyridine
CH2Cl2, 0 °C
O
MeO2CPhSe
2.153
O
MeO2CPhSe
2.153
H2O2 (30%)
CH2Cl2, 0 °C
O
MeO2C
2.7
OMe
OTBS
O
MeO2C+
MeOCO2Me
H
O
OTBS
Toluene, 100 °C
2.72.5 2.154

! (&!
# 211.9, 171.0, 128.0, 124.3, 73.7, 63.8, 63.3, 58.7, 52.7, 39.4, 38.6, 36.7, 25.8, 20.6, 18.2, -5.4, -5.5; IR (thin film) "max cm-1 2954, 2930, 2896, 2857, 1757, 1730, 1253, 1109, 1085, 838, 777; HRMS (ESI) calc’d for [C19H32O5NaSi]+: m/z 391.1911, found 391.1914. Hydrindenone exo-2.154 (Minor, exo product): The exo diastereomer of hydrindenone 2.154 was also isolated as a white solid (923 mg, 5% yield). Rf 0.17 (10:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 5.98 – 5.89 (m, 2H), 4.28 (dd, J = 4.4, 1.4 Hz, 1H), 3.75 (s, 3H), 3.74 – 3.68 (m, 1H), 3.57 (dd, J = 9.6, 7.4 Hz, 1H), 3.31 (s, 3H), 2.77 (t, J = 5.8 Hz, 1H), 2.36 – 2.27 (m, 1H), 2.20 – 2.08 (m, 1H), 1.96 – 1.85 (m, 2H), .90 (s, 9H), .06 (s, 6H); 13C NMR (150 MHz, CDCl3) # 209.8, 167.4, 132.2, 123.8, 72.4, 66.6, 66.0, 56.2, 52.5, 40.2, 35.3, 33.6, 25.9, 25.7, 18.1, -5.58, -5.62; IR (film) "max 2953, 1732, 1471 cm-1; HRMS (EI) calc’d for [C19H32O5NaSi]+ m/z 391.1911, found 391.1916.
Hydrindanone 2.8: Inside a Parr hydrogenation bomb, hydrindenone 2.154 (19.6 g, 53.2 mmol) and 10% Pd/C (3.00 g) in EtOAc (200 mL) were pressurized to 550 psi and stirred at room temperature for 12 h. After this time, the mixture was filtered through celite and concentrated under reduced pressure to deliver hydrindanone 2.8 (19.6 g, 53.0 mmol, 99% yield) as a white solid. Rf = 0.42 (9:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3): # 4.08 (s, 1H), 3.69 (s, 3H), 3.53 (dt, J = 24, 10 Hz, 2H), 3.17 (s, 3H), 3.02 (q, J = 6.6 Hz, 1H), 2.39 (dd, J = 19, 8.4 Hz, 1H), 2.25 (q, J = 9.0 Hz, 1H), 2.08 – 2.00 (m, 2H), 1.82 – 1.75 (m, 2H), 1.44 – 1.38 (m, 3H), 0.89 (s, 9H), 0.05 (s, 6H); 13C NMR (150 MHz, CDCl3): # 214.7, 170.9, 78.7, 65.6, 63.1, 57.2, 52.5, 41.2, 39.2, 38.2, 25.9, 25.4, 20.4, 18.3, 17.1, -5.36, -5.42; IR (thin film) "max cm-1 2953, 2930, 2896, 2857, 1755, 1728, 1252, 1092, 837, 776; m.p.: 37.4 – 39.2 °C; HRMS (ESI) calc’d for [C19H34O5
23Na28Si]+: m/z 393.2068, found 393.2070.
Vinyl Nitrile 2.101: Hydrindanone 2.8 (20.0 g, 54.0 mmol) was dissolved in THF (250 mL) in a 1.0 L round bottom flask equipped with a magnetic stir bar. The reaction vessel was then cooled to -78 °C and LiHMDS (1.0 M in THF, 70 mL, 70 mmol) was added dropwise via syringe and stirred at that temperature for 1 h, at which time N-phenyl-bis(trifluoromethanesulfonimide) (26.6 g, 75.6 mmol) was added in THF (120 mL) and the reaction vessel warmed to room temperature. The reaction proceeds at that temperature for 12 h before being cooled to 0 °C and quenched with saturated NaHCO3 solution and extracted with EtOAc (3 X 70 mL). The combined organic phases
MeOCO2Me
H
O
OTBS
MeOCO2Me
H
O
OTBS
Pd/C, H2
EtOAc
2.154 2.8
MeOCO2Me
H
CN
OTBS2.101
MeOCO2Me
H
O
OTBS2.8
1. LiHMDS, PhNTf2, THF, –78 ! 23 °C2. NaCN, CuI, Pd(PPh3)4, MeCN, 82 °C

! ('!
were dried over MgSO4, filtered and concentrated under reduced pressure to give a crude vinyl triflate, which was brought to sufficient purity by column chromatography (5% EtOAc/hexanes). The vinyl triflate was diluted with MeCN (250 mL) and sodium cyanide (5.80 g, 119 mmol), Pd(PPh3)4 (3.70 g, 3.20 mmol) and CuI (1.20 g, 6.5 mmol) were sequentially added to the reaction vessel which was then fitted with a reflux condenser and heated to reflux temperature for 2 h, at which time the vessel was cooled to room temperature and the reaction mixture filtered through a pad of celite (rinsed 2 X 30 mL EtOAc), washed with water (2 X 40 mL) and brine (2 X 40 mL) and concentrated under reduced pressure. The crude mixture was purified by column chromatography to furnish vinyl nitrile 2.101 (14.3 g, 37.7 mmol, 70% yield over two steps). Rf 0.22 (10:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) # 6.89 (t, J = 2.6 Hz, 1H), 3.88 (dd, J = 6.2, 2.5 Hz, 1H), 3.79 (s, 3H), 3.48 (dd, J = 10.1, 6.1 Hz, 1 H) 3.40 (dd, J = 10.1, 7.8 Hz, 1H), 3.29 (s, 3H), 2.82 (td, J = 9.3, 5.1 Hz, 1H), 2.42 – 2.32 (m, 2H), 2.07 – 2.00 (m, 1H), 1.81 (dq, J = 14.1, 5.4 Hz, 1H), 1.66 – 1.60 (m, 1H), 1.49 – 1.42 (m, 1H), 1.32 (ddt, J = 13.1, 9.8, 6.5 Hz, 1H), 0.87 (s, 9H), 0.024 (s, 3H), 0.021 (s, 3H); 13C NMR (150 MHz, CDCl3) # 173.0, 151.9, 116.0, 115.7, 66.0, 63.6, 57.6, 52.6, 43.4, 36.4, 33.2, 25.8, 24.1, 18.1, 17.8, -5.45, -5.51 (note: one sp3 carbon signal missing, possibly due to signal overlap); IR (film) "max 2956, 2223, 1735 cm-1; HRMS (ESI) calc’d for [C20H33NO4SiNa]+ m/z 402.2071, found 402.2075.
Alkyl Nitrile 2.110: Vinyl nitrile 2.101 (3.74 g, 9.85 mmol) was diluted with ethanol (100 mL) in a 250 mL round bottom flask equipped with a magnetic stir bar. Boronic acid 2.109 (10.5 g, 49.5 mmol) was then added followed by [Rh(cod)(OH)]2 (905 mg, 1.97 mmol) and the reaction vessel was fitted with a reflux condenser and heated to reflux temperature for 1.5 h, at which time nmr analysis indicated complete consumption of the starting material. The dark black mixture was filtered through a pad of celite (filter cake rinsed 3 X 40 mL EtOAc) and concentrated. Bulk impurities were removed by column chromatography (5'20% EtOAc/hexanes) and the majority of the !-diastereomer isolated containing slight impurity. The "-diastereomer, containing small amounts of the !-diastereomer and slight impurity, was then diluted with MeOH (40 mL) in a pressure tube and K2CO3 (673 mg, 4.87 mmol) was added. The heterogeneous mixture was heated to 80 °C with rapid stirring for 6 h at which time, the vessel was cooled to room temperature and brine (30 ml) and EtOAc (40 mL) were added. The two phases were separated and the aqueous phase extracted with EtOAc (3 X 30 mL). The combined organic phases were dried over MgSO4, filtered and concentrated under reduced pressure. The crude material was combined with slightly impure !-diastereomer isolated from initial conjugate addition and purified by column chromatography (5'10'20% EtOAc/hexanes) to furnish alkyl nitrile 2.110 (3.87 g, 7.07 mmol, 72% yield over two steps). Rf 0.46 (4:1 hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) # 7.03 (d, J = 8.1 Hz, 1H), 6.97 (t, J = 7.9 Hz, 1H), 6.88 (d, J = 6.88 Hz, 1H), 5.23 – 5.18 (m,
MeOCO2Me
HOTBS
CN
MeO OMOM
MeOCO2Me
H
CN
OTBS
(HO)2BOMe
OMOM
2.109
2.1102.101
1. 2.109, [Rh(cod)(OH)]2 EtOH, 78 °C2. K2CO3, MeOH, 80 °C

! ((!
2H), 4.04 (t, J = 9.6 Hz, 1H), 3.98 (s, 1 H), 3.94 (s, 3H), 3.74 (s, 3H), 3.51 (s, 3H), 3.44 (s, 3H), 3.43 – 3.36 (m, 2H), 3.25 (d, J = 8.4 Hz, 1H), 3.14 – 3.08 (m, 1H), 2.40 (q, J = 12.5 Hz, 1H), 2.09 (d, J = 14.4 Hz, 1H), 1.99 (bs, 1H), 1.62 (t, J = 10.4 Hz, 1H), 1.47 – 1.32 (m, 3H), 0.87 (s, 9H), 0.02 (s, 3H), -0.01 (s, 3H); 13C NMR (150 MHz, CDCl3) # 173.2, 150.2, 147.8, 138.8, 124.2, 121.5, 120.2, 115.3, 95.1, 66.2, 61.3, 60.4, 56.9, 56.2, 52.6, 45.7, 43.6, 42.7, 38.1, 33.1, 25.8, 24.8, 18.2, 16.5, -5.4, -5.5 (note: one sp3 carbon signal missing, possibly due to signal overlap); IR (film) "max 2952, 2239, 1732 cm-1; HRMS (ESI) calc’d for [C29H45NO7SiNa]+ m/z 570.2858, found 570.2863. A small amount of the "-diastereomer of alkyl nitrile was isolated and purified for characterization purposes: Akyl Nitrile (!-diastereomer) !-2.110: Rf 0.24 (4:1 hexanes/EtOAc); 1H NMR (300 MHz, CDCl3) # 7.06 – 7.01 (m, 3H), 5.21 (aq, J = 6.6 Hz, 2H), 4.26 (td, J = 10.6, 2.9 Hz, 1H), 3.92 (bs, 1H), 3.89 (s, 3H), 3.76 (s, 3H), 3.72 (d, J = 9.7 Hz, 1H), 3.51 (s, 3H), 3.47 (d, J = 7.4 Hz, 2H), 3.40 (s, 3H), 3.22 (ddd, J = 13.6, 8.6, 5.0 Hz, 1H), 2.28 (q, J = 13 Hz, 1H), 2.20 – 2.01 (m, 2H), 1.89 (ddd, J = 12.4, 8.7, 3.0 Hz, 1H), 1.41 – 1.28 (m, 2H), 1.28 – 1.10 (m, 2H), 0.92 (s, 9H), 0.07 (s, 3H), 0.05 (s, 3H); 13C NMR (100 MHz, CDCl3) # 172.6, 149.8, 147.8, 136.5, 124.0, 121.3, 119.1, 115.5, 95.2, 79.6, 66.4, 62.8, 60.9, 57.3, 56.2, 52.8, 44.2, 41.1, 38.0, 37.9, 30.9, 25.9, 25.4, 18.2, 16.3, -5.37, -5.39; IR (film) "max 2930, 2237, 1740 cm-1; HRMS (ESI) calc’d for [C29H45NO7SiNa]+ m/z 570.2858, found 570.2867.
Primary Alcohol 2.155: alkyl nitrile 2.110 (6.40 g, 11.7 mmol) was diluted with CH2Cl2 (100 mL) in a 250 mL round bottom flask equipped with a magnetic stir bar. The reaction vessel was cooled to -78 °C and Red-Al® (65 wt% in toluene, 38 mL, 117 mmol) was added dropwise. Following addition, the reaction mixture warmed to room temperature and stirred for an additional 1 h, at which time the reaction was cooled to 0 °C and quenched with isopropanol (20 mL) and saturated NH4Cl solution and the layers separated. The aqueous phase was extracted with CH2Cl2 and the combined organic phases dried over MgSO4, filtered and concentrated under reduced pressure. Purification by column chromatography (20'33% EtOAc/hexanes) to furnish primary alcohol 2.155 (5.00 g, 9.62 mmol, 82% yield) as a clear, viscous oil. Rf 0.11 (4:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) # 7.01 (dd, J = 8.3, 1.7 Hz, 1H), 6.96 (t, = 7.9 Hz, 1H), 6.87 (dd, J = 7.5, 1.7 Hz, 1H), 5.23 – 5.18 (m, 2H), 4.01 (ddd, J = 11.4, 8.7, 2.9 Hz, 1 H), 3.92 (s, 3H), 3.62 (q, J = 11.3 Hz, 2H), 3.52 (s, 3H), 3.42 – 3.36 (m, 5H), 3.16 (d, J = 8.7 Hz, 1H), 2.54 – 2.46 (m, 1H), 2.39 (q, J = 11.0 Hz, 1H), 2.09 (bs, 1H), 2.06 – 1.99 (m, 1H), 1.86 – 1.76 (m, 1H), 1.54 (ddd, J = 12.0, 8.4, 3.0 Hz, 1H), 1.45 – 1.32 (m, 3H), 0.87 (s, 9H), 0.01 (s, 3H), -0.01 (s, 3H); 13C NMR (100 MHz, CDCl3) # 150.2, 147.7, 139.8, 124.2, 121.6, 121.4, 114.9, 95.0, 77.5, 66.5, 64.3, 61.4, 56.8, 56.2, 55.5, 43.8, 42.2, 39.7, 37.8, 33.9, 25.9, 24.1, 18.3, 17.0, -5.4; IR (film) "max 3475, 2932, 2237 cm-1; HRMS (ESI) calc’d for [C28H45NO6SiNa]+ m/z 542.2908, found 542.2905.
MeO
HOTBS
CN
MeO OMOM
2.155
MeOCO2Me
HOTBS
CN
MeO OMOM
2.110
Red-Al®
CH2Cl2
OH

! ()!
Aldehyde 2.119: In a 100 mL round bottom flask equipped with a stir bar, primary alcohol 2.155 (2.16, 4.16 mmol) was diluted with CH2Cl2 (40 mL) and NaHCO3 (1.75 g, 20.8 mmol) was added. The reaction vessel was cooled to 0 °C and Dess-Martin periodinane (3.52 g, 8.30 mmol) was added portionwise and the reaction proceeds at that temperature for 1.5 h at which time TLC analysis indicated complete consumption of the starting material. The reaction was quenched by slow addition of saturated NaHCO3 solution (10 mL) and saturated Na2S2O3 solution (10 mL). The layers were separated and the aqueous phase was extracted with CH2Cl2 (3 X 20 mL) and the combined organic phases dried over Na2SO4, filtered and concentrated under reduced pressure. The crude mixture was purified by column chromatography (20% EtOAc/hexanes) to furnish aldehyde 2.119 (1.95 g, 3.77 mmol, 91% yield). Rf 0.41 (4:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) # 9.48 (s, 1H), 7.06 (dd, J = 8.2, 1.5 Hz, 1H), 6.97 (t, J = 7.9 Hz, 1H), 6.87 (dd, J = 7.8, 1.5 Hz, 1H), 5.24 – 5.20 (m, 2H), 4.04 (ddd, J = 11.9, 8.7, 3.2 Hz, 1H), 3.96 (s, 3H), 3.87 (bs, 1H), 3.52 (s, 3H), 3.47 – 3.35 (m, 5H), 3.16 (d, 8.7 Hz, 1H), 2.93 (ddd, J = 13.1, 8.7, 4.9 Hz, 1H), 2.44 (q, J = 12.3 Hz, 1H), 2.19 – 2.12 (m, 1H), 1.80 – 1.67 (m, 2H), 1.49 – 1.23 (m, 3H), 0.87 (s, 9H), 0.01 (s, 3H), -0.01 (s, 3H); 13C NMR (100 MHz, CDCl3) # 200.3, 150.4, 147.7, 137.9, 124.2, 121.9, 119.3, 115.6, 95.1, 77.2, 75.3, 66.0, 63.9, 61.3, 56.9, 56.3, 44.9, 42.2, 41.8, 38.7, 33.0, 25.9, 24.7, 18.3, 16.3, -5.4, -5.5; IR (film) "max 2932, 2239, 1727 cm-1; HRMS (ESI) calc’d for [C28H43NO6NaSi]+ m/z 540.2752, found 540.2751.
Olefin 2.118: Methyltriphenylphosphonium bromide (6.60 g, 18.5 mmol) was suspended in THF (60 mL) in a 250 mL, two neck round bottom flask fitted with a reflux condenser and a rubber septum. The reaction vessel was cooled to 0 °C and LiHMDS (1.0 M in THF, 15.5 mL, 15.5 mmol) was added dropwise to produce a yellow solution which was heated to reflux temperature for 30 min, at which time the vessel was cooled back to 0 °C and aldehyde 2.119 (3.20 g, 6.18 mmol) was added in THF (20 mL). Following addition, the reaction vessel was allowed to warm to room temperature and stirred at that temperature for 1 h before being cooled to 0 °C and quenched by the addition of saturated NH4Cl solution (20 mL). The reaction mixture was extracted with EtOAc (3 X 30 mL) and the combined organic phases dried over MgSO4, filtered and concentrated under reduced pressure. The crude mixture was purified by column
MeO
HOTBS
CN
MeO OMOM
2.119
MeO
HOTBS
CN
MeO OMOM
2.155
DMP, NaHCO3
CH2Cl2
OOH
MeO
HOTBS
CN
MeO OMOM
MeO
HOTBS
CN
MeO OMOM
LiHMDS, PPh3MeBr
THF
O
2.1182.119

! (*!
chromatography (10'20% EtOAc/hexanes) to furnish olefin 2.118 (3.00 g, 5.82 mmol, 94% yield). Rf 0.50 (4:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) # 7.02 (dd, J = 8.2, 1.7 Hz, 1H), 6.96 (t, J = 7.9 Hz, 1H), 6.84 (dd, J = 7.6, 1.7 Hz, 1H), 5.91 (dd, J = 17.9, 11.1 Hz, 1H), 5.29 (d, J = 17.7 Hz, 1H), 5.24 – 5.18 (m, 3H), 4.02 (ddd, J = 11.7, 8.8, 3.1 Hz, 1H), 3.93 (s, 3H), 3.52 (as, 4H), 3.45 – 3.36 (m, 5H), 2.85 (d, J = 8.9 Hz, 1H), 2.64 – 2.56 (m ,1H), 2.41 (q, J = 12.3 Hz, 1H), 2.03 – 1.88 (m ,2H), 1.64 – 1.56 (m, 1H), 1.48 – 1.22 (m, 3H), 0.88 (s, 9H), 0.20 (s, 3H), 0.0 (s, 3H); 13C NMR (100 MHz, CDCl3) # 150.5, 148.0, 141.5, 139.7, 124.3, 121.8, 121.3, 115.3, 95.3, 80.1, 66.6, 61.6, 56.9, 56.4, 55.6, 48.8, 44.2, 43.3, 37.9, 33.7, 26.1, 23.9, 18.5, 17.1, -5.21, -5.24; IR (film) "max 2931, 2235, 1479 cm-1; HRMS (ESI) calc’d for [C29H45NO5SiNa]+ m/z 538.2959, found 538.2955.
Amide 2.120: Olefin 2.118 (1.74 g, 3.37 mmol) was diluted with toluene (3.5 mL) and acetaldoxime (6.2 mL) in a 25 mL round bottom flask equipped with a magnetic stir bar. Wilkinson’s catalyst (624 mg, 0.67 mmol) was then added and the reaction vessel was fitted with a reflux condenser and heated to reflux temperature for 12 h followed by a second addition of Wilkinson’s catalyst (312 mg, 0.34 mmol) and continued heating for an additional 3 h, at which time nmr analysis indicated complete consumption of the starting material and the reaction mixture was concentrated under reduced pressure and purified via column chromatography (10'20% EtOAc/hexanes) to produce amide 2.120 (1.46 g, 2.74 mmol, 81% yield) as a white solid. Rf 0.38 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) # 7.03 – 6.91 (m, 3H), 6.80 (bs, 1H), 6.18 (dd, J = 17.8, 11.1 Hz, 1H), 5.78 (bs, 1H), 5.23 – 5.16 (m, 3H), 5.13 (d, J = 11.4 Hz, 1H), 3.96 – 3.85 (m, 4H), 3.68 (bs, IH), 3.48 (s, 3H), 3.39 (7.6, 2.8 Hz, 2H), 3.23 (s, 3H), 2.78 – 2.68, 2H), 2.44 (q, J = 12.3 Hz, 1H), 2.00 – 1.86 (m, 2H), 1.56 (ddd, J = 11.9, 8.9, 2.6 Hz, 1H), 1.44 – 1.21 (m, 3H), 0.89 (s, 9H), 0.01 (s, 3H), -0.01 (s, 3H); 13C NMR (100 MHz, CDCl3) # 175.1, 149.7, 145.8, 144.7, 142.5, 125.1, 121.1, 113.5, 112.4, 94.9, 79.3, 66.2, 61.3, 56.3, 56.1, 55.2, 3.1, 38.2, 37.4, 34.4, 25.8, 23.7, 18.1, 17.1, -5.5, -5.6; IR (film) "max 3427, 3188, 2929, 1668 cm-1; HRMS (ESI) calc’d for [C29H48NO6Si]+ m/z 534.3245, found 534.3253.
Carbamate 2.156: Amide 2.120 (203 mg, 0.38 mmol) was suspended in MeOH (4 mL) in a 25 mL round bottom flask equipped with a magnetic stir bar. Reaction vessel was
MeO
HOTBS
CONH2
MeO OMOM
MeO
HOTBS
CN
MeO OMOM
RhCl(PPh3)3
2.1202.118
N
HMe
OH
PhMe, 110 °C
MeO
HOH
NHCO2Me
MeO OMOM
MeO
HOTBS
CONH2
MeO OMOM
PIFA, MeOH;
2.1562.120
1% HCl/MeOH

! )+!
cooled to 0 °C and PIFA (245 mg, 0.57 mmol) was added portionwise and reaction warmed to room temperature after 15 min. 14 h after addition of the oxidant, NMR analysis indicated complete rearrangement of the amide and 1% HCl/MeOH was added and the reaction stirred for 10 min before being cooled to 0 °C and quenched with saturated NaHCO3 (3 mL) and extracted with EtOAc (3 X 5 mL). The combined organic phases were dried over MgSO4, filtered and concentrated under reduced pressure. The crude mixture was purified by column chromatography (50% EtOAc/hexanes) to produce carbamate 2.156 (123 mg, 0.27 mmol, 72% yield). Rf 0.26 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) # 6.96 – 6.79 (m, 3H), 5.85 – 5.69 (m, 1H), 5.20 – 5.05 (m, 4H), 5.01 (d, J = 10.5 Hz, 1H), 4.91 (d, J = 10.7 Hz, 1H)*, 4.22 (t, J = 9.6 Hz, 1H), 4.06 (t, J = 9.6 Hz, 1H)*, 3.82 (s, 3H), 3.53 – 3.35 (m, 10H), 3.33 (s, 3H), 2.62 – 2.50 (m, 1H), 2.28 (q, J = 12.4 Hz, 1H), 2.16 (bs, 1H), 2.00 (J = 11.1, 1H), 1.94 – 1.83 (m, 1H), 1.47 – 1.26 (m, 4H); 13C NMR (100 MHz, CDCl3) # 157.3*, 156.6, 149.9, 147.8*, 147.6, 142.7*, 142.5, 140.3*, 140.2, 124.0, 123.7*, 122.0*, 121.6, 114.4*, 114.2, 113.7, 113.5*, 94.9, 78.6, 78.2*, 68.6*, 67.6, 65.8, 60.8, 56.0, 55.3, 53.8, 53.5*, 51.8*, 51.7, 46.3*, 45.4, 41.8*, 41.5, 37.7, 32.1, 31.6*, 22.9, 17.0; IR (film) "max 3446, 2940, 1716 cm-1; HRMS (ESI) calc’d for [C24H36NO7]+ m/z 450.2486, found 450.2495.
Mesylate 2.158: Carbamate 2.157 (123 mg, 0.27 mmol) was diluted with CH2Cl2 (3 mL) and Et3N (1 mL) in a 10 mL round bottom flask equipped with a stir bar. The reaction vessel was cooled to 0 °C and methanesulfonyl chloride (50 µL, 0.65 mmol) was added dropwise and the reaction proceeded at that temperature. 3 h after addition, LCMS analysis indicates complete reaction and the reaction was quenched with saturated NaHCO3 (2 mL) and the layers separated. The aqueous phase was extracted with CH2Cl2 (3 X 3 mL) and the combined organic phases were charged with silica gel and the solvent removed under reduced pressure. The dried silica gel was immediately purified via column chromatography (50% EtOAc/hexanes) and mesylate 2.158 (107 mg, 0.20 mmol, 74% yield) was isolated as a clear oil. Rf 0.30 (1:1 hexanes/EtOAc); 1H NMR (400 MHz, CDCl3) # 6.98 – 6.76 (m, 3H), 5.86 – 5.68 (m, 1H), 5.20 – 4.82 (m 5H), 4.24 (t, J = 9.7 Hz, 1H), 4.09 (t, J = 10 Hz, 1H)*, 4.01 (d, J = 7.4 Hz, 1H), 3.84 (s, 3H), 3.82 (s, 3H)*, 3.54 – 3.31 (m, 12H), 3.07 (s, 3H)*, 2.94 (s, 3H), 2.67 – 2.53 (m, 1H), 2.31 (q, J = 12.2 Hz, 1H), 2.23 – 2.10 (m 1H), 2.09 – 1.96 (m 1H), 1.52 – 1.30 (m, 4H); 13C NMR (100 MHz, CDCl3) # 157.1*, 156.5, 150.0, 149.9*, 147.7, 142.5*, 141.9, 140.2*, 139.6, 123.92*, 123.86, 123.6*, 114.5, 114.2*, 114.0, 113.9*, 113.6*, 94.9, 78.5*, 78.0, 77.7*, 72.7, 67.6*, 67.5, 65.9*, 60.8*, 60.7, 56.0, 55.4, 55.3*, 53.8, 51.74, 51.68*, 46.7*, 45.7, 45.3*, 41.5*, 40.8, 37.7*, 37.0, 34.9, 32.1*, 31.9, 31.4, 22.9*, 22.5, 17.0*, 16.8; IR (film) "max 3447, 2943, 1724, 1354, 1175, 835, 776 cm-1; HRMS (ESI) calc’d for [C25H37NO9SNa]+ m/z 550.2081, found 550.2092.
MeO
HOH
NHCO2Me
MeO OMOM
2.157
MsCl
MeO
HOMs
NHCO2Me
MeO OMOM
2.158
CH2Cl2:Et3N

! )"!
Tricycle 2.108: Mesylate 2.158 (107 mg, 0.20 mmol) was diluted in THF (4 mL) in a 10 mL round bottom flask and the reaction vessel cooled to 0 °C. To the cooled solution, KOtBu (1.0 M in THF, 0.56 mL, 0.56 mmol) was added dropwise to produce a light yellow solution which was warmed to room temperature and stirred for 2 h at which time LCMS analysis indicated complete consumption of the starting material and the reaction vessel cooled back to 0 °C and quenched with saturated NaHCO3 (2 mL) and extracted with EtOAc (3 mL). The combined organic phases were dried over Na2SO4, filtered and concentrated. The crude mixture was purified by column chromatography (50% EtOAc/hexanes) to produce tricycle 2.108 (58 mg, 0.13mmol, 66% yield). Rf 0.43 (1:1 hexanes/EtOAc); 1H NMR (300 MHz, CDCl3) # 7.01 – 6.80 (m, 3H), 5.95 (dd, J = 17.8, 10.9 Hz, 1H)*, 5.79 (dd, 17.8, 10.8 Hz, 1H), 5.19 – 4.74 (m, 5H), 3.90 -3.66 (m, 2H), 3.53 – 3.43 (m, 4H), 3.42 – 3.17 (m, 5H), 2.34 – 2.11 (m, 2H), 2.01 – 1.07 (m 4H), 1.61 – 1.51 (m, 1H); 13C NMR (100 MHz, CDCl3) # 155.2, 149.97, 149.94*, 148.4, 145.2*, 145.1, 136.6, 122.33, 122.25*, 122.2*, 121.8, 114.6, 113.4, 113.1*, 95.2, 85.9*, 85.7, 63.7, 62.3*, 60.3*, 59.9, 57.7*, 57.1, 56.2, 52.9*, 52.8, 52.5, 52.4*, 44.1, 43.6*, 43.4*, 43.2*, 43.12, 43.07, 33.2*, 33.15*, 33.11, 31.8, 30.0*, 29.8, 25.0*, 24.8; IR (film) "max 2932, 1691, 1470 cm-1; HRMS (ESI) calc’d for [C24H34NO6]+ m/z 432.2381, found 432.2386.
Phenol 2.159: Tricycle 2.108 (108 mg, 0.13 mmol) was diluted with a 0 °C solution of 2N HCl/isopropanol (2 mL) in a 10 mL round bottom flask and stirred at room temperature for 3.5 h before being cooled to 0 °C and slowly quenched with saturated NaHCO3 solution (3 mL) and extracted with EtOAc (3 X 2 mL). The combined organic phases were dried over Na2SO4, filtered and concentrated. The crude product was purified by column chromatography (50% EtOAc/hexanes) to furnish phenol 2.159 (52 mg, 0.13 mmol, 99% yield). Rf 0.33 (1:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 6.91 – 6.76 (m, 3H), 5.81 (dd, J = 17.9, 11.0 Hz, 1H)*, 5.73 (dd, J = 17.8, 10.9 Hz, 1H), 5.58 (bs, 1H), 5.11 – 4.89 (m, 3H), 3.91 (d, J = 13.8 Hz, 1H), 3.75 (s, 3H), 3.74 (s, 3H)*, 3.72 (s, 3H)*, 3.70 (s, 3H), 3.47 – 3.30 (m, 2H), 3.28 – 3.21 (m, 4H), 2.40 – 2.29 (m, 2H), 2.02 – 1.86 (m, 3H), 1.83 (bs, 1H), 1.63 – 1.51 (m, 2H); 13C NMR (125 MHz, CDCl3) # 155.2, 154.0*, 149.1, 149.0*, 145.7, 145.6*, 144.8*, 144.7, 123.3, 119.9*, 119.7, 113.8, 113.7*, 113.3, 113.1*, 85.7*, 85.5, 63.6, 62.6*, 59.7*, 59.6, 57.7*, 57.0, 52.63, 52.56, 52.5*, 43.9, 43.5*, 43.3*, 43.2*, 43.1, 43.0, 33.0*, 32.9, 32.2*, 31.5*, 29.9*,
MeO
HOMs
NHCO2Me
MeO OMOM
2.158
OH Me
H
N
O OMeOMOMMeO
2.108
KOtBu
THF
OH Me
H
N
O OMeOMOMMeO
2.108
2N HCl/iPrOHO
H Me
H
N
O OMeOHMeO
2.159

! )#!
29.6, 24.9*, 24.6 (note: 2 sp3 carbons missing, possibly due to signal overlap); IR (film) "max 3306, 2933, 1674, 1586 cm-1; HRMS (ESI) calc’d for [C22H28NO5]+ m/z 386.1973, found 386.1969.
Ortho-Quinone Dimethyl Ketal 2.121: Phenol 2.159 (160 mg, 0.41 mmol) was diluted with MeOH (4 mL) in a 25 mL round bottom flask which was then cooled to 0 °C. To this mixture, NaHCO3 (173 mg, 2.06 mmol) was added followed by PIDA (200 mg, 0.62 mmol) to produce a bright green solution. The reaction proceeded for 1 h at that temperature before being concentrated under reduced pressure and the crude mixture purified by column chromatography (20'50% EtOAc/hexanes) to furnish ortho-quinone dimethyl ketal 2.121 (170 mg, 0.41 mmol, 99% yield) as a bright green solid. Rf 0.38 (1:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 6.92 – 6.86 (m, 1H), 6.40 (d, J = 6.5 Hz, 1H)*, 6.28 (d, J = 6.4 Hz, 1H), 6.11 (dd, J = 17.9, 10.9 Hz, 1H)*, 6.01 (dd, J = 17.9, 10.9 Hz, 1H), 5.94 (d, J = 5.94 Hz, 1H), 5.18 – 5.10 (m, 2H), 5.03 (s, 1H)*, 4.98 (s, 1H), 3.83 (d, J = 13.9 Hz, 1H), 3.71 (s, 3H), 3.69 (s, 3H)*, 3.66 (d, J = 13.9 Hz, 1H)*, 3.30 – 3.19 (m, 8H), 3.14 (s, 3H)*, 3.07 (s, 3H)*, 3.04 (t, J = 8.6 Hz, 1H), 2.99 (t, J = 8.6 Hz, 1H)*, 2.30 – 2.25 (m, 1H), 2.22 – 2.08 (m, 1H), 1.99 – 1.93 (m, 1H), 1.92 – 1.84 (m, 1H), 1.81 – 1.75 (m, 2H), 1.68 – 1.49 (m, 5H); 13C NMR (100 MHz, CDCl3) # 197.0, 154.9, 153.1*, 152.6, 145.4, 145.1*, 141.0*, 140.7, 124.0*, 123.9, 123.4, 123.3*, 113.3, 94.8, 85.7, 62.2, 61.8*, 57.0, 53.1, 52.9*, 52.4, 52.3*, 50.9, 50.3*, 49.9, 43.6, 43.4, 43.1*, 43.0*, 42.6, 32.8, 31.8, 29.9*, 29.7, 24.7*, 24.6; IR (film) "max 2937, 1683, 1575, 1450 cm-1; HRMS (ESI) calc’d for [C23H32NO6]+ m/z 418.2224, found 418.2230.
Hexacycle 2.107: Ortho-quinone dimethyl ketal 2.121 (5.6 mg, 0.013 mmol) was diluted with p-xylene (0.6 mL) in a pressure tube and the reaction mixture was degassed by the freeze-pump-thaw method. The reaction vessel was sealed and heated to 150 °C for 17.5 h at which time the solvent was removed under reduced pressure and the crude material purified by column chromatography (20'30'50% EtOAc/hexanes) to furnish hexacycle 2.107 (3.1 mg, 0.0074 mmol, 55% yield) as a clear film. Rf 0.39 (1:1 hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) # 6.39 (dd, J = 8.0, 6.5 Hz, 1H), 6.35 (dd, J = 8.1, 6.5 Hz, 1H)*, 6.11 (d, J = 6.6 Hz, 1H)*, 5.99 (d, J = 6.5 Hz, 1H), 4.38 (bs, 1H)*, 4.20 (bs, 1H), 3.73 (s, 3H), 3.70 (s, 3H)*, 3.45 (s, 3H), 3.40 – 3.37 (m, 1H), 3.29 – 3.24 (m, 4H), 3.21 (s, 3H), 3.18 (s, 3H)*, 3.02 – 2.95 (m, 1H), 2.88 (ddd, J = 13.6, 6.9, 2.8 Hz, 1H), 2.57 (d, J = 5.2 Hz, 1H)*, 2.50 (d, J = 5.3 Hz, 1H), 2.36 (q, J = 10.2 Hz, 1H),
OH Me
H
N
O OMeOHMeO
2.159
PIDA, NaHCO3
MeOH, 0 °C
OH Me
N
O OMeOMeO OMe
2.121
H
N
H OH
OMe
OMe
O
OMeO
MeO
H Me
N
O OMeOMeO OMe
2.121
p-xylene, 150 °C
2.107

! )$!
2.16 – 1.99 (m, 3H), 1.94 – 1.73 (m, 3H), 1.48 – 1.22 (m, 4H); 13C NMR (125 MHz, CDCl3) # 206.9*, 206.6, 164.5, 156.32*, 156.25, 135.9*, 135.7, 129.8, 129.4*, 99.5*, 99.4, 77.8, 77.4*, 62.5*, 62.3, 55.3, 55.2*, 52.8, 52.4*, 51.70*, 51.66, 50.9, 50.8*, 50.5, 50.3*, 49.1, 48.84, 48.82*, 48.5*, 45.94, 45.91*, 45.7*, 45.6, 45.3*, 45.2, 44.5, 44.0*, 32.8*, 32.6, 30.1*, 30.0, 29.43, 29.36*, 25.12*, 25.1, 24.4*, 24.3; IR (film) "max 2940, 1731, 1696, 1448 cm-1; HRMS (ESI) calc’d for [C23H31NO6Na]+ m/z 440.2044, found 440.2051.
p-Nitrobenzoate 2.122: Hexacycle 2.107 (29 mg, 0.069 mmol) was diluted in MeOH (1 mL) in a 5 mL round bottom flask and cooled to 0 °C. Sodium borohydride (5.3 mg, 0.14 mmol) was then added and reaction proceeds at that temperature for 2 h, at which time it was diluted with water (1 mL) and methanol removed under reduced pressure. The resulting aqueous solution was extracted with EtOAc (3 x 2 mL) and the combined organic phases dried over MgSO4, filtered and concentrated. The resulting crude alcohol was diluted with CH2Cl2 (0.5 mL) and Et3N (0.1 mL) and DMAP (8.4 mg, 0.069 mmol) was added followed by p-nitrobenzoyl chloride (64 mg, 0.34 mmol) and the resulting mixture stirred at room temperature for 12 h at which point, saturated NH4Cl solution (0.5 mL) was added and the mixture extracted with EtOAc (3 X 1 mL). The combined organic phases were dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (20'30% EtOAc/hexanes) to furnish p-nitrobenzoate 2.122 (22 mg, 0.039 mmol) as viscous oil, which could produce a solid when exposed to hexanes. An X-ray quality crystal could be obtained by slow diffusion of hexanes into a nearly saturated solution of the product in EtOAc. Rf 0.61 (1:1 hexanes/EtOAc); 1H NMR (600 MHz, p-xylene) # 7.84 (t, J = 9.0 Hz, 2H), 7.52 (dd, J = 11.0, 8.3 Hz, 2H), 6.29 (t, J = 7.0 Hz, 1H), 6.23 (t, J = 7.1 Hz, 1H)*, 6.12 (d, J = 7.9 Hz, 1H)*, 5.28 (d, J = 5.4 Hz, 1H), 4.64 (s, 1H)*, 4.40 (s, 1H), 3.56 – 3.49 (m, 4H), 3.43 (dt, J = 12.1, 6.0, 1H), 3.29 (bs, 1H), 3.15 (s, 3H), 3.13 (s, 3H)*, 3.10 – 3.07 (m, 3H), 2.97 (s, 3H), 2.51 (d, 5.1 Hz, 1H)*, 2.41 (s, 1H), 2.38 (d, J = 5.3 Hz, 1H), 1.90 – 1.81 (m, 1H), 1.80 – 1.71 (m, 3H), 1.56 – 1.45 (m, 5H), 0.88 – 0.99 (m, 3H); 13C NMR (125 MHz, CDCl3) # 163.9*, 163.8, 156.4, 150.5, 135.9, 135.8*, 132.6*, 132.4, 132.3*, 131.8*, 130.93*, 130.91, 123.4, 108.1*, 108.0, 78.5*, 78.3, 77.9, 77.5*, 61.9*, 61.7, 60.4, 55.3, 55.2*, 52.8, 52.4*, 50.75*, 50.70, 50.67*, 50.03, 49.7, 49.6*, 49.4*, 46.2, 46.1*, 45.4*, 45.3, 44.6*, 44.1*, 43.6*, 43.5, 37.91, 37.88*, 32.8*, 32.6, 30.2*, 30.1, 29.7, 28.65*, 28.61, 25.13*, 25.11, 24.5*, 24.3, 14.2; IR (film) "max 2938, 1721, 1693, 1606, 1529, 1345 cm-1; HRMS (ESI) calc’d for [C30H36N2O9]+ m/z 568.2426, found 568.2421.
N
H OH
OMe
OMe
O
OMeO
Me
2.107
1. NaBH4, MeOH, 0 °C
2. p-nitrobenzoyl chloride, DMAP, CH2Cl2:Et3N
N
H OH
OMe
OMe
OMeO
Me
H
O
O
NO2
2.122

! )%!
Bromo-Arene 2.161: Phenol 2.160 (24.4 g, 105 mmol) was diluted with DMF (300 mL) in a 1 L round bottom flask equipped with a magnetic stir bar and the reaction vessel was cooled to 0 °C. Sodium hydride (60% wt in mineral oil, 5.4 g, 136 mmol) was then added portionwise and the reaction proceeds at that temperature for 30 min, at which time methyl iodide (8.5 mL, 136 mmol) was added and the vessel warmed to room temperature. After 2 h, TLC analysis indicated complete consumption of the starting material and the reaction was carefully quenched by the addition of saturated NH4Cl solution (100 mL) and extracted with diethyl ether (3 X 200 mL). Combined organic phases were washed with water (2 X 200 mL) and brine (1 X 200 mL), dried over MgSO4, filtered and concentrated. The crude mixture was purified by column chromatography (10% EtOAc/hexanes) to deliver bromo-arene 2.160 (22.6 g, 91.5 mmol, 87% yield) as a clear oil. Rf 0.56 (4:1 hexanes/EtOAc); 1H NMR (300 MHz, CDCl3) # 7.20 (dd, J = 8.1, 1.5 Hz, 1H), 7.10 (dd, J = 8.3, 1.5 Hz, 1H), 6.91 (t, J = 7.1 Hz, 1H), 5.22 (s, 2H), 3.88 (s, 3H), 3.51 (s, 3H); 13C NMR (100 MHz, CDCl3) # 151.4, 147.3, 126.2, 125.0, 117.8, 116.0, 95.2, 60.6, 56.3; IR (film) "max 2934, 1000, cm-1; HRMS (EI) calc’d for [C9H11O3Br]+ m/z 245.9892, found 245.9896.
Boronic Acid 2.109: Bromo-arene 2.161 (22.6 g, 91.5 mmol) was diluted in THF (500 mL) in a 1 L round bottom flask equipped with a magnetic stir bar and the reaction vessel was cooled to -78 °C. n-BuLi (2.5 M in hexanes, 51 mL, 128 mmol) was then added dropwise and the reaction proceeded at that temperature for 30 min, at which time triisopropyl borate (38 mL, 164 mmol) was added and the reaction vessel was warmed to room temperature and stirred for 12 h. After this period, the reaction was quenched by slow addition of NH4Cl (55 g) in water (380 mL), which was stirred for 4 h. The layers were then separated and the aqueous phase was extracted with EtOAc (3 X 200 mL) and the combined organic phases were dried over MgSO4, filtered and concentrated under reduced pressure. Boronic acid 2.109 (16.0 g, 75.5 mmol, 83% yield) was isolated by addition of hexanes and filtration and repeating of this process with the filtrate until no further solids formed upon addition of hexanes. 1H NMR (400 MHz, CDCl3) # 7.48 (dd, J = 7.5, 1.7 Hz, 1H), 7.27 (dd, J = 1.6, 8.1 Hz, 1H), 7.10 (t, J = 7.9 Hz, 1H), 5.96 (br s, 2H), 5.23 (s, 2H), 3.97 (s, 3H), 3.53 (s, 3H); 13C NMR (100 MHz, CDCl3) # 154.9, 149.0, 129.1, 124.7, 120.0, 95.0, 61.5, 56.2; IR (film) "max 3374, 1350 cm-1. 2.9 References and Notes (1) Pelletier, S. W.; Mody, N. V. J. Nat. Prod. 1980, 43, 41-71. (2) Singhuber, J.; Zhu, M.; Prinz, S.; Kopp, B. J. Enthopharmacol. 2009, 126, 18-30.
OHOMOM NaH, MeI
2.160
BrDMF, 0 ! 23 °C
OMeOMOM
2.161
Br
OMeOMOM
nBuLi, B(OiPr)3
2.161
BrOMe
OMOM
2.109
(HO)2BTHF, –78 ! 23 °C;
NH4Cl, H2O

! )&!
(3) Friese, J.; Gleitz, J.; Gutser, U. T.; Heubach, J. F.; Matthiesen, T.; Wilffert, B.; Selve, N. Eur. J. of Pharmacol. 1997, 337, 165-174. (4) Ameri, A. Prog. Neurobiol. 1998, 56, 211-235. (5) Kim, D. K.; Kwon, H. Y.; Lee, K. R.; Rhee, D. K.; Zee, O. P. Arch. Pharmacal. Res. 1998, 21, 344 –347. (6) Zhao, D.-K.; Ai, H.-L.; Zi, S.-H.; Zhang, L.-M.; Yang, S.-C.; Guo, H.-C.; Shen, Y.; Chen, Y.-P.; Chen, J.-J. Fitoterapia 2013, 91, 280-283. (7) Ahmad, M.; Ahmad, W.; Ahmad, M.; Zeeshan, M.; Shaheen, F. J. Enzyme Inhib. Med. Chem. 2008, 23, 1018 – 1022. (8) Ren, Y.; Houghton, P. J.; Hider, R. C.; Howes, M.-J. R. Planta Med. 2004, 70, 201-204. (9) Wang, F.-P.; Chen, Q.-H.; Liu, X.-Y. Nat. Prod. Rep. 2010, 27, 529-570. (10) Mericli, A. H.; Mericli, F.; Seyhan, G. V.; Bahar, M.; Desai, H. K.; Ozcelik, H.; Ulubelen, A. Pharmazie, 2002, 57, 761-762. (11) Pelletier, S. W.; Jacobs, W. A. J. Am. Chem. Soc. 1956, 78, 4144-4145. (12) Nagata, W.; Sugasawa, T.; Narisada, M.; Wakabayashi, T.; Hayase, Y. J. Am. Chem. Soc. 1963, 85, 2342-2343. (13) Masamune, S. J. Am. Chem. Soc. 1964, 86, 291-292. (14) Guthrie, R. W.; Valenta, Z.; Wiesner, K. Tetrahedron Lett. 1966, 7, 4645-4654. (15) Nagata, W.; Sugasawa, T.; Narisada, M.; Wakabayashi, T.; Hayase, Y. J. Am. Chem. Soc. 1967, 89, 1483-1499. (16) Ihara, M.; Suzuki, M.; Fukumoto, K.; Kametani, T.; Kabuto, C. J. Am. Chem. Soc. 1988, 110, 1963-1964. (17) Liu, X.-Y.; Cheng, H.; Li, X.-H.; Chen, Q.-H.; Xu, L.; Wang, F.-P. Or.g Biomol. Chem. 2012, 10, 1411-1417. (18) Edwards, O. E.; Singh, T. Can. J. Chem. 1955, 33, 448-451. (19) Wiesner, K.; Edwards, J.A. Experientia 1955, 11, 255-259. (20) Wiesner, K.; Tsai, T. Y. R.; Huber, K.; Bolton, S. E.; Vlahov, R. J. Am. Chem. Soc. 1974, 96, 4990-4992. (21) Wiesner, K.; Tsai, T. Y. R.; Nambiar, K. P. Can. J. Chem. 1978, 56, 1451-1454. (22) Wiesner, K.; Uyeo, S.; Philipp, A.; Valenta, Z. Tetrahedron Lett. 1968, 9, 6279-6282. (23) For relevant examples, see: (a) Edwards, O. E.; Kolt, R. J.; Purushothaman, K. K. Can. J. Chem. 1983, 61, 1194-1196. (b) Masamune, S. J. Am. Chem. Soc. 1964, 86, 290-291. (c) Masamune, S. J. Am. Chem. Soc. 1964, 86, 289-290. (d) Williams, C. M.; Mander, L. N. Org. Lett. 2003, 5, 3499-3502. (e) Muratake, H.; Natsume, M. Angew. Chem. Int. Ed. 2004, 43, 4646-4649. (f) Cheng, H.; Xu, L.; Chen, D.-L.; Chen, Q.-H.; Wang, F.-P. Tetrahedron 2012, 68, 1171-1176. (g) Mei, R.-H.; Liu, Z.-G.; Cheng, H.; Xu, L.; Wang, F.-P. Org. Lett. 2013, 15, 2206-2209. (24) Peese, K. M.; Gin, D. Y. J. Am. Chem. Soc. 2006, 128, 8734-8735. (25) Shi, Y.; Wilmot, J. T.; Nordstrøm, L. U.; Tan, D. S.; Gin, D. Y. J. Am. Chem. Soc. 2013, 135, 14313-14320. (26) Tambar, U. K.; Stoltz, B. M. J. Am. Chem. Soc. 2005, 127, 5340-5341. (27) Cornelisse, J. Chem. Rev. 1993, 93, 615-669. (28) Pinhey, J. T. Aust. J. Chem. 1991, 44, 1353-1382. (29) Zhai, H.; Zlotorzynska, M.; Sammis, G. Chem. Commun. 2009, 5716-5718.

! )'!
(30) Becker, K. B.; Gabutti, C. A. Tetrahedron Lett. 1982, 23, 1883-1886. (31) Sasaki, T.; Eguchi, S.; Okano, T. J. Am. Chem. Soc. 1983, 105, 5912-5913. (32) Xu, F.; Simmons, B.; Reamer, R. A.; Corley, E.; Murry, J.; Tschaen, D. J. Org. Chem. 2007, 73, 312-315. (33) Anderson, J. C.; Smith, S. C. Synlett 1990, 1990, 107-108. (34) Lebel, H.; Paquet, V. J. Am. Chem. Soc. 2003, 126, 320-328. (35) Kürti, L.; Szilágyi, L.; Antus, S.; Nógrádi, M. Eur. J. Org. Chem. 1999, 1999, 2579-2581. (36) Tamura, Y.; Yakura, T.; Haruta, J.; Kita, Y. J. Org. Chem. 1987, 52, 3927-3930. (37) Hassner, A.; Murthy, K. Tetrahedron Lett. 1987, 28, 683-684. (38) Li, P.; Majireck, M. M.; Witek, J. A.; Weinreb, S. M. Tetrahedron Lett. 2010, 51, 2032-2035. (39) Corey, E. J.; Estreicher, H. Tetrahedron Lett. 1980, 21, 1113-1116. (40) Moriarty, R. M.; Chany, C. J.; Vaid, R. K.; Prakash, O.; Tuladhar, S. M. J. Org. Chem. 1993, 58, 2478-2482. (41) Ghaffar, T.; Parkins, A. W. Tetrahedron Lett. 1995, 36, 8657-8660. (42) Lee, J.; Kim, M.; Chang, S.; Lee, H.-Y. Org. Lett. 2009, 11, 5598-5601. (43) Jarvo, E. R.; Boothroyd, S. R.; Kerr, M. A. Synlett 1996, 1996, 897-899. (44) Stork, G.; Dowd, S. R. J. Am. Chem. Soc. 1963, 85, 2178-2180. (45) Lu, P.; Gu, Z.; Zakarian, A. J. Am. Chem. Soc. 2013, 135, 14552-14555. (46) Girard, P.; Namy, J. L.; Kagan, H. B. J. Am. Chem. Soc. 1980, 102, 2693-2698. (47) (a) Ley, S. V.; Morray, P. J.; Palmer, B. D. Tetrahedron 1985, 41, 4765-4769. (b) Wang, C.; Gu, X.; Yu, M. S.; Curran, D. P. Tetrahedron 1998, 54, 8355-8370.!!!

! )(!
APPENDIX TWO
Spectra and Crystallographic Data Relevant to Chapter Two:
Diterpenoid Alkaloids
!!!!!!!!!!!!!!!!!!

! ))!
!
Figure A2.1 Proton and carbon NMR spectra for compound 2.154
MeOCO2Me
H
O
OTBS

! )*!
Figure A2.2 Proton and carbon NMR spectra for compound exo-2.154
MeOCO2Me
H
O
OTBS

! *+!
Figure A2.3 Proton and carbon NMR spectra for compound 2.8
MeOCO2Me
H
O
OTBS
! *"!
Figure A2.4 Proton and carbon spectra for compound 2.101
MeOCO2Me
H
CN
OTBS

! *#!
Figure A2.5 Proton and carbon NMR spectra for compound !-2.110
MeOCO2Me
H
OTBS
CN
MeO OMOM

! *$!
Figure A2.6 Proton and carbon NMR spectra for compound 2.110
MeOCO2Me
H
OTBS
CN
MeO OMOM

! *%!
Figure A2.7 Proton and carbon NMR spectra for compound 2.155
MeO
H
OTBS
CN
MeO OMOM
HO

! *&!
Figure A2.8 Proton and carbon NMR spectra for compound 2.119
MeOCHO
H
OTBS
CN
MeO OMOM

! *'!
Figure A2.9 Proton and carbon NMR spectra for compound 2.118
MeO
H
OTBS
CN
MeO OMOM

! *(!
Figure A2.10 Proton and carbon NMR spectra for compound 2.120
MeO
H
OTBS
C(O)NH2
MeO OMOM

! *)!
Figure A2.11 Proton and carbon NMR spectra for compound 2.156
MeO
H
OH
NHCO2Me
MeO OMOM

! **!
Figure A2.12 Proton and carbon NMR spectra for compound 2.158
MeO
H
OMs
NHCO2Me
MeO OMOM

! "++!
Figure A2.13 Proton and carbon NMR spectra for compound 2.108
OH Me
H
N
O OMeOMOMMeO
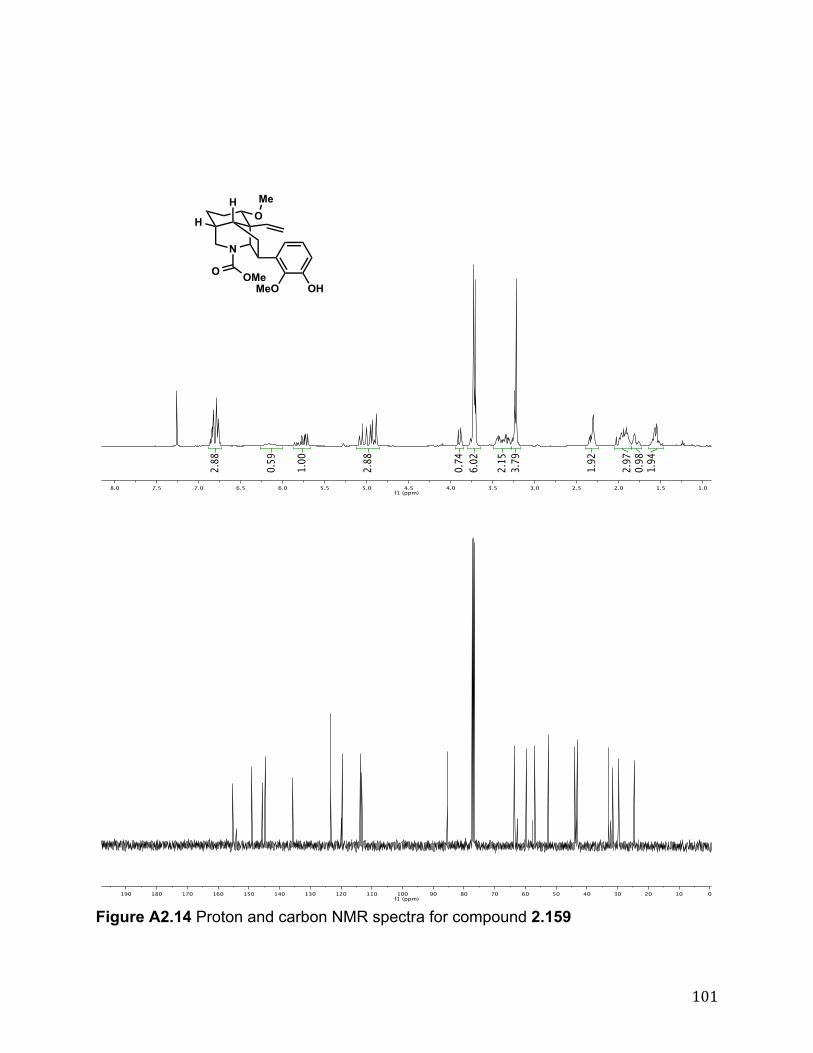
! "+"!
Figure A2.14 Proton and carbon NMR spectra for compound 2.159
OH Me
H
N
O OMeOHMeO

! "+#!
Figure A2.15 Proton and carbon NMR spectra for compound 2.121
OH Me
H
N
O OMeOMeO OMe

! "+$!
Figure A2.16 Proton and carbon NMR spectra for compound 2.107
N
H OH
OMe
OMe
O
OMeO
Me

! "+%!
Figure A2.17 Proton and carbon NMR spectra for compound 2.122
N
H OH
OMe
OMe
OMeO
Me
H
O
O
NO2
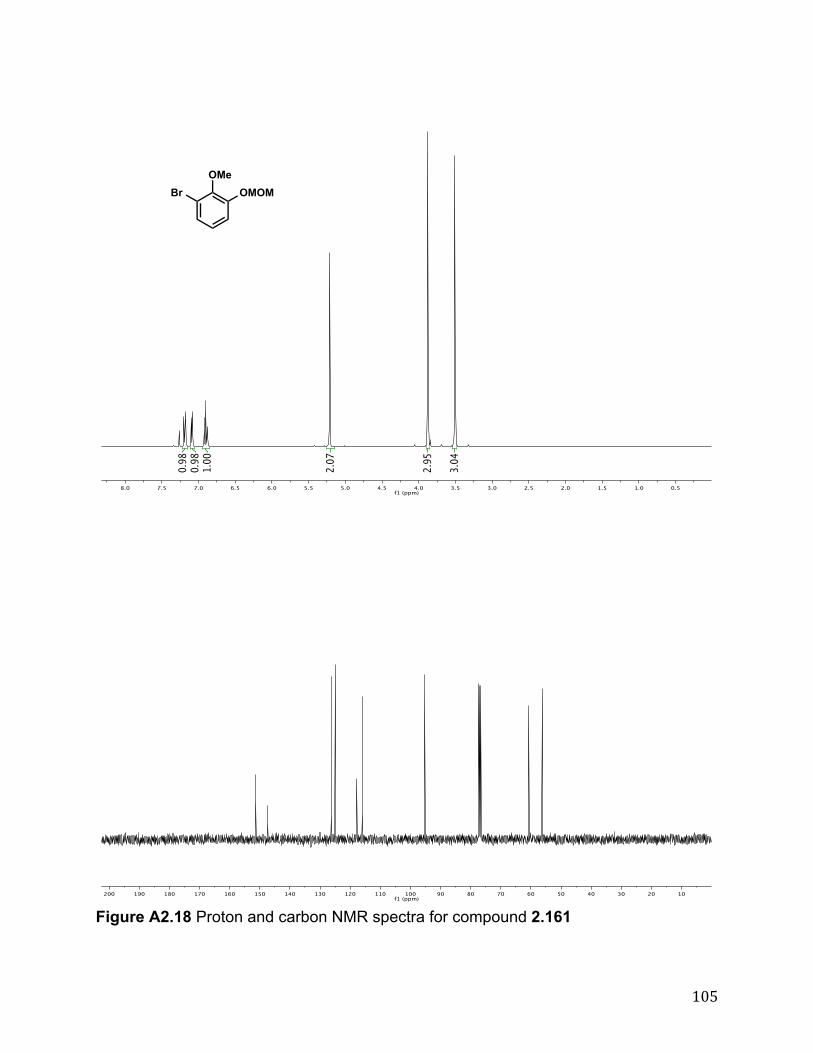
! "+&!
Figure A2.18 Proton and carbon NMR spectra for compound 2.161
BrOMe
OMOM
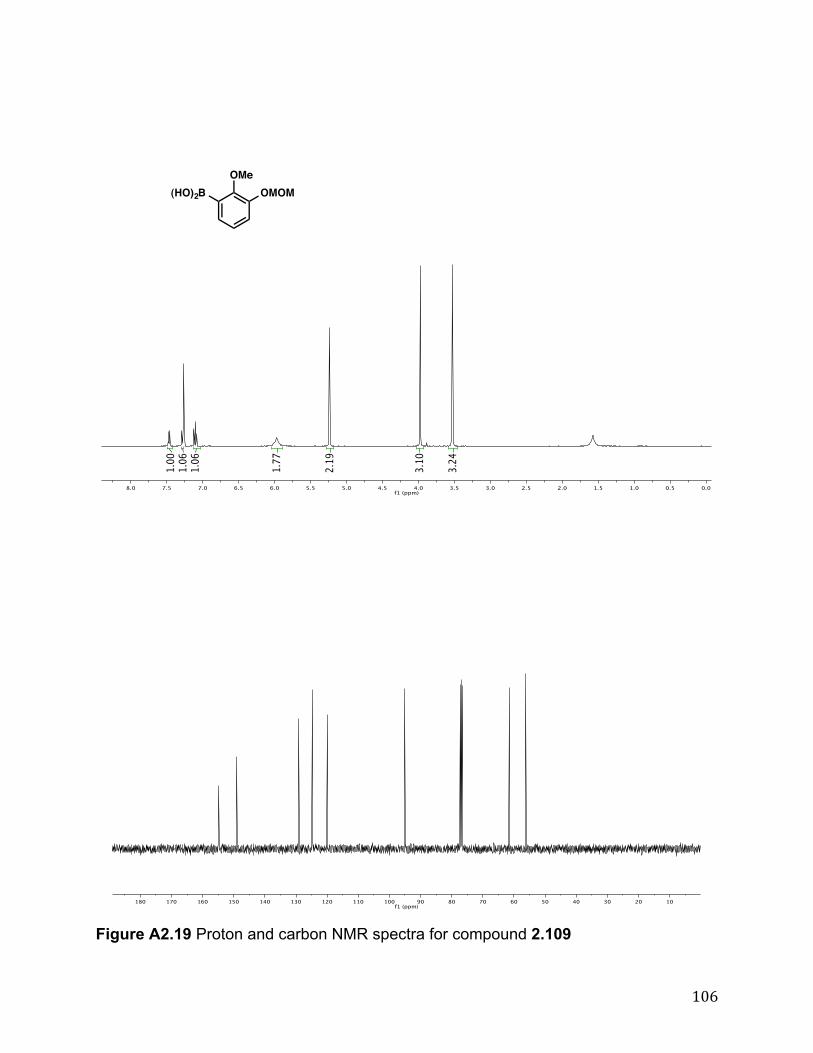
! "+'!
Figure A2.19 Proton and carbon NMR spectra for compound 2.109
OMeOMOM(HO)2B

! "+(!
Crystallographic data for 2.86: A colorless plate 0.090 x 0.070 x 0.050 mm in size was mounted on a Cryoloop with Paratone oil. Data were collected in a nitrogen gas stream at 100(2) K using phi and omega scans. Crystal-to-detector distance was 60 mm and exposure time was 5 seconds per frame using a scan width of 1.0°. Data collection was 100.0% complete to 67.000° in q. A total of 63803 reflections were collected covering the indices, -12<=h<=12, -18<=k<=17, -27<=l<=27. 6386 reflections were found to be symmetry independent, with an Rint of 0.0269. Indexing and unit cell refinement indicated a primitive, monoclinic lattice. The space group was found to be P 21/n (No. 14). The data were integrated using the Bruker SAINT software program and scaled using the SADABS software program. Solution by direct methods (SIR-2011) produced a complete heavy-atom phasing model consistent with the proposed structure. All non-hydrogen atoms were refined anisotropically by full-matrix least-squares (SHELXL-2012). All hydrogen atoms were placed using a riding model. Their positions were constrained relative to their parent atom using the appropriate HFIX command in SHELXL-2012.

! "+)!
Table 1. Crystal data and structure refinement for sarpong37. X-ray ID sarpong37
Sample/notebook ID GG08-26B
Empirical formula C35 H49 Br O8 Si
Formula weight 705.74
Temperature 100(2) K
Wavelength 1.54178 Å
Crystal system Monoclinic
Space group P 21/n
Unit cell dimensions a = 10.0372(7) Å a= 90°.
b = 15.1873(11) Å b= 92.899(2)°.
c = 22.9188(17) Å g = 90°.
Volume 3489.2(4) Å3
Z 4
Density (calculated) 1.343 Mg/m3
Absorption coefficient 2.336 mm-1
F(000) 1488
Crystal size 0.090 x 0.070 x 0.050 mm3
Crystal color/habit colorless plate
Theta range for data collection 3.492 to 68.287°.
Index ranges -12<=h<=12, -18<=k<=17, -27<=l<=27
Reflections collected 63803
Independent reflections 6386 [R(int) = 0.0269]
Completeness to theta = 67.000° 100.0 %
Absorption correction Semi-empirical from equivalents
Max. and min. transmission 0.864 and 0.772
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 6386 / 0 / 415
Goodness-of-fit on F2 1.042
Final R indices [I>2sigma(I)] R1 = 0.0289, wR2 = 0.0720
R indices (all data) R1 = 0.0296, wR2 = 0.0725
Extinction coefficient n/a
Largest diff. peak and hole 0.803 and -0.793 e.Å-3

! "+*!
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2x 103)
for sarpong37. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________ C(1) 10430(1) 3674(1) 9164(1) 12(1)
C(2) 10378(1) 2782(1) 9477(1) 13(1)
C(3) 11286(2) 2012(1) 9352(1) 17(1)
C(4) 10466(2) 1175(1) 9498(1) 20(1)
C(5) 9708(2) 1343(1) 10053(1) 18(1)
C(6) 8558(2) 2055(1) 9976(1) 16(1)
C(7) 8856(1) 2518(1) 9393(1) 13(1)
C(8) 8198(1) 3419(1) 9222(1) 13(1)
C(9) 8377(1) 3583(1) 8564(1) 13(1)
C(10) 9911(1) 3552(1) 8515(1) 12(1)
C(11) 10532(2) 4208(1) 8090(1) 14(1)
C(12) 10828(2) 5103(1) 8374(1) 16(1)
C(13) 11821(2) 5021(1) 8907(1) 18(1)
C(14) 11787(2) 4111(1) 9200(1) 14(1)
C(15) 9404(2) 1089(1) 9012(1) 20(1)
C(16) 8616(2) 1786(1) 8947(1) 16(1)
C(17) 9243(1) 4094(1) 9475(1) 12(1)
C(18) 8174(2) 2426(1) 10984(1) 24(1)
C(19) 7063(2) 807(1) 10112(1) 25(1)
C(20) 6791(1) 3572(1) 9402(1) 15(1)
C(21) 5139(2) 3174(1) 8649(1) 17(1)
C(22) 4320(2) 2417(1) 8417(1) 17(1)
C(23) 4402(2) 1583(1) 8669(1) 20(1)
C(24) 3565(2) 913(1) 8459(1) 24(1)
C(25) 2653(2) 1086(1) 7998(1) 24(1)
C(26) 2573(2) 1908(1) 7735(1) 25(1)
C(27) 3420(2) 2572(1) 7944(1) 22(1)
C(28) 9672(2) 4336(1) 7532(1) 18(1)
C(29) 8661(2) 3966(1) 6072(1) 36(1)
C(30) 6592(2) 3877(2) 6985(1) 34(1)
C(31) 8037(2) 2172(1) 6631(1) 28(1)

! ""+!
C(32) 9370(2) 1791(2) 6447(1) 46(1)
C(33) 7657(3) 1718(2) 7197(1) 61(1)
C(34) 6961(2) 1992(1) 6144(1) 36(1)
C(35) 13649(2) 4171(1) 9892(1) 23(1)
O(1) 9973(1) 979(1) 10513(1) 25(1)
O(2) 8669(1) 2679(1) 10432(1) 18(1)
O(3) 7255(1) 1698(1) 9939(1) 19(1)
O(4) 8934(1) 4973(1) 9316(1) 16(1)
O(5) 5899(1) 2926(1) 9120(1) 16(1)
O(6) 5089(1) 3907(1) 8448(1) 23(1)
O(7) 9378(1) 3504(1) 7266(1) 19(1)
O(8) 12238(1) 4165(1) 9807(1) 15(1)
Si(1) 8180(1) 3390(1) 6749(1) 19(1)
Br(1) 1470(1) 187(1) 7724(1) 35(1)
________________________________________________________________________________
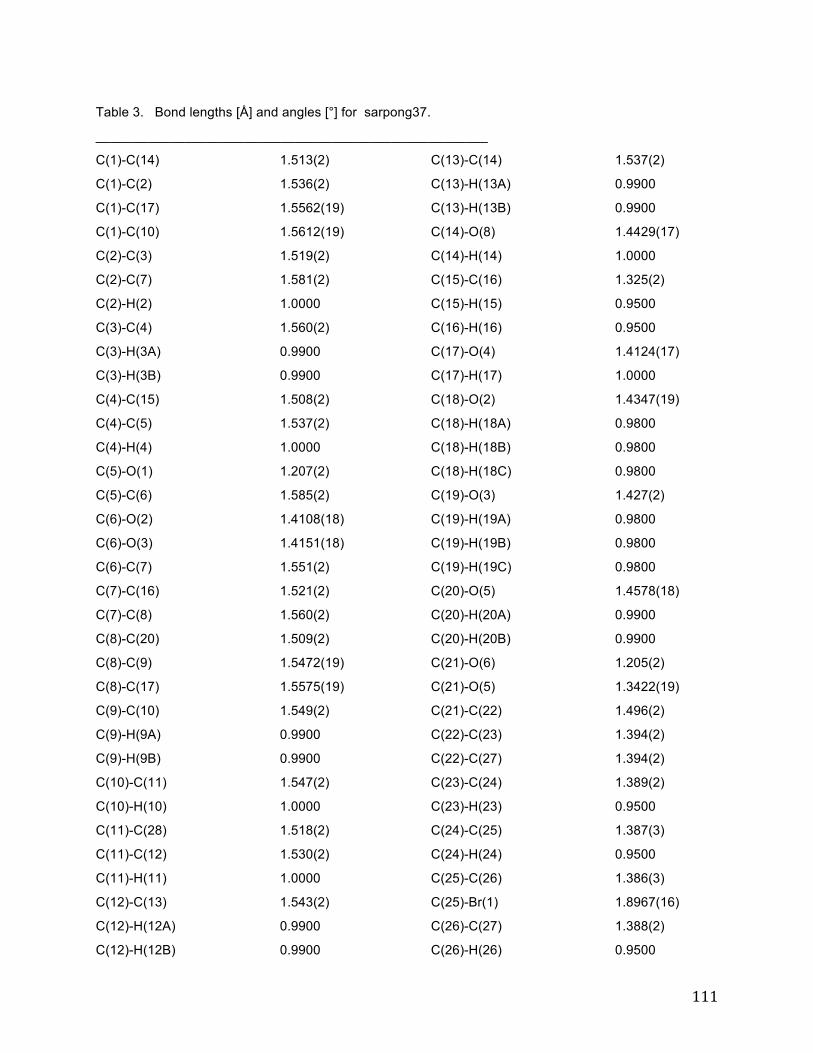
! """!
Table 3. Bond lengths [Å] and angles [°] for sarpong37.
_____________________________________________________
C(1)-C(14) 1.513(2)
C(1)-C(2) 1.536(2)
C(1)-C(17) 1.5562(19)
C(1)-C(10) 1.5612(19)
C(2)-C(3) 1.519(2)
C(2)-C(7) 1.581(2)
C(2)-H(2) 1.0000
C(3)-C(4) 1.560(2)
C(3)-H(3A) 0.9900
C(3)-H(3B) 0.9900
C(4)-C(15) 1.508(2)
C(4)-C(5) 1.537(2)
C(4)-H(4) 1.0000
C(5)-O(1) 1.207(2)
C(5)-C(6) 1.585(2)
C(6)-O(2) 1.4108(18)
C(6)-O(3) 1.4151(18)
C(6)-C(7) 1.551(2)
C(7)-C(16) 1.521(2)
C(7)-C(8) 1.560(2)
C(8)-C(20) 1.509(2)
C(8)-C(9) 1.5472(19)
C(8)-C(17) 1.5575(19)
C(9)-C(10) 1.549(2)
C(9)-H(9A) 0.9900
C(9)-H(9B) 0.9900
C(10)-C(11) 1.547(2)
C(10)-H(10) 1.0000
C(11)-C(28) 1.518(2)
C(11)-C(12) 1.530(2)
C(11)-H(11) 1.0000
C(12)-C(13) 1.543(2)
C(12)-H(12A) 0.9900
C(12)-H(12B) 0.9900
C(13)-C(14) 1.537(2)
C(13)-H(13A) 0.9900
C(13)-H(13B) 0.9900
C(14)-O(8) 1.4429(17)
C(14)-H(14) 1.0000
C(15)-C(16) 1.325(2)
C(15)-H(15) 0.9500
C(16)-H(16) 0.9500
C(17)-O(4) 1.4124(17)
C(17)-H(17) 1.0000
C(18)-O(2) 1.4347(19)
C(18)-H(18A) 0.9800
C(18)-H(18B) 0.9800
C(18)-H(18C) 0.9800
C(19)-O(3) 1.427(2)
C(19)-H(19A) 0.9800
C(19)-H(19B) 0.9800
C(19)-H(19C) 0.9800
C(20)-O(5) 1.4578(18)
C(20)-H(20A) 0.9900
C(20)-H(20B) 0.9900
C(21)-O(6) 1.205(2)
C(21)-O(5) 1.3422(19)
C(21)-C(22) 1.496(2)
C(22)-C(23) 1.394(2)
C(22)-C(27) 1.394(2)
C(23)-C(24) 1.389(2)
C(23)-H(23) 0.9500
C(24)-C(25) 1.387(3)
C(24)-H(24) 0.9500
C(25)-C(26) 1.386(3)
C(25)-Br(1) 1.8967(16)
C(26)-C(27) 1.388(2)
C(26)-H(26) 0.9500

! ""#!
C(27)-H(27) 0.9500
C(28)-O(7) 1.4283(18)
C(28)-H(28A) 0.9900
C(28)-H(28B) 0.9900
C(29)-Si(1) 1.8667(19)
C(29)-H(29A) 0.9800
C(29)-H(29B) 0.9800
C(29)-H(29C) 0.9800
C(30)-Si(1) 1.862(2)
C(30)-H(30A) 0.9800
C(30)-H(30B) 0.9800
C(30)-H(30C) 0.9800
C(31)-C(33) 1.533(3)
C(31)-C(32) 1.536(3)
C(31)-C(34) 1.537(2)
C(31)-Si(1) 1.8742(18)
C(32)-H(32A) 0.9800
C(32)-H(32B) 0.9800
C(32)-H(32C) 0.9800
C(33)-H(33A) 0.9800
C(33)-H(33B) 0.9800
C(33)-H(33C) 0.9800
C(34)-H(34A) 0.9800
C(34)-H(34B) 0.9800
C(34)-H(34C) 0.9800
C(35)-O(8) 1.4195(18)
C(35)-H(35A) 0.9800
C(35)-H(35B) 0.9800
C(35)-H(35C) 0.9800
O(4)-H(4A) 0.8400
O(7)-Si(1) 1.6529(11)
C(14)-C(1)-C(2) 114.42(12)
C(14)-C(1)-C(17) 120.22(12)
C(2)-C(1)-C(17) 95.88(11)
C(14)-C(1)-C(10) 111.12(12)
C(2)-C(1)-C(10) 108.82(11)
C(17)-C(1)-C(10) 104.95(11)
C(3)-C(2)-C(1) 123.81(13)
C(3)-C(2)-C(7) 111.60(12)
C(1)-C(2)-C(7) 102.85(11)
C(3)-C(2)-H(2) 105.8
C(1)-C(2)-H(2) 105.8
C(7)-C(2)-H(2) 105.8
C(2)-C(3)-C(4) 105.01(12)
C(2)-C(3)-H(3A) 110.7
C(4)-C(3)-H(3A) 110.7
C(2)-C(3)-H(3B) 110.7
C(4)-C(3)-H(3B) 110.7
H(3A)-C(3)-H(3B) 108.8
C(15)-C(4)-C(5) 105.33(13)
C(15)-C(4)-C(3) 105.88(13)
C(5)-C(4)-C(3) 109.27(13)
C(15)-C(4)-H(4) 112.0
C(5)-C(4)-H(4) 112.0
C(3)-C(4)-H(4) 112.0
O(1)-C(5)-C(4) 123.38(15)
O(1)-C(5)-C(6) 122.49(15)
C(4)-C(5)-C(6) 114.12(13)
O(2)-C(6)-O(3) 109.84(12)
O(2)-C(6)-C(7) 108.74(12)
O(3)-C(6)-C(7) 109.89(12)
O(2)-C(6)-C(5) 110.21(12)
O(3)-C(6)-C(5) 114.28(12)
C(7)-C(6)-C(5) 103.62(12)
C(16)-C(7)-C(6) 102.44(12)
C(16)-C(7)-C(8) 114.90(12)
C(6)-C(7)-C(8) 121.16(12)
C(16)-C(7)-C(2) 112.66(12)
C(6)-C(7)-C(2) 103.73(11)

! ""$!
C(8)-C(7)-C(2) 101.67(11)
C(20)-C(8)-C(9) 113.43(12)
C(20)-C(8)-C(17) 114.72(12)
C(9)-C(8)-C(17) 98.51(11)
C(20)-C(8)-C(7) 117.13(12)
C(9)-C(8)-C(7) 108.54(11)
C(17)-C(8)-C(7) 102.44(11)
C(8)-C(9)-C(10) 103.31(11)
C(8)-C(9)-H(9A) 111.1
C(10)-C(9)-H(9A) 111.1
C(8)-C(9)-H(9B) 111.1
C(10)-C(9)-H(9B) 111.1
H(9A)-C(9)-H(9B) 109.1
C(11)-C(10)-C(9) 117.37(12)
C(11)-C(10)-C(1) 113.44(12)
C(9)-C(10)-C(1) 102.30(11)
C(11)-C(10)-H(10) 107.7
C(9)-C(10)-H(10) 107.7
C(1)-C(10)-H(10) 107.7
C(28)-C(11)-C(12) 109.55(12)
C(28)-C(11)-C(10) 112.53(12)
C(12)-C(11)-C(10) 112.33(12)
C(28)-C(11)-H(11) 107.4
C(12)-C(11)-H(11) 107.4
C(10)-C(11)-H(11) 107.4
C(11)-C(12)-C(13) 111.66(13)
C(11)-C(12)-H(12A) 109.3
C(13)-C(12)-H(12A) 109.3
C(11)-C(12)-H(12B) 109.3
C(13)-C(12)-H(12B) 109.3
H(12A)-C(12)-H(12B) 107.9
C(14)-C(13)-C(12) 112.97(12)
C(14)-C(13)-H(13A) 109.0
C(12)-C(13)-H(13A) 109.0
C(14)-C(13)-H(13B) 109.0
C(12)-C(13)-H(13B) 109.0
H(13A)-C(13)-H(13B) 107.8
O(8)-C(14)-C(1) 108.38(11)
O(8)-C(14)-C(13) 110.90(12)
C(1)-C(14)-C(13) 114.23(12)
O(8)-C(14)-H(14) 107.7
C(1)-C(14)-H(14) 107.7
C(13)-C(14)-H(14) 107.7
C(16)-C(15)-C(4) 114.15(14)
C(16)-C(15)-H(15) 122.9
C(4)-C(15)-H(15) 122.9
C(15)-C(16)-C(7) 115.86(14)
C(15)-C(16)-H(16) 122.1
C(7)-C(16)-H(16) 122.1
O(4)-C(17)-C(1) 115.62(12)
O(4)-C(17)-C(8) 113.04(12)
C(1)-C(17)-C(8) 94.36(11)
O(4)-C(17)-H(17) 110.9
C(1)-C(17)-H(17) 110.9
C(8)-C(17)-H(17) 110.9
O(2)-C(18)-H(18A) 109.5
O(2)-C(18)-H(18B) 109.5
H(18A)-C(18)-H(18B) 109.5
O(2)-C(18)-H(18C) 109.5
H(18A)-C(18)-H(18C) 109.5
H(18B)-C(18)-H(18C) 109.5
O(3)-C(19)-H(19A) 109.5
O(3)-C(19)-H(19B) 109.5
H(19A)-C(19)-H(19B) 109.5
O(3)-C(19)-H(19C) 109.5
H(19A)-C(19)-H(19C) 109.5
H(19B)-C(19)-H(19C) 109.5
O(5)-C(20)-C(8) 109.74(12)
O(5)-C(20)-H(20A) 109.7
C(8)-C(20)-H(20A) 109.7
O(5)-C(20)-H(20B) 109.7
C(8)-C(20)-H(20B) 109.7

! ""%!
H(20A)-C(20)-H(20B) 108.2
O(6)-C(21)-O(5) 125.30(15)
O(6)-C(21)-C(22) 124.17(14)
O(5)-C(21)-C(22) 110.51(13)
C(23)-C(22)-C(27) 119.78(15)
C(23)-C(22)-C(21) 122.08(14)
C(27)-C(22)-C(21) 118.10(15)
C(24)-C(23)-C(22) 120.01(15)
C(24)-C(23)-H(23) 120.0
C(22)-C(23)-H(23) 120.0
C(25)-C(24)-C(23) 119.31(16)
C(25)-C(24)-H(24) 120.3
C(23)-C(24)-H(24) 120.3
C(26)-C(25)-C(24) 121.48(15)
C(26)-C(25)-Br(1) 118.85(13)
C(24)-C(25)-Br(1) 119.67(14)
C(25)-C(26)-C(27) 118.89(16)
C(25)-C(26)-H(26) 120.6
C(27)-C(26)-H(26) 120.6
C(26)-C(27)-C(22) 120.49(16)
C(26)-C(27)-H(27) 119.8
C(22)-C(27)-H(27) 119.8
O(7)-C(28)-C(11) 110.10(12)
O(7)-C(28)-H(28A) 109.6
C(11)-C(28)-H(28A) 109.6
O(7)-C(28)-H(28B) 109.6
C(11)-C(28)-H(28B) 109.6
H(28A)-C(28)-H(28B) 108.2
Si(1)-C(29)-H(29A) 109.5
Si(1)-C(29)-H(29B) 109.5
H(29A)-C(29)-H(29B) 109.5
Si(1)-C(29)-H(29C) 109.5
H(29A)-C(29)-H(29C) 109.5
H(29B)-C(29)-H(29C) 109.5
Si(1)-C(30)-H(30A) 109.5
Si(1)-C(30)-H(30B) 109.5
H(30A)-C(30)-H(30B) 109.5
Si(1)-C(30)-H(30C) 109.5
H(30A)-C(30)-H(30C) 109.5
H(30B)-C(30)-H(30C) 109.5
C(33)-C(31)-C(32) 108.7(2)
C(33)-C(31)-C(34) 109.78(18)
C(32)-C(31)-C(34) 108.85(15)
C(33)-C(31)-Si(1) 110.00(13)
C(32)-C(31)-Si(1) 110.46(14)
C(34)-C(31)-Si(1) 109.03(13)
C(31)-C(32)-H(32A) 109.5
C(31)-C(32)-H(32B) 109.5
H(32A)-C(32)-H(32B) 109.5
C(31)-C(32)-H(32C) 109.5
H(32A)-C(32)-H(32C) 109.5
H(32B)-C(32)-H(32C) 109.5
C(31)-C(33)-H(33A) 109.5
C(31)-C(33)-H(33B) 109.5
H(33A)-C(33)-H(33B) 109.5
C(31)-C(33)-H(33C) 109.5
H(33A)-C(33)-H(33C) 109.5
H(33B)-C(33)-H(33C) 109.5
C(31)-C(34)-H(34A) 109.5
C(31)-C(34)-H(34B) 109.5
H(34A)-C(34)-H(34B) 109.5
C(31)-C(34)-H(34C) 109.5
H(34A)-C(34)-H(34C) 109.5
H(34B)-C(34)-H(34C) 109.5
O(8)-C(35)-H(35A) 109.5
O(8)-C(35)-H(35B) 109.5
H(35A)-C(35)-H(35B) 109.5
O(8)-C(35)-H(35C) 109.5
H(35A)-C(35)-H(35C) 109.5
H(35B)-C(35)-H(35C) 109.5
C(6)-O(2)-C(18) 117.14(12)
C(6)-O(3)-C(19) 118.95(12)

! ""&!
C(17)-O(4)-H(4A) 109.5
C(21)-O(5)-C(20) 118.56(12)
C(28)-O(7)-Si(1) 121.77(10)
C(35)-O(8)-C(14) 113.34(11)
O(7)-Si(1)-C(30) 110.71(7)
O(7)-Si(1)-C(29) 109.67(8)
C(30)-Si(1)-C(29) 108.42(10)
O(7)-Si(1)-C(31) 104.75(7)
C(30)-Si(1)-C(31) 111.93(10)
C(29)-Si(1)-C(31) 111.34(9)

! ""'!
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms:

! ""(!
Table 4. Anisotropic displacement parameters (Å2x 103) for sarpong37. The anisotropic
displacement factor exponent takes the form: -2p2[ h2 a*2U11 + ... + 2 h k a* b* U12 ]
______________________________________________________________________________
U11 U22 U33 U23 U13 U12
______________________________________________________________________________
C(1) 12(1) 12(1) 12(1) -2(1) 1(1) 1(1)
C(2) 12(1) 14(1) 14(1) 0(1) 1(1) 1(1)
C(3) 16(1) 16(1) 20(1) 1(1) 1(1) 4(1)
C(4) 22(1) 14(1) 25(1) 1(1) 3(1) 5(1)
C(5) 20(1) 13(1) 22(1) 1(1) -2(1) -2(1)
C(6) 16(1) 15(1) 16(1) 0(1) 0(1) -2(1)
C(7) 13(1) 13(1) 13(1) -1(1) 1(1) 0(1)
C(8) 13(1) 13(1) 12(1) -1(1) 0(1) 0(1)
C(9) 13(1) 14(1) 12(1) 0(1) 0(1) -1(1)
C(10) 13(1) 12(1) 12(1) -1(1) 0(1) -1(1)
C(11) 15(1) 16(1) 13(1) -1(1) 2(1) -2(1)
C(12) 20(1) 15(1) 15(1) 1(1) 2(1) -3(1)
C(13) 19(1) 18(1) 17(1) -2(1) 1(1) -6(1)
C(14) 13(1) 18(1) 12(1) -4(1) 1(1) -1(1)
C(15) 25(1) 14(1) 20(1) -3(1) 4(1) -2(1)
C(16) 18(1) 15(1) 16(1) -2(1) 2(1) -3(1)
C(17) 13(1) 12(1) 12(1) 0(1) 1(1) 0(1)
C(18) 30(1) 27(1) 15(1) 2(1) 6(1) -2(1)
C(19) 26(1) 20(1) 31(1) 5(1) 2(1) -7(1)
C(20) 12(1) 16(1) 17(1) -3(1) 1(1) -1(1)
C(21) 12(1) 21(1) 18(1) -1(1) 3(1) 3(1)
C(22) 12(1) 21(1) 20(1) -4(1) 3(1) 1(1)
C(23) 16(1) 21(1) 22(1) -2(1) 3(1) 2(1)
C(24) 21(1) 19(1) 31(1) -5(1) 9(1) 0(1)
C(25) 17(1) 27(1) 30(1) -15(1) 7(1) -5(1)
C(26) 18(1) 34(1) 24(1) -10(1) -1(1) -1(1)
C(27) 18(1) 25(1) 22(1) -2(1) 1(1) 2(1)
C(28) 23(1) 16(1) 15(1) 0(1) -1(1) -4(1)
C(29) 45(1) 39(1) 22(1) 8(1) -7(1) -13(1)
C(30) 27(1) 45(1) 30(1) -6(1) -7(1) 7(1)
C(31) 39(1) 22(1) 21(1) -2(1) -6(1) -7(1)

! "")!
C(32) 46(1) 38(1) 51(1) -20(1) -22(1) 11(1)
C(33) 121(3) 30(1) 33(1) 4(1) 2(1) -30(1)
C(34) 34(1) 34(1) 37(1) -13(1) -7(1) -9(1)
C(35) 12(1) 36(1) 21(1) -4(1) -2(1) -2(1)
O(1) 30(1) 21(1) 24(1) 6(1) -3(1) 2(1)
O(2) 24(1) 18(1) 12(1) 0(1) 3(1) -2(1)
O(3) 17(1) 17(1) 23(1) 4(1) 2(1) -4(1)
O(4) 21(1) 11(1) 15(1) -2(1) 4(1) 2(1)
O(5) 12(1) 18(1) 19(1) 0(1) -1(1) -2(1)
O(6) 23(1) 20(1) 27(1) 4(1) -5(1) 0(1)
O(7) 23(1) 18(1) 16(1) -3(1) -4(1) -2(1)
O(8) 11(1) 22(1) 13(1) -4(1) -1(1) 0(1)
Si(1) 22(1) 20(1) 14(1) 0(1) -3(1) -3(1)
Br(1) 22(1) 35(1) 49(1) -22(1) 9(1) -10(1)
______________________________________________________________________________

! ""*!
Table 5. Hydrogen coordinates ( x 104) and isotropic displacement parameters (Å2x 10 3)
for sarpong37.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________
H(2) 10534 2914 9902 16
H(3A) 12120 2042 9602 21
H(3B) 11517 2010 8938 21
H(4) 11041 638 9534 24
H(9A) 8013 4164 8442 16
H(9B) 7932 3118 8322 16
H(10) 10153 2944 8392 15
H(11) 11401 3954 7978 17
H(12A) 9986 5366 8499 20
H(12B) 11203 5503 8083 20
H(13A) 12735 5134 8781 21
H(13B) 11613 5477 9198 21
H(14) 12419 3720 8998 17
H(15) 9308 580 8773 23
H(16) 7944 1826 8642 20
H(17) 9367 4034 9909 15
H(18A) 7219 2296 10935 36
H(18B) 8315 2909 11264 36
H(18C) 8652 1901 11130 36
H(19A) 7551 414 9859 38
H(19B) 6110 663 10079 38
H(19C) 7397 730 10518 38
H(20A) 6759 3521 9831 18
H(20B) 6500 4174 9287 18
H(23) 5030 1472 8984 24
H(24) 3617 343 8629 28
H(26) 1950 2014 7418 30
H(27) 3385 3135 7764 26
H(28A) 8831 4636 7623 21
H(28B) 10148 4713 7258 21

! "#+!
H(29A) 8782 4595 6154 54
H(29B) 7956 3888 5765 54
H(29C) 9497 3718 5942 54
H(30A) 6314 3573 7336 52
H(30B) 5898 3811 6671 52
H(30C) 6726 4503 7071 52
H(32A) 10071 1926 6747 69
H(32B) 9601 2054 6075 69
H(32C) 9289 1152 6402 69
H(33A) 7598 1081 7133 92
H(33B) 6793 1941 7312 92
H(33C) 8339 1842 7507 92
H(34A) 7232 2256 5778 53
H(34B) 6113 2250 6252 53
H(34C) 6853 1355 6092 53
H(35A) 14007 3601 9774 35
H(35B) 13893 4275 10306 35
H(35C) 14021 4640 9656 35
H(4A) 8627 5236 9603 23
________________________________________________________________________________

! "#"!
Crystallographic data for 2.106: A colorless plate 0.040 x 0.030 x 0.030 mm in size was mounted on a Cryoloop with Paratone oil. Data were collected in a nitrogen gas stream at 100(2) K using phi and omega scans. Crystal-to-detector distance was 60 mm and exposure time was 10 seconds per frame using a scan width of 1.0°. Data collection was 99.3% complete to 67.000° in q. A total of 23099 reflections were collected covering the indices, -17<=h<=17, -8<=k<=11, -17<=l<=17. 3314 reflections were found to be symmetry independent, with an Rint of 0.0228. Indexing and unit cell refinement indicated a primitive, monoclinic lattice. The space group was found to be P 21/c (No. 14). The data were integrated using the Bruker SAINT software program and scaled using the SADABS software program. Solution by iterative methods (SHELXT) produced a complete heavy-atom phasing model consistent with the proposed structure. All non-hydrogen atoms were refined anisotropically by full-matrix least-squares (SHELXL-2013). All hydrogen atoms were placed using a riding model. Their positions were constrained relative to their parent atom using the appropriate HFIX command in SHELXL-2013.

! "##!
Table 1. Crystal data and structure refinement for sarpong60. X-ray ID sarpong60
Sample/notebook ID GG09-66B
Empirical formula C21 H27 N O5
Formula weight 373.43
Temperature 100(2) K
Wavelength 1.54178 Å
Crystal system Monoclinic
Space group P 21/c
Unit cell dimensions a = 14.3864(12) Å a= 90°.
b = 9.9205(8) Å b= 115.337(4)°.
c = 14.1698(12) Å g = 90°.
Volume 1827.8(3) Å3
Z 4
Density (calculated) 1.357 Mg/m3
Absorption coefficient 0.788 mm-1
F(000) 800
Crystal size 0.040 x 0.030 x 0.030 mm3
Crystal color/habit colorless plate
Theta range for data collection 3.399 to 68.280°.
Index ranges -17<=h<=17, -8<=k<=11, -17<=l<=17
Reflections collected 23099
Independent reflections 3314 [R(int) = 0.0228]
Completeness to theta = 67.000° 99.3 %
Absorption correction Semi-empirical from equivalents
Max. and min. transmission 0.929 and 0.872
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 3314 / 0 / 247
Goodness-of-fit on F2 1.052
Final R indices [I>2sigma(I)] R1 = 0.0319, wR2 = 0.0842
R indices (all data) R1 = 0.0338, wR2 = 0.0861
Extinction coefficient n/a
Largest diff. peak and hole 0.298 and -0.178 e.Å-3
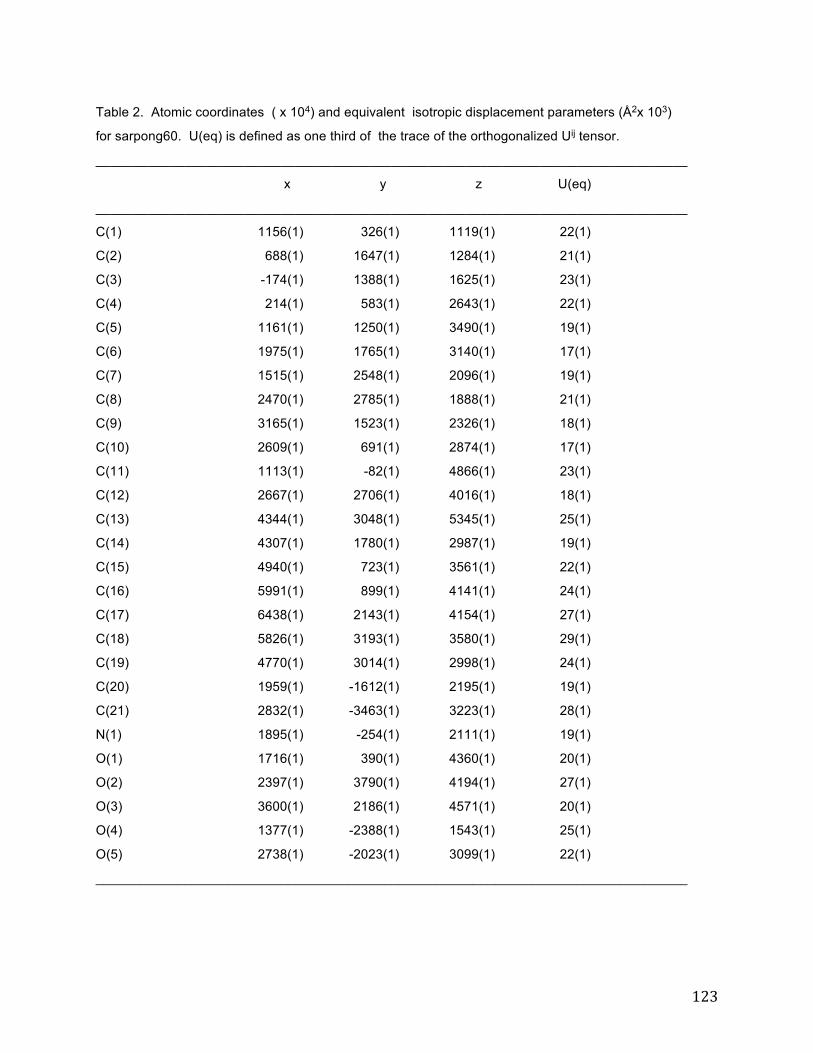
! "#$!
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2x 103)
for sarpong60. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________ C(1) 1156(1) 326(1) 1119(1) 22(1)
C(2) 688(1) 1647(1) 1284(1) 21(1)
C(3) -174(1) 1388(1) 1625(1) 23(1)
C(4) 214(1) 583(1) 2643(1) 22(1)
C(5) 1161(1) 1250(1) 3490(1) 19(1)
C(6) 1975(1) 1765(1) 3140(1) 17(1)
C(7) 1515(1) 2548(1) 2096(1) 19(1)
C(8) 2470(1) 2785(1) 1888(1) 21(1)
C(9) 3165(1) 1523(1) 2326(1) 18(1)
C(10) 2609(1) 691(1) 2874(1) 17(1)
C(11) 1113(1) -82(1) 4866(1) 23(1)
C(12) 2667(1) 2706(1) 4016(1) 18(1)
C(13) 4344(1) 3048(1) 5345(1) 25(1)
C(14) 4307(1) 1780(1) 2987(1) 19(1)
C(15) 4940(1) 723(1) 3561(1) 22(1)
C(16) 5991(1) 899(1) 4141(1) 24(1)
C(17) 6438(1) 2143(1) 4154(1) 27(1)
C(18) 5826(1) 3193(1) 3580(1) 29(1)
C(19) 4770(1) 3014(1) 2998(1) 24(1)
C(20) 1959(1) -1612(1) 2195(1) 19(1)
C(21) 2832(1) -3463(1) 3223(1) 28(1)
N(1) 1895(1) -254(1) 2111(1) 19(1)
O(1) 1716(1) 390(1) 4360(1) 20(1)
O(2) 2397(1) 3790(1) 4194(1) 27(1)
O(3) 3600(1) 2186(1) 4571(1) 20(1)
O(4) 1377(1) -2388(1) 1543(1) 25(1)
O(5) 2738(1) -2023(1) 3099(1) 22(1)
________________________________________________________________________________

! "#%!
Table 3. Bond lengths [Å] and angles [°] for sarpong60.
_____________________________________________________
C(1)-N(1) 1.4692(14)
C(1)-C(2) 1.5361(16)
C(1)-H(1A) 0.9900
C(1)-H(1B) 0.9900
C(2)-C(3) 1.5325(17)
C(2)-C(7) 1.5391(16)
C(2)-H(2) 1.0000
C(3)-C(4) 1.5309(17)
C(3)-H(3A) 0.9900
C(3)-H(3B) 0.9900
C(4)-C(5) 1.5283(15)
C(4)-H(4A) 0.9900
C(4)-H(4B) 0.9900
C(5)-O(1) 1.4286(14)
C(5)-C(6) 1.5420(15)
C(5)-H(5) 1.0000
C(6)-C(12) 1.5309(15)
C(6)-C(7) 1.5464(15)
C(6)-C(10) 1.5507(15)
C(7)-C(8) 1.5416(16)
C(7)-H(7) 1.0000
C(8)-C(9) 1.5560(15)
C(8)-H(8A) 0.9900
C(8)-H(8B) 0.9900
C(9)-C(14) 1.5251(16)
C(9)-C(10) 1.5676(15)
C(9)-H(9) 1.0000
C(10)-N(1) 1.4674(14)
C(10)-H(10) 1.0000
C(11)-O(1) 1.4212(14)
C(11)-H(11A) 0.9800
C(11)-H(11B) 0.9800
C(11)-H(11C) 0.9800
C(12)-O(2) 1.2064(15)
C(12)-O(3) 1.3357(14)
C(13)-O(3) 1.4406(14)
C(13)-H(13A) 0.9800
C(13)-H(13B) 0.9800
C(13)-H(13C) 0.9800
C(14)-C(19) 1.3900(17)
C(14)-C(15) 1.3984(16)
C(15)-C(16) 1.3889(17)
C(15)-H(15) 0.9500
C(16)-C(17) 1.3883(19)
C(16)-H(16) 0.9500
C(17)-C(18) 1.3812(19)
C(17)-H(17) 0.9500
C(18)-C(19) 1.3951(18)
C(18)-H(18) 0.9500
C(19)-H(19) 0.9500
C(20)-O(4) 1.2193(14)
C(20)-N(1) 1.3517(16)
C(20)-O(5) 1.3541(14)
C(21)-O(5) 1.4383(15)
C(21)-H(21A) 0.9800
C(21)-H(21B) 0.9800
C(21)-H(21C) 0.9800
N(1)-C(1)-C(2) 111.93(9)
N(1)-C(1)-H(1A) 109.2
C(2)-C(1)-H(1A) 109.2
N(1)-C(1)-H(1B) 109.2
C(2)-C(1)-H(1B) 109.2
H(1A)-C(1)-H(1B) 107.9
C(3)-C(2)-C(1) 111.84(10)
C(3)-C(2)-C(7) 109.46(10)

! "#&!
C(1)-C(2)-C(7) 111.10(9)
C(3)-C(2)-H(2) 108.1
C(1)-C(2)-H(2) 108.1
C(7)-C(2)-H(2) 108.1
C(4)-C(3)-C(2) 111.50(9)
C(4)-C(3)-H(3A) 109.3
C(2)-C(3)-H(3A) 109.3
C(4)-C(3)-H(3B) 109.3
C(2)-C(3)-H(3B) 109.3
H(3A)-C(3)-H(3B) 108.0
C(5)-C(4)-C(3) 110.92(10)
C(5)-C(4)-H(4A) 109.5
C(3)-C(4)-H(4A) 109.5
C(5)-C(4)-H(4B) 109.5
C(3)-C(4)-H(4B) 109.5
H(4A)-C(4)-H(4B) 108.0
O(1)-C(5)-C(4) 113.59(9)
O(1)-C(5)-C(6) 104.71(8)
C(4)-C(5)-C(6) 115.85(10)
O(1)-C(5)-H(5) 107.4
C(4)-C(5)-H(5) 107.4
C(6)-C(5)-H(5) 107.4
C(12)-C(6)-C(5) 105.16(9)
C(12)-C(6)-C(7) 109.43(9)
C(5)-C(6)-C(7) 113.65(9)
C(12)-C(6)-C(10) 111.73(9)
C(5)-C(6)-C(10) 117.21(9)
C(7)-C(6)-C(10) 99.68(9)
C(2)-C(7)-C(8) 114.39(10)
C(2)-C(7)-C(6) 107.50(9)
C(8)-C(7)-C(6) 102.01(9)
C(2)-C(7)-H(7) 110.8
C(8)-C(7)-H(7) 110.8
C(6)-C(7)-H(7) 110.8
C(7)-C(8)-C(9) 105.91(9)
C(7)-C(8)-H(8A) 110.6
C(9)-C(8)-H(8A) 110.6
C(7)-C(8)-H(8B) 110.6
C(9)-C(8)-H(8B) 110.6
H(8A)-C(8)-H(8B) 108.7
C(14)-C(9)-C(8) 116.73(10)
C(14)-C(9)-C(10) 115.31(9)
C(8)-C(9)-C(10) 104.14(9)
C(14)-C(9)-H(9) 106.7
C(8)-C(9)-H(9) 106.7
C(10)-C(9)-H(9) 106.7
N(1)-C(10)-C(6) 108.61(9)
N(1)-C(10)-C(9) 108.42(9)
C(6)-C(10)-C(9) 103.68(9)
N(1)-C(10)-H(10) 111.9
C(6)-C(10)-H(10) 111.9
C(9)-C(10)-H(10) 111.9
O(1)-C(11)-H(11A) 109.5
O(1)-C(11)-H(11B) 109.5
H(11A)-C(11)-H(11B) 109.5
O(1)-C(11)-H(11C) 109.5
H(11A)-C(11)-H(11C) 109.5
H(11B)-C(11)-H(11C) 109.5
O(2)-C(12)-O(3) 123.60(11)
O(2)-C(12)-C(6) 123.89(10)
O(3)-C(12)-C(6) 112.51(9)
O(3)-C(13)-H(13A) 109.5
O(3)-C(13)-H(13B) 109.5
H(13A)-C(13)-H(13B) 109.5
O(3)-C(13)-H(13C) 109.5
H(13A)-C(13)-H(13C) 109.5
H(13B)-C(13)-H(13C) 109.5
C(19)-C(14)-C(15) 117.67(11)
C(19)-C(14)-C(9) 122.61(10)
C(15)-C(14)-C(9) 119.60(10)
C(16)-C(15)-C(14) 121.52(11)
C(16)-C(15)-H(15) 119.2

! "#'!
C(14)-C(15)-H(15) 119.2
C(17)-C(16)-C(15) 120.00(11)
C(17)-C(16)-H(16) 120.0
C(15)-C(16)-H(16) 120.0
C(18)-C(17)-C(16) 119.18(11)
C(18)-C(17)-H(17) 120.4
C(16)-C(17)-H(17) 120.4
C(17)-C(18)-C(19) 120.71(12)
C(17)-C(18)-H(18) 119.6
C(19)-C(18)-H(18) 119.6
C(14)-C(19)-C(18) 120.89(12)
C(14)-C(19)-H(19) 119.6
C(18)-C(19)-H(19) 119.6
O(4)-C(20)-N(1) 124.34(11)
O(4)-C(20)-O(5) 123.25(11)
N(1)-C(20)-O(5) 112.41(10)
O(5)-C(21)-H(21A) 109.5
O(5)-C(21)-H(21B) 109.5
H(21A)-C(21)-H(21B) 109.5
O(5)-C(21)-H(21C) 109.5
H(21A)-C(21)-H(21C) 109.5
H(21B)-C(21)-H(21C) 109.5
C(20)-N(1)-C(10) 124.89(9)
C(20)-N(1)-C(1) 117.97(10)
C(10)-N(1)-C(1) 116.50(9)
C(11)-O(1)-C(5) 113.31(9)
C(12)-O(3)-C(13) 116.84(9)
C(20)-O(5)-C(21) 114.31(9)

! "#(!
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms:

! "#)!
Table 4. Anisotropic displacement parameters (Å2x 103) for sarpong60. The anisotropic
displacement factor exponent takes the form: -2p2[ h2 a*2U11 + ... + 2 h k a* b* U12 ]
______________________________________________________________________________
U11 U22 U33 U23 U13 U12
______________________________________________________________________________
C(1) 21(1) 19(1) 19(1) -1(1) 3(1) 2(1)
C(2) 19(1) 18(1) 21(1) 2(1) 4(1) 3(1)
C(3) 17(1) 20(1) 27(1) 0(1) 4(1) 1(1)
C(4) 17(1) 18(1) 29(1) 1(1) 8(1) -1(1)
C(5) 17(1) 16(1) 23(1) 1(1) 9(1) 1(1)
C(6) 16(1) 14(1) 21(1) 0(1) 7(1) 1(1)
C(7) 19(1) 15(1) 22(1) 2(1) 8(1) 2(1)
C(8) 21(1) 18(1) 23(1) 4(1) 9(1) 3(1)
C(9) 20(1) 16(1) 19(1) 1(1) 9(1) 2(1)
C(10) 15(1) 15(1) 18(1) 0(1) 6(1) 0(1)
C(11) 24(1) 22(1) 27(1) -2(1) 15(1) -6(1)
C(12) 18(1) 16(1) 22(1) 2(1) 10(1) -1(1)
C(13) 22(1) 28(1) 23(1) -7(1) 7(1) -6(1)
C(14) 21(1) 20(1) 18(1) 0(1) 11(1) 1(1)
C(15) 21(1) 19(1) 25(1) 1(1) 11(1) 1(1)
C(16) 21(1) 26(1) 24(1) 3(1) 9(1) 4(1)
C(17) 19(1) 33(1) 28(1) -2(1) 9(1) -2(1)
C(18) 26(1) 24(1) 40(1) 1(1) 16(1) -6(1)
C(19) 24(1) 21(1) 30(1) 5(1) 13(1) 1(1)
C(20) 17(1) 18(1) 22(1) -1(1) 8(1) 1(1)
C(21) 26(1) 14(1) 35(1) 3(1) 6(1) 1(1)
N(1) 18(1) 15(1) 19(1) -1(1) 4(1) 1(1)
O(1) 18(1) 21(1) 23(1) 4(1) 10(1) 0(1)
O(2) 27(1) 17(1) 33(1) -5(1) 10(1) 1(1)
O(3) 18(1) 20(1) 21(1) -3(1) 6(1) -1(1)
O(4) 24(1) 17(1) 28(1) -5(1) 5(1) -2(1)
O(5) 21(1) 13(1) 25(1) 1(1) 5(1) 1(1)
______________________________________________________________________________

! "#*!
Table 5. Hydrogen coordinates ( x 104) and isotropic displacement parameters (Å2x 10 3)
for sarpong60.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________
H(1A) 599 -332 759 26
H(1B) 1508 501 665 26
H(2) 387 2144 605 25
H(3A) -455 2261 1722 28
H(3B) -737 885 1069 28
H(4A) -338 522 2883 26
H(4B) 389 -344 2517 26
H(5) 917 2043 3758 23
H(7) 1213 3424 2178 23
H(8A) 2838 3612 2248 25
H(8B) 2269 2883 1131 25
H(9) 3111 983 1708 22
H(10) 3110 218 3513 20
H(11A) 737 674 4984 35
H(11B) 1563 -488 5538 35
H(11C) 622 -759 4426 35
H(13A) 4370 3917 5027 38
H(13B) 5024 2621 5617 38
H(13C) 4146 3194 5918 38
H(15) 4644 -134 3555 26
H(16) 6405 167 4527 29
H(17) 7156 2272 4554 32
H(18) 6127 4046 3582 35
H(19) 4362 3745 2603 29
H(21A) 2159 -3852 3081 41
H(21B) 3321 -3680 3939 41
H(21C) 3080 -3838 2733 41 ________________________________________________________________________________

! "$+!
Crystallographic data for 2.122: A colorless plate 0.050 x 0.040 x 0.020 mm in size was mounted on a Cryoloop with Paratone oil. Data were collected in a nitrogen gas stream at 100(2) K using and scans. Crystal-to-detector distance was 60 mm and exposure time was 20 seconds per frame using a scan width of 1.0°. Data collection was 93.1% complete to 67.000° in q. A total of 47090 reflections were collected covering the indices, -9<=h<=10, -62<=k<=62, -13<=l<=14. 47090 reflections were found to be symmetry independent, with an Rint of 0.0729. Indexing and unit cell refinement indicated a primitive, monoclinic lattice. The space group was found to be P 2(1)/a (No. 14). The data were integrated using the Bruker SAINT software program and scaled using the TWINABS software program. Solution by iterative methods (SHELXT) produced a complete heavy-atom phasing model consistent with the proposed structure. All non-hydrogen atoms were refined anisotropically by full-matrix least-squares (SHELXL-2013). All hydrogen atoms were placed using a riding model. Their positions were constrained relative to their parent atom using the appropriate HFIX command in SHELXL-2013.

! "$"!
Table 1. Crystal data and structure refinement for sarpong64. X-ray ID sarpong64
Sample/notebook ID JLII-050B
Empirical formula C30 H36 N2 O9
Formula weight 568.61
Temperature 100(2) K
Wavelength 1.54178 Å
Crystal system Monoclinic
Space group P 2(1)/a
Unit cell dimensions a = 9.0579(6) Å a= 90°.
b = 51.983(3) Å b= 90.896(4)°.
c = 11.7360(8) Å g = 90°.
Volume 5525.3(6) Å3
Z 8
Density (calculated) 1.367 Mg/m3
Absorption coefficient 0.840 mm-1
F(000) 2416
Crystal size 0.050 x 0.040 x 0.020 mm3
Crystal color/habit colorless plate
Theta range for data collection 3.401 to 68.876°.
Index ranges -9<=h<=10, -62<=k<=62, -13<=l<=14
Reflections collected 47090
Independent reflections 47090 [R(int) = 0.0729]
Completeness to theta = 67.000° 93.1 %
Absorption correction Semi-empirical from equivalents
Max. and min. transmission 0.929 and 0.815
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 47090 / 0 / 749
Goodness-of-fit on F2 1.423
Final R indices [I>2sigma(I)] R1 = 0.1549, wR2 = 0.4185
R indices (all data) R1 = 0.1825, wR2 = 0.4365
Extinction coefficient n/a
Largest diff. peak and hole 1.274 and -0.770 e.Å-3

! "$#!
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2x 103)
for sarpong64. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________ C(1) -197(8) 883(1) 14473(7) 28(2)
C(2) -164(8) 1182(1) 14264(6) 26(2)
C(3) -1526(9) 1339(1) 13879(7) 30(2)
C(4) -941(9) 1552(1) 13078(7) 30(2)
C(5) 402(9) 1681(1) 13656(7) 31(2)
C(6) 1799(8) 1495(1) 13672(6) 26(2)
C(7) 1132(8) 1221(1) 13427(7) 28(2)
C(8) 1984(8) 965(1) 13617(7) 29(2)
C(9) 2420(8) 925(1) 14878(7) 30(2)
C(10) 966(9) 829(1) 15435(7) 29(2)
C(11) 1012(9) 541(1) 15684(6) 30(2)
C(12) -492(10) 448(1) 16073(7) 34(2)
C(13) -1662(9) 490(1) 15142(7) 34(2)
C(14) -1712(9) 769(1) 14732(7) 31(2)
C(15) 658(8) 776(1) 13478(7) 28(2)
C(16) 1570(9) 395(1) 14636(7) 31(2)
C(17) -472(10) 1418(1) 12007(7) 32(2)
C(18) 565(9) 1240(1) 12212(7) 29(2)
C(19) 626(10) 2137(1) 13562(7) 32(2)
C(20) 1186(9) 2351(1) 12832(7) 32(2)
C(21) 784(10) 2601(1) 13164(7) 34(2)
C(22) 1278(10) 2811(1) 12539(8) 38(2)
C(23) 2166(10) 2765(1) 11625(7) 36(2)
C(24) 2621(11) 2522(1) 11297(8) 39(2)
C(25) 2086(10) 2312(1) 11924(8) 37(2)
C(26) 4064(10) 1436(1) 12673(8) 39(2)
C(27) 3104(11) 1737(1) 15105(7) 39(2)
C(28) 1274(9) 378(1) 12561(7) 33(2)
C(29) 1944(12) -20(1) 11787(9) 46(2)
C(30) -4120(9) 798(1) 13936(8) 38(2)
C(31) 5923(8) 902(1) 9471(6) 26(2)

! "$$!
C(32) 5788(9) 1199(1) 9293(7) 28(2)
C(33) 7094(9) 1366(1) 8919(7) 31(2)
C(34) 6440(9) 1574(1) 8121(7) 31(2)
C(35) 5053(9) 1690(1) 8663(6) 28(2)
C(36) 3722(9) 1495(1) 8677(7) 29(2)
C(37) 4454(8) 1228(1) 8406(7) 27(2)
C(38) 3688(8) 966(1) 8581(7) 29(2)
C(39) 3295(9) 919(1) 9817(7) 30(2)
C(40) 4787(9) 835(1) 10390(6) 28(2)
C(41) 4838(10) 543(1) 10631(7) 33(2)
C(42) 6387(10) 467(1) 11057(7) 33(2)
C(43) 7536(9) 526(1) 10159(7) 34(2)
C(44) 7479(9) 803(1) 9738(7) 31(2)
C(45) 5084(8) 794(1) 8432(7) 28(2)
C(46) 4338(9) 392(1) 9569(7) 32(2)
C(47) 5988(9) 1440(1) 7041(6) 28(2)
C(48) 4989(9) 1254(1) 7215(7) 30(2)
C(49) 4821(10) 2147(1) 8577(8) 36(2)
C(50) 4281(9) 2365(1) 7859(7) 30(2)
C(51) 3360(10) 2329(1) 6912(7) 35(2)
C(52) 2864(11) 2539(1) 6269(8) 41(2)
C(53) 3363(10) 2783(1) 6608(7) 35(2)
C(54) 4289(10) 2822(1) 7523(8) 38(2)
C(55) 4739(10) 2613(1) 8171(8) 38(2)
C(56) 2384(11) 1725(1) 10094(7) 39(2)
C(57) 1465(10) 1426(1) 7610(7) 37(2)
C(58) 4710(9) 389(1) 7490(7) 29(2)
C(59) 4328(13) -13(1) 6587(8) 48(2)
C(60) 9824(10) 822(1) 8924(8) 40(2)
N(1) 1047(7) 505(1) 13562(6) 31(1)
N(2) 2660(10) 2989(1) 10952(7) 41(2)
N(3) 4805(7) 515(1) 8494(6) 30(2)
N(4) 2890(9) 3007(1) 5914(7) 40(2)
O(1) 757(7) 1910(1) 13007(5) 33(1)
O(2) 148(9) 2166(1) 14492(6) 50(2)
O(3) 2035(8) 3192(1) 11098(6) 47(2)
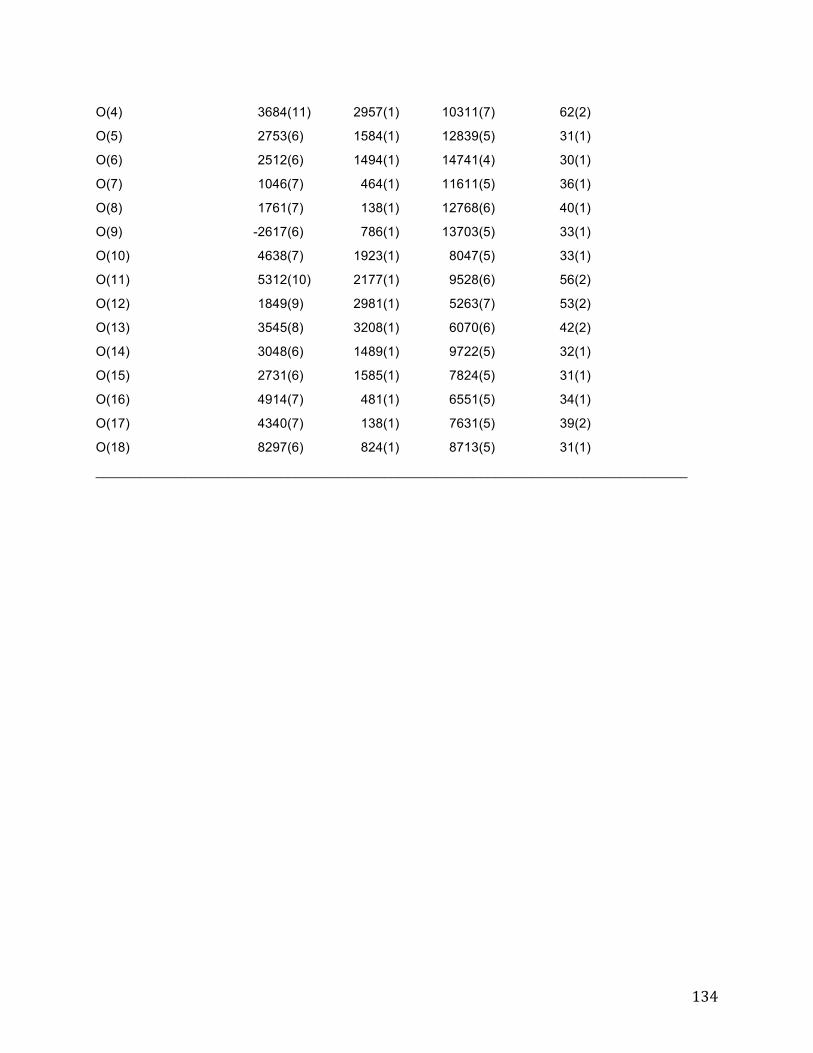
! "$%!
O(4) 3684(11) 2957(1) 10311(7) 62(2)
O(5) 2753(6) 1584(1) 12839(5) 31(1)
O(6) 2512(6) 1494(1) 14741(4) 30(1)
O(7) 1046(7) 464(1) 11611(5) 36(1)
O(8) 1761(7) 138(1) 12768(6) 40(1)
O(9) -2617(6) 786(1) 13703(5) 33(1)
O(10) 4638(7) 1923(1) 8047(5) 33(1)
O(11) 5312(10) 2177(1) 9528(6) 56(2)
O(12) 1849(9) 2981(1) 5263(7) 53(2)
O(13) 3545(8) 3208(1) 6070(6) 42(2)
O(14) 3048(6) 1489(1) 9722(5) 32(1)
O(15) 2731(6) 1585(1) 7824(5) 31(1)
O(16) 4914(7) 481(1) 6551(5) 34(1)
O(17) 4340(7) 138(1) 7631(5) 39(2)
O(18) 8297(6) 824(1) 8713(5) 31(1)
________________________________________________________________________________

! "$&!
Table 3. Bond lengths [Å] and angles [°] for sarpong64.
_____________________________________________________
C(1)-C(15) 1.516(11)
C(1)-C(14) 1.528(11)
C(1)-C(10) 1.557(10)
C(1)-C(2) 1.573(8)
C(2)-C(3) 1.542(9)
C(2)-C(7) 1.556(11)
C(2)-H(2) 1.0000
C(3)-C(4) 1.554(10)
C(3)-H(3A) 0.9900
C(3)-H(3B) 0.9900
C(4)-C(17) 1.506(10)
C(4)-C(5) 1.535(10)
C(4)-H(4) 1.0000
C(5)-O(1) 1.452(8)
C(5)-C(6) 1.590(10)
C(5)-H(5) 1.0000
C(6)-O(5) 1.394(9)
C(6)-O(6) 1.402(9)
C(6)-C(7) 1.573(8)
C(7)-C(18) 1.511(10)
C(7)-C(8) 1.551(8)
C(8)-C(9) 1.540(11)
C(8)-C(15) 1.559(9)
C(8)-H(8) 1.0000
C(9)-C(10) 1.561(11)
C(9)-H(9A) 0.9900
C(9)-H(9B) 0.9900
C(10)-C(11) 1.529(8)
C(10)-H(10) 1.0000
C(11)-C(12) 1.522(12)
C(11)-C(16) 1.538(11)
C(11)-H(11) 1.0000
C(12)-C(13) 1.526(12)
C(12)-H(12A) 0.9900
C(12)-H(12B) 0.9900
C(13)-C(14) 1.533(9)
C(13)-H(13A) 0.9900
C(13)-H(13B) 0.9900
C(14)-O(9) 1.451(9)
C(14)-H(14) 1.0000
C(15)-N(1) 1.456(8)
C(15)-H(15) 1.0000
C(16)-N(1) 1.457(10)
C(16)-H(16A) 0.9900
C(16)-H(16B) 0.9900
C(17)-C(18) 1.335(11)
C(17)-H(17) 0.9500
C(18)-H(18) 0.9500
C(19)-O(2) 1.190(11)
C(19)-O(1) 1.357(8)
C(19)-C(20) 1.496(10)
C(20)-C(25) 1.368(13)
C(20)-C(21) 1.408(10)
C(21)-C(22) 1.390(11)
C(21)-H(21) 0.9500
C(22)-C(23) 1.372(14)
C(22)-H(22) 0.9500
C(23)-C(24) 1.384(11)
C(23)-N(2) 1.480(9)
C(24)-C(25) 1.409(11)
C(24)-H(24) 0.9500
C(25)-H(25) 0.9500
C(26)-O(5) 1.433(10)
C(26)-H(26A) 0.9800
C(26)-H(26B) 0.9800
C(26)-H(26C) 0.9800
C(27)-O(6) 1.435(8)
C(27)-H(27A) 0.9800

! "$'!
C(27)-H(27B) 0.9800
C(27)-H(27C) 0.9800
C(28)-O(7) 1.215(11)
C(28)-O(8) 1.345(9)
C(28)-N(1) 1.365(10)
C(29)-O(8) 1.428(10)
C(29)-H(29A) 0.9800
C(29)-H(29B) 0.9800
C(29)-H(29C) 0.9800
C(30)-O(9) 1.394(11)
C(30)-H(30A) 0.9800
C(30)-H(30B) 0.9800
C(30)-H(30C) 0.9800
C(31)-C(44) 1.528(10)
C(31)-C(45) 1.534(9)
C(31)-C(40) 1.542(11)
C(31)-C(32) 1.564(8)
C(32)-C(33) 1.536(10)
C(32)-C(37) 1.589(10)
C(32)-H(32) 1.0000
C(33)-C(34) 1.541(9)
C(33)-H(33A) 0.9900
C(33)-H(33B) 0.9900
C(34)-C(47) 1.496(10)
C(34)-C(35) 1.541(11)
C(34)-H(34) 1.0000
C(35)-O(10) 1.455(7)
C(35)-C(36) 1.575(10)
C(35)-H(35) 1.0000
C(36)-O(14) 1.379(10)
C(36)-O(15) 1.413(9)
C(36)-C(37) 1.576(8)
C(37)-C(48) 1.493(11)
C(37)-C(38) 1.541(8)
C(38)-C(39) 1.519(11)
C(38)-C(45) 1.562(9)
C(38)-H(38) 1.0000
C(39)-C(40) 1.561(10)
C(39)-H(39A) 0.9900
C(39)-H(39B) 0.9900
C(40)-C(41) 1.547(8)
C(40)-H(40) 1.0000
C(41)-C(42) 1.534(11)
C(41)-C(46) 1.535(9)
C(41)-H(41) 1.0000
C(42)-C(43) 1.525(12)
C(42)-H(42A) 0.9900
C(42)-H(42B) 0.9900
C(43)-C(44) 1.521(9)
C(43)-H(43A) 0.9900
C(43)-H(43B) 0.9900
C(44)-O(18) 1.427(10)
C(44)-H(44) 1.0000
C(45)-N(3) 1.471(8)
C(45)-H(45) 1.0000
C(46)-N(3) 1.483(10)
C(46)-H(46A) 0.9900
C(46)-H(46B) 0.9900
C(47)-C(48) 1.341(11)
C(47)-H(47) 0.9500
C(48)-H(48) 0.9500
C(49)-O(11) 1.205(11)
C(49)-O(10) 1.331(9)
C(49)-C(50) 1.490(9)
C(50)-C(51) 1.392(11)
C(50)-C(55) 1.401(10)
C(51)-C(52) 1.396(11)
C(51)-H(51) 0.9500
C(52)-C(53) 1.403(11)
C(52)-H(52) 0.9500
C(53)-C(54) 1.368(13)
C(53)-N(4) 1.482(9)
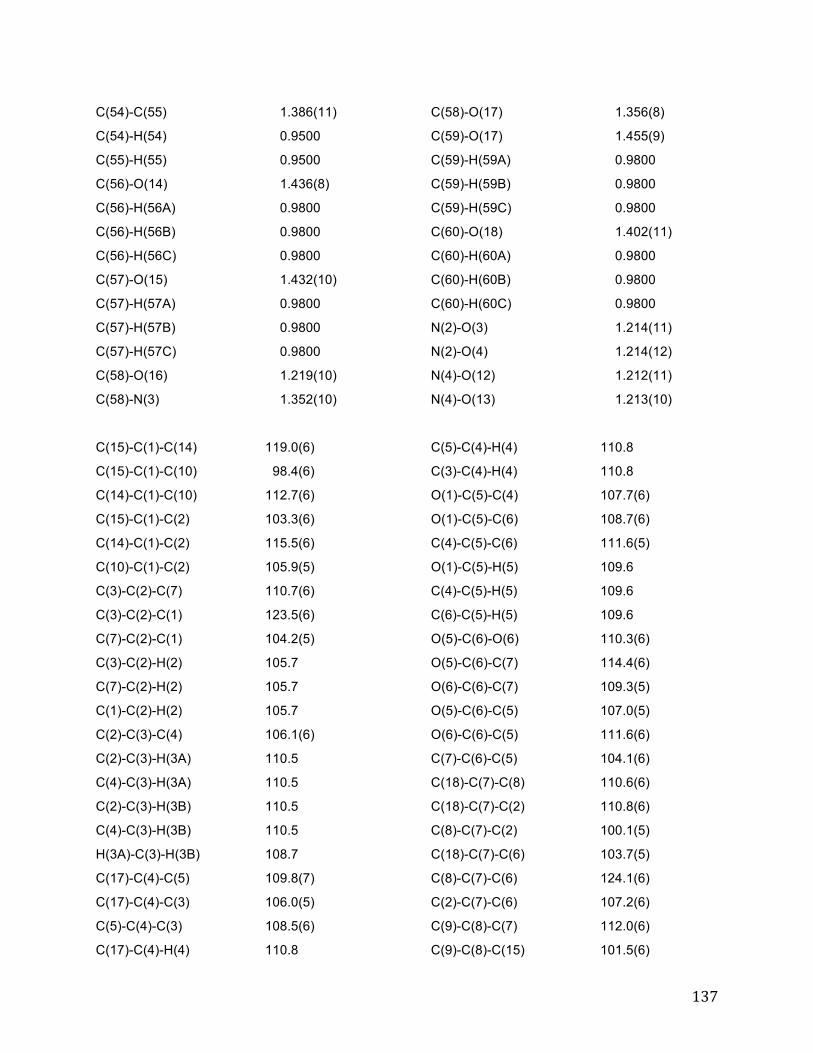
! "$(!
C(54)-C(55) 1.386(11)
C(54)-H(54) 0.9500
C(55)-H(55) 0.9500
C(56)-O(14) 1.436(8)
C(56)-H(56A) 0.9800
C(56)-H(56B) 0.9800
C(56)-H(56C) 0.9800
C(57)-O(15) 1.432(10)
C(57)-H(57A) 0.9800
C(57)-H(57B) 0.9800
C(57)-H(57C) 0.9800
C(58)-O(16) 1.219(10)
C(58)-N(3) 1.352(10)
C(58)-O(17) 1.356(8)
C(59)-O(17) 1.455(9)
C(59)-H(59A) 0.9800
C(59)-H(59B) 0.9800
C(59)-H(59C) 0.9800
C(60)-O(18) 1.402(11)
C(60)-H(60A) 0.9800
C(60)-H(60B) 0.9800
C(60)-H(60C) 0.9800
N(2)-O(3) 1.214(11)
N(2)-O(4) 1.214(12)
N(4)-O(12) 1.212(11)
N(4)-O(13) 1.213(10)
C(15)-C(1)-C(14) 119.0(6)
C(15)-C(1)-C(10) 98.4(6)
C(14)-C(1)-C(10) 112.7(6)
C(15)-C(1)-C(2) 103.3(6)
C(14)-C(1)-C(2) 115.5(6)
C(10)-C(1)-C(2) 105.9(5)
C(3)-C(2)-C(7) 110.7(6)
C(3)-C(2)-C(1) 123.5(6)
C(7)-C(2)-C(1) 104.2(5)
C(3)-C(2)-H(2) 105.7
C(7)-C(2)-H(2) 105.7
C(1)-C(2)-H(2) 105.7
C(2)-C(3)-C(4) 106.1(6)
C(2)-C(3)-H(3A) 110.5
C(4)-C(3)-H(3A) 110.5
C(2)-C(3)-H(3B) 110.5
C(4)-C(3)-H(3B) 110.5
H(3A)-C(3)-H(3B) 108.7
C(17)-C(4)-C(5) 109.8(7)
C(17)-C(4)-C(3) 106.0(5)
C(5)-C(4)-C(3) 108.5(6)
C(17)-C(4)-H(4) 110.8
C(5)-C(4)-H(4) 110.8
C(3)-C(4)-H(4) 110.8
O(1)-C(5)-C(4) 107.7(6)
O(1)-C(5)-C(6) 108.7(6)
C(4)-C(5)-C(6) 111.6(5)
O(1)-C(5)-H(5) 109.6
C(4)-C(5)-H(5) 109.6
C(6)-C(5)-H(5) 109.6
O(5)-C(6)-O(6) 110.3(6)
O(5)-C(6)-C(7) 114.4(6)
O(6)-C(6)-C(7) 109.3(5)
O(5)-C(6)-C(5) 107.0(5)
O(6)-C(6)-C(5) 111.6(6)
C(7)-C(6)-C(5) 104.1(6)
C(18)-C(7)-C(8) 110.6(6)
C(18)-C(7)-C(2) 110.8(6)
C(8)-C(7)-C(2) 100.1(5)
C(18)-C(7)-C(6) 103.7(5)
C(8)-C(7)-C(6) 124.1(6)
C(2)-C(7)-C(6) 107.2(6)
C(9)-C(8)-C(7) 112.0(6)
C(9)-C(8)-C(15) 101.5(6)

! "$)!
C(7)-C(8)-C(15) 98.3(5)
C(9)-C(8)-H(8) 114.4
C(7)-C(8)-H(8) 114.4
C(15)-C(8)-H(8) 114.4
C(8)-C(9)-C(10) 104.0(6)
C(8)-C(9)-H(9A) 111.0
C(10)-C(9)-H(9A) 111.0
C(8)-C(9)-H(9B) 111.0
C(10)-C(9)-H(9B) 111.0
H(9A)-C(9)-H(9B) 109.0
C(11)-C(10)-C(1) 109.2(5)
C(11)-C(10)-C(9) 111.8(6)
C(1)-C(10)-C(9) 101.8(6)
C(11)-C(10)-H(10) 111.2
C(1)-C(10)-H(10) 111.2
C(9)-C(10)-H(10) 111.2
C(12)-C(11)-C(10) 110.2(6)
C(12)-C(11)-C(16) 113.0(6)
C(10)-C(11)-C(16) 109.8(6)
C(12)-C(11)-H(11) 107.9
C(10)-C(11)-H(11) 107.9
C(16)-C(11)-H(11) 107.9
C(11)-C(12)-C(13) 110.8(7)
C(11)-C(12)-H(12A) 109.5
C(13)-C(12)-H(12A) 109.5
C(11)-C(12)-H(12B) 109.5
C(13)-C(12)-H(12B) 109.5
H(12A)-C(12)-H(12B) 108.1
C(12)-C(13)-C(14) 112.1(6)
C(12)-C(13)-H(13A) 109.2
C(14)-C(13)-H(13A) 109.2
C(12)-C(13)-H(13B) 109.2
C(14)-C(13)-H(13B) 109.2
H(13A)-C(13)-H(13B) 107.9
O(9)-C(14)-C(1) 108.0(6)
O(9)-C(14)-C(13) 109.4(5)
C(1)-C(14)-C(13) 113.9(6)
O(9)-C(14)-H(14) 108.5
C(1)-C(14)-H(14) 108.5
C(13)-C(14)-H(14) 108.5
N(1)-C(15)-C(1) 115.3(6)
N(1)-C(15)-C(8) 114.8(6)
C(1)-C(15)-C(8) 95.2(5)
N(1)-C(15)-H(15) 110.2
C(1)-C(15)-H(15) 110.2
C(8)-C(15)-H(15) 110.2
N(1)-C(16)-C(11) 113.1(6)
N(1)-C(16)-H(16A) 109.0
C(11)-C(16)-H(16A) 109.0
N(1)-C(16)-H(16B) 109.0
C(11)-C(16)-H(16B) 109.0
H(16A)-C(16)-H(16B) 107.8
C(18)-C(17)-C(4) 112.3(7)
C(18)-C(17)-H(17) 123.9
C(4)-C(17)-H(17) 123.9
C(17)-C(18)-C(7) 116.3(7)
C(17)-C(18)-H(18) 121.9
C(7)-C(18)-H(18) 121.9
O(2)-C(19)-O(1) 125.8(7)
O(2)-C(19)-C(20) 124.3(6)
O(1)-C(19)-C(20) 109.9(7)
C(25)-C(20)-C(21) 121.0(7)
C(25)-C(20)-C(19) 123.1(6)
C(21)-C(20)-C(19) 115.9(8)
C(22)-C(21)-C(20) 119.4(8)
C(22)-C(21)-H(21) 120.3
C(20)-C(21)-H(21) 120.3
C(23)-C(22)-C(21) 118.3(7)
C(23)-C(22)-H(22) 120.8
C(21)-C(22)-H(22) 120.8
C(22)-C(23)-C(24) 123.8(7)
C(22)-C(23)-N(2) 117.8(7)
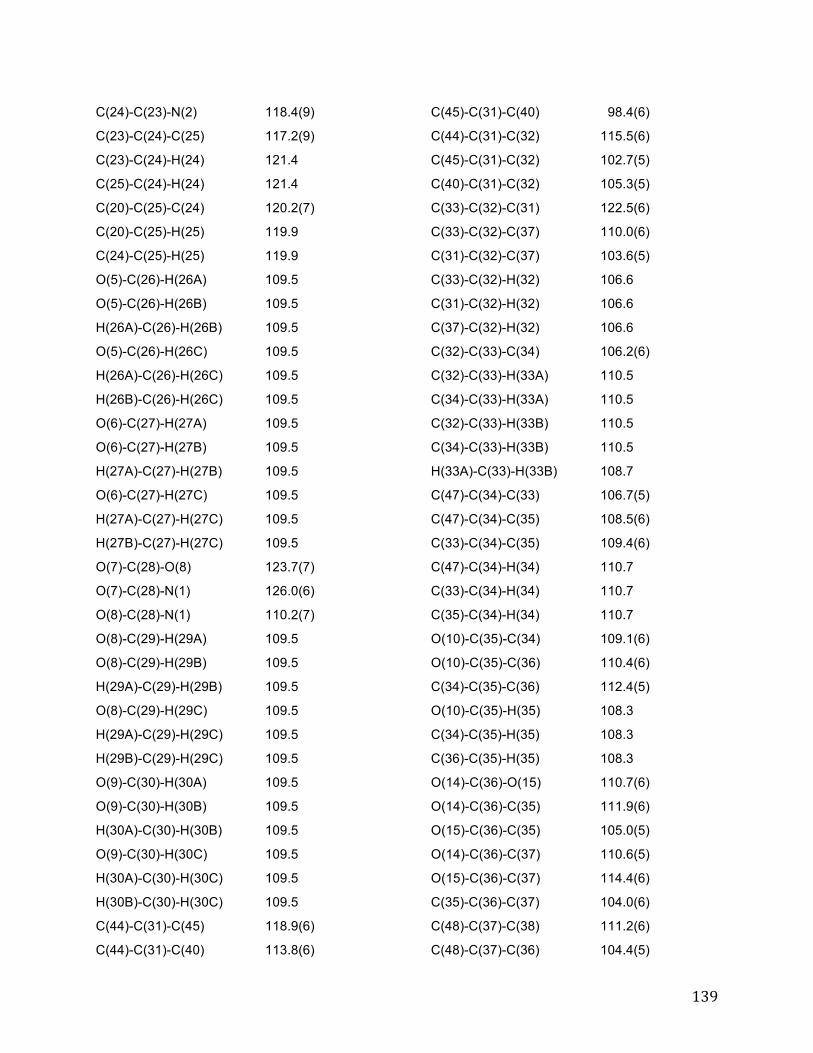
! "$*!
C(24)-C(23)-N(2) 118.4(9)
C(23)-C(24)-C(25) 117.2(9)
C(23)-C(24)-H(24) 121.4
C(25)-C(24)-H(24) 121.4
C(20)-C(25)-C(24) 120.2(7)
C(20)-C(25)-H(25) 119.9
C(24)-C(25)-H(25) 119.9
O(5)-C(26)-H(26A) 109.5
O(5)-C(26)-H(26B) 109.5
H(26A)-C(26)-H(26B) 109.5
O(5)-C(26)-H(26C) 109.5
H(26A)-C(26)-H(26C) 109.5
H(26B)-C(26)-H(26C) 109.5
O(6)-C(27)-H(27A) 109.5
O(6)-C(27)-H(27B) 109.5
H(27A)-C(27)-H(27B) 109.5
O(6)-C(27)-H(27C) 109.5
H(27A)-C(27)-H(27C) 109.5
H(27B)-C(27)-H(27C) 109.5
O(7)-C(28)-O(8) 123.7(7)
O(7)-C(28)-N(1) 126.0(6)
O(8)-C(28)-N(1) 110.2(7)
O(8)-C(29)-H(29A) 109.5
O(8)-C(29)-H(29B) 109.5
H(29A)-C(29)-H(29B) 109.5
O(8)-C(29)-H(29C) 109.5
H(29A)-C(29)-H(29C) 109.5
H(29B)-C(29)-H(29C) 109.5
O(9)-C(30)-H(30A) 109.5
O(9)-C(30)-H(30B) 109.5
H(30A)-C(30)-H(30B) 109.5
O(9)-C(30)-H(30C) 109.5
H(30A)-C(30)-H(30C) 109.5
H(30B)-C(30)-H(30C) 109.5
C(44)-C(31)-C(45) 118.9(6)
C(44)-C(31)-C(40) 113.8(6)
C(45)-C(31)-C(40) 98.4(6)
C(44)-C(31)-C(32) 115.5(6)
C(45)-C(31)-C(32) 102.7(5)
C(40)-C(31)-C(32) 105.3(5)
C(33)-C(32)-C(31) 122.5(6)
C(33)-C(32)-C(37) 110.0(6)
C(31)-C(32)-C(37) 103.6(5)
C(33)-C(32)-H(32) 106.6
C(31)-C(32)-H(32) 106.6
C(37)-C(32)-H(32) 106.6
C(32)-C(33)-C(34) 106.2(6)
C(32)-C(33)-H(33A) 110.5
C(34)-C(33)-H(33A) 110.5
C(32)-C(33)-H(33B) 110.5
C(34)-C(33)-H(33B) 110.5
H(33A)-C(33)-H(33B) 108.7
C(47)-C(34)-C(33) 106.7(5)
C(47)-C(34)-C(35) 108.5(6)
C(33)-C(34)-C(35) 109.4(6)
C(47)-C(34)-H(34) 110.7
C(33)-C(34)-H(34) 110.7
C(35)-C(34)-H(34) 110.7
O(10)-C(35)-C(34) 109.1(6)
O(10)-C(35)-C(36) 110.4(6)
C(34)-C(35)-C(36) 112.4(5)
O(10)-C(35)-H(35) 108.3
C(34)-C(35)-H(35) 108.3
C(36)-C(35)-H(35) 108.3
O(14)-C(36)-O(15) 110.7(6)
O(14)-C(36)-C(35) 111.9(6)
O(15)-C(36)-C(35) 105.0(5)
O(14)-C(36)-C(37) 110.6(5)
O(15)-C(36)-C(37) 114.4(6)
C(35)-C(36)-C(37) 104.0(6)
C(48)-C(37)-C(38) 111.2(6)
C(48)-C(37)-C(36) 104.4(5)

! "%+!
C(38)-C(37)-C(36) 124.1(6)
C(48)-C(37)-C(32) 111.5(6)
C(38)-C(37)-C(32) 99.8(5)
C(36)-C(37)-C(32) 105.5(5)
C(39)-C(38)-C(37) 112.5(6)
C(39)-C(38)-C(45) 102.5(6)
C(37)-C(38)-C(45) 97.1(6)
C(39)-C(38)-H(38) 114.3
C(37)-C(38)-H(38) 114.3
C(45)-C(38)-H(38) 114.3
C(38)-C(39)-C(40) 104.0(6)
C(38)-C(39)-H(39A) 111.0
C(40)-C(39)-H(39A) 111.0
C(38)-C(39)-H(39B) 111.0
C(40)-C(39)-H(39B) 111.0
H(39A)-C(39)-H(39B) 109.0
C(31)-C(40)-C(41) 109.3(6)
C(31)-C(40)-C(39) 102.7(6)
C(41)-C(40)-C(39) 112.0(6)
C(31)-C(40)-H(40) 110.9
C(41)-C(40)-H(40) 110.9
C(39)-C(40)-H(40) 110.9
C(42)-C(41)-C(46) 112.9(6)
C(42)-C(41)-C(40) 109.7(6)
C(46)-C(41)-C(40) 110.3(6)
C(42)-C(41)-H(41) 107.9
C(46)-C(41)-H(41) 107.9
C(40)-C(41)-H(41) 107.9
C(43)-C(42)-C(41) 110.7(6)
C(43)-C(42)-H(42A) 109.5
C(41)-C(42)-H(42A) 109.5
C(43)-C(42)-H(42B) 109.5
C(41)-C(42)-H(42B) 109.5
H(42A)-C(42)-H(42B) 108.1
C(44)-C(43)-C(42) 113.4(6)
C(44)-C(43)-H(43A) 108.9
C(42)-C(43)-H(43A) 108.9
C(44)-C(43)-H(43B) 108.9
C(42)-C(43)-H(43B) 108.9
H(43A)-C(43)-H(43B) 107.7
O(18)-C(44)-C(43) 109.2(6)
O(18)-C(44)-C(31) 106.9(6)
C(43)-C(44)-C(31) 114.3(6)
O(18)-C(44)-H(44) 108.8
C(43)-C(44)-H(44) 108.8
C(31)-C(44)-H(44) 108.8
N(3)-C(45)-C(31) 113.9(6)
N(3)-C(45)-C(38) 114.7(6)
C(31)-C(45)-C(38) 95.3(5)
N(3)-C(45)-H(45) 110.7
C(31)-C(45)-H(45) 110.7
C(38)-C(45)-H(45) 110.7
N(3)-C(46)-C(41) 112.6(6)
N(3)-C(46)-H(46A) 109.1
C(41)-C(46)-H(46A) 109.1
N(3)-C(46)-H(46B) 109.1
C(41)-C(46)-H(46B) 109.1
H(46A)-C(46)-H(46B) 107.8
C(48)-C(47)-C(34) 112.4(7)
C(48)-C(47)-H(47) 123.8
C(34)-C(47)-H(47) 123.8
C(47)-C(48)-C(37) 116.1(6)
C(47)-C(48)-H(48) 122.0
C(37)-C(48)-H(48) 122.0
O(11)-C(49)-O(10) 125.9(6)
O(11)-C(49)-C(50) 122.7(7)
O(10)-C(49)-C(50) 111.4(7)
C(51)-C(50)-C(55) 120.3(6)
C(51)-C(50)-C(49) 122.4(6)
C(55)-C(50)-C(49) 117.3(7)
C(50)-C(51)-C(52) 120.7(7)
C(50)-C(51)-H(51) 119.7
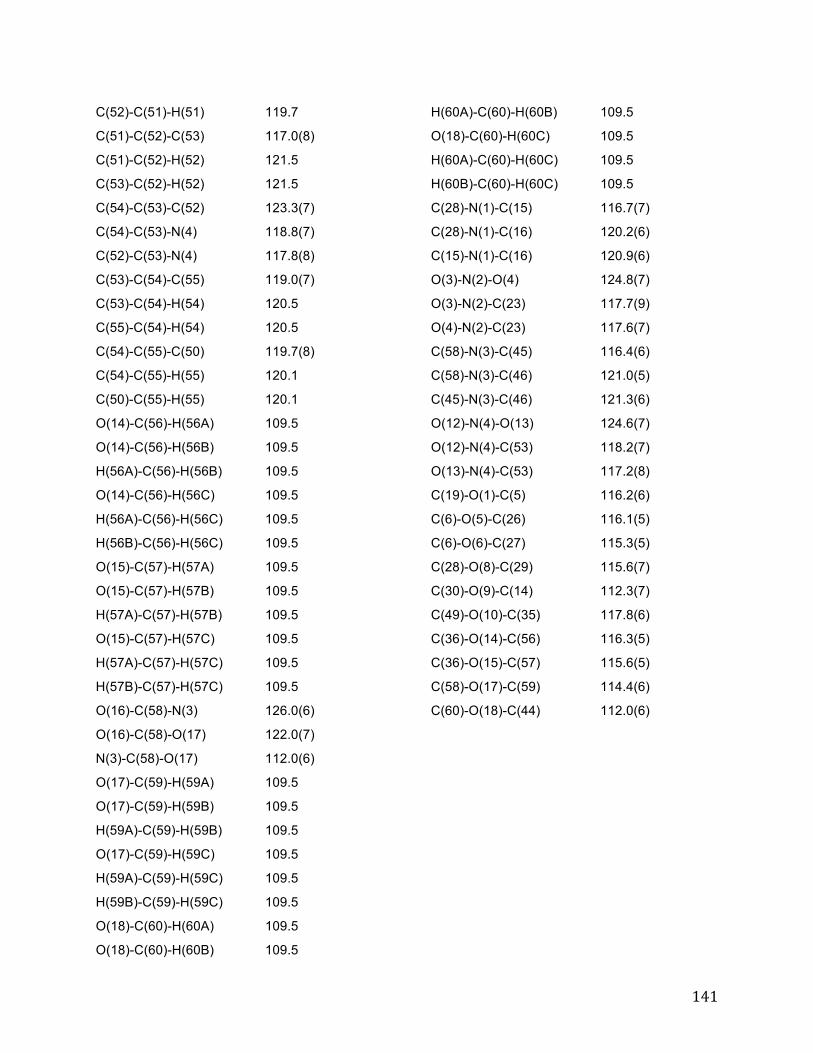
! "%"!
C(52)-C(51)-H(51) 119.7
C(51)-C(52)-C(53) 117.0(8)
C(51)-C(52)-H(52) 121.5
C(53)-C(52)-H(52) 121.5
C(54)-C(53)-C(52) 123.3(7)
C(54)-C(53)-N(4) 118.8(7)
C(52)-C(53)-N(4) 117.8(8)
C(53)-C(54)-C(55) 119.0(7)
C(53)-C(54)-H(54) 120.5
C(55)-C(54)-H(54) 120.5
C(54)-C(55)-C(50) 119.7(8)
C(54)-C(55)-H(55) 120.1
C(50)-C(55)-H(55) 120.1
O(14)-C(56)-H(56A) 109.5
O(14)-C(56)-H(56B) 109.5
H(56A)-C(56)-H(56B) 109.5
O(14)-C(56)-H(56C) 109.5
H(56A)-C(56)-H(56C) 109.5
H(56B)-C(56)-H(56C) 109.5
O(15)-C(57)-H(57A) 109.5
O(15)-C(57)-H(57B) 109.5
H(57A)-C(57)-H(57B) 109.5
O(15)-C(57)-H(57C) 109.5
H(57A)-C(57)-H(57C) 109.5
H(57B)-C(57)-H(57C) 109.5
O(16)-C(58)-N(3) 126.0(6)
O(16)-C(58)-O(17) 122.0(7)
N(3)-C(58)-O(17) 112.0(6)
O(17)-C(59)-H(59A) 109.5
O(17)-C(59)-H(59B) 109.5
H(59A)-C(59)-H(59B) 109.5
O(17)-C(59)-H(59C) 109.5
H(59A)-C(59)-H(59C) 109.5
H(59B)-C(59)-H(59C) 109.5
O(18)-C(60)-H(60A) 109.5
O(18)-C(60)-H(60B) 109.5
H(60A)-C(60)-H(60B) 109.5
O(18)-C(60)-H(60C) 109.5
H(60A)-C(60)-H(60C) 109.5
H(60B)-C(60)-H(60C) 109.5
C(28)-N(1)-C(15) 116.7(7)
C(28)-N(1)-C(16) 120.2(6)
C(15)-N(1)-C(16) 120.9(6)
O(3)-N(2)-O(4) 124.8(7)
O(3)-N(2)-C(23) 117.7(9)
O(4)-N(2)-C(23) 117.6(7)
C(58)-N(3)-C(45) 116.4(6)
C(58)-N(3)-C(46) 121.0(5)
C(45)-N(3)-C(46) 121.3(6)
O(12)-N(4)-O(13) 124.6(7)
O(12)-N(4)-C(53) 118.2(7)
O(13)-N(4)-C(53) 117.2(8)
C(19)-O(1)-C(5) 116.2(6)
C(6)-O(5)-C(26) 116.1(5)
C(6)-O(6)-C(27) 115.3(5)
C(28)-O(8)-C(29) 115.6(7)
C(30)-O(9)-C(14) 112.3(7)
C(49)-O(10)-C(35) 117.8(6)
C(36)-O(14)-C(56) 116.3(5)
C(36)-O(15)-C(57) 115.6(5)
C(58)-O(17)-C(59) 114.4(6)
C(60)-O(18)-C(44) 112.0(6)

! "%#!
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms:

! "%$!
Table 4. Anisotropic displacement parameters (Å2x 103) for sarpong64. The anisotropic
displacement factor exponent takes the form: -2p2[ h2 a*2U11 + ... + 2 h k a* b* U12 ]
______________________________________________________________________________
U11 U22 U33 U23 U13 U12
______________________________________________________________________________
C(1) 40(4) 23(3) 21(4) 3(2) -1(3) 0(2)
C(2) 43(4) 21(3) 16(4) 2(2) -4(3) 1(2)
C(3) 41(4) 24(3) 25(4) -3(2) -2(3) 1(2)
C(4) 43(4) 23(3) 22(4) 2(2) 0(3) 4(2)
C(5) 47(5) 23(3) 22(4) 1(2) 1(3) 3(2)
C(6) 45(4) 16(3) 16(4) 3(2) 0(3) -5(2)
C(7) 42(4) 17(3) 24(4) 1(2) -4(3) -2(2)
C(8) 33(4) 19(3) 33(5) 2(2) -3(3) -1(2)
C(9) 40(4) 21(3) 29(4) 0(2) -5(3) -1(2)
C(10) 44(4) 23(3) 21(4) 0(2) -5(3) 1(2)
C(11) 48(5) 26(3) 17(4) 5(2) -2(3) -2(2)
C(12) 57(5) 25(3) 21(4) 1(2) 1(4) -5(3)
C(13) 44(5) 32(3) 27(5) 1(3) 1(4) -7(3)
C(14) 39(4) 28(3) 26(4) -2(2) -1(3) -1(2)
C(15) 34(4) 22(3) 29(4) -3(2) -2(3) 1(2)
C(16) 42(4) 25(3) 26(4) 6(2) 0(3) 1(2)
C(17) 51(5) 28(3) 16(4) 3(2) -4(3) -4(3)
C(18) 46(4) 22(3) 19(4) -1(2) -3(3) -2(2)
C(19) 55(5) 21(3) 22(5) -5(2) 0(4) 2(2)
C(20) 44(4) 26(3) 27(5) 3(3) 0(4) -2(2)
C(21) 55(5) 23(3) 23(4) -4(2) 1(4) 2(3)
C(22) 65(5) 19(3) 29(5) 0(2) -4(4) -3(3)
C(23) 55(5) 28(3) 26(5) 4(3) -10(4) -9(3)
C(24) 61(6) 28(3) 27(5) 2(3) 6(4) -4(3)
C(25) 59(5) 22(3) 30(5) -4(3) -1(4) -1(3)
C(26) 42(5) 33(3) 42(5) 1(3) 8(4) -1(3)
C(27) 66(6) 25(3) 25(4) 0(2) -9(4) -10(3)
C(28) 37(4) 29(3) 32(5) -3(3) 4(4) -2(2)
C(29) 72(6) 28(3) 39(6) -4(3) 9(5) 3(3)
C(30) 38(4) 34(4) 42(5) -2(3) -5(4) -2(3)
C(31) 39(4) 21(3) 19(4) -3(2) -1(3) 1(2)

! "%%!
C(32) 42(4) 22(3) 20(4) -1(2) -1(3) -1(2)
C(33) 43(4) 22(3) 28(4) 3(2) -5(3) -2(2)
C(34) 44(4) 23(3) 26(4) 1(2) -3(3) -2(2)
C(35) 55(5) 14(3) 16(4) 3(2) 0(3) 1(2)
C(36) 48(5) 22(3) 19(4) 0(2) -1(3) 1(2)
C(37) 36(4) 22(3) 23(4) 1(2) 3(3) 0(2)
C(38) 40(4) 16(3) 30(4) 0(2) 1(3) -1(2)
C(39) 42(4) 26(3) 22(4) 1(2) 0(3) -1(2)
C(40) 45(4) 20(3) 18(4) 2(2) 2(3) 3(2)
C(41) 56(5) 21(3) 22(4) -1(2) -5(4) 0(2)
C(42) 53(5) 28(3) 20(4) 6(2) 0(4) 4(3)
C(43) 41(4) 28(3) 33(5) 1(3) 0(4) 6(2)
C(44) 43(4) 30(3) 21(4) -1(2) -3(3) 0(2)
C(45) 34(4) 26(3) 23(4) 1(2) 1(3) -1(2)
C(46) 53(5) 19(3) 23(4) -2(2) -4(3) -3(2)
C(47) 49(5) 21(3) 15(4) 1(2) 1(3) 0(2)
C(48) 46(4) 22(3) 21(4) -1(2) -6(3) -2(2)
C(49) 61(5) 22(3) 25(5) 7(3) -9(4) 0(3)
C(50) 50(5) 23(3) 17(4) -3(2) -2(3) 3(2)
C(51) 58(5) 25(3) 23(4) -6(2) -6(4) 1(3)
C(52) 61(6) 33(4) 28(5) -2(3) -7(4) 4(3)
C(53) 50(5) 29(3) 25(5) 5(3) 3(4) 6(3)
C(54) 61(5) 18(3) 36(5) -2(3) 0(4) 3(3)
C(55) 55(5) 27(3) 32(5) -1(3) -11(4) 1(3)
C(56) 65(6) 29(3) 23(4) -1(3) 6(4) 10(3)
C(57) 51(5) 30(3) 30(5) 0(3) -12(4) 1(3)
C(58) 44(4) 19(3) 24(4) 1(2) -1(3) 1(2)
C(59) 94(7) 25(3) 26(5) -8(3) -10(5) -3(3)
C(60) 42(5) 36(4) 42(6) 0(3) 3(4) -1(3)
N(1) 41(4) 21(3) 30(4) 0(2) 0(3) 0(2)
N(2) 66(6) 33(3) 23(4) 6(2) -5(4) -15(3)
N(3) 45(4) 17(3) 28(4) -1(2) -2(3) 2(2)
N(4) 55(5) 33(3) 33(5) 3(3) 7(4) 13(3)
O(1) 61(4) 18(2) 21(3) 0(2) 2(3) 2(2)
O(2) 98(5) 23(2) 31(4) -3(2) 24(4) -1(2)
O(3) 83(5) 23(3) 35(4) 2(2) -20(3) -10(2)

! "%&!
O(4) 97(6) 40(3) 49(5) 8(3) 9(5) -18(3)
O(5) 47(3) 24(2) 23(3) 2(2) 3(2) -1(2)
O(6) 53(3) 22(2) 14(3) 2(2) -4(2) -5(2)
O(7) 58(4) 30(2) 19(3) -4(2) 1(3) 1(2)
O(8) 64(4) 22(2) 33(4) -2(2) 10(3) 4(2)
O(9) 40(3) 35(2) 24(3) -5(2) -1(2) -2(2)
O(10) 59(4) 18(2) 22(3) 1(2) -3(3) 2(2)
O(11) 110(6) 25(3) 33(4) 2(2) -29(4) 2(3)
O(12) 74(5) 42(3) 43(4) 9(2) -13(4) 11(3)
O(13) 71(4) 22(2) 33(4) 3(2) 11(3) 7(2)
O(14) 51(3) 21(2) 23(3) 2(2) 4(2) 5(2)
O(15) 51(3) 21(2) 20(3) 5(2) -3(2) 1(2)
O(16) 56(3) 20(2) 27(3) 2(2) -3(3) -2(2)
O(17) 65(4) 23(2) 29(4) -4(2) -6(3) -3(2)
O(18) 43(3) 29(2) 21(3) 1(2) 1(2) 2(2)
______________________________________________________________________________
! "%'!
Table 5. Hydrogen coordinates ( x 104) and isotropic displacement parameters (Å2x 10 3)
for sarpong64.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________
H(2) 173 1259 15003 32
H(3A) -2016 1416 14546 36
H(3B) -2246 1228 13469 36
H(4) -1729 1682 12906 35
H(5) 153 1730 14453 37
H(8) 2803 936 13070 34
H(9A) 2763 1088 15230 36
H(9B) 3211 794 14956 36
H(10) 743 930 16138 35
H(11) 1736 512 16323 36
H(12A) -774 543 16768 41
H(12B) -436 263 16266 41
H(13A) -2641 441 15439 41
H(13B) -1449 376 14489 41
H(14) -2178 877 15336 37
H(15) 114 810 12746 34
H(16A) 2663 396 14650 37
H(16B) 1242 213 14679 37
H(17) -867 1454 11271 39
H(18) 925 1132 11626 35
H(21) 181 2627 13810 41
H(22) 1008 2981 12740 45
H(24) 3267 2498 10676 47
H(25) 2351 2141 11714 45
H(26A) 4492 1388 13416 58
H(26B) 4780 1538 12249 58
H(26C) 3817 1280 12242 58
H(27A) 3878 1791 14582 58
H(27B) 3524 1720 15876 58
H(27C) 2315 1866 15105 58

! "%(!
H(29A) 1002 -34 11368 70
H(29B) 2267 -192 12026 70
H(29C) 2689 56 11293 70
H(30A) -4401 646 14382 57
H(30B) -4688 800 13218 57
H(30C) -4328 954 14371 57
H(32) 5442 1273 10030 34
H(33A) 7828 1261 8513 37
H(33B) 7584 1447 9589 37
H(34) 7187 1711 7966 37
H(35) 5303 1738 9468 34
H(38) 2869 929 8024 35
H(39A) 2546 781 9878 36
H(39B) 2909 1077 10173 36
H(40) 4983 936 11103 33
H(41) 4127 505 11253 40
H(42A) 6630 562 11765 40
H(42B) 6407 281 11234 40
H(43A) 8532 492 10483 41
H(43B) 7382 409 9502 41
H(44) 7946 917 10329 37
H(45) 5602 838 7712 33
H(46A) 4755 216 9609 38
H(46B) 3249 377 9564 38
H(47) 6366 1482 6314 34
H(48) 4641 1146 6616 36
H(51) 3065 2160 6702 42
H(52) 2218 2517 5631 49
H(54) 4618 2991 7711 46
H(55) 5356 2638 8824 46
H(56A) 3159 1848 10308 58
H(56B) 1763 1691 10753 58
H(56C) 1777 1797 9473 58
H(57A) 1749 1278 7147 56
H(57B) 707 1526 7202 56
H(57C) 1071 1366 8336 56

! "%)!
H(59A) 3619 62 6041 73
H(59B) 4040 -190 6757 73
H(59C) 5316 -12 6258 73
H(60A) 10097 972 9390 60
H(60B) 10345 829 8199 60
H(60C) 10099 664 9331 60 ________________________________________________________________________________

! "%*!
Chapter 3 !Phragmalin-Type Limonoids A portion of the work described below has been previously published: Lebold, T. P.; Gallego, G. M.; Marth, C. J.; Sarpong, R. Org. Lett. 2012, 14, 2110-2113. 3.1 Introduction ! As discussed in Section 2.1, our efforts to access several complex natural products have centered on a key hydrindanone intermediate. We recognized the implementation of this strategy to the synthesis of the phragmalin-type limonoids as a valuable extension of this approach for two reasons. Synthetic approaches to these highly complex natural products is quite limited, largely owing the their daunting molecular complexity. A key contributor to the architectural complexity within these compounds is the octahydro-1H-2,4-methanoindene framework (highlighted in Figure 3.1). A lack of effective approaches to this motif has largely hampered synthetic efforts. We envisioned access to this scaffold using hydrindanone 3.3.
Figure 3.1 Proposed precursor to the bridging framework of phragmalin-type limonoids. A second driving force behind these studies concerned gaining insight into the conformational implications of performing a C–C bond reaction to gain access to the methanoindene framework (Figure 3.2). As discussed in Chapter 2, the formation of the key C–N bond within the diterpenoid tricyclic component was a persistent challenge (the transformation of 3.6 into 3.7 is representative). We reasoned that any insight derived from a productive C–C bond-forming event (3.4 forming 3.7) should translate to the diterpenoid chemistry.
Figure 3.2 Representation of conformational implications of C-C bond formation.
Me
O
AcO CO2MeMe
HORO
O Me
OH
O
O
Me
AcO
AcO
Xyloccensin O (R = H) (3.1)Xyloccensin P (R = OAc) (3.2)
ROCO2Me
HOTBS
O
3.3
ORCO2Me
O
H
H
LG
ORCO2Me
O
H
H
NHR
ORCO2MeH
H
H NR
ORCO2Me
O
H
H
H
3.4 3.5 3.6 3.7

! "&+!
This chapter details our approach to the bridging framework of the phragmalin-type limonoids, with a key hydridanone precursor. A brief discussion of the reactivity of the core framework is also discussed along with classification and previous synthetic efforts. ! 3.2 Isolation and Classification of Limonoids The limonoid family of natural products have been found only in plants belonging to the order Rutales. These triterpenes are characterized by a parent 4,4,8-trimethyl-17-furanylstereoid (Figure 3.3). Various natural products defined as limonoids are derived from oxidation and/or rearrangement of 3.8. These compounds have demonstrated anticancer,1-3 anti-HIV3 antibiotic and anti-inflammatory4 activity. These activities offer the opportunity, through efficient syntheses, for the identification of more potent derivatives.
Figure 3.3 Parent limonoid framework.
Limonoids are divided into four general classes according to their relationship to the parent limonoid framework as described by Heasley5 (representatives of each class are shown in Figure 3.4). Those that maintain the connectivity and parent limonoid framework (3.8) are referred to as intact limonoids. Even within this class, the level of oxygenation and unsaturation varies greatly between natural products, although they all maintain the C17 furyl group. The degraded class of limonoids are structurally simplified natural products typically containing fused (- or #-lactones. These molecules are often used as a platform for the total synthesis of other, more complex natural products.6,7 Limonoids that have undergone oxidative fission of one or more rings within the parent limonoid framework (3.8) are referred to as seco-limonoids. The remainder of the limonoid natural products are categorized as highly oxidatively modified limonoids. This large class of unique natural products lacks a general classification of all derivatives, as they typically undergo significant oxygenation, skeletal rearrangements and oxidative cleavages.
O
O
Me
H
HMeMe
HO
MeMe
48
17
Parent LimonoidFramework (3.8)
H

! "&"!
Figure 3.4 Representative examples of the limonoid classes. The phragmalin-type limonoids (which are the focus of the work presented in this chapter), are classified as those congeners maintain the integrity of the parent four rings and have a characteristic tricyclo[3.3.1]decane or tricyclo[4.2.1]decane ring system. Of the phragmalin-type limonoids isolated, the majority are orthoesters.8 These molecules are further classified according to the position of the hydroxyl from which the orthoester
O
Me
MeMeO
MeMe
OAc
Chisosiamensin (3.9)
O
Me
Me
Me
OHO
OAc
OHO
O
HOAcO
Toosendanin (3.10)
O
Me O
O
Me
Fraxinellone (3.11)
O
MeO
Me
Pyroangolensolide (3.12)
O
O
Me
Me
H H
OHMe
OO
Me
H
Tecleanin (3.13)
O
MeMeMe
O
O
Me
Me
Me HO
OOOMe
Desacetylsalannin (3.14)
O
O O
O
O
OMe
MeH
Me
O
MeOH
Trichillin A (3.15)
O
OAcO
O
OAc
O
OO
OH
O
Me
OAcOHMe
Me
HMeO2C
OMe
O
OMe
Me
Tabularisin A (3.16)
O
Intact Limonoids
Degraded Limonoids
seco-Limonoids
Highly Oxidatively Modified Limonoids

! "&#!
is formed. The compound from which the class derives its name is shown in Figure 3.5 along with the adapted numbering system. Phragmalin (3.17), for example, is a 1,8,9 orthoester. Numerous other natural products of this type, however, have been isolated, including 8,9,14-, 8,9,30- and 8,9,11- orthoesters.9-11
Figure 3.5 Phragmalin and the general numbering system. 3.3 Syntheses and Approaches Given the extraordinary complexity of these natural products, relatively few syntheses or approaches have been reported. This section will briefly cover major advances in the synthesis of these natural products. For a compressive review see ref. 5. The simplest compounds within the limonoid family, as described above, are the degraded limonoids. A representative example of the syntheses of these compounds is shown in Scheme 3.1. In 1972, Fukuyama and coworkers described a concise synthesis of fraxinellone (3.11) in a total of three steps.12 Commencing with a Diels-Alder cycloaddition between diene 3.18 and 3.19, a highly substituted cyclohexene (3.20) was accessed. Lithiated furan was then added into the aldehyde group which spontaneously lactonized to generate 3.21 as a mixture of diastereomers. Following separation, the natural product was accessed by isomerization of the double bond into conjugation with the carbonyl group by the action of basic methanol at refluxing temperature. Since this synthesis, the Morken group and others have reported elegant syntheses of this and related limonoid natural products.13,14
O
O
O
Me
Me
Phragmalin (3.17)
Me
OOO
MeOMe
O
OH
HOH
OH
1234
5
6 8910
1112
1314
15
1617
18
19
202122
23
28
2931
32
7 30

! "&$!
Scheme 3.1 Fukuyama’s synthesis of fraxinellone.
Most notable in the realm of synthetic strategy toward the limonoid natural products comes the synthesis of azadiradione (3.31) by Corey and coworkers.15 This synthesis represents the only reported total synthesis of the fully intact pentacyclic limonoid framework. A summary of the synthesis of 3.31 is shown in Scheme 3.2. The synthesis commenced with akylation of dianion 3.23 with farnesyl bromide (3.22) and phosphonate ester formation, which formed 3.24. Generation of the mercurium ion (3.25) initiates a truly impressive polyene cyclization to rapidly construct the tricyclic core in a stereocontrolled manner, generating the all trans-fused tricycle (3.26). The final ring (D-ring) was appended by elaboration of 3.26 to !-furyl ketone 3.27, which undergoes aldol condensation under basic conditions at elevated temperature. More traditional conjugate addition protocols failed to install the requisite C13 methyl group (see 3.30) due to steric encumbrance. In a clever solution, Corey effects a cyclopropanation (directed by the D-ring alcohol group) generating 3.29, which, after oxidation, undergoes reductive ring-opening to generate 3.30 following re-oxidation. Intermediate 3.30 was then taken on to prepare the natural product (3.31) through oxidation level adjustment and protective group removal.
O
Me O
O
Me
CO2EtMe MeOHC
MeCO2Et
CHOMeK2CO3
Hydroquinone
120 °C+
3-Br-Furan, nBuLiEt2O
O
Me O
O
Me
Fraxinellone (3.11)
H
3.21
1. Seperation
2. KOH, MeOH 65 °C
3.193.18 3.20

! "&%!
Scheme 3.2 Corey’s synthesis of azadiradione.
Br OLi
OMe
ONa
2. Diethylcholoro- phosphate
1. CO2Me
OP(O)(OEt)2
CO2Me
OP(O)(OEt)2
HgCF3CO2
Hg(TFA)2, MeNO2;NaCl(aq)
HMeMe
ClHg
MeMe
HCO2Me
O
HMeMe
MeMe
H
O
O
OO 1. 1M NaOEt,
EtOH, 70 °C2. MOMBr, TBAI, DIPEA, MeCN, 70 °COH
HMeMe
MeMe
HO OMOM
O
H
O
HMeMe
MeMe
HHO OMOM
OH
H
O
1.DMP2. Li, NH3:THF3. DMP
HMeMe
MeMe
HO OMOM
O
H
O
Me
HMeMe
MeMe
HO OAc
O
O
Me
3.22 3.24
3.253.26
3.27 3.28
3.30 3.29
Azadiradione (3.31)
13
3.23

! "&&!
In a truly landmark synthetic campaign, Steven Ley has reported an elegant synthesis of the highly oxidatively modified, seco limonoid azadirachtin (3.38, Scheme 3.3). This natural product boasts notable biological activity including antifeedant and growth disrupting effects on a range of insects, while remaining innocuous to higher order organisms. Additionally, the complexity of the molecule itself is quite notable, containing 16 contiguous stereogenic centers, of which seven are tetrasubstituted. Intermediate 3.32 was the focus of study in his preliminary reports16-18 and can be accessed by ‘pure’ synthesis or by degradation of the natural product. Propargylation of 3.32 with mesylate 3.33 furnished 3.34, which set the stage for a [3,3] sigmatropic rearrangement. Not only would this maneuver generate the key quaternary center within the natural product at C8, but also the resulting allene would serve as a precursor to an anticipated radical cyclization. Rearrangement of 3.34 occurs at 185 °C under microwave irradiation to generate allene 3.35. Replacing the p-methoxybenozate in 3.35 with a xanthate group gave radical precursor 3.36. The final ring-forming step (excluding epoxidation) occurs by heating xanthate 3.36 in the presence of AIBN and a hydride source, furnishing bridging cycle 3.37 by means of a 5-exo-dig cyclization. Further manipulation of 3.37 furnished the natural product, which in total required 48 steps in the longest linear sequence.19

! "&'!
Scheme 3.3 Ley’s synthesis of azadirachtin.
Despite the impressive tactics described for the synthesis of these highly complex triterpenes, it is clear that improvements can be made in the general approaches to these compounds. Below, we disclose our approach toward the complex phragmalin-type limonoids, which exploits our hydrindanone strategy. 3.4 Construction of the Bridging Framework of the Phragmalin-Type Limonoids Our synthetic investigations into the phragmalin-type limonoids commenced with the synthesis of the hydrindanone that would serve as a synthetic starting point. We selected benzyl hydrindanone 3.45 (Scheme 3.4) as our target since the benzyl group could be more easily removed and would be potentially useful in structural elucidation of our late stage intermediates. Thus, alcohol 3.39 was oxidized according to the Ley protocol20 to deliver aldehyde 3.40. Wittig homologation then delivered diene 3.41 as a separable mixture of diastereomers, ultimately leading to the isolation of the E,E-isomer
TESO
TESOO
H
Me
OMeO2C
OBnCO2Me
O
O O
PMBO
OMe
OBn
H3.32
3.33
HO
HOO
H
Me
MeO2C
OBnCO2Me
O
3.34
OO O
PMBO
OMe
OBn
H
1. TBAF2. 1,2-Dichlorobenzene 185 °C (µW)
NaH, 15-C-5
OMs
HO
HOO
H
Me
OMeO2C
OBnCO2Me
O
3.35
C
O
O
OPMBOBn
H
OMe
HO
TBSOO
H
Me
OMeO2C
OBnCO2Me
O
3.36
C
O
O
OOBn
H
OMe
S SMe
nBu3SnH, AIBNPhMe, 100 °C
HO
TBSOO
H
Me
OMeO2C
OBnCO2Me
O
O
Me
OH
OMe
BnO
3.37
O
AcOO
H
Me
OHMeO2C
OHCO2Me
O
OMe OHHO
Azadirachtin (3.38)
O
O
Me
Me
8
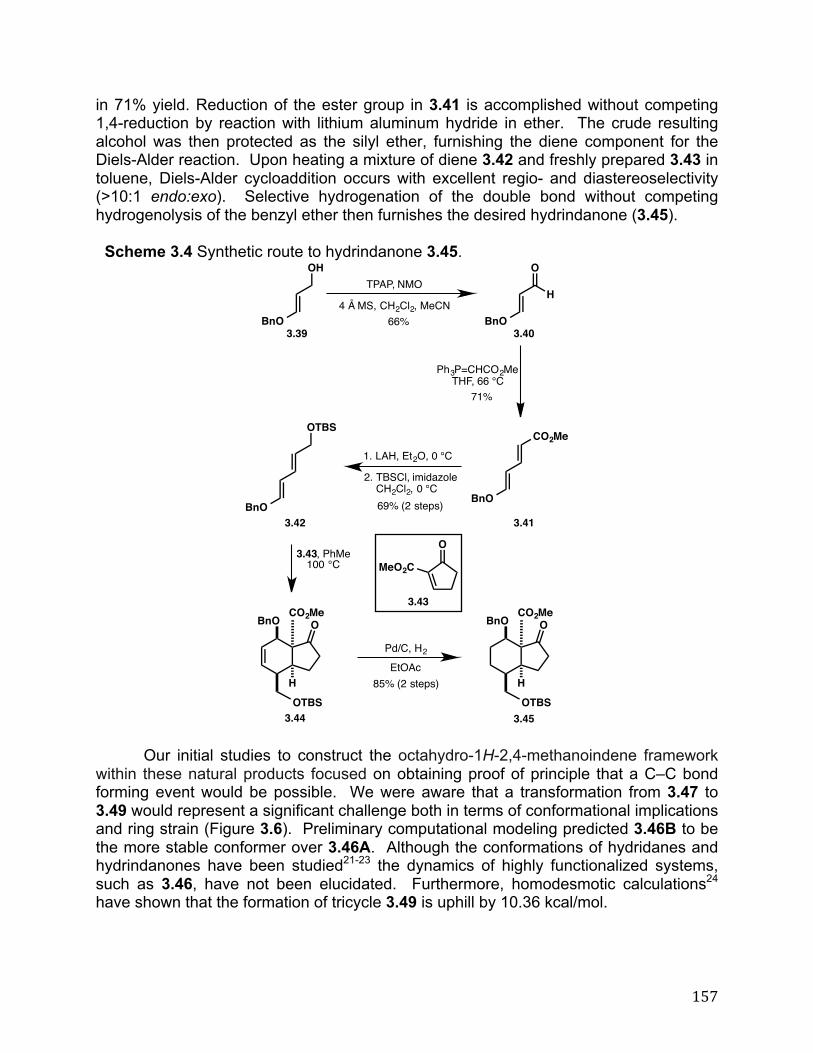
! "&(!
in 71% yield. Reduction of the ester group in 3.41 is accomplished without competing 1,4-reduction by reaction with lithium aluminum hydride in ether. The crude resulting alcohol was then protected as the silyl ether, furnishing the diene component for the Diels-Alder reaction. Upon heating a mixture of diene 3.42 and freshly prepared 3.43 in toluene, Diels-Alder cycloaddition occurs with excellent regio- and diastereoselectivity (>10:1 endo:exo). Selective hydrogenation of the double bond without competing hydrogenolysis of the benzyl ether then furnishes the desired hydrindanone (3.45). Scheme 3.4 Synthetic route to hydrindanone 3.45.
Our initial studies to construct the octahydro-1H-2,4-methanoindene framework within these natural products focused on obtaining proof of principle that a C–C bond forming event would be possible. We were aware that a transformation from 3.47 to 3.49 would represent a significant challenge both in terms of conformational implications and ring strain (Figure 3.6). Preliminary computational modeling predicted 3.46B to be the more stable conformer over 3.46A. Although the conformations of hydridanes and hydrindanones have been studied21-23 the dynamics of highly functionalized systems, such as 3.46, have not been elucidated. Furthermore, homodesmotic calculations24 have shown that the formation of tricycle 3.49 is uphill by 10.36 kcal/mol.
BnO
OHTPAP, NMO
4 Å MS, CH2Cl2, MeCNBnO
O
H
66%
Ph3P=CHCO2MeTHF, 66 °C
BnO
CO2Me
3.39 3.40
3.41
71%
BnO3.42
OTBS
1. LAH, Et2O, 0 °C
2. TBSCl, imidazole CH2Cl2, 0 °C
69% (2 steps)
3.43, PhMe100 °C
BnOCO2Me
H
O
OTBS3.44
Pd/C, H2
EtOAc
BnOCO2Me
H
O
OTBS3.45
85% (2 steps)
O
MeO2C
3.43

! "&)!
Figure 3.6 conformational and ring strain considerations in framework formation. With the implications described above in mind, we first chose to investigate an irreversible, intramolecular alkylative C-C bond-forming reaction before attempting bond construction through a potentially more challenging, reversible Michael addition (vide infra). In that regard, the primary alcohol of 3.51 was unveiled by treatment of TBS ether 3.45 with dilute acid in methanol (Scheme 3.5). Activation of the alcohol group as the benzenesulfonate was accomplished under standard sulfonating conditions. Scheme 3.5 Synthesis of alkylation precursor 3.46.
Having keto-benzenesulfonate 3.46 in hand, the stage was set to test the key alkylation reaction to build the bridging framework of the phragmalin-type limonoids (Table 3.1). The reaction of benzenesulfonate 3.46 with base was found to be quite sensitive. For example, excess base (entry 1) largely led to decomposition products, with no recovery of 3.49. Reducing the amount of base led to the formation of 3.49, albeit in variable and inconsistent yields. The remainder of the mass was determined to be 3.52. Ultimately, we found that a carefully controlled reaction of 3.46 in the presence of TBAI led to more consistent reactivity and to the formation of the product in 74% yield.
OBnCO2Me
O
H
H
OBs
3.46ACO2Me
OBn
OBs
OH
3.46B
CO2Me
OBn
OH
OBnCO2Me
O
H
H
HO O
+ +
Endothermic by 10.36 kcal/mol
3.47 3.48 3.49 3.50
BnOCO2Me
H
O
OTBS3.45
1% HCl/MeOH
BnOCO2Me
H
O
OH3.51
BnOCO2Me
H
O
OBs3.46
BsCl, DMAP
Pyridine: CH2Cl283% (two steps)

! "&*!
Table 3.1 Optimization of alkylation reaction.
We were able to unambiguously confirm the connectivity of the product by deprotection of the alcohol group (i.e., removal of the benzyl group) and installation of a p-bromobenzoyl group for X-ray analysis (Scheme 3.6). Scheme 3.6 Unambiguous confirmation of relative connectivity of 3.49.
We did not anticipate the formation of 3.52 (Table 3.1) under the reaction conditions. We reasoned that it was likely arising from decomposition of 3.49 under the reaction conditions. A proposed mechanism for the formation of cis-hydrindene 3.52 is shown in Figure 3.7. Trace alkoxide (possibly generated from E2 elimination) could add into the ketone group in a 1,2-fashion to generate tetrahedral intermediate 3.54. Collapse of 3.54 then generates ester enolate 3.55, which can eject a molecule of benzyl alkoxide via an E1cB-type mechanism. Support for this hypothesis may be gleaned from the fact that exposing 3.49 to potassium benzyl alkoxide generates 3.52. The generation of benzyl alkoxide as a product of this decomposition (which is initiated by benzyl alkoxide) explains the rapid formation of this by-product if these reactions are not carefully controlled.
BnOCO2Me
H
O
OBs
ConditionsOBn
CO2Me
O
H
H
H
Entry Yield of 3.49aConditions
1 DecompositionKHMDS (2 equiv), THF, –78 ! 23 °C
2 38 - 84%KHMDS (1.1 equiv), THF, –78 ! 23 °C
3 74%
H
H
BnO2C
MeO2C
+
KHMDS (1.1 equiv), TBAI (1 equiv)THF:Et3N, –78 ! 23 °C
aIsolated yield.
3.46 3.49 3.52
OBnCO2Me
O
H
H
H1. H2, Pd/C, MeOH
2. p-Bromobenzoyl chloride, Et3N, DMAP, CH2Cl2
OCO2Me
O
H
H
H
O
Br
64% (2 steps)3.49 3.53

! "'+!
Figure 3.7 Proposed mechanism for the formation of 3.52 from 3.49. After establishing the feasibility of the formation of the highly caged octahydro-1H-2,4-methanoindene skeleton, we then focused on the potentially more challenging Michael addition, which would incorporate the methylene ester moiety found in the natural products (3.1 and 3.2, Figure 3.1). Our forward synthetic route is shown in Scheme 3.7. Oxidation of alcohol 3.51 with Dess-Martin periodinane followed by Wittig olefination furnishes enoate 3.57. With Michael acceptor 3.57 in hand, we were gratified to find that exposure of 3.57 to catalytic base at low temperatures furnishes the conjugate addition product (3.58) in 79% yield as a single diastereomer. Scheme 3.7 Conjugate addition approach toward the limonoid core.
Finally, our analysis of the core-bridging framework of the phragmalin-type limonoids, unveiled two other components that we could readily install (Figure 3.8). The first was achieving the correct oxidation level at the carbon marked “B”. We understood the potential challenge of a hydride donor acting as a nucleophile in a 1,2-addition, as possible fragmentation could occur (vide supra). Secondly, the quaternary carbon center at the position marked “A” would translate to a more hindered nucleophile for alkylation and thus, also represented a challenge.
Figure 3.8 Final components of the limonoid core.
OBnCO2Me
O
H
H
H
OBnCO2MeH
H
HOBn
O
CO2Me
CO2Bn
H
H
H
OBnCO2Me
CO2Bn
H
H
HH
H
BnO2C
MeO2C
BnO
3.49 3.54 3.55 3.56 3.52
BnOCO2Me
H
O
OH
3.51
1. DMP, NaHCO3 CH2Cl22. Ph3P=CHCO2Me CH2Cl2
BnOCO2Me
H
O
CO2Me
3.57
KOtBuTHF, –78 ! 0 °C
OBnCO2Me
O
H
H
3.58
CO2Me
79%49% (2 steps)
Me
O
AcO CO2MeMe
HORO
O Me
OH
O
O
Me
AcO
AcO
Xyloccensin O (R = H) (3.1)Xyloccensin P (R = OAc) (3.2)
A B

! "'"!
Our investigation into these transformations began with position “B” and achieving the correct oxidation level found within the natural products (Scheme 3.8). We were delighted to find that reduction of the ketone group provided the secondary alcohol (3.59) as a single diastereomer with no product derived from ring opening. We attribute this stability to the hydrogen bonding of the protic solvent to the oxygen atom of the carbonyl, thus protonating the incipient alkoxide. Scheme 3.8 Introducing final components onto the limonoid core.
Introduction of the quaternary carbon center was accomplished by first methylating hydrindanone 3.45 under standard conditions. Cleavage of the TBS ether followed by activation of the newly unveiled alcohol group furnished benzenesulfonate 3.60. Under our optimized conditions for the alkylation of 3.46 (see Table 3.1), we found that the methylated bridging framework could be obtained, albeit in modest yield. 3.5 Conclusion We have constructed the bridging framework of the phragmalin-type limonoids. The accomplishment of this goal further highlights the synthetic utility of the hydrindanone precursor. Central to the success of this approach was a diastereoselective, intramolecular conjugate addition to assemble the architecturally complex core of these natural products. 3.6 Experimental Methods General: Unless otherwise stated, reactions were performed in flame-dried glassware or dried in an oven overnight. All reaction vessels were fitted with rubber septa or Teflon screw caps and kept under an atmosphere of nitrogen. Liquid reagents and solvents were transferred via syringe under nitrogen using standard Schlenk techniques. Tetrahydrofuran, toluene, and benzene were sparged with argon and passed through an alumina column. Dichloromethane was distilled over calcium hydride. All other solvents were used as received unless otherwise noted. Reaction temperatures above 23 °C refer to oil bath temperature which was controlled by an IKA® temperature
OBnCO2Me
O
H
H
HNaBH4
OBnCO2Me
OH
H
H
H
93%
MeOH:CH2Cl2
BnOCO2Me
H
O
OTBS
1. KHMDS, MeI THF, 0 °C 2. 1% HCl/MeOH
3. BsCl, Pyridine CH2Cl2
BnOCO2Me
H
O
OBs
Me
59% (3 steps)
KHMDS, TBAI
THF, –78 ! 23 °C OBnCO2Me
O
H
H
Me
10%
3.593.49
3.603.45 3.61

! "'#!
modulator. Reactions were monitored by thin layer chromatography using SiliCycle silica gel 60 F254 precoated plates (0.25 mm), which were visualized using UV irradiation, p-anisaldehyde stain or KMnO4 stain. Sorbent Technologies silica gel (particle size 40-63 µm) was used for column chromatography. 1H and 13C NMR were recorded on Bruker AVB-400, AVQ-400, DRX-500 or AV-600 spectrometers with 13C operating frequencies at 100, 125 and 150 MHz, respectively, in deuterated chloroform, benzene or p-xylene at 23 °C. Chemical shifts are reported relative to residual solvent signal (! = 7.26 for 1H NMR and 77.00 for 13C NMR in chloroform, 7.16 for 1H NMR in benzene, 6.91 for 1H NMR in p-xylene). Data for 1H NMR are reported as follows: chemical shift (multiplicity, coupling constant, number of hydrogens). Multiplicity is abbreviated as follows: s (singlet), br s (broad singlet), d (doublet), dd (doublet of doublets), t (triplet), tt (triplet of triplets), q (quartet), aq (apparent quartet), ap (apparent pentent), hept (heptet), m (multiplet). Signals marked by an asterisk (*) denote the minor rotamer. IR spectra were recorded on a Nicolet MAGNA-IR 850 spectrometer and are reported in frequency of absorption (cm-1). Only selected IR absorbencies are reported. Enantiomeric excess (ee) was determined by HPLC analysis on a Waters chromatography system (1525 binary pump, 717+ autosampler, 2487 dual wavelength detector) using a Chiralcel OD-H (0.46 cm x 25 cm)(from Daicel Chemical Ind., Ltd.) stationary phase and 97:3 hexanes/isopropanol mobile phase (1 mL/min) at 220 nm. Mass spectra were recorded on an LTQ Orbitrap XL (ThermoFisher Scientific) for ESI and AutoSpec Premier (Waters) for EI through the mass spectral facility at the University of California, Berkeley.
Aldehyde 3.40: Alcohol 3.39 (1.672 g, 10.18 mmol) and NMO (2.386 g, 20.37 mmol) were dissolved in DCM (40 mL). Finely powdered 4 Å molecular sieves (7.67 g) were then added followed by TPAP (179 mg, 0.509 mmol). Upon complete consumption of the starting material by TLC analysis, the reaction mixture was filtered through a short pad of silica, rinsed with DCM (40 mL) and concentrated. The crude reaction mixture was purified by column chromatography on silica gel (20'50% EtOAc/hexanes) to yield aldehyde 3.40 (1.087 g, 66% yield) as a colorless oil. Rf = 0.33 (30% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) d 9.38 (d, J = 8.0 Hz, 1H), 7.45 (d, J = 12.0 Hz, 1H), 7.44 – 7.35 (m, 5H), 5.72 (dd, J = 12.0, 8.0 Hz, 1H), 4.99 (s, 2H); 13C NMR (100 MHz, CDCl3) d 191.1, 169.8, 134.5, 128.88, 128.86, 127.8, 110.9, 73.5; IR (thin film) "max 3034, 2831, 2753, 1680, 1635, 1615, 1456, 1375, 1331, 1217, 1143, 956, 823, 741, 698 cm-1; HRMS (ESI) calc’d for [C10H10O2]+: m/z 162.0681, found 162.0682.
O
H
BnO
TPAP, NMO
4 Å MS, CH2Cl2
OH
BnO3.39 3.40

! "'$!
Dienoate 3.41: Aldehyde 3.40 (4.165 g, 25.68 mmol) was dissolved in THF (50 mL). Methyl (triphenylphosphoranylidene)acetate (17.174 g, 51.37 mmol) was then added and the mixture brought to reflux for 16 h after which NMR analysis showed complete consumption of the starting material. The reaction mixture was allowed to cool to room temperature, diluted with ether (50 mL) and the resulting solids filtered off. The filter cake was washed with ether (50 mL) and the solvent removed under reduced pressure to give a crude mixture that was purified by column chromatography on silica gel (5'20% EtOAc/hexanes) to yield trans-diene 3.41 (3.982 g, 18.24 mmol, 71% yield) and cis-diene (0.574 g, 2.63 mmol, 10% yield) as a waxy cream colored solid. Rf = 0.57 (30% EtOAc/hexanes); 1H NMR (500 MHz, C6D6) d 7.40 (dd, J = 15.0, 12.0 Hz, 1H), 7.12 – 7.01 (m, 5H), 6.36 (d, J = 12.0 Hz, 1H), 5.86 (d, J = 15.0 Hz, 1H), 5.84 (dd, J = 12.0, 12.0 Hz, 1H), 4.22 (s, 2H), 3.50 (s, 3H); 13C NMR (125 MHz, C6D6) d 167.5, 157.4, 143.2, 136.4, 128.7, 128.4, 127.7, 116.3, 106.1, 72.4, 50.9; IR (thin film) "max
2951, 1723, 1631, 1435, 1311, 1257, 1134, 993, 739 cm-1.
Diene 3.42: Dienoate 3.41 (840.0 mg, 3.85 mmol) was dissolved ether (50 mL) and the solution cooled to 0 °C. LiAlH4 (146 mg, 3.85 mmol) was then added in one portion and the reaction mixture was stirred for 45 min, after which TLC analysis showed complete consumption of the starting material. The reaction was carefully quenched by the sequential dropwise addition of water (0.15 mL), 15% KOH solution (0.15 mL), and water (0.30 mL). After 15 min MgSO4 was added and the mixture was stirred vigorously. After an additional 15 min the heterogeneous mixture was filtered through a sintered glass funnel to remove all solids. The filter cake was washed with DCM (50 mL) and the combined organics concentrated under reduced pressure to give a crude alcohol as a pale yellow sticky solid. The crude alcohol was dissolved in DCM (15 mL) and the solution cooled to 0 °C. Imidazole (524 mg, 7.70 mmol) was then added followed TBSCl (738 mg, 4.90 mmol) and the mixture was stirred for 1.5 h, after which TLC analysis showed complete consumption of the starting material. The reaction mixture was poured into saturated NaHCO3 solution and extracted with DCM (3 X 20 mL). The combined organic phases were dried with MgSO4 and the solvent was removed under reduced pressure to give a crude mixture that was purified by column chromatography on silica gel (3'5% EtOAc/hexanes) to yield diene 3.42 (813 mg, 69% yield over two steps) as a colorless viscous oil. Rf = 0.80 (30% EtOAc/hexanes); 1H NMR (500 MHz, C6D6) # 7.14 – 7.05 (m, 5H), 6.45 (d, J = 15.0 Hz, 1H), 6.07 (dd, J = 15.0, 10.0 Hz, 1H), 5.61 (dd, J = 12.4, 10.8 Hz, 1H), 5.54 (dt, J = 15.2, 5.5 Hz, 1H), 4.42 (s, 2H), 4.13 (d, J
O
H
BnO
3.40
THF, 66 °C
PPh3
OMe
O
BnO3.41
O
OMe+
BnO3.41
O
OMe 1. LiAlH4, Et2O, 0 °C
2. TBSCl, Imidazole CH2Cl2, 0 °C
BnO
3.42
OTBS

! "'%!
= 10.0 Hz, 2H), 0.96 (s, 9H), 0.07 (s, 6H); 13C NMR (125 MHz, C6D6) # 150.3, 137.3, 128.69, 128.65, 127.7, 127.6, 126.9, 107.3, 71.6, 64.2, 26.3, 18.6, -4.9; IR (thin film) "max 3089, 3065, 3031, 2956, 2929, 2885, 2857, 1657, 1620, 1497, 1472, 1463, 1455, 1377, 1254, 1207, 1097, 1070, 1028, 1006, 972, 838, 776, 729, 695, 667 cm-1; HRMS (EI) calc’d for [C18H28O2Si]+: m/z 304.1859, found 304.1852.
Hydrindenone 3.44: Diene 3.42 (2.060 g, 6.765 mmol) and enone 3.43 (1.375 g, 9.812 mmol) were diluted with toluene (45 mL) and the mixture heated to 100 °C. After 16 h NMR analysis showed complete consumption of the diene. The reaction mixture was concentrated under reduced pressure to give a crude mixture that was purified by column chromatography on silica gel (hexanes'5% EtOAc/hexanes) to yield endo adduct 3.44 (2.553 g, 85% yield) as a viscous, pale yellow oil. Rf = 0.47 (10% EtOAc/hexanes X 2); 1H NMR (500 MHz, CDCl3) d 7.33 – 7.29 (m, 2H), 7.28-7.24 (m, 3H), 5.91 (ddd, J = 10.4, 4.3, 2.7 Hz, 1H), 5.70 (ddd, J = 10.4, 1.1, 1.1 Hz, 1H), 4.65 – 4.63 (m, 1H), 4.54 – 4.49 (m, 2H), 3.75 (s, 3H), 3.67 – 3.60 (m, 2H), 3.02 (ddd, J = 13.0, 6.5, 6.5 Hz, 1H), 2.52 – 2.46 (m, 1H), 2.42 – 2.36 (m, 1H), 2.29 – 2.22 (m, 1H), 2.07 – 1.98 (m, 1H), 1.91 – 1.85 (m, 1H), 0.90 (s, 9H), 0.06 (s, 6H); 13C NMR (150 MHz, CDCl3) d 212.0, 171.0, 137.9, 128.3, 128.0, 127.9, 127.6, 124.9, 73.2, 71.8, 63.8, 63.4, 54.7, 39.4, 38.7, 36.7, 25.8, 20.6, 18.2, -5.43, -5.46; IR (thin film) )max 3031, 2953, 2859, 1753, 1727, 1598, 1497, 1455, 1360, 1254, 1213, 1189, 1111, 1088, 1069, 960, 835, 775, 737, 698, 667 cm-1; HRMS (ESI) calc’d for [C25H36O5SiLi]+: m/z 451.2492, found 451.2487. The exo adduct was also obtained (117 mg, 0.263 mmol, 4% yield) as a viscous, pale yellow oil. Rf = 0.55 (20% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) # 7.36 – 7.21 (m, 5H), 5.96 – 5.88 (m, 2H), 4.57 (d, J = 11.5 Hz, 1H), 4.52 – 4.51 (m, 1H), 4.48 (d, J = 11.5 Hz, 1H), 3.72 (dd, J = 9.6, 6.1 Hz, 1H), 3.67 (s, 3H), 3.58 (dd, J = 9.5, 7.4 Hz, 1H), 2.89 – 2.81 (m, 1H), 2.37 – 2.25 (m, 1H), 2.22 – 2.11 (m, 2H), 2.00 – 1.91 (m, 1H), 1.93 – 1.84 (m, 1H), 0.89 (s, 9H), 0.05 (s, 6H); 13C NMR (100 MHz, CDCl3) d 210.3, 167.6, 138.3, 132.5, 128.2, 127.6, 127.5, 124.6, 71.3, 70.8, 66.8, 66.4, 52.7, 40.6, 35.5, 34.0, 26.3, 25.9, 18.3, -5.38, -5.42; IR (thin film) "max 3031, 2953, 2929, 2894, 2857, 1755, 1738, 1730, 1462, 1433, 1399, 1249, 1211, 1101, 1061, 938, 837, 777, 735, 698 cm-1.
Benzenesulfonate 3.46: Alkene 3.44 (1.248 g, 2.807 mmol) was dissolved in EtOAc (30 mL). 10% Pd/C (325 mg) was then added and the reaction flask was evacuated and backfilled with H2 (3X). The reaction was carefully monitored and upon completion by
OBn
OTBS
O
MeO2C+
BnOCO2Me
H
O
OTBS
Toluene, 100 °C
3.433.42 3.44
BnOCO2Me
H
O
OTBS
3.44
1. H2, Pd/C, EtOAc
2. 1% HCl/MeOH3. BsCl, DMAP CH2Cl2:Pyridine
BnOCO2Me
H
O
OBs
3.46

! "'&!
NMR analysis (indicated by disappearance of olefinic protons) the reaction mixture was filtered through Celite, the filter cake washed with EtOAc, and the solvent removed under reduced pressure to produce crude alkane 3.45 as a colorless oil which was carried on without purification. Crude alkane 3.45 was dissolved in 1% HCl/MeOH (25 mL) and stirred for 30 minutes after which TLC showed complete consumption of the starting material. The reaction was poured into saturated NaHCO3 solution and extracted with DCM (4 X 10 mL). The combined organics were dried with MgSO4 and the solvent was removed under reduced pressure to produce crude alcohol 3.51 as a light yellow oil which was carried on without purification. Crude alcohol 3.51 was dissolved in DCM (15 mL) and pyridine (2 mL). Benzenesulfonyl chloride (1.43 mL, 11.2 mmol) was then added dropwise along with a few crystals of DMAP. The reaction was allowed to stir for 17 h after which TLC showed complete consumption of the starting material. The reaction was poured into 5% HCl and extracted with DCM (4 X 10 mL). The combined organics were dried with MgSO4 and the solvent was removed under reduced pressure to give a crude mixture that was purified by column chromatography on silica gel (20'30% EtOAc/hexanes) to yield benzenesulfonate 3.46 (1.106 g, 83% yield over three steps) as a colorless viscous oil. Rf = 0.52 (40% EtOAc/hexanes); 1H NMR (600 MHz, CDCl3) d 7.91 (dd, J = 8.3, 1.3 Hz, 2H), 7.69 – 7.64 (m, 1H), 7.56 (t, J = 7.8 Hz, 2H), 7.32 – 7.27 (m, 2H), 7.27 – 7.22 (m, 1H), 7.18 (dd, J = 8.0, 1.5 Hz, 2H), 4.40 (d, J = 11.1 Hz, 1H), 4.37 (s, 1H), 4.26 (d, J = 11.1 Hz, 1H), 3.98 – 3.92 (m, 2H), 3.71 (s, 3H), 3.02 (ddd, J = 13.2, 6.0, 6.0 Hz, 1H), 2.35 – 2.30 (m, 1H), 2.25 – 2.18 (m, 1H), 2.11 – 1.96 (m, 3H), 1.60 (dt, J = 11.3, 7.9 Hz, 1H), 1.56 – 1.43 (m, 2H), 1.37-1.33 (m, 1H); 13C NMR (150 MHz, CDCl3) # 213.1, 170.2, 137.8, 135.8, 133.8, 129.2, 128.2, 127.8, 127.5, 127.3, 76.8, 72.2, 72.0, 62.6, 52.7, 40.1, 38.8, 35.0, 25.8, 20.1, 16.9; IR (thin film) "max 2952, 1752, 1725, 1449, 1361, 1255, 1216, 1188, 1112, 1095, 1071, 1049, 1028, 958, 896, 827, 756, 736, 719, 689 cm-1; HRMS (ESI) calc’d for [C25H28O7SNa]+: m/z 495.1453, found 495.1454.
Tricycle 3.49: Benzenesulfonate 3.46 (224 mg, 0.474 mmol) was dissolved in THF (12 mL) in a 25 mL round bottom flask equipped with a magnetic stir bar. The reaction vessel was cooled to -78 °C and KHMDS was added dropwise (0.5 M in toluene, 1.05 mL, 0.525 mmol) to produce a light yellow solution. The reaction stirred at that temperature for 15 minutes at which time triethylamine (4.0 mL) and tetrabutylammonium iodide (176 mg, 0.476 mmol) were added and the reaction vessel warmed to room temperature. After 20 min, the reaction was quenched with saturated NaHCO3 solution (4.0 mL), poured into water (5.0 mL) and extracted with diethyl ether (3 X 7.0 mL). The combined organic phases were washed with water then brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (30% EtOAc/hexanes) to deliver octahydro-1H-2,4-methanoindene 3.49 (110 mg, 0.350 mmol, 74% yield). Rf 0.48 (40% EtOAc/hexanes);
BnOCO2Me
H
O
OBs
3.46
LiHMDS, TBAI
THF:Et3N, 0 ! 23 °C
O
CO2MeOBn
H
H
3.49

! "''!
1H NMR (300 MHz, CDCl3) # 7.33 – 7.25 (m, 5H), 4.63 (d, J = 12.6 Hz, 1H), 4.53 (d, J = 12.6 Hz, 1H), 3.80 (dd, J = 11.7, 4.5 Hz, 1H), 3.69 (s, 3H), 2.70 (d, J = 4.2 Hz, 1H), 2.57 (dd, J = 2.4, 1.8 Hz, 1H), 2.29 (dt, J = 11.8, 3.7 Hz, 1H), 1.97 (td, J = 12.6, 4.8 Hz, 1H), 1.92 – 1.78 (m, 2H), 1.75 – 1.65 (m, 1H), 1.65 – 1.49 (m, 2H), 1.36 – 1.20 (m, 2H); 13C NMR (100 MHz, CDCl3) # 211.4, 171.6, 138.5, 128.2, 127.7, 127.4, 78.3, 71.4, 64.8, 52.5, 51.7, 47.7, 36.1, 32.0, 29.5, 25.5, 22.1; IR (film) "max 2950, 1751, 1727, 1245 cm-1; HRMS (ESI) calcd for [C19H23O4]+: m/z 315.1591, found 315.1591.
Secondary alcohol 3.62: Octahydro-1H-2,4-methanoindene 3.49 (46.6 mg, 0.148 mmol) was diluted in wet methanol (2 mL) in 5 mL round bottom flask equipped with a magnetic stir bar. Pd/C (10%, 7.0 mg) was added and the reaction vessel was evacuated/backfilled (3X) with hydrogen. The reaction proceeded under a balloon of hydrogen for 19 h, at which time TLC analysis indicated complete consumption of the starting material and the heterogeneous mixture was filtered through celite and concentrated under reduced pressure. The crude product was purified over silica gel (33% EtOAc/hexanes) to deliver secondary alcohol 3.62 (25.0 mg, 0.111 mmol, 75% yield) as a colorless oil. Rf 0.32 (40% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3) # 3.95 (dd, J = 11.8, 4.8 Hz, 1H), 3.76 (s, 3H), 3.20 (br s, 1H), 2.72 (d, J = 4.6 Hz, 1H), 2.64 (dd, J = 3.9, 1.6 Hz, 1H), 2.31 (dq, J = 11.5, 3.8 Hz, 1H), 2.02 (dt, J = 12.8, 4.8 Hz, 1H), 1.98 – 1.88 (m, 2H), 1.75 – 1.60 (m, 3H), 1.31 (ddd, J = 13.2, 4.1, 2.4 Hz, 1H), 1.22 (qd, J = 13.1, 5.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) # 216.8, 170.9, 71.9, 65.0, 52.7, 51.4, 46.9, 36.6, 31.5, 29.8, 26.3, 25.2; IR (film) "max 3507, 2952, 1731, 1241, 1064 cm-
1; HRMS (EI) calc’d for [C12H16O4]+: m/z 224.1049, found 224.1044.
Benzoate 3.53: Secondary alcohol 3.62 (27.6 mg, 0.123 mmol) was dissolved in DCM (3.0 mL). Triethylamine was then added (0.12 mL, 0.369 mmol) followed by DMAP (15.0 mg, 0.123 mmol) and 4-bromobenzoyl chloride (81.0 mg, 0.369 mmol) and the resulting mixture was stirred at room temperature for 5 h at which time, TLC indicated completion and the reaction was quenched with saturated NaHCO3 solution (2.0 mL) and extracted with DCM (3 X 4.0 mL). The combined organic phases were dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (20% EtOAc/hexanes) to furnish benzoate 3.53 (42.4 mg, 0.104 mmol, 85% yield) as a colorless solid. Rf 0.43 (33% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3) # 7.98 (d, J = 8.6 Hz, 2H), 7.57 (d, J = 8.6 Hz, 2H), 5.39 (dd, J = 12.0, 4.7 Hz, 1H), 3.61 (s, 3H), 2.76 – 2.72 (m, 2H), 2.41 (ddt, J = 11.4, 7.5, 3.6 Hz, 1H), 2.10 – 2.01 (m, 3H), 1.88 – 1.74 (m, 2H), 1.69 (d, J = 10.3 Hz, 1H), 1.45 (qd, J = 13.0, 4.7 Hz, 1H), 1.35 (ddd, J = 13.3, 4.3, 2.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) # 210.4, 170.4, 164.9, 131.7, 131.5, 129.0, 128.2, 73.4, 63.1, 52.6, 51.6, 57.1, 36.2, 31.9,
O
CO2MeOBn
H
H
3.49
H2, Pd/C
MeOH
O
CO2MeOH
H
H
3.62
O
CO2MeOH
H
H
3.62
p-Bromobenzoyl Chloride
DMAP
O
CO2MeO
H
H
3.53
O
BrCH2Cl2:Et3N

! "'(!
29.4, 25.0, 22.4; IR (film) "max 2953, 1752, 1731, 1590, 1270, 1105, 1066, 1012, 758 cm-
1; m.p.: 125 – 127 °C; HRMS (ESI) calc’d for [C19H20O5Br]+: m/z 407.0489, found 407.0507.
Enoate 3.57: Alcohol 3.51 (74.5 mg, 0.224 mmol) was dissolved in DCM (2.0 mL) in a 20 mL screw cap vial equipped with a stir bar. Sodium bicarbonate (94.0 mg, 1.12 mmol) was then added, followed Dess-Martin periodinane (143 mg, 0.336 mmol). The reaction was allowed to proceed over 1 h, at which time TLC analysis indicated complete consumption of the starting material. The mixture was quenched with saturated Na2SO3 solution (1.0 mL) followed by saturated NaHCO3 (1.0 mL), extracted with DCM (3 X 2 mL). The combined organic phases were dried over MgSO4, filtered and concentrated under reduced pressure. The crude aldehyde was diluted with DCM (2.0 mL) in a 20 mL screw cap vial equipped with magnetic stir bar and methyl (triphenylphosphoranylidene)acetate (300 mg, 0.897 mmol) was added. The resulting solution was stirred at rt for 18 h at which time TLC analysis indicated complete consumption of the starting material. Silica gel was added to the reaction mixture, which was subsequently concentrated under reduced pressure and purified by column chromatography (20'33% EtOAc/hexanes) to furnish enoate 3.57 (42.4 mg, 0.110 mmol, 49% yield). Rf 0.41 (33% EtOAc/hexanes); 1H NMR (600 MHz, CDCl3) # 7.31 (t, J = 7.3 Hz, 2H), 7.27 (d, J = 7.3 Hz, 1H), 7.22 (d, J = 7.3 Hz, 2H), 6.99 (dd, J = 15.7, 6.8 Hz, 1H), 5.88 (d, J = 15.7 Hz, 1H), 4.46 (d, J = 11.1 Hz, 1H), 4.41 (s, 1H), 4.31 (d, J = 11.1 Hz, 1H), 3.74 (s, 3H), 3.73 (s, 3H), 3.04 (dt, J = 12.4, 5.9 Hz, 1H), 2.57 – 2.56 (m, 1H), 2.41 – 2.34 (m, 1H), 2.28 – 2.24 (m, 1H), 2.10 (d, J = 11.3 Hz, 2H), 1.87 (q, J = 12.2, 11.8 Hz, 1H), 1.81 – 1.74 (m, 1H), 1.52 (d, J = 14.6 Hz, 1H), 1.46 (dd, J = 13.3, 3.7 Hz, 1H). 13C NMR (150 MHz, CDCl3) # 213.3, 170.4, 166.8, 150.7, 137.9, 128.3, 127.6, 127.3, 120.7, 76.5, 72.1, 62.9, 52.8, 51.5, 43.0, 39.0, 38.4, 26.0, 21.2, 18.7; IR (film) "max 2951, 1751, 1717, 1655, 1435, 1255, 1070, 738 cm-1; HRMS (ESI) calc’d for [C22H26O6K]+: m/z 425.1361, found 425.1374.
Tricycle 3.58: Enoate 3.57 (29.5 mg, .0763 mmol) was dissolved in THF (3.8 mL) in a 10 mL round bottom flask equipped with a stir bar and cooled to -78 °C. Potassium t-butoxide (5.1 mg, 0.045 mmol) was then added in one portion and the reaction proceed at that temperature for 30 min to produce fluorescent blue solution. The mixture was
BnOCO2Me
H
O
OH
3.51
1. DMP, NaHCO3, CH2Cl2, 0 °C
2. (Triphenylphosphoranylidene)acetate, CH2Cl2
BnOCO2Me
H
O
CO2Me
3.57
BnOCO2Me
H
O
CO2Me
3.57
O
CO2MeOBn
H
H
3.58
CO2Me
KOtBu
THF, —78 ! 0 °C

! "')!
then warmed to 0 °C and allowed to proceed for an additional 45 min at which time TLC analysis indicated complete consumption of the starting material. The reaction was quenched with saturated NH4Cl solution, poured into water and extracted with EtOAc (3 X 2 mL). The combined organic phases were dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (20'33% EtOAc/hexanes) to deliver 3.58 (23.4 mg, 0.0610, 79% yield). Rf 0.07 (20% EtOAc/hexanes); 1H NMR (600 MHz, CDCl3) # 7.33 – 7.28 (m, 3H), 7.27 – 7.24 (m, 2H), 4.63 (d, J = 12.6 Hz, 1H), 4.52 (d, J = 12.6 Hz, 1H), 3.81 (dd, J = 12.0, 4.3 Hz, 1H), 3.68 (s, 3H), 3.67 (s, 3H), 2.59 (dd, J = 4.1, 2.0 Hz, 1H), 2.52 (s, 1H), 2.45 (dd, J = 15.0, 6.6 Hz, 1H), 2.32 (dd, J = 15.2, 8.4 Hz, 1H), 2.01 (dd, J = 8.9, 3.8 Hz, 1H), 1.90 – 1.81 (m, 4H), 1.76 (d, J = 11.4 Hz, 1H), 1.54 (tt, J = 4.2 Hz, 1H), 1.34 – 1.24 (m, 1H); 13C NMR (150 MHz, CDCl3) # 210.0, 171.9, 171.4, 138.4, 128.2, 127.7, 127.5, 78.6, 71.4, 64.2, 56.3, 52.6, 51.7, 48.1, 40.5, 39.1, 37.2, 32.9, 24.8, 22.8; IR (film) "max 2951, 1758, 1730, 1247, 738 cm-1; HRMS (EI) calc’d for [C22H26O6K]+: m/z 425.1361, found 425.1369.
Secondary Alcohol 3.59: Octahydro-1H-2,4-methanoindene 3.49 (20.2 mg, 0.064 mmol) was dissolved in DCM:MeOH (1:1, 4 mL) in a 10 mL round bottom flask equipped with a stir bar and NaBH4 (17 mg, 0.45 mmol) was added in one portion and the reaction proceed at room temperature for 6 h at which time TLC analysis indicated complete consumption of the starting material and was quenched with water, with DCM (3 x 2 mL). The combined organic phases were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography (30% hexanes/EtOAc) to deliver alcohol 3.59 as a colorless oil (18.8 mg, 0.060, 93% yield). Rf 0.67 (40% hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) # 7.35 – 7.22 (m, 5H), 5.06 (d, J = 4.1 Hz, 1H), 4.64 (d, J = 11.8 Hz, 1H), 4.39 (d, J = 11.8 Hz, 1H), 4.01 (dd, J = 11.9, 5.7 Hz, 1H), 3.61 (s, 3H), 3.21 (s, 1H), 2.41 (s, 1H), 2.18 (d, J = 3.9 Hz, 1H), 2.14 (td, J = 13.2, 4.5 Hz, 1H), 2.10 – 2.04 (m, 1H), 1.94 – 1.89 (m, 1H), 1.82 (dd, J = 13.5, 3.7 Hz, 1H), 1.71 (ddd, 12.6, 4.8, 1.2 Hz, 1H), 1.53 (tt, J = 13.9, 4.1 Hz, 1H), 1.46 (td, J = 12.3, 4.4 Hz, 1H), 1.34 (d, J = 10.8, 1H), 1.28 (d, J = 10.8 Hz, 1H); 13C NMR (150 MHz, CDCl3) # 176.5, 138.0, 128.3, 127.72, 127.68, 80.01, 75.5, 70.7, 55.2, 52.1, 48.3, 42.6, 35.9, 33.7, 25.1, 23.7, 21.9; IR (film) "max 3530, 2951, 1728, 1235, 1194, 1047 cm-1; HRMS (ESI) calc’d for [C19H24O4Li]+: m/z 323.1829, found 323.1841. 3.7 References and Notes (1) Brandt, G. E. L.; Schmidt, M. D.; Prisinzano, T. E.; Blagg, B. S. J. J. Med. Chem. 2008, 51, 6495-6502. (2) Cui, J.; Deng, Z.; Xu, M.; Proksch, P.; Li, Q.; Lin, W. Helv. Chim. Acta. 2009, 92, 139-150.
O
CO2MeOBn
H
H
3.49
MeOH:CH2Cl2
OH
CO2MeOBn
H
H
3.59
NaBH4

! "'*!
(3) Kuo, R.-Y.; Qian, K.; Morris-Natschke, S. L.; Lee, K.-H. Nat. Prod. Rep. 2009, 26, 1321-1344. (4) Ravangpai, W.; Sommit, D.; Teerawatananond, T.; Sinpranee, N.; Palaga, T.; Pengpreecha, S.; Muangsin, N.; Pudhom, K. Bioorg. Med. Chem. Lett. 2011, 21, 4485-4489. (5) Heasley, B. Eur. J. Org. Chem. 2011, 2011, 19-46. (6) Okamura, H.; Yamauchi, K.; Miyawaki, K.; Iwagawa, T.; Nakatani, M. Tetrahedron Lett. 1997, 38, 263-266. (7) Baker, L. A.; Williams, C. M.; Bernhardt, P. V.; Yanik, G. W. Tetrahedron 2006, 62, 7355-7360. (8) For a review of plant orthoesters, including limonoid orthoesters, see: Liao, S.-G.; Chen, H.-D.; Yue, J.-M. Chem. Rev. 2009, 109, 1092-1140. (9) Wu, J.; Xiao, Q.; Huang, J.; Xiao, Z.; Qi, S.; Li, Q.; Zhang, S. Org. Lett. 2004, 6, 1841-1844. (10) Taylor, D. A. H. J. Chem. Soc., Perkin. Trans. 1 1974, 437-441. (11) Zhang, C.-R.; Yang, S.-P.; Zhu, Q.; Liao, S.-G.; Wu, Y.; Yue, J.-M. J. Nat. Prod. 2007, 70, 1616-1619. (12) Fukuyama, Y.; Tokoroyama, T.; Kubota, T. Tetrahedron Lett. 1972, 13, 3401-3404. (13) Trudeau, S.; Morken, J. P. Org. Lett. 2005, 7, 5465-5468. (14) Miyawaki, K.; Iwagawa, T.; Nakatani, M. Tetrahedron Lett. 1997, 38, 263-266. (15) Corey, E. J.; Hahl, R. W. Tetrahedron Lett. 1989, 30, 3023-3026. (16) Kolb, H. C.; Ley, S. V. Tetrahedron Lett. 1991, 32, 6187-6190. (17) Koot, W.-J.; Ley, S. V. Tetrahedron 1995, 51, 2077-2090. (18) Kolb, H. C.; Ley, S. V.; Slawin, A. M. Z.; Williams, D. J. J. Chem. Soc., Perkin. Trans. 1 1992, 2735-272. (19) Ley, S. V.; Abad-Somovilla, A.; Anderson, J. C.; Ayats, C.; Bänteli, R.; Beckmann, E.; Boyer, A.; Brasca, M. G.; Brice, A.; Broughton, H. B.; Burke, B. J.; Cleator, E.; Craig, D.; Denholm, A. A.; Denton, R. M.; Durand-Reville, T.; Gobbi, L. B.; Göbel, M.; Gray, B. L.; Grossmann, R. B.; Gutteridge, C. E.; Hahn, N.; Harding, S. L.; Jennens, D. C.; Jennens, L.; Lovell, P. J.; Lovell, H. J.; de la Puente, M. L.; Kolb, H. C.; Koot, W.-J.; Maslen, S. L.; McCusker, C. F.; Mattes, A.; Pape, A. R.; Pinto, A.; Santafianos, D.; Scott, J. S.; Smith, S. C.; Somers, A. Q.; Spilling, C. D.; Stelzer, F.; Toogood, P. L.; Turner, R. M.; Veitch, G. E.; Wood, A.; Zumbrunn, C. Chem. – Eur. J. 2008, 14, 10683-10704. (20) Griffith, W. P.; Ley, S. V.; Whitcombe, G. P.; White, A. D. J. Chem. Soc., Chem. Commun. 1987, 1625-1627. (21) Lack, R. E.; Roberts, J. D. J. Am. Chem. Soc. 1968, 90, 6997-7001. (22) Moniz, W. B.; Dixon, J. A. J. Am. Chem. Soc. 1961, 83, 1671-1675. (23) Schneider, H.-J.; Nguyen-ba, N. Org. Magn. Resonance 1982, 18, 38-41. (24) Wheeler, S. E.; Houk, K. N.; Schleyer, P. v. R.; Allen, W. D. J. Am. Chem. Soc. 2009, 131, 2547-2560. !

! "(+!
APPENDIX THREE
Spectra and Crystallographic Data Relevant to Chapter Three:
Phragmalin-Type Limonoids

! "("!
Figure A3.1 Proton and carbon NMR spectra for compound 3.40
BnO
O
! "(#!
Figure A3.2 Proton and carbon NMR spectra for compound 3.41
BnO
CO2Me

! "($!
Figure A3.3 Proton and carbon NMR spectra for compound 3.63
BnO
OH

! "(%!
Figure A3.4 Proton and carbon NMR spectra for compound 3.42
BnO
OTBS

! "(&!
Figure A3.5 Proton and carbon spectra of compound 3.44
BnOCO2Me
H
O
OTBS

! "('!
Figure A3.6 Proton and carbon spectra of compound exo-3.44
BnOCO2Me
H
O
OTBS
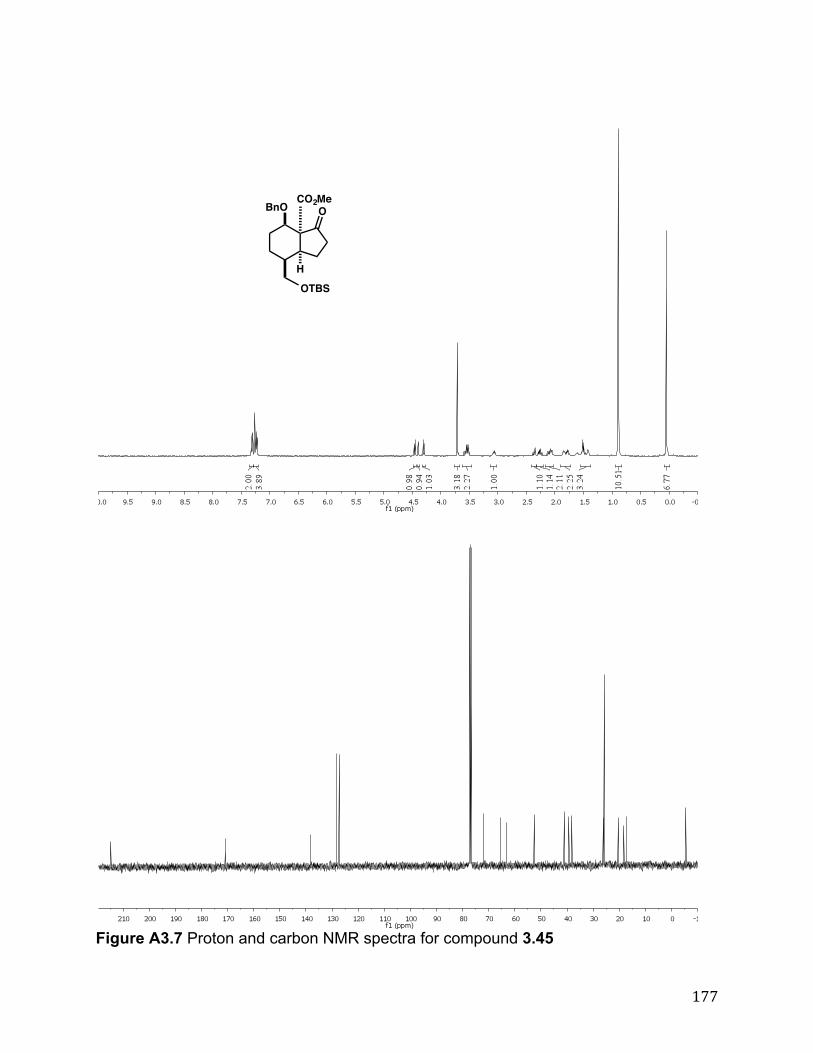
! "((!
Figure A3.7 Proton and carbon NMR spectra for compound 3.45
BnOCO2Me
H
O
OTBS

! "()!
Figure A3.8 Proton and carbon NMR spectra for compound 3.51
BnOCO2Me
H
O
OH

! "(*!
!Figure A3.9 Proton and carbon NMR spectra for compound 3.46
BnOCO2Me
H
O
OBs

! ")+!
Figure A3.10 Proton and carbon NMR spectra for compound 3.49
OBnCO2Me
O
H
H
H
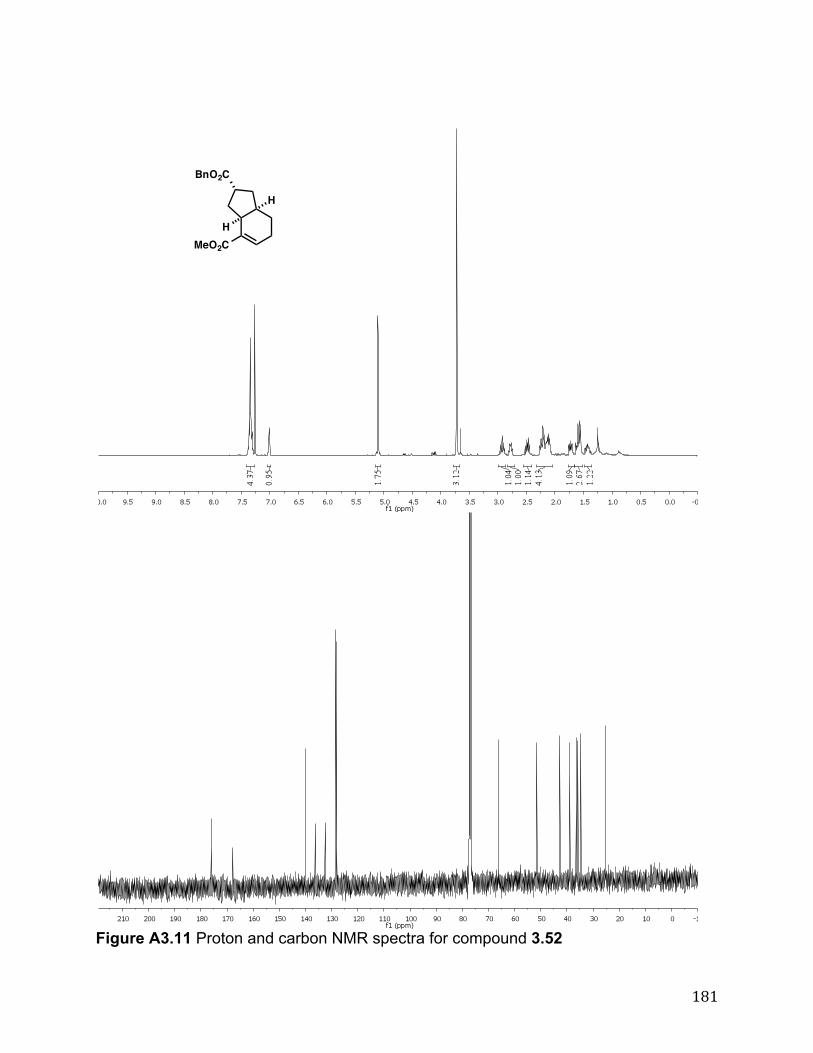
! ")"!
Figure A3.11 Proton and carbon NMR spectra for compound 3.52
H
H
BnO2C
MeO2C

! ")#!
Figure A3.12 Proton and carbon NMR spectra for compound 3.62
OHCO2Me
O
H
H
H
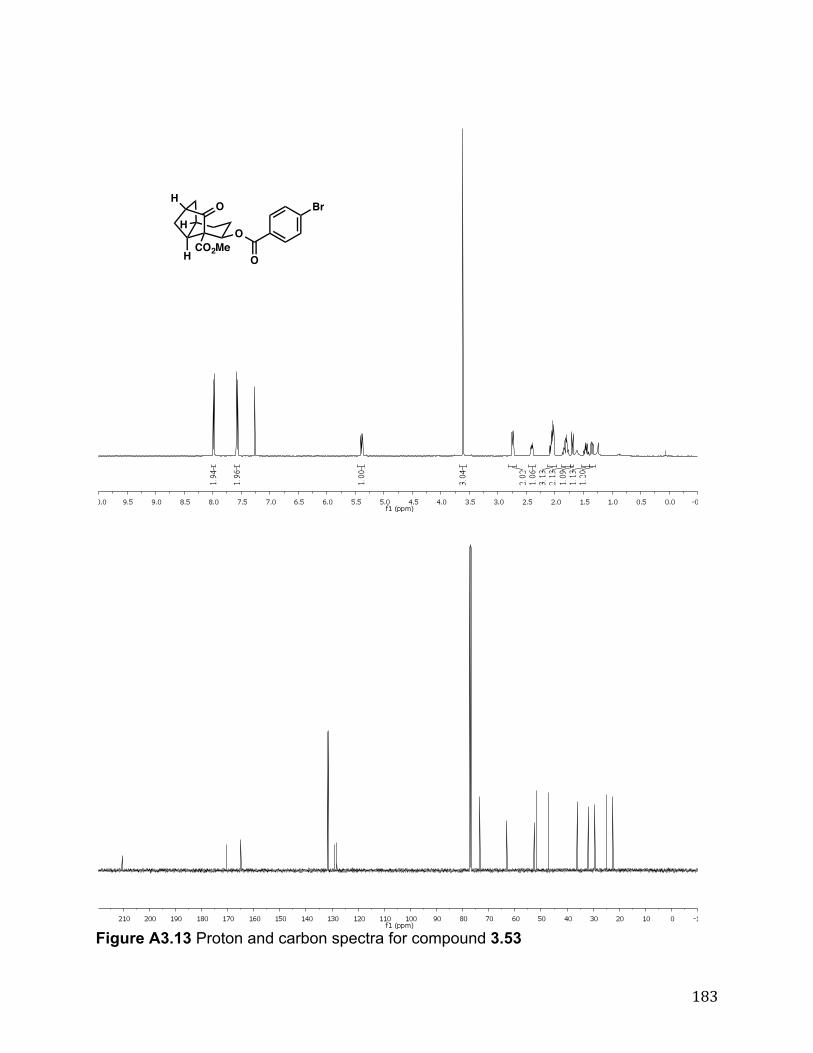
! ")$!
Figure A3.13 Proton and carbon spectra for compound 3.53
OCO2Me
O
H
H
H
O
Br

! ")%!
!Figure A3.14 Proton and carbon spectra for compound 3.57
BnOCO2Me
H
O
CO2Me

! ")&!
!Figure A3.15 Proton and carbon NMR spectra for compound 3.58
OBnCO2Me
O
H
H
CO2Me

! ")'!
!Figure A3.16 Proton and carbon NMR spectra for compound 3.59
OBnCO2Me
OH
H
H
H

! ")(!
Crystallographic data for 3.53: A colorless prism 0.08 x 0.06 x 0.04 mm in size was mounted on a Cryoloop with Paratone oil. Data were collected in a nitrogen gas stream at 100(2) K using phi and omega scans. Crystal-to-detector distance was 60 mm and exposure time was 5 seconds per frame using a scan width of 1.0°. Data collection was 97.8% complete to 67.00° in q. A total of 33120 reflections were collected covering the indices, -14<=h<=14, -15<=k<=13, -14<=l<=16. 6052 reflections were found to be symmetry independent, with an Rint of 0.0212. Indexing and unit cell refinement indicated a primitive, triclinic lattice. The space group was found to be P-1 (No. 2). The data were integrated using the Bruker SAINT software program and scaled using the SADABS software program. Solution by direct methods (SIR-2008) produced a complete heavy-atom phasing model consistent with the proposed structure. All non-hydrogen atoms were refined anisotropically by full-matrix least-squares (SHELXL-97). All hydrogen atoms were placed using a riding model. Their positions were constrained relative to their parent atom using the appropriate HFIX command in SHELXL-97.

! "))!
Table 1. Crystal data and structure refinement for sarpong25. X-ray ID sarpong25
Sample/notebook ID TL5-mullet
Empirical formula C19 H19 Br O5
Formula weight 407.25
Temperature 100(2) K
Wavelength 1.54178 Å
Crystal system Triclinic
Space group P-1
Unit cell dimensions a = 11.8721(11) Å a= 118.274(2)°.
b = 13.2210(12) Å b= 101.488(2)°.
c = 13.4918(12) Å g = 102.795(2)°.
Volume 1700.9(3) Å3
Z 4
Density (calculated) 1.590 Mg/m3
Absorption coefficient 3.537 mm-1
F(000) 832
Crystal size 0.08 x 0.06 x 0.04 mm3
Crystal color/habit colorless prism
Theta range for data collection 3.86 to 68.24°.
Index ranges -14<=h<=14, -15<=k<=13, -14<=l<=16
Reflections collected 33120
Independent reflections 6052 [R(int) = 0.0212]
Completeness to theta = 67.00° 97.8 %
Absorption correction Semi-empirical from equivalents
Max. and min. transmission 0.8715 and 0.7651
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 6052 / 0 / 453
Goodness-of-fit on F2 1.026
Final R indices [I>2sigma(I)] R1 = 0.0340, wR2 = 0.0875
R indices (all data) R1 = 0.0343, wR2 = 0.0878
Largest diff. peak and hole 1.319 and -0.952 e.Å-3

! ")*!
Table 2. Atomic coordinates ( x 104) and equivalent isotropic displacement parameters (Å2x 103)
for sarpong25. U(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________
C(1) 4020(2) 2735(2) 7101(2) 20(1)
C(2) 5319(2) 2693(2) 7445(2) 22(1)
C(3) 5021(2) 1440(2) 6292(2) 23(1)
C(4) 4456(2) 1622(2) 5309(2) 21(1)
C(5) 3810(2) 2559(2) 5839(2) 19(1)
C(6) 2436(2) 2108(2) 5131(2) 22(1)
C(7) 1692(2) 901(2) 4944(2) 27(1)
C(8) 1863(2) 1043(2) 6162(2) 27(1)
C(9) 3221(2) 1515(2) 6912(2) 23(1)
C(10) 3910(2) 629(2) 6355(2) 26(1)
C(11) 4474(2) 3770(2) 5978(2) 21(1)
C(12) 6412(2) 5199(2) 6471(2) 29(1)
C(13) 1373(2) 2155(2) 3476(2) 22(1)
C(14) 1305(2) 1886(2) 2257(2) 21(1)
C(15) 372(2) 2070(2) 1633(2) 29(1)
C(16) 242(2) 1815(3) 491(2) 31(1)
C(17) 1053(2) 1364(2) -31(2) 23(1)
C(18) 1980(2) 1155(2) 562(2) 29(1)
C(19) 2105(2) 1422(2) 1711(2) 26(1)
C(20) 4059(2) 7237(2) 7308(2) 21(1)
C(21) 4708(2) 8644(2) 7985(2) 23(1)
C(22) 3559(2) 8940(2) 8182(2) 24(1)
C(23) 3182(2) 8266(2) 8776(2) 22(1)
C(24) 3547(2) 7103(2) 8233(2) 20(1)
C(25) 2459(2) 5878(2) 7662(2) 22(1)
C(26) 1421(2) 5649(2) 6630(2) 24(1)
C(27) 1910(2) 5752(2) 5707(2) 24(1)
C(28) 2962(2) 6978(2) 6271(2) 22(1)
C(29) 2611(2) 8144(2) 6875(2) 24(1)
C(30) 4560(2) 7211(2) 9215(2) 21(1)
C(31) 5870(2) 6229(2) 9600(2) 28(1)

! "*+!
C(32) 2118(2) 5289(2) 9064(2) 25(1)
C(33) 1797(2) 5747(2) 10177(2) 24(1)
C(34) 1459(2) 4959(2) 10566(2) 26(1)
C(35) 1188(2) 5387(2) 11617(2) 27(1)
C(36) 1280(2) 6603(2) 12267(2) 27(1)
C(37) 1625(2) 7404(2) 11898(2) 27(1)
C(38) 1880(2) 6966(2) 10844(2) 25(1)
O(1) 4447(2) 1126(2) 4291(2) 28(1)
O(2) 3982(2) 4451(2) 5923(2) 26(1)
O(3) 5683(2) 3999(2) 6194(2) 26(1)
O(4) 2323(1) 1944(2) 3968(1) 22(1)
O(5) 661(2) 2511(2) 3957(2) 29(1)
O(6) 2678(2) 8550(2) 9507(2) 27(1)
O(7) 4930(2) 7986(2) 10268(2) 37(1)
O(8) 4977(2) 6291(2) 8747(2) 27(1)
O(9) 1975(1) 5974(2) 8603(2) 23(1)
O(10) 2468(2) 4441(2) 8652(2) 40(1)
Br(1) 875(1) 997(1) -1605(1) 31(1)
Br(2) 931(1) 7212(1) 13718(1) 37(1)
________________________________________________________________________________

! "*"!
Table 3. Bond lengths [Å] and angles [°] for sarpong25.
_____________________________________________________
C(1)-C(2) 1.541(3)
C(1)-C(9) 1.550(3)
C(1)-C(5) 1.564(3)
C(1)-H(1) 1.0000
C(2)-C(3) 1.539(3)
C(2)-H(2A) 0.9900
C(2)-H(2B) 0.9900
C(3)-C(4) 1.513(3)
C(3)-C(10) 1.550(3)
C(3)-H(3) 1.0000
C(4)-O(1) 1.205(3)
C(4)-C(5) 1.560(3)
C(5)-C(11) 1.523(3)
C(5)-C(6) 1.529(3)
C(6)-O(4) 1.451(3)
C(6)-C(7) 1.516(3)
C(6)-H(6) 1.0000
C(7)-C(8) 1.527(3)
C(7)-H(7A) 0.9900
C(7)-H(7B) 0.9900
C(8)-C(9) 1.524(3)
C(8)-H(8A) 0.9900
C(8)-H(8B) 0.9900
C(9)-C(10) 1.562(3)
C(9)-H(9) 1.0000
C(10)-H(10A) 0.9900
C(10)-H(10B) 0.9900
C(11)-O(2) 1.198(3)
C(11)-O(3) 1.339(3)
C(12)-O(3) 1.451(3)
C(12)-H(12A) 0.9800
C(12)-H(12B) 0.9800
C(12)-H(12C) 0.9800
C(13)-O(5) 1.210(3)
C(13)-O(4) 1.344(3)
C(13)-C(14) 1.489(3)
C(14)-C(19) 1.390(3)
C(14)-C(15) 1.391(3)
C(15)-C(16) 1.379(4)
C(15)-H(15) 0.9500
C(16)-C(17) 1.380(3)
C(16)-H(16) 0.9500
C(17)-C(18) 1.386(3)
C(17)-Br(1) 1.895(2)
C(18)-C(19) 1.385(3)
C(18)-H(18) 0.9500
C(19)-H(19) 0.9500
C(20)-C(21) 1.537(3)
C(20)-C(28) 1.548(3)
C(20)-C(24) 1.556(3)
C(20)-H(20) 1.0000
C(21)-C(22) 1.539(3)
C(21)-H(21A) 0.9900
C(21)-H(21B) 0.9900
C(22)-C(23) 1.509(3)
C(22)-C(29) 1.552(3)
C(22)-H(22) 1.0000
C(23)-O(6) 1.207(3)
C(23)-C(24) 1.564(3)
C(24)-C(30) 1.527(3)
C(24)-C(25) 1.543(3)
C(25)-O(9) 1.458(3)
C(25)-C(26) 1.518(3)
C(25)-H(25) 1.0000
C(26)-C(27) 1.523(3)
C(26)-H(26A) 0.9900
C(26)-H(26B) 0.9900
C(27)-C(28) 1.524(3)

! "*#!
C(27)-H(27A) 0.9900
C(27)-H(27B) 0.9900
C(28)-C(29) 1.562(3)
C(28)-H(28) 1.0000
C(29)-H(29A) 0.9900
C(29)-H(29B) 0.9900
C(30)-O(7) 1.198(3)
C(30)-O(8) 1.337(3)
C(31)-O(8) 1.449(3)
C(31)-H(31A) 0.9800
C(31)-H(31B) 0.9800
C(31)-H(31C) 0.9800
C(32)-O(10) 1.204(3)
C(32)-O(9) 1.341(3)
C(32)-C(33) 1.494(3)
C(33)-C(38) 1.393(4)
C(33)-C(34) 1.392(4)
C(34)-C(35) 1.391(4)
C(34)-H(34) 0.9500
C(35)-C(36) 1.384(4)
C(35)-H(35) 0.9500
C(36)-C(37) 1.388(4)
C(36)-Br(2) 1.905(2)
C(37)-C(38) 1.385(4)
C(37)-H(37) 0.9500
C(38)-H(38) 0.9500
C(2)-C(1)-C(9) 100.59(18)
C(2)-C(1)-C(5) 102.76(17)
C(9)-C(1)-C(5) 108.14(17)
C(2)-C(1)-H(1) 114.6
C(9)-C(1)-H(1) 114.6
C(5)-C(1)-H(1) 114.6
C(3)-C(2)-C(1) 95.30(17)
C(3)-C(2)-H(2A) 112.7
C(1)-C(2)-H(2A) 112.7
C(3)-C(2)-H(2B) 112.7
C(1)-C(2)-H(2B) 112.7
H(2A)-C(2)-H(2B) 110.2
C(4)-C(3)-C(2) 102.66(18)
C(4)-C(3)-C(10) 104.73(18)
C(2)-C(3)-C(10) 100.05(18)
C(4)-C(3)-H(3) 115.8
C(2)-C(3)-H(3) 115.8
C(10)-C(3)-H(3) 115.8
O(1)-C(4)-C(3) 128.3(2)
O(1)-C(4)-C(5) 124.9(2)
C(3)-C(4)-C(5) 106.69(18)
C(11)-C(5)-C(6) 108.75(18)
C(11)-C(5)-C(4) 110.50(17)
C(6)-C(5)-C(4) 115.41(18)
C(11)-C(5)-C(1) 110.40(17)
C(6)-C(5)-C(1) 111.31(17)
C(4)-C(5)-C(1) 100.25(17)
O(4)-C(6)-C(7) 109.25(18)
O(4)-C(6)-C(5) 107.72(17)
C(7)-C(6)-C(5) 112.76(19)
O(4)-C(6)-H(6) 109.0
C(7)-C(6)-H(6) 109.0
C(5)-C(6)-H(6) 109.0
C(6)-C(7)-C(8) 109.67(19)
C(6)-C(7)-H(7A) 109.7
C(8)-C(7)-H(7A) 109.7
C(6)-C(7)-H(7B) 109.7
C(8)-C(7)-H(7B) 109.7
H(7A)-C(7)-H(7B) 108.2
C(9)-C(8)-C(7) 111.89(19)
C(9)-C(8)-H(8A) 109.2
C(7)-C(8)-H(8A) 109.2
C(9)-C(8)-H(8B) 109.2
C(7)-C(8)-H(8B) 109.2

! "*$!
H(8A)-C(8)-H(8B) 107.9
C(8)-C(9)-C(1) 114.12(19)
C(8)-C(9)-C(10) 114.7(2)
C(1)-C(9)-C(10) 102.80(17)
C(8)-C(9)-H(9) 108.3
C(1)-C(9)-H(9) 108.3
C(10)-C(9)-H(9) 108.3
C(3)-C(10)-C(9) 103.87(18)
C(3)-C(10)-H(10A) 111.0
C(9)-C(10)-H(10A) 111.0
C(3)-C(10)-H(10B) 111.0
C(9)-C(10)-H(10B) 111.0
H(10A)-C(10)-H(10B) 109.0
O(2)-C(11)-O(3) 123.8(2)
O(2)-C(11)-C(5) 124.2(2)
O(3)-C(11)-C(5) 111.92(18)
O(3)-C(12)-H(12A) 109.5
O(3)-C(12)-H(12B) 109.5
H(12A)-C(12)-H(12B) 109.5
O(3)-C(12)-H(12C) 109.5
H(12A)-C(12)-H(12C) 109.5
H(12B)-C(12)-H(12C) 109.5
O(5)-C(13)-O(4) 124.4(2)
O(5)-C(13)-C(14) 124.3(2)
O(4)-C(13)-C(14) 111.33(19)
C(19)-C(14)-C(15) 119.3(2)
C(19)-C(14)-C(13) 122.5(2)
C(15)-C(14)-C(13) 118.1(2)
C(16)-C(15)-C(14) 121.0(2)
C(16)-C(15)-H(15) 119.5
C(14)-C(15)-H(15) 119.5
C(15)-C(16)-C(17) 118.8(2)
C(15)-C(16)-H(16) 120.6
C(17)-C(16)-H(16) 120.6
C(16)-C(17)-C(18) 121.5(2)
C(16)-C(17)-Br(1) 119.11(18)
C(18)-C(17)-Br(1) 119.33(18)
C(19)-C(18)-C(17) 119.1(2)
C(19)-C(18)-H(18) 120.5
C(17)-C(18)-H(18) 120.5
C(18)-C(19)-C(14) 120.3(2)
C(18)-C(19)-H(19) 119.9
C(14)-C(19)-H(19) 119.9
C(21)-C(20)-C(28) 101.87(18)
C(21)-C(20)-C(24) 101.61(18)
C(28)-C(20)-C(24) 108.21(18)
C(21)-C(20)-H(20) 114.6
C(28)-C(20)-H(20) 114.6
C(24)-C(20)-H(20) 114.6
C(20)-C(21)-C(22) 95.32(17)
C(20)-C(21)-H(21A) 112.7
C(22)-C(21)-H(21A) 112.7
C(20)-C(21)-H(21B) 112.7
C(22)-C(21)-H(21B) 112.7
H(21A)-C(21)-H(21B) 110.2
C(23)-C(22)-C(21) 101.16(18)
C(23)-C(22)-C(29) 105.32(19)
C(21)-C(22)-C(29) 101.42(18)
C(23)-C(22)-H(22) 115.6
C(21)-C(22)-H(22) 115.6
C(29)-C(22)-H(22) 115.6
O(6)-C(23)-C(22) 127.2(2)
O(6)-C(23)-C(24) 126.5(2)
C(22)-C(23)-C(24) 106.32(18)
C(30)-C(24)-C(25) 108.28(18)
C(30)-C(24)-C(20) 111.02(18)
C(25)-C(24)-C(20) 113.62(18)
C(30)-C(24)-C(23) 109.68(18)
C(25)-C(24)-C(23) 113.61(18)
C(20)-C(24)-C(23) 100.49(18)
O(9)-C(25)-C(26) 107.95(18)
O(9)-C(25)-C(24) 106.71(17)

! "*%!
C(26)-C(25)-C(24) 112.60(19)
O(9)-C(25)-H(25) 109.8
C(26)-C(25)-H(25) 109.8
C(24)-C(25)-H(25) 109.8
C(25)-C(26)-C(27) 110.78(19)
C(25)-C(26)-H(26A) 109.5
C(27)-C(26)-H(26A) 109.5
C(25)-C(26)-H(26B) 109.5
C(27)-C(26)-H(26B) 109.5
H(26A)-C(26)-H(26B) 108.1
C(26)-C(27)-C(28) 112.30(19)
C(26)-C(27)-H(27A) 109.1
C(28)-C(27)-H(27A) 109.1
C(26)-C(27)-H(27B) 109.1
C(28)-C(27)-H(27B) 109.1
H(27A)-C(27)-H(27B) 107.9
C(27)-C(28)-C(20) 113.50(19)
C(27)-C(28)-C(29) 114.86(19)
C(20)-C(28)-C(29) 102.80(18)
C(27)-C(28)-H(28) 108.5
C(20)-C(28)-H(28) 108.5
C(29)-C(28)-H(28) 108.5
C(22)-C(29)-C(28) 103.74(18)
C(22)-C(29)-H(29A) 111.0
C(28)-C(29)-H(29A) 111.0
C(22)-C(29)-H(29B) 111.0
C(28)-C(29)-H(29B) 111.0
H(29A)-C(29)-H(29B) 109.0
O(7)-C(30)-O(8) 123.3(2)
O(7)-C(30)-C(24) 126.0(2)
O(8)-C(30)-C(24) 110.65(18)
O(8)-C(31)-H(31A) 109.5
O(8)-C(31)-H(31B) 109.5
H(31A)-C(31)-H(31B) 109.5
O(8)-C(31)-H(31C) 109.5
H(31A)-C(31)-H(31C) 109.5
H(31B)-C(31)-H(31C) 109.5
O(10)-C(32)-O(9) 125.0(2)
O(10)-C(32)-C(33) 125.0(2)
O(9)-C(32)-C(33) 110.0(2)
C(38)-C(33)-C(34) 120.2(2)
C(38)-C(33)-C(32) 120.2(2)
C(34)-C(33)-C(32) 119.6(2)
C(35)-C(34)-C(33) 120.0(2)
C(35)-C(34)-H(34) 120.0
C(33)-C(34)-H(34) 120.0
C(36)-C(35)-C(34) 118.8(2)
C(36)-C(35)-H(35) 120.6
C(34)-C(35)-H(35) 120.6
C(35)-C(36)-C(37) 122.0(2)
C(35)-C(36)-Br(2) 119.54(19)
C(37)-C(36)-Br(2) 118.46(19)
C(38)-C(37)-C(36) 118.7(2)
C(38)-C(37)-H(37) 120.6
C(36)-C(37)-H(37) 120.6
C(37)-C(38)-C(33) 120.3(2)
C(37)-C(38)-H(38) 119.8
C(33)-C(38)-H(38) 119.8
C(11)-O(3)-C(12) 115.55(18)
C(13)-O(4)-C(6) 116.06(17)
C(30)-O(8)-C(31) 116.06(18)
C(32)-O(9)-C(25) 120.52(18)

! "*&!
_____________________________________________________________
Symmetry transformations used to generate equivalent atoms:
! "*'!
Table 4. Anisotropic displacement parameters (Å2x 103)for sarpong25. The anisotropic
displacement factor exponent takes the form: -2p2[ h2a*2U11 + ... + 2 h k a* b* U12 ]
______________________________________________________________________________
U11 U22 U33 U23 U13 U12
______________________________________________________________________________
C(1) 20(1) 24(1) 16(1) 11(1) 6(1) 6(1)
C(2) 20(1) 24(1) 23(1) 15(1) 5(1) 6(1)
C(3) 23(1) 24(1) 26(1) 16(1) 9(1) 9(1)
C(4) 19(1) 19(1) 25(1) 12(1) 9(1) 4(1)
C(5) 19(1) 22(1) 17(1) 11(1) 7(1) 7(1)
C(6) 19(1) 30(1) 17(1) 13(1) 6(1) 7(1)
C(7) 20(1) 33(1) 22(1) 15(1) 4(1) 2(1)
C(8) 21(1) 36(1) 26(1) 19(1) 8(1) 4(1)
C(9) 21(1) 28(1) 21(1) 15(1) 7(1) 5(1)
C(10) 26(1) 25(1) 28(1) 17(1) 9(1) 7(1)
C(11) 22(1) 24(1) 16(1) 11(1) 7(1) 7(1)
C(12) 27(1) 27(1) 33(1) 20(1) 9(1) 3(1)
C(13) 20(1) 22(1) 20(1) 11(1) 5(1) 4(1)
C(14) 19(1) 20(1) 20(1) 11(1) 4(1) 3(1)
C(15) 27(1) 42(1) 27(1) 20(1) 12(1) 19(1)
C(16) 26(1) 45(2) 30(1) 26(1) 8(1) 17(1)
C(17) 23(1) 24(1) 18(1) 14(1) 5(1) 2(1)
C(18) 29(1) 39(1) 26(1) 20(1) 14(1) 18(1)
C(19) 25(1) 34(1) 24(1) 19(1) 9(1) 14(1)
C(20) 19(1) 24(1) 21(1) 13(1) 7(1) 7(1)
C(21) 22(1) 24(1) 24(1) 14(1) 8(1) 5(1)
C(22) 24(1) 21(1) 26(1) 14(1) 9(1) 8(1)
C(23) 19(1) 21(1) 22(1) 11(1) 4(1) 6(1)
C(24) 19(1) 22(1) 20(1) 12(1) 7(1) 7(1)
C(25) 20(1) 24(1) 22(1) 14(1) 8(1) 7(1)
C(26) 19(1) 25(1) 26(1) 15(1) 4(1) 5(1)
C(27) 23(1) 25(1) 21(1) 13(1) 3(1) 6(1)
C(28) 22(1) 26(1) 21(1) 14(1) 7(1) 9(1)
C(29) 24(1) 26(1) 26(1) 18(1) 7(1) 9(1)
C(30) 21(1) 22(1) 23(1) 14(1) 7(1) 5(1)
C(31) 25(1) 29(1) 26(1) 15(1) 3(1) 10(1)
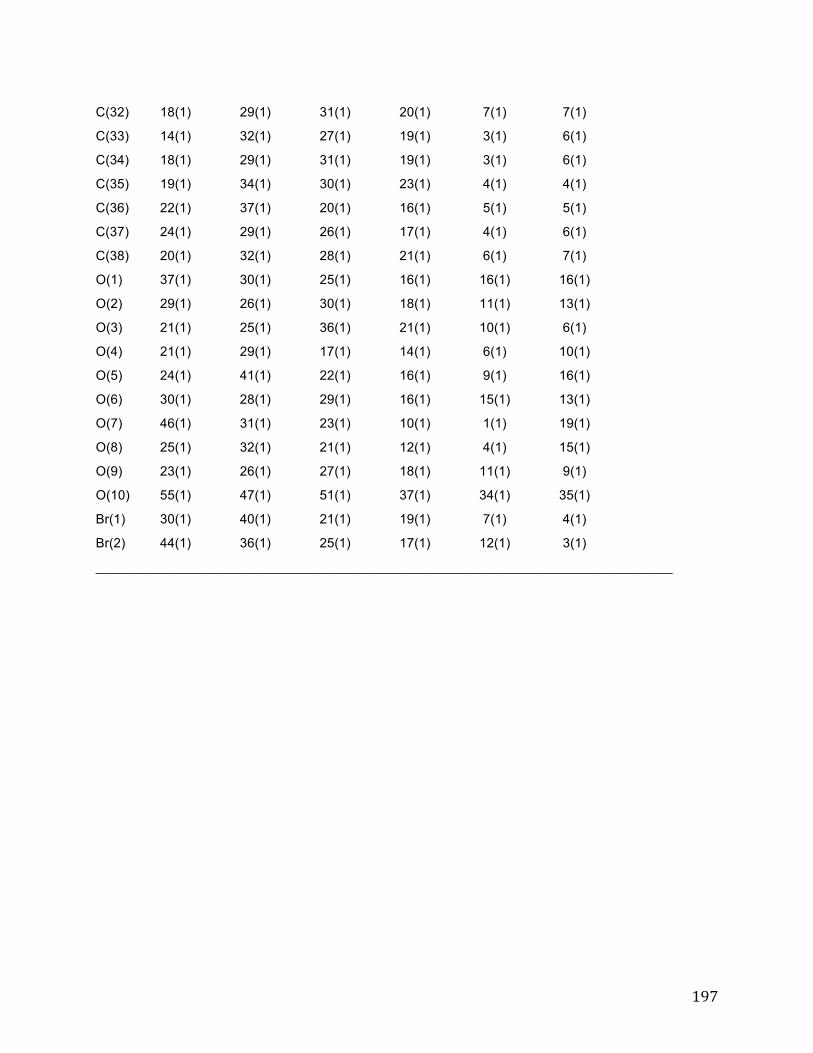
! "*(!
C(32) 18(1) 29(1) 31(1) 20(1) 7(1) 7(1)
C(33) 14(1) 32(1) 27(1) 19(1) 3(1) 6(1)
C(34) 18(1) 29(1) 31(1) 19(1) 3(1) 6(1)
C(35) 19(1) 34(1) 30(1) 23(1) 4(1) 4(1)
C(36) 22(1) 37(1) 20(1) 16(1) 5(1) 5(1)
C(37) 24(1) 29(1) 26(1) 17(1) 4(1) 6(1)
C(38) 20(1) 32(1) 28(1) 21(1) 6(1) 7(1)
O(1) 37(1) 30(1) 25(1) 16(1) 16(1) 16(1)
O(2) 29(1) 26(1) 30(1) 18(1) 11(1) 13(1)
O(3) 21(1) 25(1) 36(1) 21(1) 10(1) 6(1)
O(4) 21(1) 29(1) 17(1) 14(1) 6(1) 10(1)
O(5) 24(1) 41(1) 22(1) 16(1) 9(1) 16(1)
O(6) 30(1) 28(1) 29(1) 16(1) 15(1) 13(1)
O(7) 46(1) 31(1) 23(1) 10(1) 1(1) 19(1)
O(8) 25(1) 32(1) 21(1) 12(1) 4(1) 15(1)
O(9) 23(1) 26(1) 27(1) 18(1) 11(1) 9(1)
O(10) 55(1) 47(1) 51(1) 37(1) 34(1) 35(1)
Br(1) 30(1) 40(1) 21(1) 19(1) 7(1) 4(1)
Br(2) 44(1) 36(1) 25(1) 17(1) 12(1) 3(1)
______________________________________________________________________________
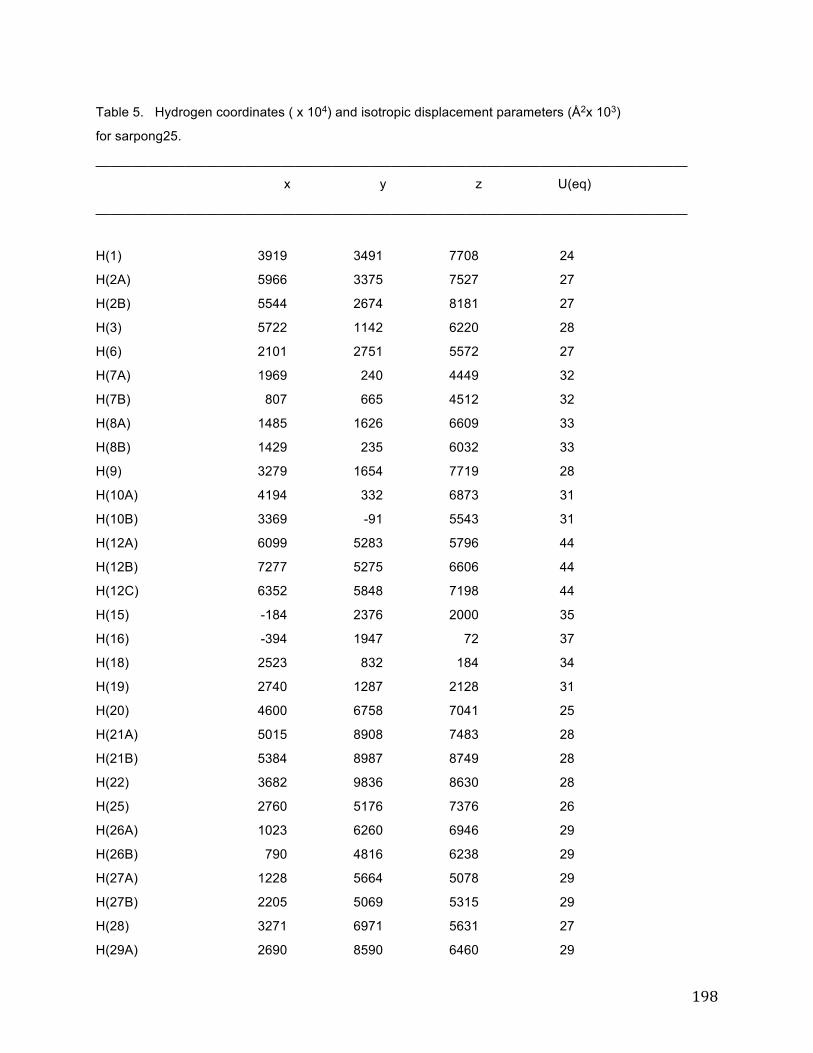
! "*)!
Table 5. Hydrogen coordinates ( x 104) and isotropic displacement parameters (Å2x 103)
for sarpong25.
________________________________________________________________________________
x y z U(eq)
________________________________________________________________________________
H(1) 3919 3491 7708 24
H(2A) 5966 3375 7527 27
H(2B) 5544 2674 8181 27
H(3) 5722 1142 6220 28
H(6) 2101 2751 5572 27
H(7A) 1969 240 4449 32
H(7B) 807 665 4512 32
H(8A) 1485 1626 6609 33
H(8B) 1429 235 6032 33
H(9) 3279 1654 7719 28
H(10A) 4194 332 6873 31
H(10B) 3369 -91 5543 31
H(12A) 6099 5283 5796 44
H(12B) 7277 5275 6606 44
H(12C) 6352 5848 7198 44
H(15) -184 2376 2000 35
H(16) -394 1947 72 37
H(18) 2523 832 184 34
H(19) 2740 1287 2128 31
H(20) 4600 6758 7041 25
H(21A) 5015 8908 7483 28
H(21B) 5384 8987 8749 28
H(22) 3682 9836 8630 28
H(25) 2760 5176 7376 26
H(26A) 1023 6260 6946 29
H(26B) 790 4816 6238 29
H(27A) 1228 5664 5078 29
H(27B) 2205 5069 5315 29
H(28) 3271 6971 5631 27
H(29A) 2690 8590 6460 29

! "**!
H(29B) 1756 7915 6867 29
H(31A) 6588 7004 10080 42
H(31B) 6134 5543 9165 42
H(31C) 5493 6098 10134 42
H(34) 1414 4131 10115 31
H(35) 943 4852 11883 32
H(37) 1686 8237 12360 33
H(38) 2112 7500 10575 30 _____________________________________________________________________________
!





























