Embed Size (px)
Citation preview
doi.org/10.26434/chemrxiv.7795484.v1
Electrochemical Tryptophan-Selective BioconjugationEisho Toyama, Katsuya Marumaya, Tomoya Sugai, Mio Kondo, Shigeyuki Masaoka, Tsuyoshi Saitoh,Kounosuke Oisaki, Motomu Kanai
Submitted date: 02/03/2019 • Posted date: 05/03/2019Licence: CC BY-NC-ND 4.0Citation information: Toyama, Eisho; Marumaya, Katsuya; Sugai, Tomoya; Kondo, Mio; Masaoka, Shigeyuki;Saitoh, Tsuyoshi; et al. (2019): Electrochemical Tryptophan-Selective Bioconjugation. ChemRxiv. Preprint.
Bioconjugation reactions are a fundamental synthetic method for generating artificial peptides and proteins.Despite the potentially superior properties of bioconjugates at hydrophobic amino acid residues comparedwith those at hydrophilic amino acids, methods to target hydrophobic amino acids with moderate reactivityunder mild and metal-free conditions are limited. Here we report the first electrochemically-promotedtryptophan (Trp)-selective bioconjugation of peptides and proteins in neutral aqueous media. The uniqueelectrochemical cooperation of two radicals, keto-ABNO and 4-oxo-TEMPO, was critical to suppress bothanodic overoxidation of the products and cross reactivity. Systematic cyclic voltammetry analysis suggestedthat these two radicals, containing similar redox potentials but contrasting steric demands, had distinctelectrochemical roles (reactant and electrochemical mediator). This new protocol will be an important advancetoward clean and scalable syntheses of chemically modified biologics.
File list (2)
download fileview on ChemRxivSI_toyama_V6499.docx (2.98 MiB)
download fileview on ChemRxivChemRxiv.pdf (658.94 KiB)

Supporting Information
Electrochemical Tryptophan-Selective Bioconjugation
Eisho Toyama, Katsuya Maruyama, Tomoki Sugai, Mio Kondo, Shigeyuki Masaoka,Tsuyoshi Saitoh, Kounosuke Oisaki* and Motomu Kanai*
*Correspondence to: [email protected] (KO); [email protected] (MK)
Contents
Material & Methods·················································································································S2
1. General Methods············································································································S2
2. Analytical Data···············································································································S5
3. Synthesis and Characterization of New Compounds·····················································S7
4. Detailed Optimization for Electrochemical Trp-selective Bioconjugation··················S11
5. Reaction Profiles··········································································································S12
6. Cyclic Voltammetry Studies·························································································S13
7. HPLC and NMR Charts·······························································································S26
8. References····················································································································S43
S1

Materials and Methods
1. General Methods
1-1. GeneralNMR spectra were recorded on JEOL ECX500 (500 MHz for 1H NMR and 125
MHz for 13C NMR) or JEOL ECS400 (400 MHz for 1H NMR and 100 MHz for 13CNMR) spectrometer. Chemical shifts were reported downfield from TMS ( = 0 ppm) for1H NMR. For 13C NMR, chemical shifts were reported in the scale relative to the solventused as an internal reference. Infrared (IR) spectra were recorded on a JASCO FT/IR 410Fourier transform infrared spectrophotometer. High resolution ESI-mass spectra (HRMS)were measured on a JEOL JMS-T100LC AccuTOF spectrometer. Columnchromatographies were performed with silica gel 60 (spherical) 40-50 μm (KantoChemicals). Analytical HPLC charts were obtained by using a JASCO HPLC system(UV-2075 spectrometer, PU-2080 pumps, DG-2080-54 degasser, and MX-2080-32mixer) or a Shimadzu HPLC system (SPD-20A UV–vis detector, LC-20AD pumps, andDGU-20A degasser). Preparative HPLC were conducted by using a JASCO HPLCsystem (UV-2075 spectrometer, PU-2086 pumps, DG-2080-53 degasser, and MX-2080-32 mixer), or a Shimadzu HPLC system (SPD-20A UV–vis detector and LC-6ADpumps). LC/MS (ESI) analyses were conducted using Agilent Technologies 6200equipped with 1260 infinity. All non-commercially available compounds were preparedand characterized as described in Section 3. Other reagents were purchased from Sigma-Aldrich, Tokyo Chemical Industry Co., Ltd. (TCI), Kanto Chemical Co., Inc., FujifilmWako Pure Chemical Corp., Peptides Institute, Inc., Watanabe Chemical Industries, Ltd.,Nacalai Tesque, Inc. and used without further purification. Water was purified using aMerck Millipore Milli-Q water purification system.
1-2. Analytical HPLCPeptide compositions were evaluated by analytical reverse phase HPLC using a
gradient of acetonitrile versus 0.1% TFA in water. Analytical HPLC was carried out asfollows: YMC-Triart-C18 (4.6 mm I.D. × 150 mm) column using a linear gradient of 0–100% acetonitrile in 0.1% aqueous TFA over 40 min at room temperature with a flow rateof 1 mL min–1. The eluent was monitored by absorbance at 230 nm.
1-3. Analytical LC/MSReactions were monitored by LC/MS spectroscopy using a gradient of acetonitrile
versus 0.1% formic acid in water. LC was carried out with YMC-Triart-C18 (4.6 mm I.D.× 150 mm) column at 40 oC with a flow rate of 0.2 mL min–1. The eluent was monitoredby on-line ESI-Qq-TOF MS. Linear gradient methods are discribed as follows: LC Method A: After washing with 0.1% aqueous formic acid over 2 min, 20–40%acetonitrile in 0.1% aqueous formic acid over 16 min LC Method B: After washing with 0.1% aqueous formic acid over 2 min, 20–60%acetonitrile in 0.1% aqueous formic acid over 38 min
S2

LC Method C: After washing with 0.1% aqueous formic acid over 2 min, 0–30%acetonitrile in 0.1% aqueous formic acid over 21 minLC Method D: After washing with 0.1% aqueous formic acid over 2 min, 0–40%acetonitrile in 0.1% aqueous formic acid over 28 min LC Method E: After washing with 0.1% aqueous formic acid over 2 min, 58–98%acetonitrile in 0.1 % aqueous formic acid over 15 min
1-4. Preparative HPLCPeptides were purified by preparative reverse phase HPLC using a gradient of
acetonitrile versus 0.1% TFA in water. Preparative HPLC was carried out as follows:YMC-Triart C18 (10 mm I.D. × 250 mm) column using a linear gradient of 0-100%acetonitrile in 0.1% aqueous TFA over 100 min at 40 oC with a flow rate of 3.0 mL min–1.The eluent was monitored by absorbance at 230 nm.
1-5. General Protocol for Peptide SynthesisPeptide syntheses were performed manually on a 0.1 mmol scale using
chlorotrityl chloride resin. Fmoc-protected amino acids were sequentially coupled using a2.5-fold excess (0.25 mmol) using a DIC–HOBt method (30 min) after removal of eachFmoc group with 20% piperidine–DMF (10 min) to obtain a peptide-resin. Treating theobtained peptide-resin with TFA–triisopropylsilane (TIS)–water (95:2.5:2.5) for 60 minat room temperature, concentrated in vacuo, precipitated with diethyl ether followed bylyophilization gave the crude peptide. The crude peptide was purified using a preparativeHPLC with 0.1% aqueous TFA–acetonitrile system and freeze-drying of the collectedfraction gave the desired peptide as a white solid.
1-6. General Protocol for Electrochemical Trp-selective Bioconjugation of Peptidesand Proteins (represented by 1a)
The ElectraSyn vial (5 mL) with a stir bar (AS ONE, 1-5409-01, 8 × 10 mm)was charged with solution A (0.1 M solution of tetrabutylammonium perchlorate (TBAP)in CH3CN, 1.5 mL), H2O (140 L), Fmoc-Gly-Ser-Asn-Trp-Gly-OH (1a, 8.5 mg, 11mol), 6.5 mM keto-ABNO (1.8 mL H2O solution, 11 mol), 5.9 mM 4-oxo-TEMPO(390 L, in solution A, 2.3 mol). The EletraSyn vial cap equipped with anode (graphite)and cathode (Pt) were inserted into the mixture. The reaction mixture was electrolyzedunder a constant voltage of 1.0 V until 1.0-3.0 F/mol of electron was charged. After theelectrolysis, PBS buffer (pH = 7.4) was added to quench the reaction. The reaction wasmonitored by LC/MS spectroscopy. The LC analysis was performed using a C18 reversephase column (4.6 × 150 mm; YMC-Triart-C18 column), detected at 230 nm to determinethe HPLC yield by the ratio between the sum of peak areas for the target products (whendiastereomers were separated) vs. the sum of peak areas of all the detected peaks.
1-7. Protocol for Electrochemical Trp-selective Bioconjugation Orthogonally toElectrochemical Tyr-selective Bioconjugation using Peptide 1e
S3

The ElectraSyn vial (5 mL) with a stir bar (AS ONE, 1-5409-01, 8 × 10 mm)was charged with solution A (1.9 mL), H2O (1.9 mL), Fmoc-Gly-Tyr-Asn-Trp-Gly-OH(1e, 9.3 mg, 11 mol), and Me-Luminol (6.0 mg, 34 mol). The EletraSyn vial capequipped with anode (graphite) and cathode (Pt) were inserted into the mixture. The firstelectrolysis was conducted to the reaction mixture under a constant voltage of 1.2 V until4.0 F/mol of electron was charged. After the first electrolysis, keto-ABNO (1.8 mg, 11mol), 4-oxo-TEMPO (1.0 mg, 5.7 mol) was added to the solution. The secondelectrolysis was conducted to the reaction mixture under a constant voltage of 1.0 V until1.0 F/mol of electron was charged. After the second electrolysis, PBS buffer (pH = 7.4)was added to quench the reaction. The reaction was monitored by LC/MS spectroscopy.The LC analysis was performed using a C18 reverse phase column (4.6 × 150 mm;YMC-Triart-C18 column), detected at 230 nm to determine the HPLC yield by the ratiobetween the sum of peak areas for the target products (when diastereomers wereseparated) vs. the sum of peak areas of all the detected peaks.
S4

2. Analytical Data
Fmoc-Gly-Ser-Asn-Trp-Gly-OH (1a). MS (ESI): m/z 742.2 (calcd [M+H]+ = 742.3).Purity: >95% (HPLC analysis at 230 nm). Retention time: 18.0 min for LC method A.Fmoc-Gly-Lys-Asn-Trp-Gly-OH (1c). MS (ESI): m/z 783.3 (calcd [M+H]+ = 783.4).Purity: >95% (HPLC analysis at 230 nm). Retention time: 13.1 min for LC method B.Fmoc-Gly-His-Asn-Trp-Gly-OH (1d). MS (ESI): m/z 792.3 (calcd [M+H]+ = 792.3).Purity: >95% (HPLC analysis at 230 nm). Retention time: 11.5 min for LC method A.Fmoc-Gly-Tyr-Asn-Trp-Gly-OH (1e). MS (ESI): m/z 818.3 (calcd [M+H]+ = 818.3).Purity: >95% (HPLC analysis at 230 nm). Retention time: 20.4 min for LC method B. Fmoc-Gly-Met-Asn-Trp-Gly-OH (1f). MS (ESI): m/z 786.3 (calcd [M+H]+ = 786.3).Purity: >95% (HPLC analysis at 230 nm). Retention time: 21.4 min for LC method B.Fmoc-Gly-Cys-Asn-Trp-Gly-OH (1g). MS (ESI): m/z 757.2 (calcd [M+H]+ = 757.3).Purity: >95% (HPLC analysis at 230 nm). Retention time: 19.5 min for LC method B.Fmoc-Cys-Gly-Trp-Arg-Ala-Cys-Gly-OH, disulfide bond (1h). After the solid phasepeptide synthesis of reduced form of 1f, the resulting crude product (10.0 mg) wasdissolved in ammonium bicarbonate solution (0.05 M, 50 mL). The mixture was stirred atr.t. under O2 atmosphere for 14 h. After aerobic oxidation, the mass decreased by 2.1 Da,indicating the formation of a disulfide bond. Then, after lyophilization of the reactionmixture, the desired product was purified by preparative HPLC. MS (ESI): m/z 972.3(calcd m/z for [M+H]+ = 972.4). Purity: >95% (HPLC analysis at 230 nm). Retentiontime: 16.7 min for LC method B.H-Gly-Ser-Asn-Trp-Gly-OH (1b). MS (ESI): m/z 520.5 (calcd [M+H]+ = 520.5).Purity: >95% (HPLC analysis at 230 nm). Retention time: 10.8 for LC method C.H-Gly-Tyr-Asn-Trp-Gly-OH (1i). MS (ESI): m/z 596.3 (calcd [M+H]+=596.2). Purity:>95% (HPLC analysis at 230 nm). Retention time: 16.2 for LC method C.H-Gly-Met-Asn-Trp-Gly-OH (1j). MS (ESI): m/z 564.3 (calcd [M+H]+=564.2). Purity:>95% (HPLC analysis at 230 nm). Retention time: 17.5 for LC method C.Leuprorelin (1k). MS (ESI): m/z 1209.5 (calcd [M+H]+ = 1209.6). Retention time: 20.1min for LC method D.keto-ABNO-adduct of Fmoc-Gly-Ser-Asn-Trp-Gly-OH (2a). MS (ESI): m/z 895.3(calcd [M+H]+ = 895.4). Retention time: 18.2 min, 18.8 min (diastereomers) for LCmethod A.keto-ABNO-adduct of Fmoc-Gly-Lys-Asn-Trp-Gly-OH (2c). MS (ESI): m/z 936.4(calcd [M+H]+ = 936.4). Retention time: 15.4 min, 15.6 min (diastereomers) for LCmethod B.keto-ABNO-adduct of Fmoc-Gly-His-Asn-Trp-Gly-OH (2d). MS (ESI): m/z 945.4(calcd [M+H]+ = 945.4). Retention time: 14.0 min, 14.2 min (diastereomers) for LCmethod A.keto-ABNO-adduct of Fmoc-Gly-Tyr-Asn-Trp-Gly-OH (2e). MS (ESI): m/z 971.4(calcd [M+H]+ = 971.4). Retention time: 20.3 min, 20.5 min (diastereomers) for LCmethod A. keto-ABNO-adduct of Fmoc-Gly-Met-Asn-Trp-Gly-OH (2f). MS (ESI): m/z 939.2(calcd [M+H]+ = 939.4). Retention time: 23.8 min, 25.0 min (diastereomers) for LCmethod B.
S5

keto-ABNO-adduct of Fmoc-Gly-Cys-Asn-Trp-Gly-OH (2g). MS (ESI): m/z 911.3(calcd [M+H]+ = 911.3). Retention time: 22.0 min, 22.4 min, 23.6 min (diastereomersand rotamers) for LC method B.keto-ABNO-adduct of Fmoc-Cys-Gly-Trp-Arg-Ala-Cys-Gly-OH, disulfide bond(2h). MS (ESI): m/z 1125.5 (calcd [M+H]+ = 1125.4). Retention time: 18.4 min, 18.8min, 20.2 min (diastereomers and rotamers) for LC method B.keto-ABNO-adduct of H-Gly-Ser-Asn-Trp-Gly-OH (2b). MS (ESI): m/z 673.5 (calcd[M+H]+ = 673.6). Retention time: 19.9 min, 20.9 min (diastereomers) for LC method C.keto-ABNO-adduct of H-Gly-Tyr-Asn-Trp-Gly-OH (2i). MS (ESI): m/z 749.3 (calcd[M+H]+ = 749.3). Retention time: 20.7 min, 21.9 min, 22.6 min (diastereomers androtamers) for LC method C.keto-ABNO-adduct of H-Gly-Met-Asn-Trp-Gly-OH (2j). MS (ESI): m/z 717.3 (calcd[M+H]+ = 717.3). Retention time: 22.6 min, 23.2 min, 24.0 min (diastereomers androtamers) for LC method C.keto-ABNO-adduct of Leuprorelin (2k). MS (ESI): m/z 1362.6 (calcd [M+H]+ =1362.7). Retention time: 21.9 min, 22.4 min (diastereomers) for LC method D.MeLum1-adduct of Fmoc-Gly-Tyr-Asn-Trp-Gly-OH (MeLum1-1e). MS (ESI): m/z992.3 (calcd [M+H]+ = 992.4). Retention time: 20.7 min for LC method B. keto-ABNO and MeLum1-adduct of Fmoc-Gly-Tyr-Asn-Trp-Gly-OH (MeLum1-keto-ABNO-1e). MS (ESI): m/z 1145.3 (calcd [M+H]+ = 1145.4). Retention time: 23.6min, 24.4 min (diastereomers) for LC method B.
S6

3. Synthesis and Characterization of New Compounds
3-1. Preparation of Reagentsketo-ABNO1and MeLum reagent2 were synthesized following the literature
procedure. Other N-oxyl radicals were commercially available and used as purchased.
3-2. Synthesis and Structural Determination of Overoxidation ProductsN-Ac-Tryptamine was used as a model substrate and increased charge was applied
to produce large amount of overoxidation products.
The ElectraSyn vial (20 mL) with a stir bar (AS ONE, 1-5409-01, 8 × 10 mm)was charged with solution A (0.1 M solution of tetrabutylammonium perchlorate inCH3CN, 8 mL), H2O (mL), N-Ac-tryptamine (101 mg, 500 mol), keto-ABNO (77 mg,500 mol). The EletraSyn vial cap equipped with anode (graphite) and cathode (Pt) wereinserted into the mixture. The reaction mixture was electrolyzed under a constant voltageof 1.2 V until 6.0 F/mol of electron was charged. After the electrolysis, PBS buffer (pH =7.4) was added to quench the reaction. According to HPLC analysis, adduct S1 was notdetected (N.D.), and over-oxidized products S2a and S2b (ca. 48%, ca. 2:1 mixture) wereobserved as the major products. AcOEt was added to the mixture and the mixture waswashed with H2O and brine, then dried over Na2SO4. After the desiccant was filtered off,the filtrate was concentrated under reduced pressure. The residue was purified bypreparative TLC (CH2Cl2/MeOH = 10/1) to give S2a and S2b as yellow oil (3.6 mg, 2 %yield). S2a and S2b existed as an equilibrium mixture (see below). These products wereacid-sensitive and easily produce dimerized products (structure undetermined).
9-((1-acetyl-2,3,8,8a-tetrahydropyrrolo[2,3-b]indol-3a(1H)-yl)oxy)-9-azabicyclo[3.3.1]nonan-3-one (S1)
N-Ac-tryptamine (202 mg, 1 mmol), keto-ABNO (154 mg, 1mmol), NaNO2 (42 mg, 0.6 mg), AcOH (100 L), H2O (50 mL) andMeCN (50 mL) were added in a flask and the mixture was stirred for12 h at room temperature. The mixture was extracted with EtOAc,the combined organic layer was washed with brine, then dried overNa2SO4. After the desiccant was filtered off, the filtrate wasconcentrated under reduced pressure. The residue was purified by flash silica gel columnchromatography (EtOAc) to give S1 as yellow oil (155 mg, 44% yield). 1H NMR(CDCl3): δ = 1.20-1.28 (m, 1H), 1.29-1.35 (m, 1H), 1.54-1.65 (m, 2H), 1.70-1.80 (m,1H), 1.84-1.94 (m, 1H), 2.02 (s, 1H), 2.08 (dd, J = 15.5, 5.2 Hz, 1H), 2.41 (ddd, J = 12.0,
S7

6.3, 2.3 Hz, 1H), 2.60-2.68 (m, 1H), 2.93-3.00 (m, 2H), 3.27 (ddd, J = 10.3, 10.3, 6.3 Hz,1H), 3.56 (brs, 2H), 3.68 (ddd, J = 10.3, 8.0, 1.7 Hz, 1H), 5.24 (brs, 1H), 5.51 (s, 1H),6.62 (d, J = 7.4 Hz, 1H), 6.75 (dd, J = 7.4, 7.4 Hz, 1H), 7.15 (dd, J = 7.4, 7.4 Hz, 1H),7.30 (d, J = 7.4 Hz, 1H); 13C NMR (CDCl3): = 210.6, 170.4, 150.6, 130.4, 124.6, 118.6,110.1, 94.6, 77.2, 59.7, 46.6, 40.8, 33.8, 31.7, 22.1; IR (KBr): 3340, 1709, 1641 cm -1;HRMS (ESI): m/z calcd for C20H25N3O3Na+ [M+Na]+ 378.1788, found 378.1774.
1-acetyl-3a-((3-oxo-9-azabicyclo[3.3.1]nonan-9-yl)oxy)-2,3,3a,8a-tetrahydropyrrolo[2,3-b]indol-5(1H)-one (S2a)9-((1-acetyl-5-hydroxy-2,3-dihydropyrrolo[2,3-b]indol-3a(1H)-yl)oxy)-9-azabicyclo[3.3.1]nonan-3-one (S2b)Because those compounds were inseparable on silica gel preparative TLC and reversephase HPLC (mobile phase; H2O/MeCN) and the ratio observed by 1H NMR differed infour solvents as shown below, we concluded that S2a and S2b exist as an equilibriummixture.
S2a/S2b = 1.6 (in CDCl3); see page S45.
S2a/S2b = 3.2 (in acetone-d6)
S8
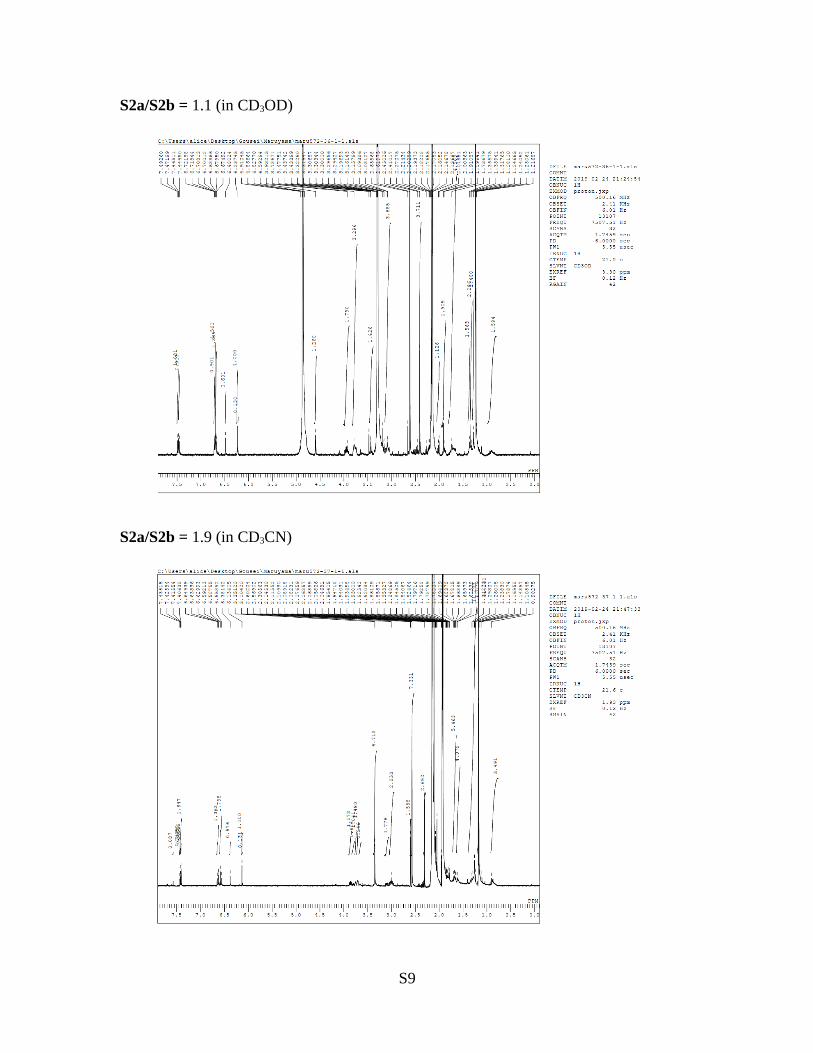
S2a/S2b = 1.1 (in CD3OD)
S2a/S2b = 1.9 (in CD3CN)
S9

1H NMR (CDCl3, underlined chemical shifts for S2a): δ = 1.22-1.32 (m, 1H + 1H), 1.32-1.42 (m, 1H + 1H), 1.56-1.82 (m, 4H + 4H), 1.83-1.94 (m, 1H + 1H), 2.07-2.13 (m, 1H +1H), 2.17 (s, 3H), 2.17-2.24 (m, 2H + 2H), 2.42 (s, 3H), 2.92-3.06 (m, 2H + 2H), 3.24(ddd, J = 12.0, 4.6 Hz, 1H), 3.42 (ddd, J = 15.4, 8.0 Hz, 1H), 3.66 (brs, 2H), 3.72 (brs,2H), 3.80 (ddd, J = 10.9, 8.6, 4.6 Hz, 1H), 4.02 (ddd, J = 12.0, 8.6, 4.6 Hz, 1H), 6.08 (s,1H), 6.52 (d, J = 2.3 Hz, 1H), 6.57 (d, J = 1.7 Hz, 1H), 6.68 (dd, J = 10.3, 1.7 Hz, 1H),6.71 (dd, J = 8.0, 1 .7 Hz, 1H), 7.42 (d, J = 10.3 Hz, 1H), 7.47 (d, J = 10.3 Hz, 1H);LRMS (ESI): m/z calcd for C20H24N3O4
+ [M+H]+ 370.2, found .370.1.
S10

4. Detailed Optimization for Electrochemical Trp-selective Bioconjugation
Table S1. Optimization for electrochemical Trp-selective bioconjugation of pentapeptide1b.
[a] Yields were calculated from the absorbance at 230 nm using LC-MS analysis (forentries 1-8) and HPLC analysis (for entries 9-15).
The conditions shown in Table S1, entry 7 were defined as optimal, based on the resultswith systematically varying six parameters A F. ‒Parameter F: Electrolysis with other electrolytes (entries 2-4) afforded desired product(2b) was lower than entry 1. In entry 2, more by-product 3b was generated. In entry 3 and4, electrolysis was suspended after applying 0.5 F/mol of electric charge.Parameter A: Increasing the amount of 4-oxo-TEMPO (entries 5-7) yielded slightlymore desired product 2b than entry 1. However, when 3 equivalents of 4-oxo-TEMPOwere used, the yield decreased to 78% (entry 8). Parameter D and E: Ni cathode (entries 10, 13 and 14) afforded desired product 2b withgood yield, but it did not show significant difference against Pt cathode (entry 7). Othercombinations of electrodes yielded less desired product 2b than entry 7.
S11
Parameter B and C: The voltage was set as low as possible at which electrolysis couldbe completed. Electrolysis with 0.5 F/mol, 1.0 F/mol, 1.5 F/mol and 2.0 F/mol of electriccharge was also conducted, and the result with best yield was shown in the table.
5. Reaction Profiles
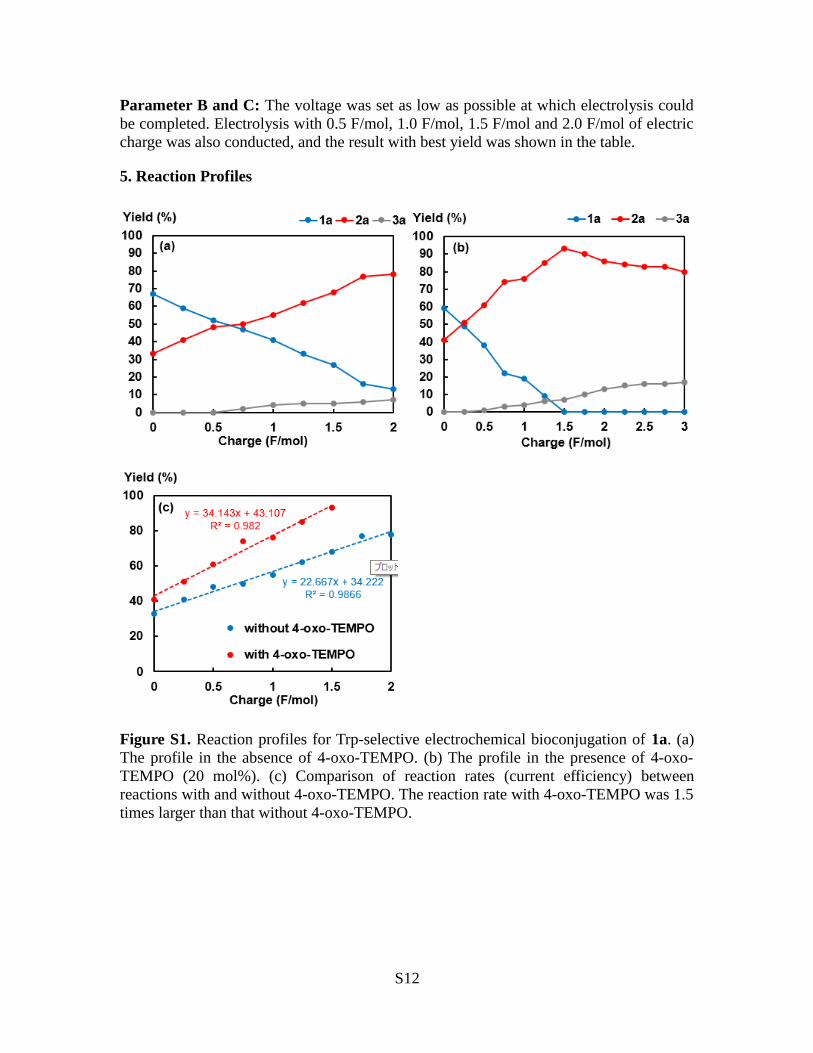
Figure S1. Reaction profiles for Trp-selective electrochemical bioconjugation of 1a. (a)The profile in the absence of 4-oxo-TEMPO. (b) The profile in the presence of 4-oxo-TEMPO (20 mol%). (c) Comparison of reaction rates (current efficiency) betweenreactions with and without 4-oxo-TEMPO. The reaction rate with 4-oxo-TEMPO was 1.5times larger than that without 4-oxo-TEMPO.
S12

6. Cyclic Voltammetry Studies
Procedure for cyclic voltammetry measurementCyclic voltammetry measurements were carried out using HSV-110 (Hokuto
Denko). A 3 mM solution of the substrate in 2 mL solvent A or B was placed in a beaker-type glass cell (Solvent A: 0.1 M TBAP/CH3CN-H2O (1:1), Solvent B: 0.1 M LiClO4
aq.). Cyclic voltammogram was measured with a scan rate of 10 or 100 mV s-1 using agrassy carbon as a working electrode (WE), a Pt wire as a counter electrode (CE), andAg/Ag+ (in 3.33 M KCl aq.) as a reference electrode (RE).Dimensions: WE to RE: 8.0 mm, WE to RE: 6.0 mm, CE to RE: 6.0 mm, Surface area of WE: 7.07 mm2 (Figure S2).
Figure S2. Experimental setup for cyclic voltammetry.
S13
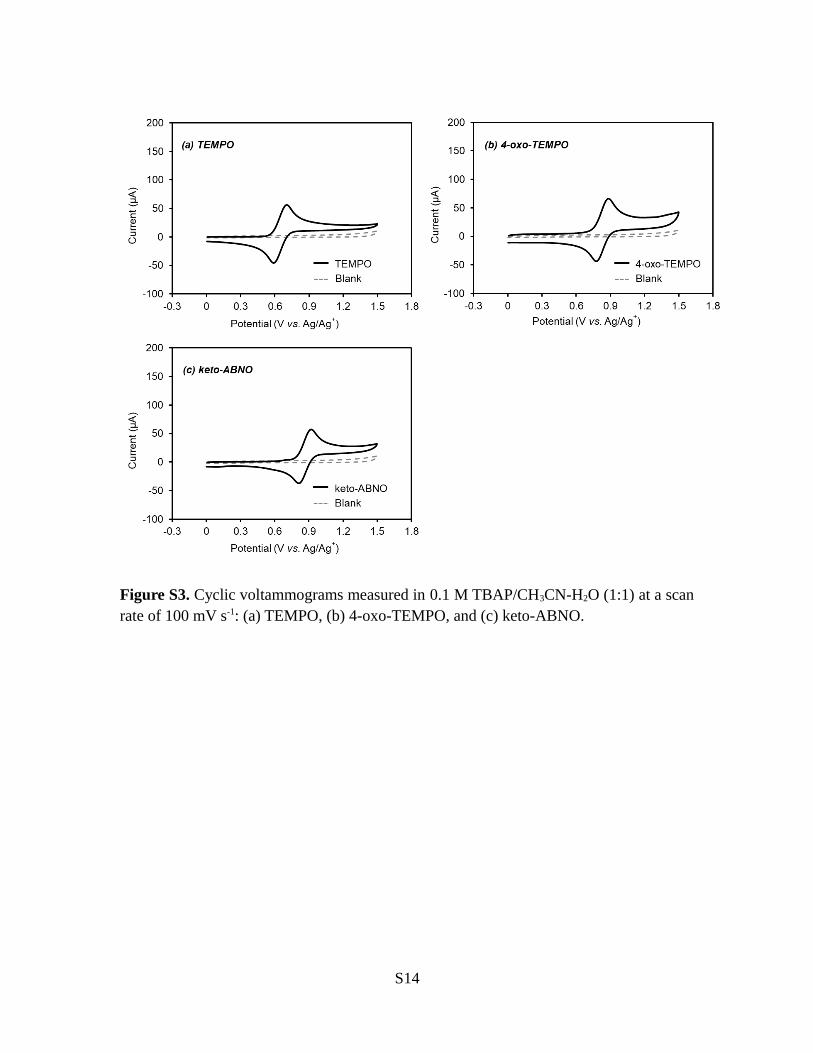
Figure S3. Cyclic voltammograms measured in 0.1 M TBAP/CH3CN-H2O (1:1) at a scanrate of 100 mV s-1: (a) TEMPO, (b) 4-oxo-TEMPO, and (c) keto-ABNO.
S14
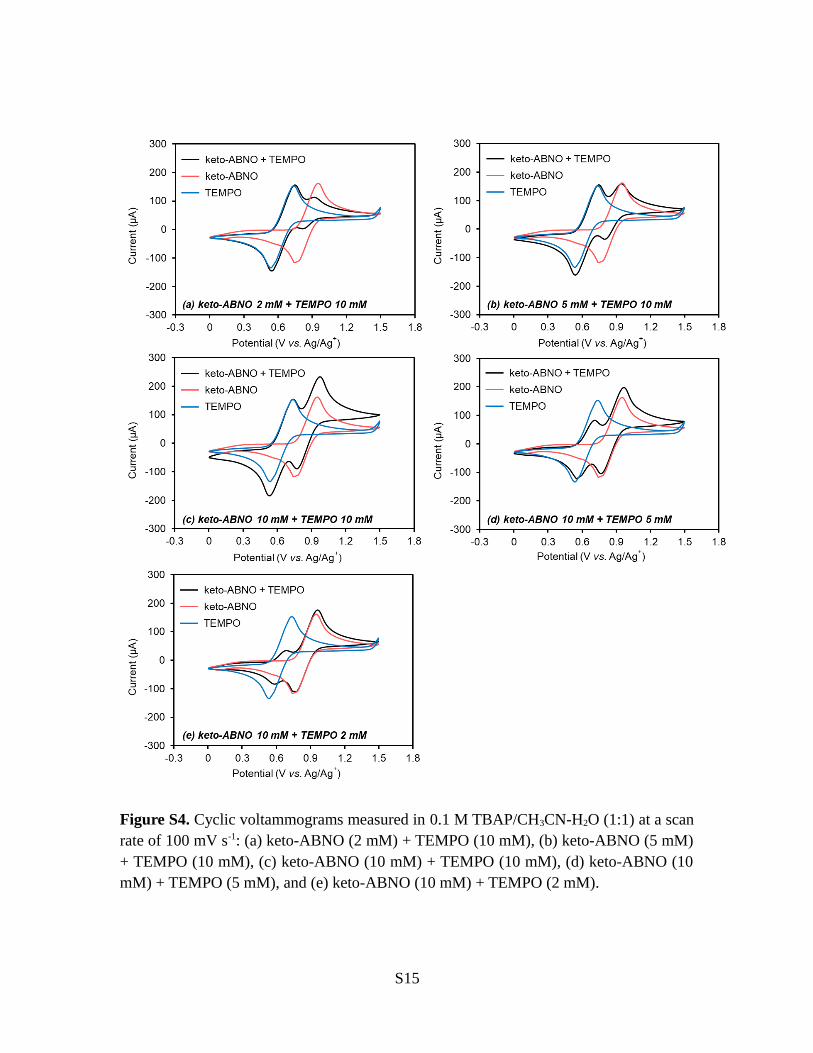
Figure S4. Cyclic voltammograms measured in 0.1 M TBAP/CH3CN-H2O (1:1) at a scanrate of 100 mV s-1: (a) keto-ABNO (2 mM) + TEMPO (10 mM), (b) keto-ABNO (5 mM)+ TEMPO (10 mM), (c) keto-ABNO (10 mM) + TEMPO (10 mM), (d) keto-ABNO (10mM) + TEMPO (5 mM), and (e) keto-ABNO (10 mM) + TEMPO (2 mM).
S15
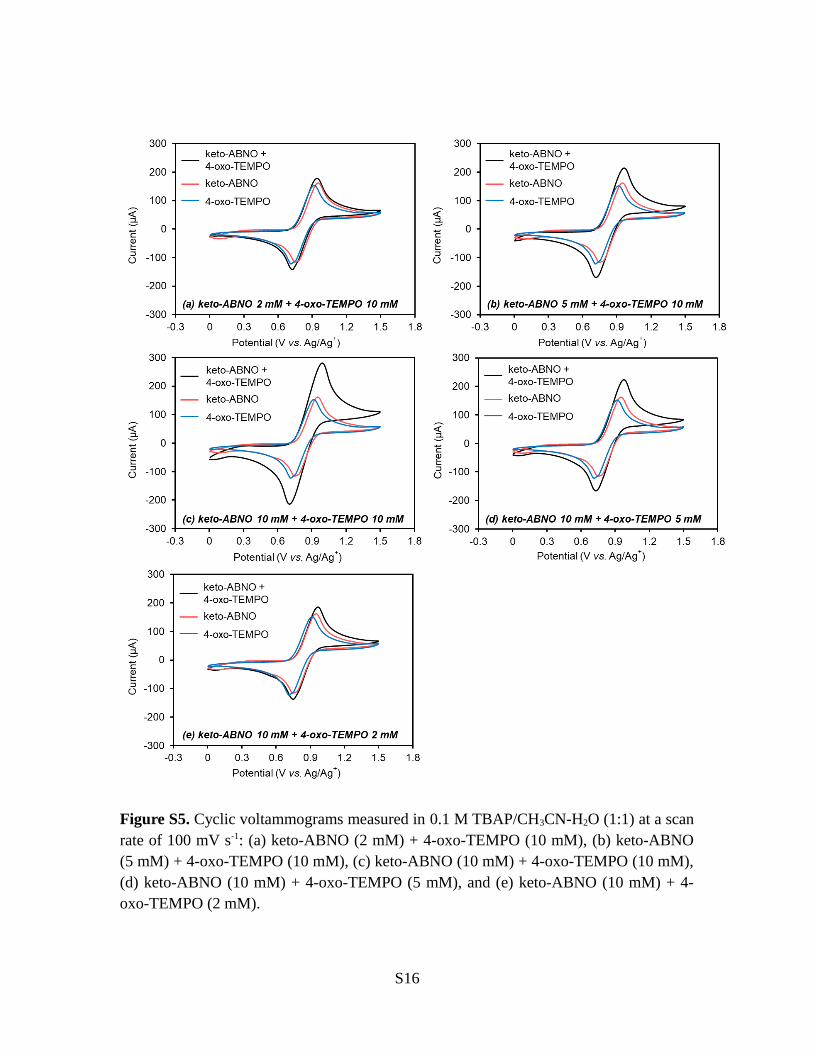
Figure S5. Cyclic voltammograms measured in 0.1 M TBAP/CH3CN-H2O (1:1) at a scanrate of 100 mV s-1: (a) keto-ABNO (2 mM) + 4-oxo-TEMPO (10 mM), (b) keto-ABNO(5 mM) + 4-oxo-TEMPO (10 mM), (c) keto-ABNO (10 mM) + 4-oxo-TEMPO (10 mM),(d) keto-ABNO (10 mM) + 4-oxo-TEMPO (5 mM), and (e) keto-ABNO (10 mM) + 4-oxo-TEMPO (2 mM).
S16
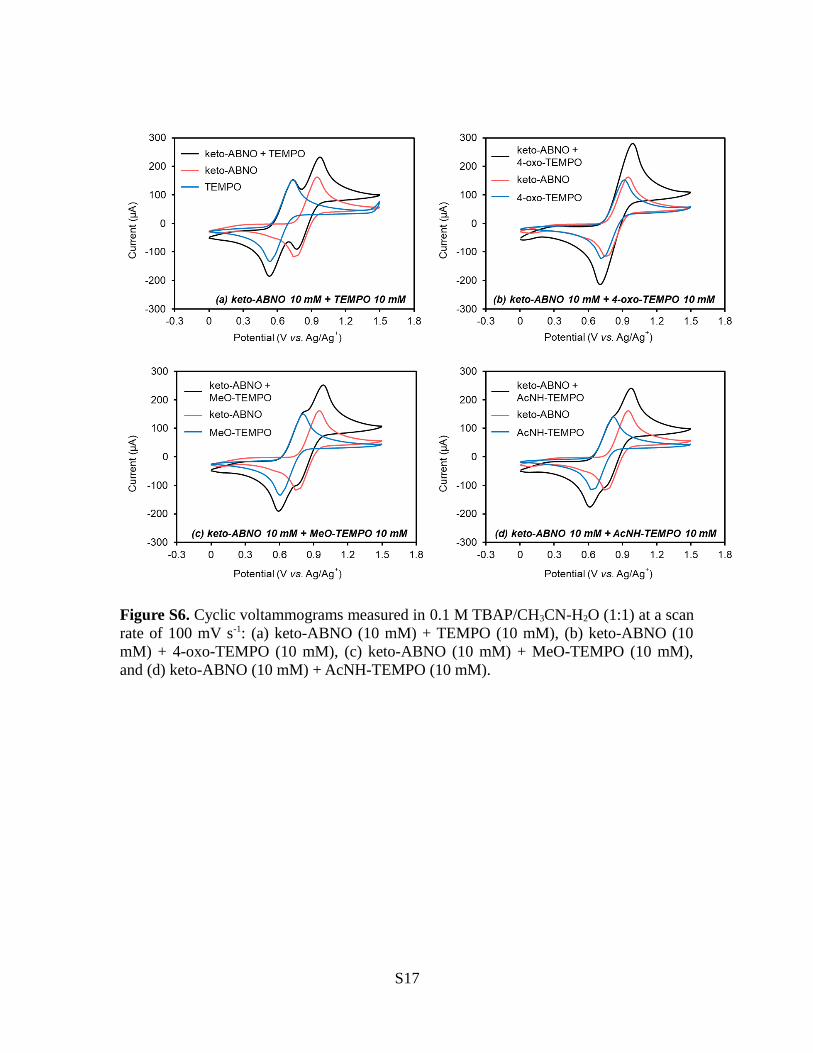
Figure S6. Cyclic voltammograms measured in 0.1 M TBAP/CH3CN-H2O (1:1) at a scanrate of 100 mV s-1: (a) keto-ABNO (10 mM) + TEMPO (10 mM), (b) keto-ABNO (10mM) + 4-oxo-TEMPO (10 mM), (c) keto-ABNO (10 mM) + MeO-TEMPO (10 mM),and (d) keto-ABNO (10 mM) + AcNH-TEMPO (10 mM).
S17
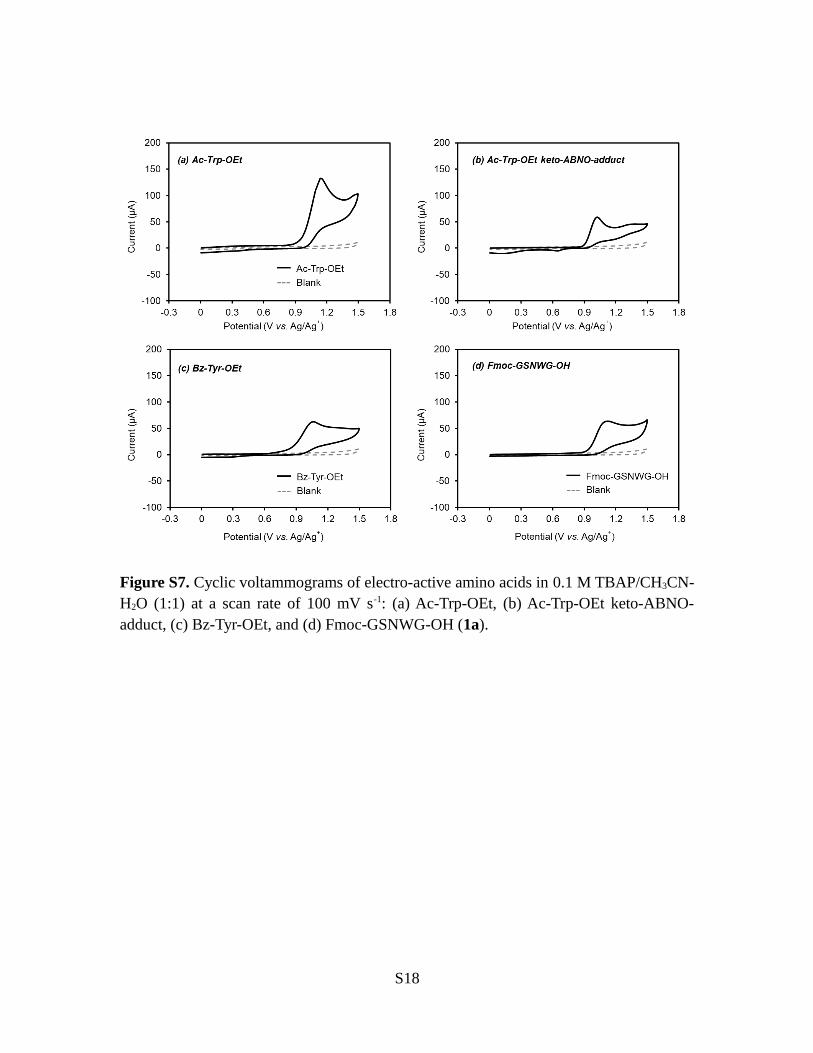
Figure S7. Cyclic voltammograms of electro-active amino acids in 0.1 M TBAP/CH3CN-H2O (1:1) at a scan rate of 100 mV s-1: (a) Ac-Trp-OEt, (b) Ac-Trp-OEt keto-ABNO-adduct, (c) Bz-Tyr-OEt, and (d) Fmoc-GSNWG-OH (1a).
S18
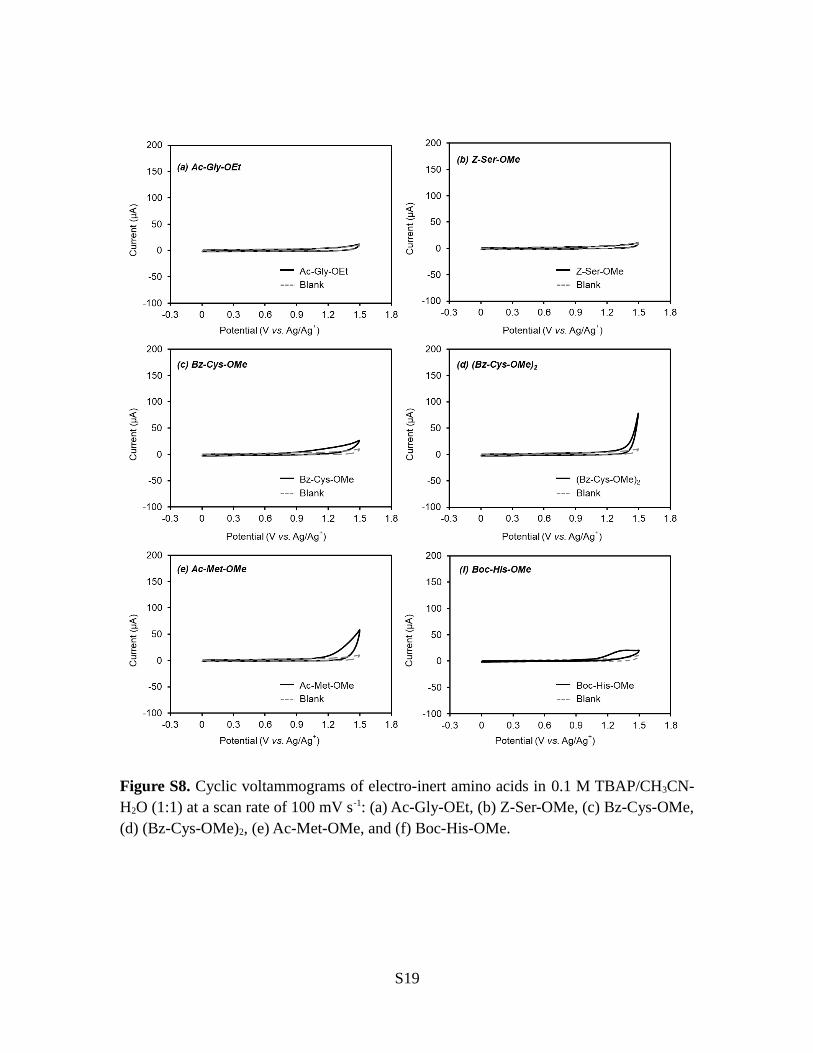
Figure S8. Cyclic voltammograms of electro-inert amino acids in 0.1 M TBAP/CH3CN-H2O (1:1) at a scan rate of 100 mV s-1: (a) Ac-Gly-OEt, (b) Z-Ser-OMe, (c) Bz-Cys-OMe,(d) (Bz-Cys-OMe)2, (e) Ac-Met-OMe, and (f) Boc-His-OMe.
S19
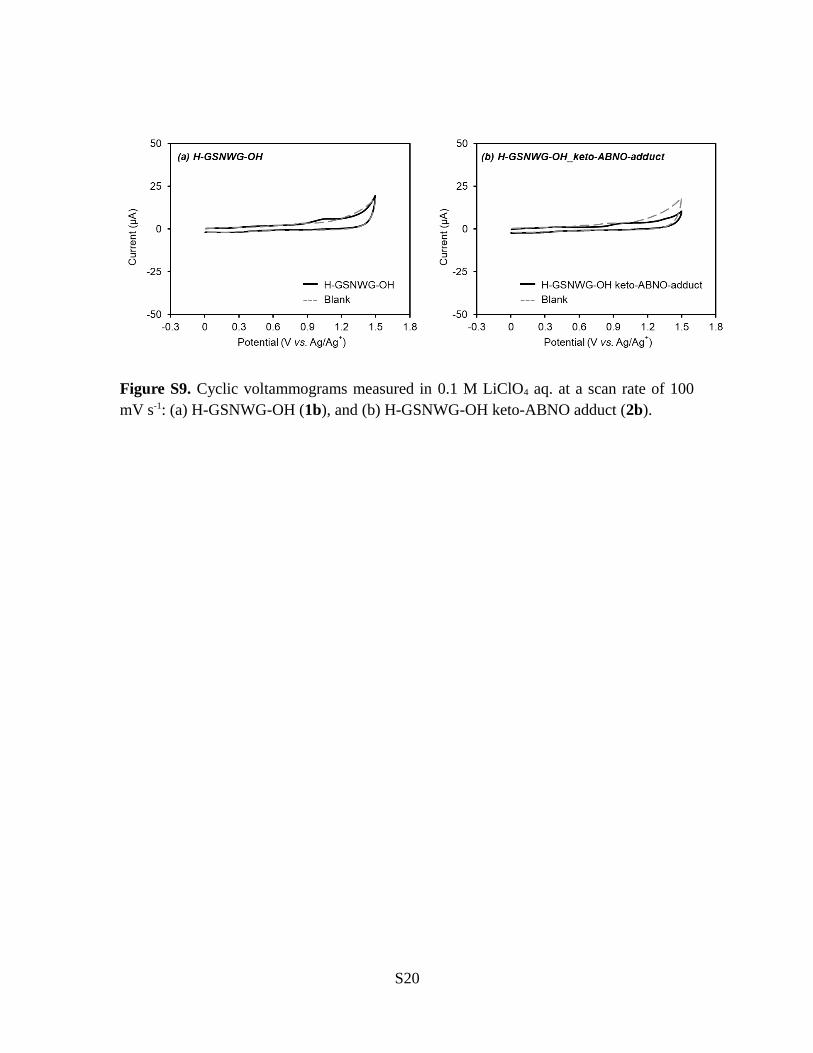
Figure S9. Cyclic voltammograms measured in 0.1 M LiClO4 aq. at a scan rate of 100mV s-1: (a) H-GSNWG-OH (1b), and (b) H-GSNWG-OH keto-ABNO adduct (2b).
S20
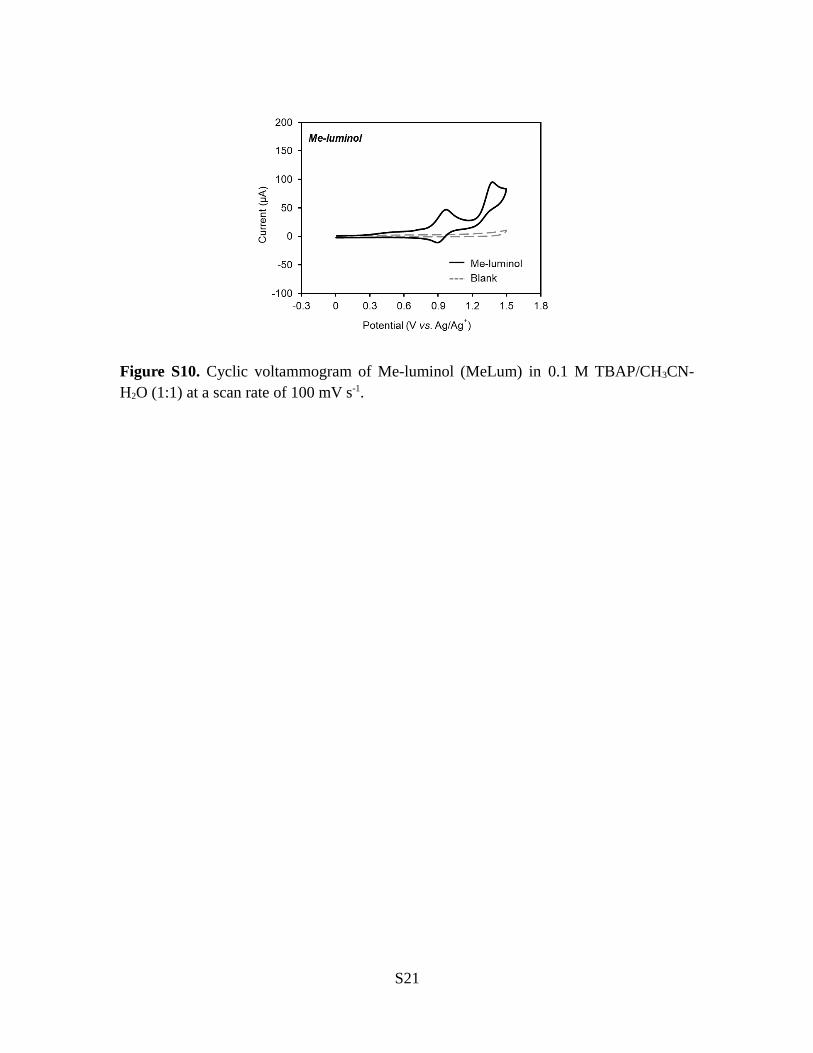
Figure S10. Cyclic voltammogram of Me-luminol (MeLum) in 0.1 M TBAP/CH3CN-H2O (1:1) at a scan rate of 100 mV s-1.
S21
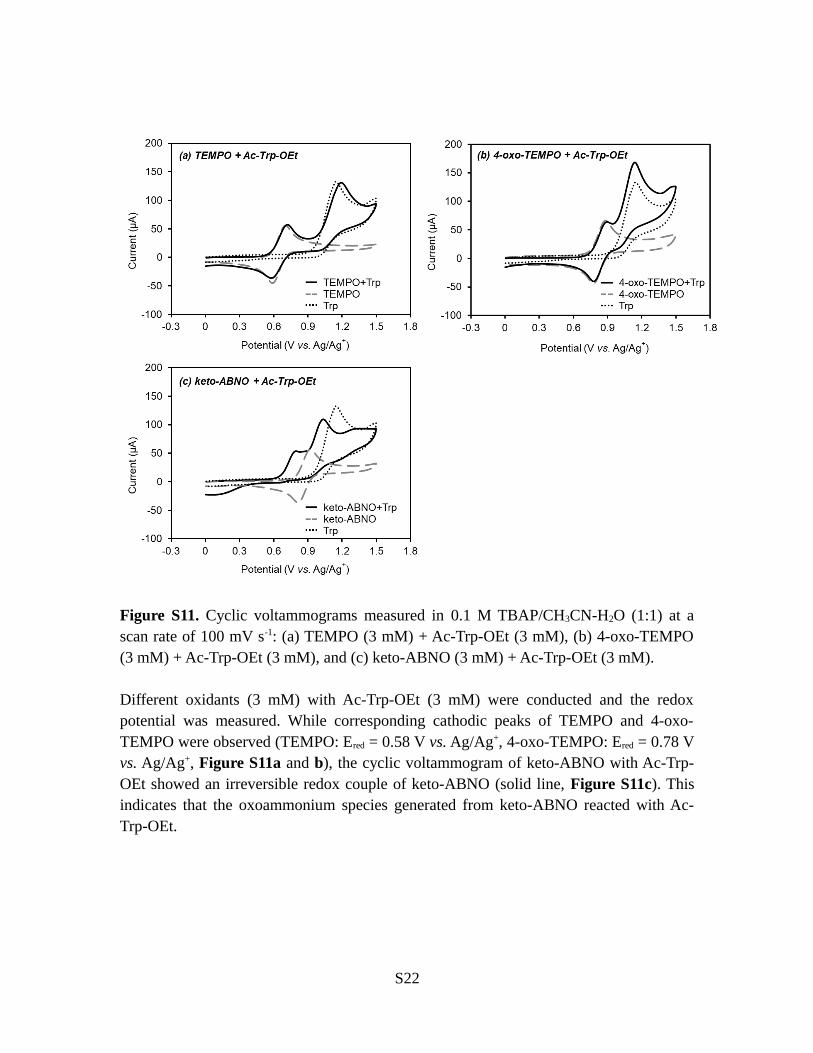
Figure S11. Cyclic voltammograms measured in 0.1 M TBAP/CH3CN-H2O (1:1) at ascan rate of 100 mV s-1: (a) TEMPO (3 mM) + Ac-Trp-OEt (3 mM), (b) 4-oxo-TEMPO(3 mM) + Ac-Trp-OEt (3 mM), and (c) keto-ABNO (3 mM) + Ac-Trp-OEt (3 mM).
Different oxidants (3 mM) with Ac-Trp-OEt (3 mM) were conducted and the redoxpotential was measured. While corresponding cathodic peaks of TEMPO and 4-oxo-TEMPO were observed (TEMPO: Ered = 0.58 V vs. Ag/Ag+, 4-oxo-TEMPO: Ered = 0.78 Vvs. Ag/Ag+, Figure S11a and b), the cyclic voltammogram of keto-ABNO with Ac-Trp-OEt showed an irreversible redox couple of keto-ABNO (solid line, Figure S11c). Thisindicates that the oxoammonium species generated from keto-ABNO reacted with Ac-Trp-OEt.
S22
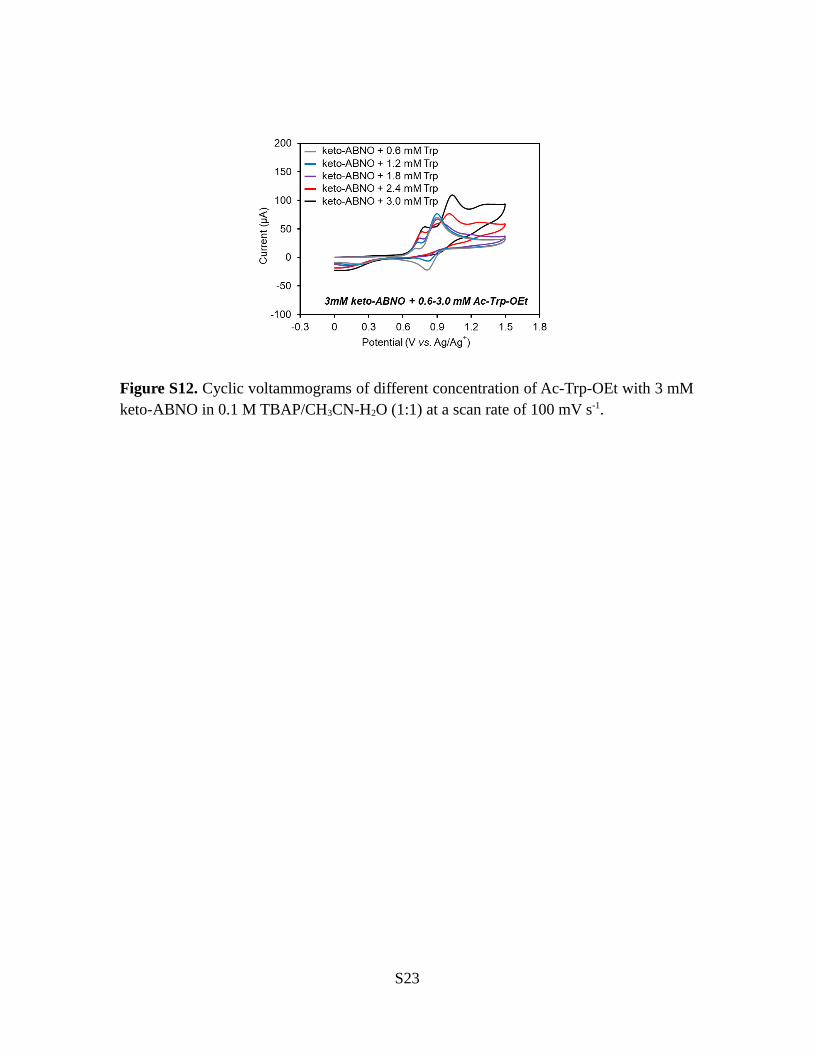
Figure S12. Cyclic voltammograms of different concentration of Ac-Trp-OEt with 3 mMketo-ABNO in 0.1 M TBAP/CH3CN-H2O (1:1) at a scan rate of 100 mV s-1.
S23

Figure S13. Cyclic voltammograms of different concentration of TEMPO with 3 mMketo-ABNO and 3 mM Ac-Trp-OEt in 0.1 M TBAP/CH3CN-H2O (1:1) at a scan rate of10 or 100 mV s-1: (a) 10 mV s-1, (b) 100 mV s-1.
S24
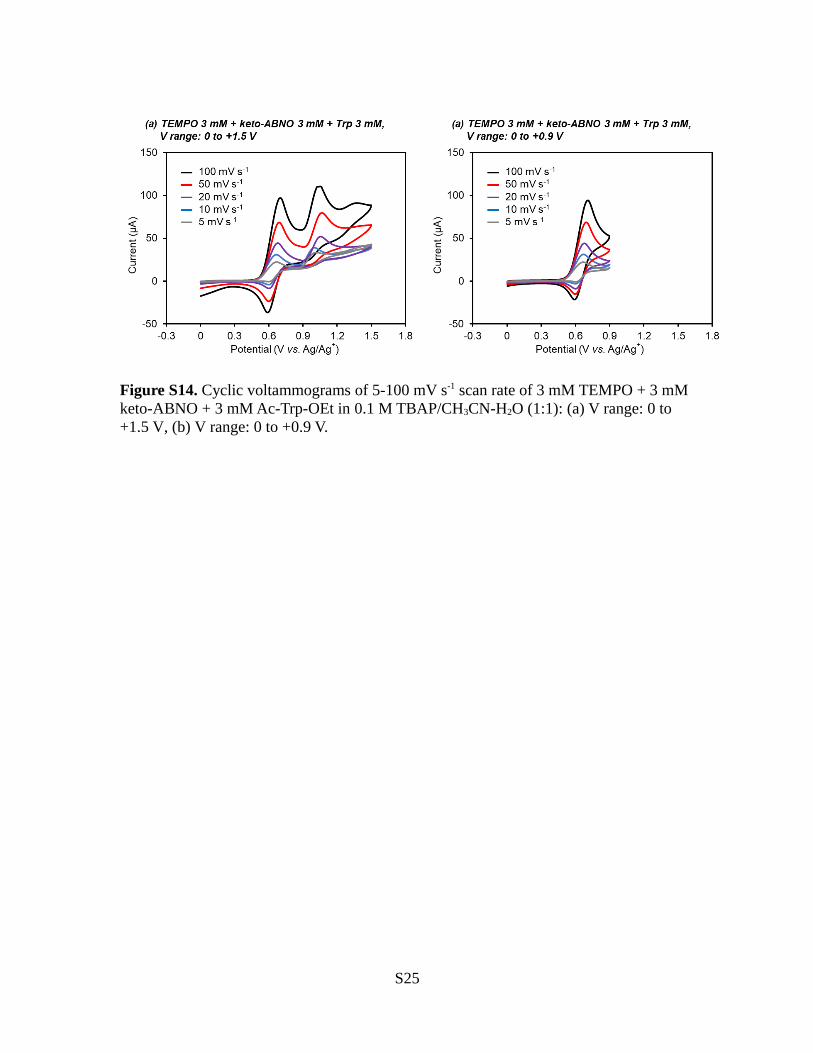
Figure S14. Cyclic voltammograms of 5-100 mV s-1 scan rate of 3 mM TEMPO + 3 mM keto-ABNO + 3 mM Ac-Trp-OEt in 0.1 M TBAP/CH3CN-H2O (1:1): (a) V range: 0 to +1.5 V, (b) V range: 0 to +0.9 V.
S25
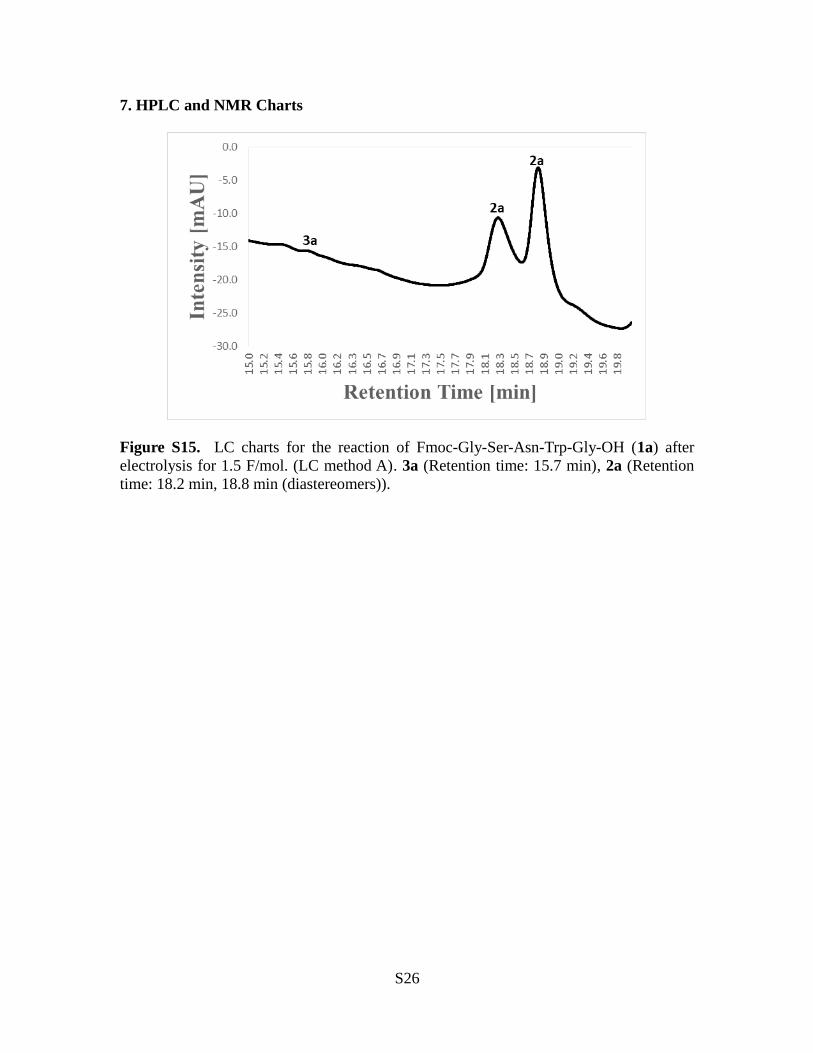
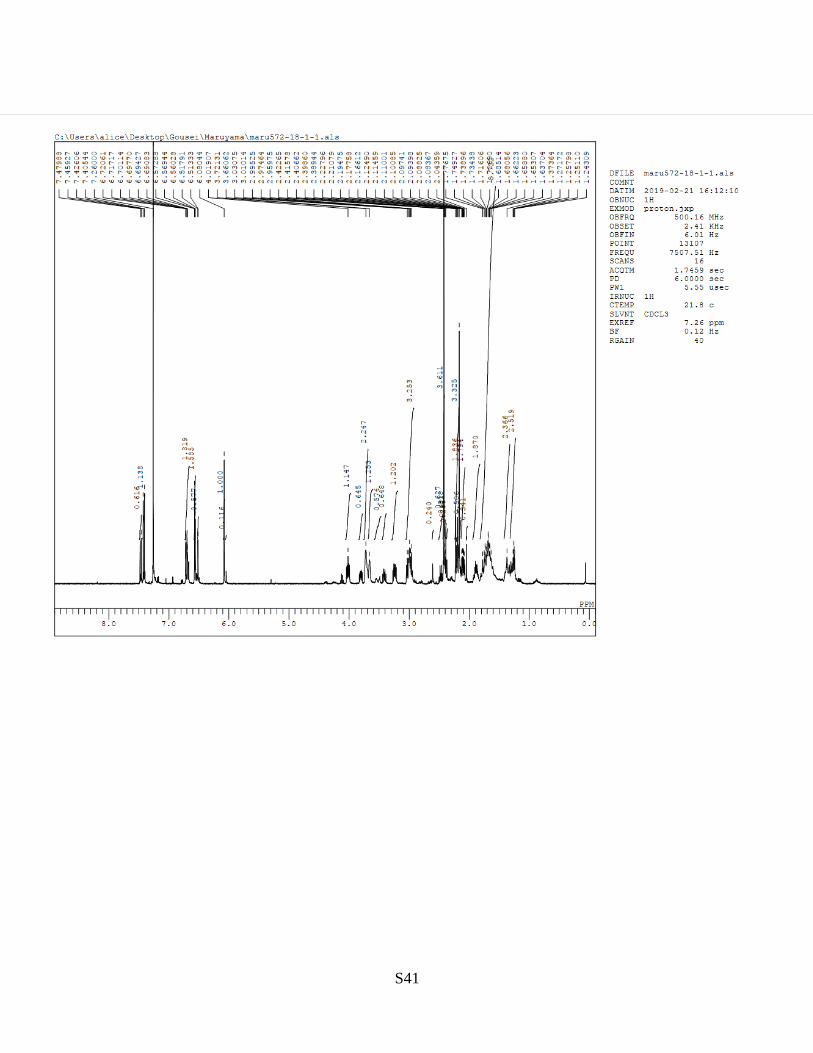
7. HPLC and NMR Charts
Figure S15. LC charts for the reaction of Fmoc-Gly-Ser-Asn-Trp-Gly-OH (1a) afterelectrolysis for 1.5 F/mol. (LC method A). 3a (Retention time: 15.7 min), 2a (Retentiontime: 18.2 min, 18.8 min (diastereomers)).
S26
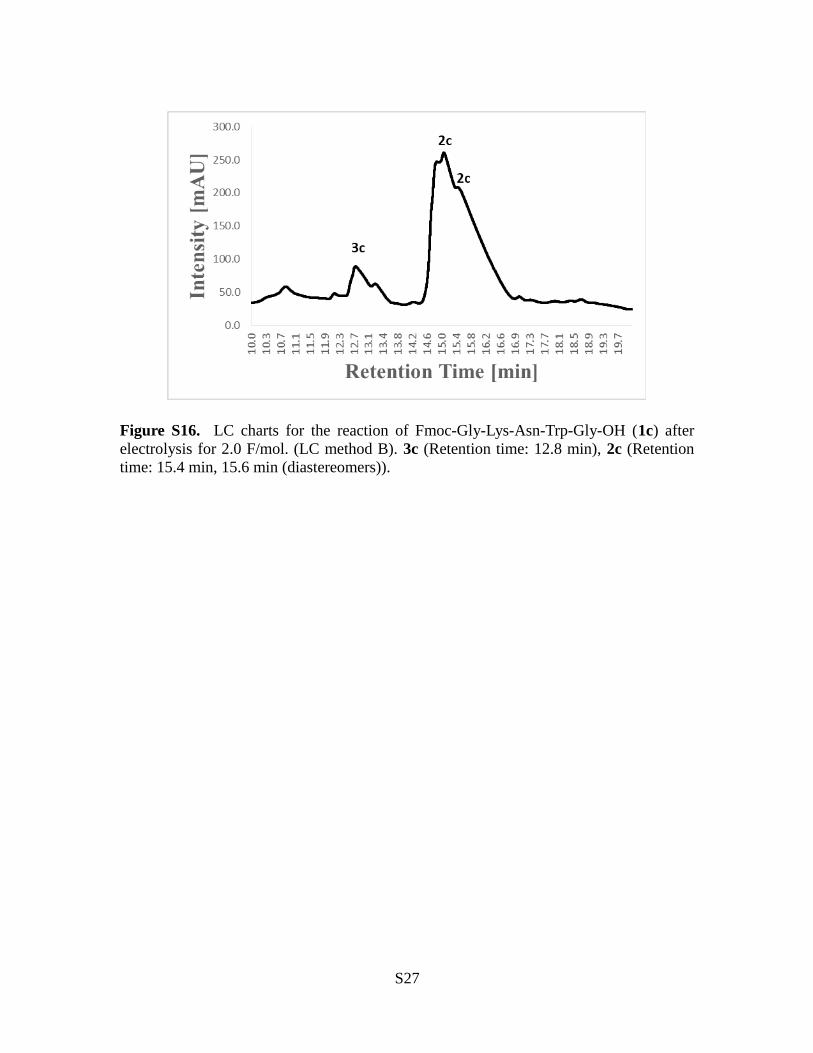
Figure S16. LC charts for the reaction of Fmoc-Gly-Lys-Asn-Trp-Gly-OH (1c) afterelectrolysis for 2.0 F/mol. (LC method B). 3c (Retention time: 12.8 min), 2c (Retentiontime: 15.4 min, 15.6 min (diastereomers)).
S27
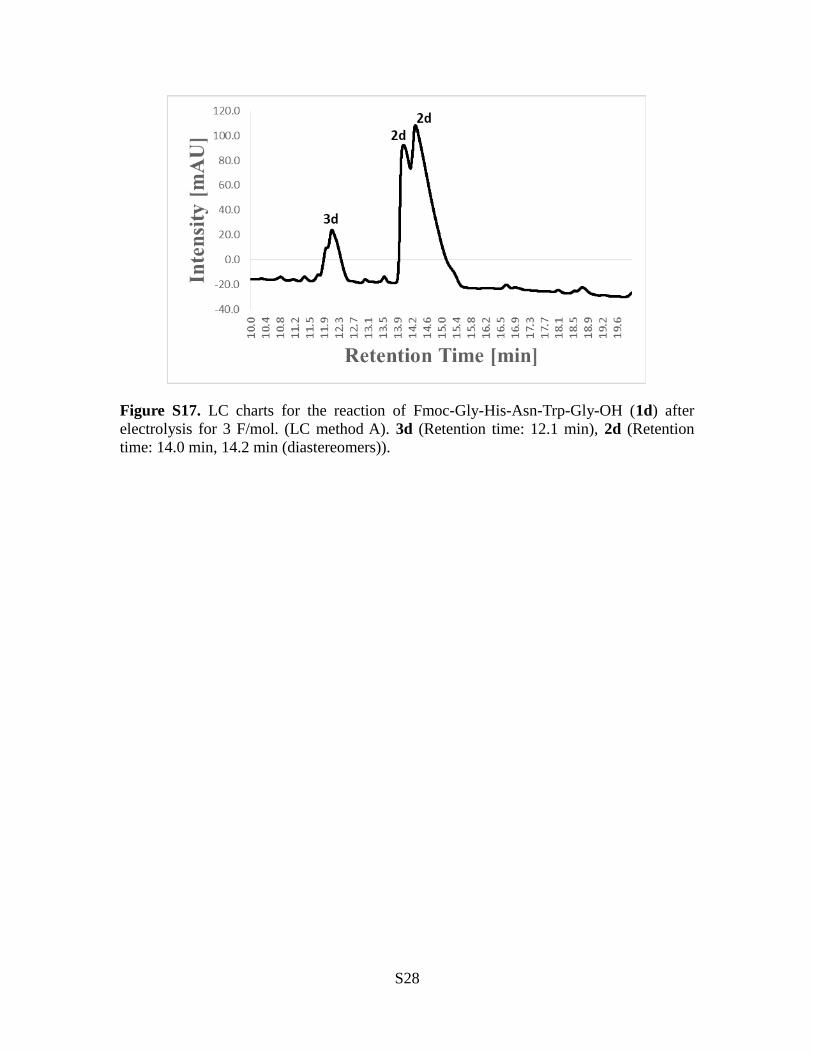
Figure S17. LC charts for the reaction of Fmoc-Gly-His-Asn-Trp-Gly-OH (1d) afterelectrolysis for 3 F/mol. (LC method A). 3d (Retention time: 12.1 min), 2d (Retentiontime: 14.0 min, 14.2 min (diastereomers)).
S28
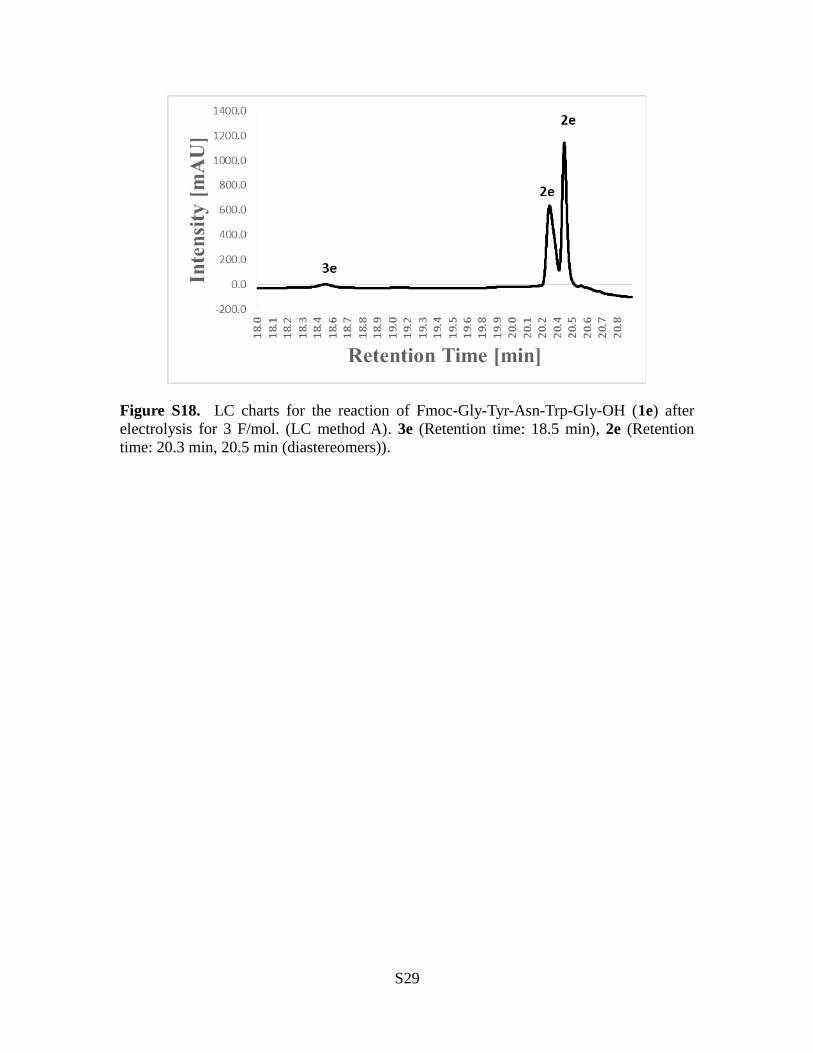
Figure S18. LC charts for the reaction of Fmoc-Gly-Tyr-Asn-Trp-Gly-OH (1e) afterelectrolysis for 3 F/mol. (LC method A). 3e (Retention time: 18.5 min), 2e (Retentiontime: 20.3 min, 20.5 min (diastereomers)).
S29
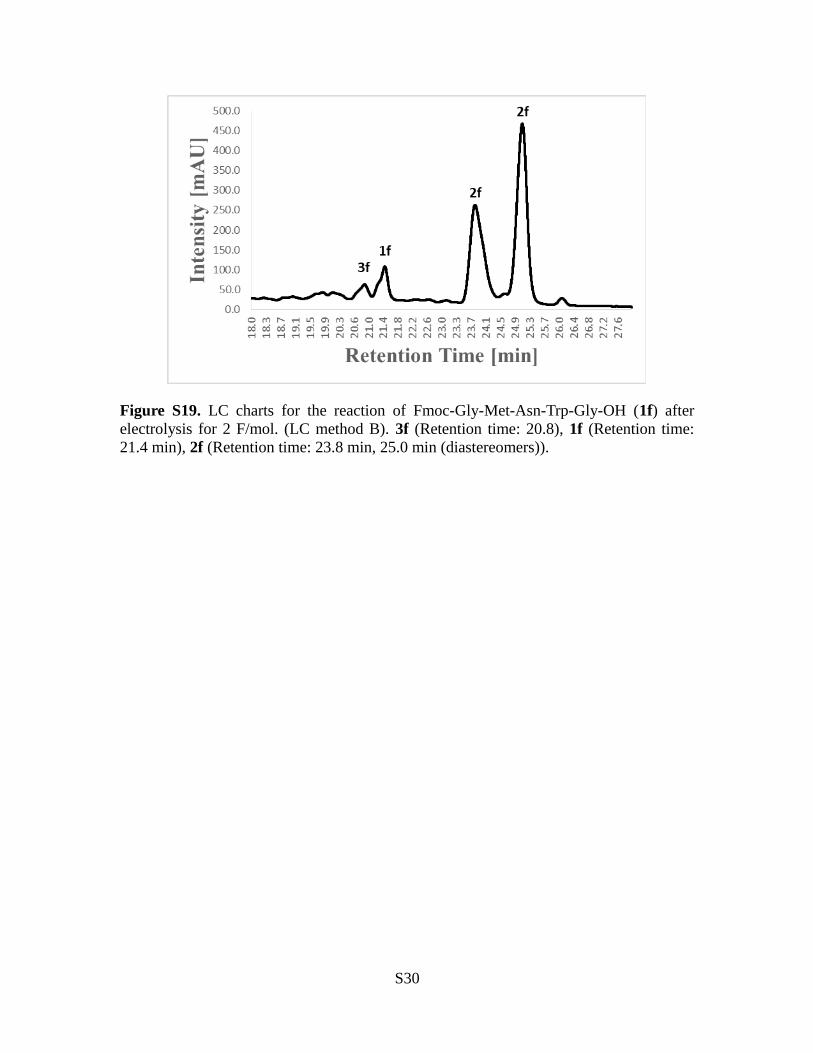
Figure S19. LC charts for the reaction of Fmoc-Gly-Met-Asn-Trp-Gly-OH (1f) afterelectrolysis for 2 F/mol. (LC method B). 3f (Retention time: 20.8), 1f (Retention time:21.4 min), 2f (Retention time: 23.8 min, 25.0 min (diastereomers)).
S30
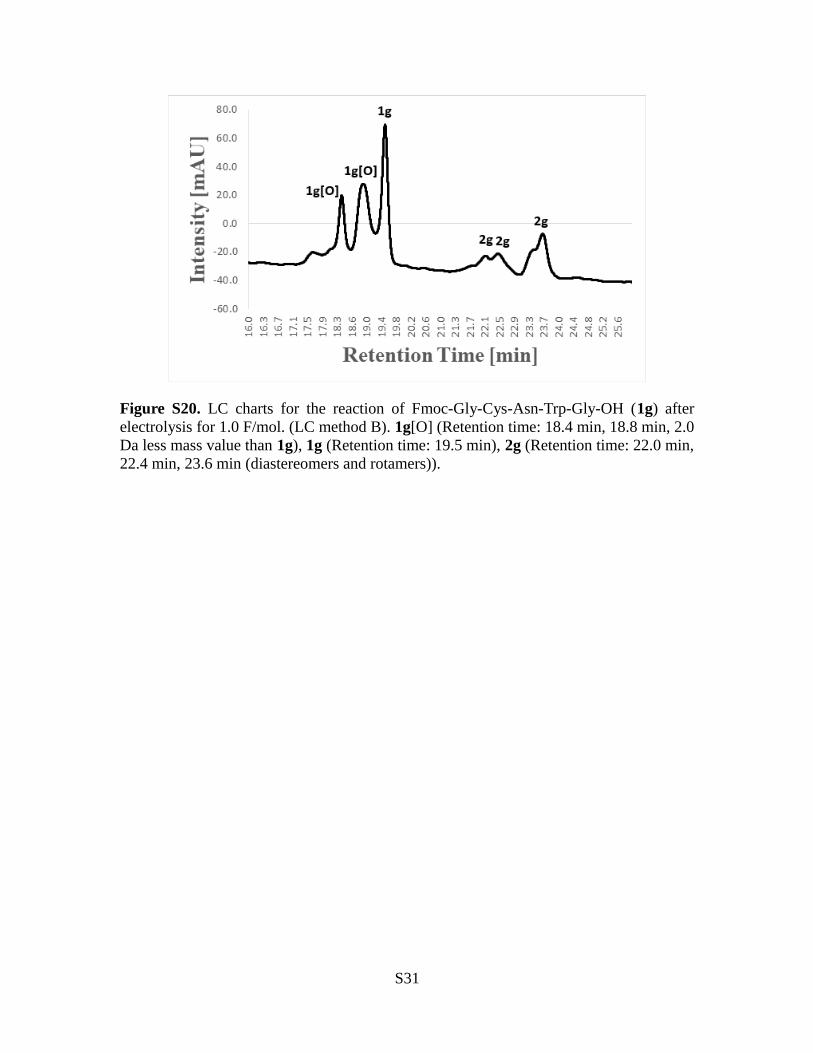
Figure S20. LC charts for the reaction of Fmoc-Gly-Cys-Asn-Trp-Gly-OH (1g) afterelectrolysis for 1.0 F/mol. (LC method B). 1g[O] (Retention time: 18.4 min, 18.8 min, 2.0Da less mass value than 1g), 1g (Retention time: 19.5 min), 2g (Retention time: 22.0 min,22.4 min, 23.6 min (diastereomers and rotamers)).
S31
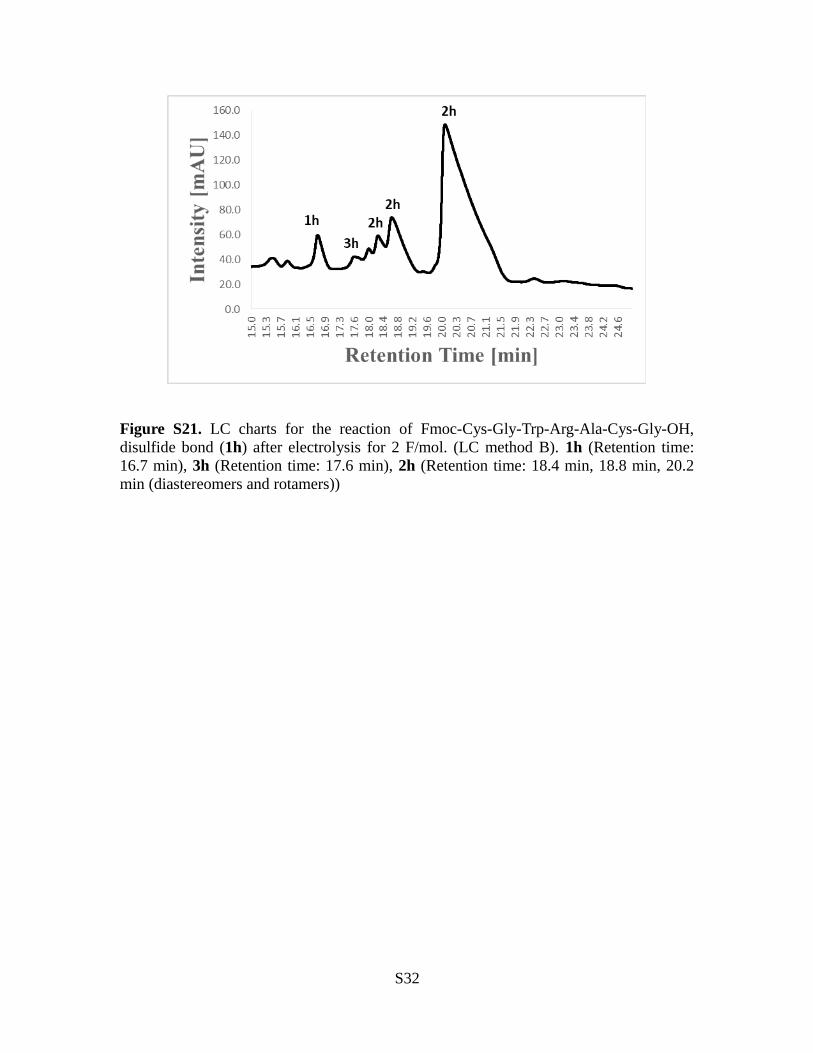
Figure S21. LC charts for the reaction of Fmoc-Cys-Gly-Trp-Arg-Ala-Cys-Gly-OH,disulfide bond (1h) after electrolysis for 2 F/mol. (LC method B). 1h (Retention time:16.7 min), 3h (Retention time: 17.6 min), 2h (Retention time: 18.4 min, 18.8 min, 20.2min (diastereomers and rotamers))
S32
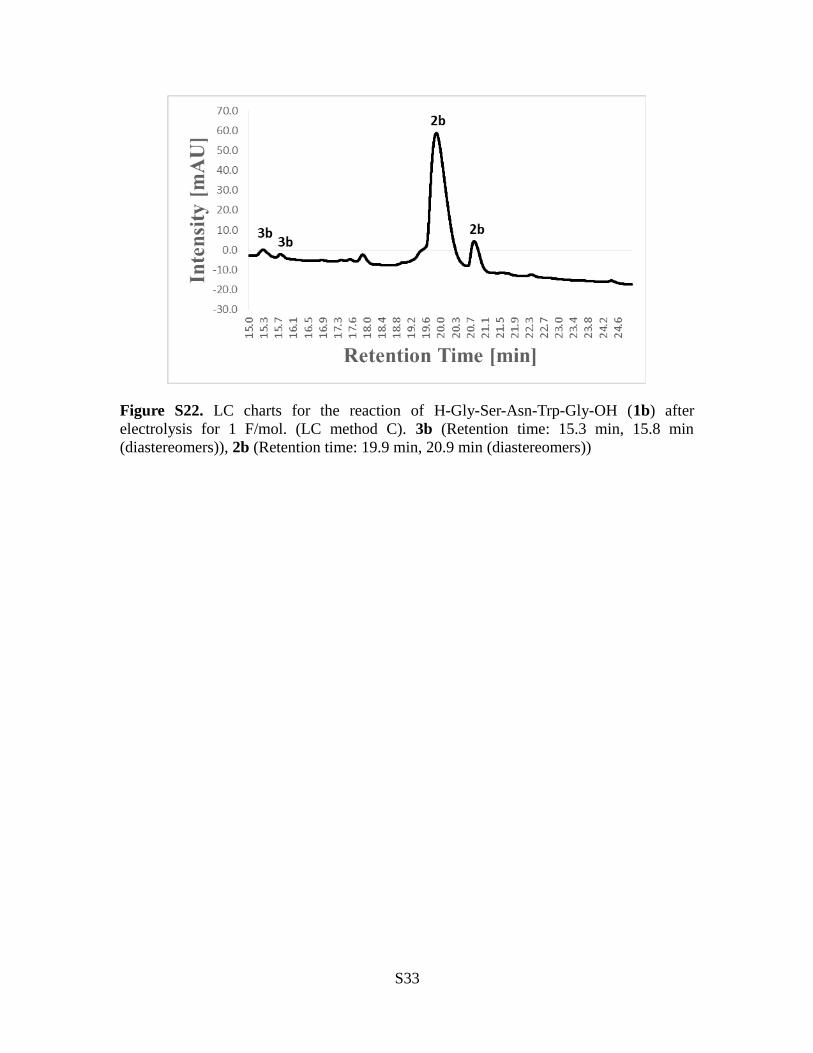
Figure S22. LC charts for the reaction of H-Gly-Ser-Asn-Trp-Gly-OH (1b) afterelectrolysis for 1 F/mol. (LC method C). 3b (Retention time: 15.3 min, 15.8 min(diastereomers)), 2b (Retention time: 19.9 min, 20.9 min (diastereomers))
S33
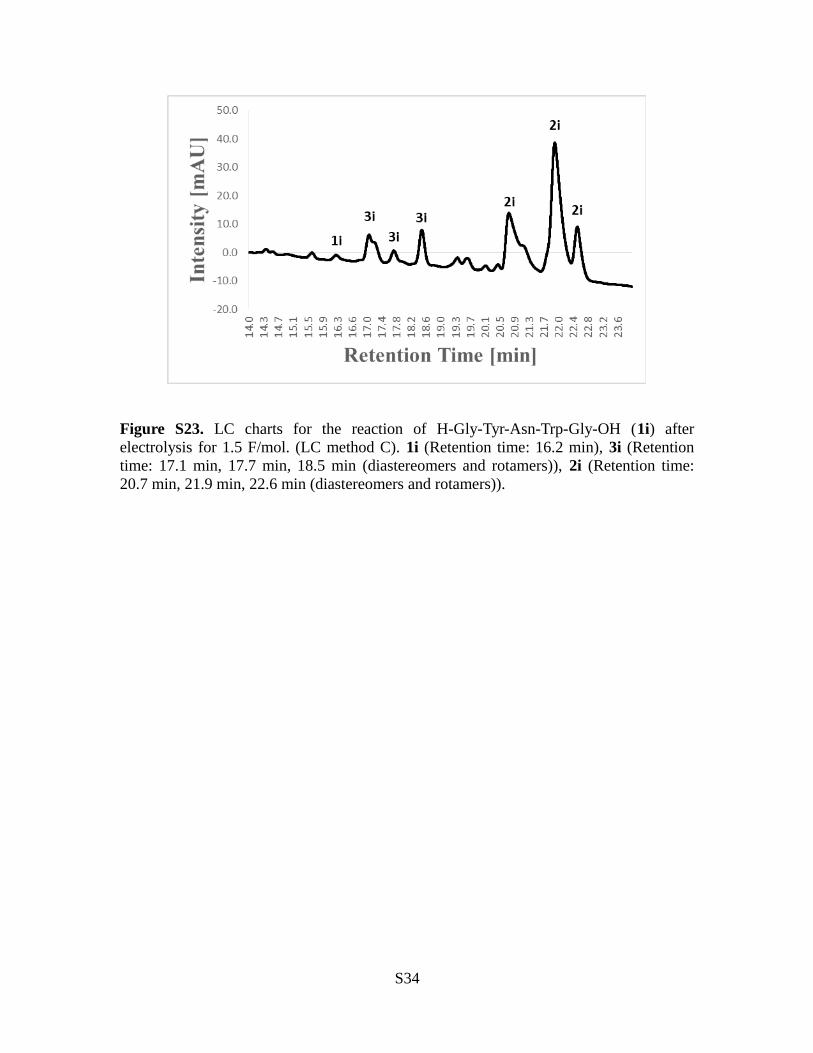
Figure S23. LC charts for the reaction of H-Gly-Tyr-Asn-Trp-Gly-OH (1i) afterelectrolysis for 1.5 F/mol. (LC method C). 1i (Retention time: 16.2 min), 3i (Retentiontime: 17.1 min, 17.7 min, 18.5 min (diastereomers and rotamers)), 2i (Retention time:20.7 min, 21.9 min, 22.6 min (diastereomers and rotamers)).
S34
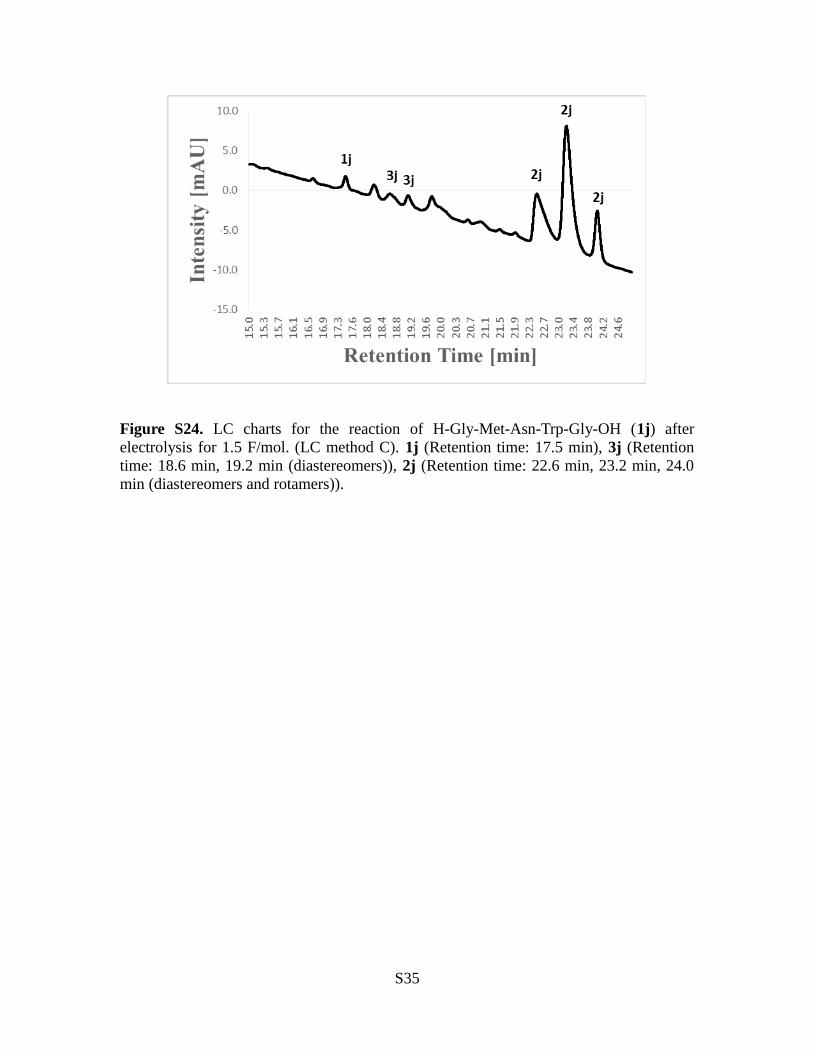
Figure S24. LC charts for the reaction of H-Gly-Met-Asn-Trp-Gly-OH (1j) afterelectrolysis for 1.5 F/mol. (LC method C). 1j (Retention time: 17.5 min), 3j (Retentiontime: 18.6 min, 19.2 min (diastereomers)), 2j (Retention time: 22.6 min, 23.2 min, 24.0min (diastereomers and rotamers)).
S35
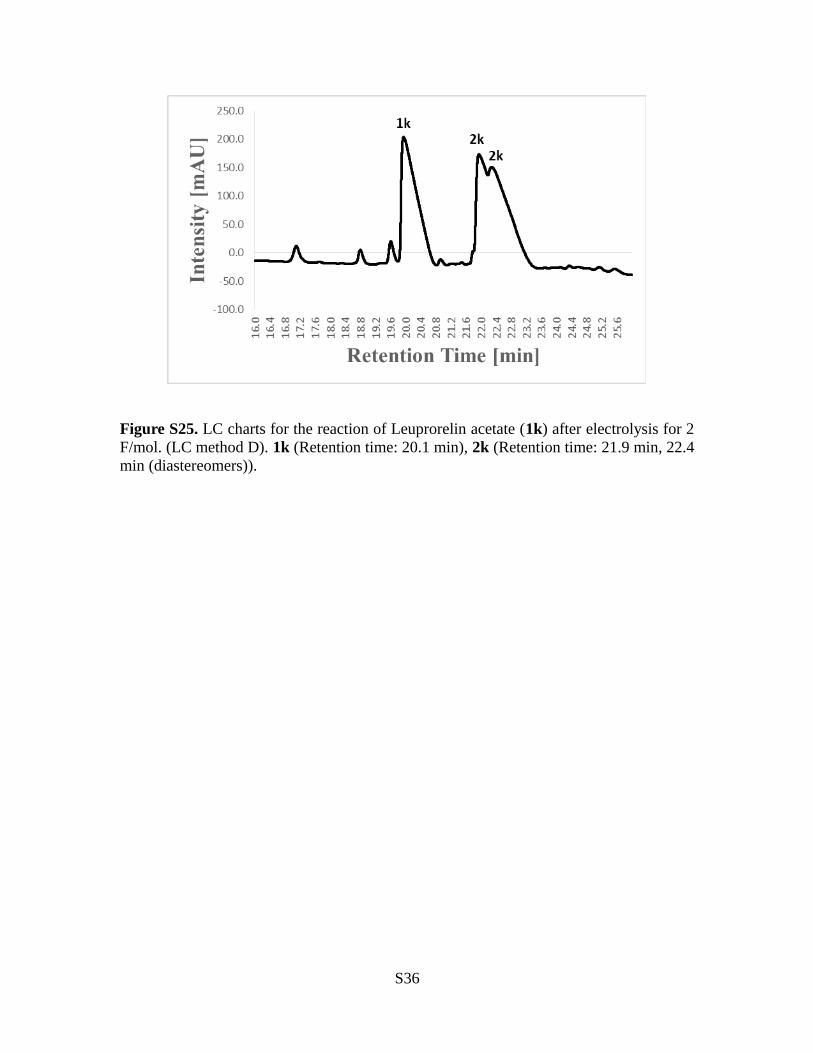
Figure S25. LC charts for the reaction of Leuprorelin acetate (1k) after electrolysis for 2F/mol. (LC method D). 1k (Retention time: 20.1 min), 2k (Retention time: 21.9 min, 22.4min (diastereomers)).
S36
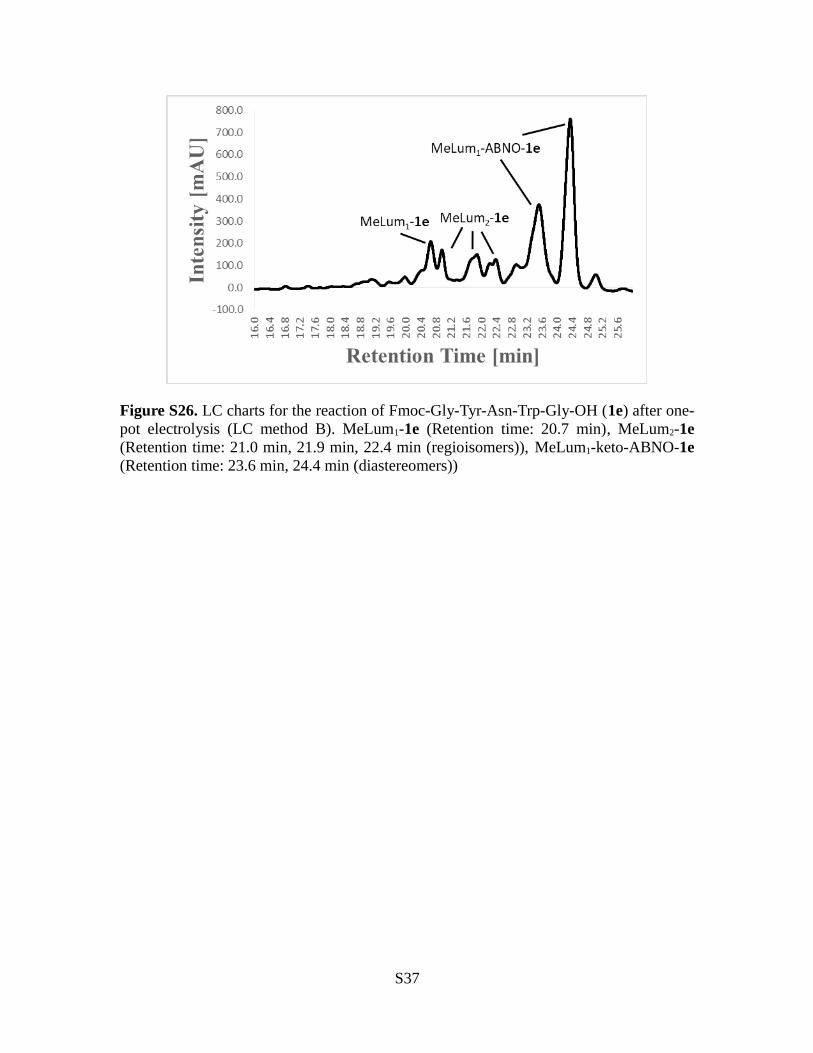
Figure S26. LC charts for the reaction of Fmoc-Gly-Tyr-Asn-Trp-Gly-OH (1e) after one-pot electrolysis (LC method B). MeLum1-1e (Retention time: 20.7 min), MeLum2-1e(Retention time: 21.0 min, 21.9 min, 22.4 min (regioisomers)), MeLum1-keto-ABNO-1e(Retention time: 23.6 min, 24.4 min (diastereomers))
S37
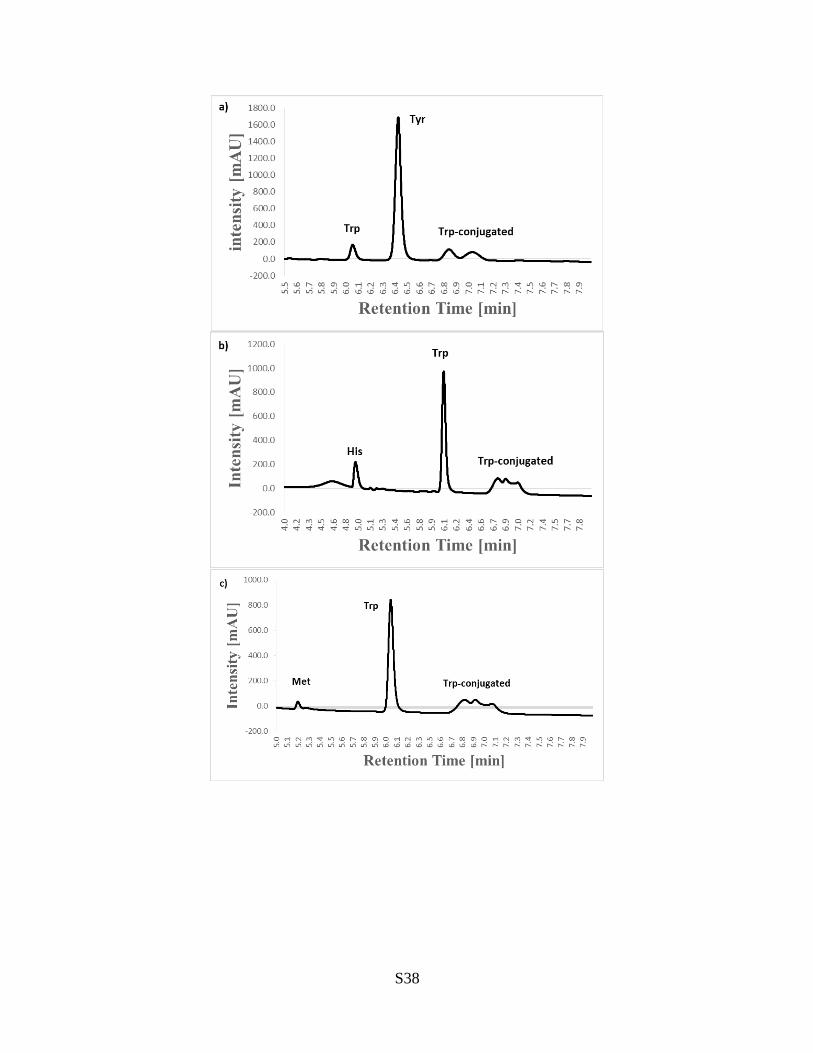
S38

Figure S27. LC charts for the electrochemical Trp-selective bioconjugation to assesscross reactivities. (a) LC charts of the reaction mixture of Ac-Trp-OEt and Bz-Tyr-OEtafter electrolysis for 3.0 F/mol. (LC method E) (b) LC charts of the reaction mixture ofAc-Trp-OEt and Boc-His-OMe after electrolysis for 1.0 F/mol. (LC method E) (c) LCcharts of the reaction mixture of Ac-Trp-OEt and Ac-Met-OMe after electrolysis for 1.0F/mol. (LC method E)
In any cases, cross-reacted bioconjugation products to redox non-innocent amino acids(Tyr, His, and Met) were not observed at all. This result has confirmed high selectivity toTrp.
S39
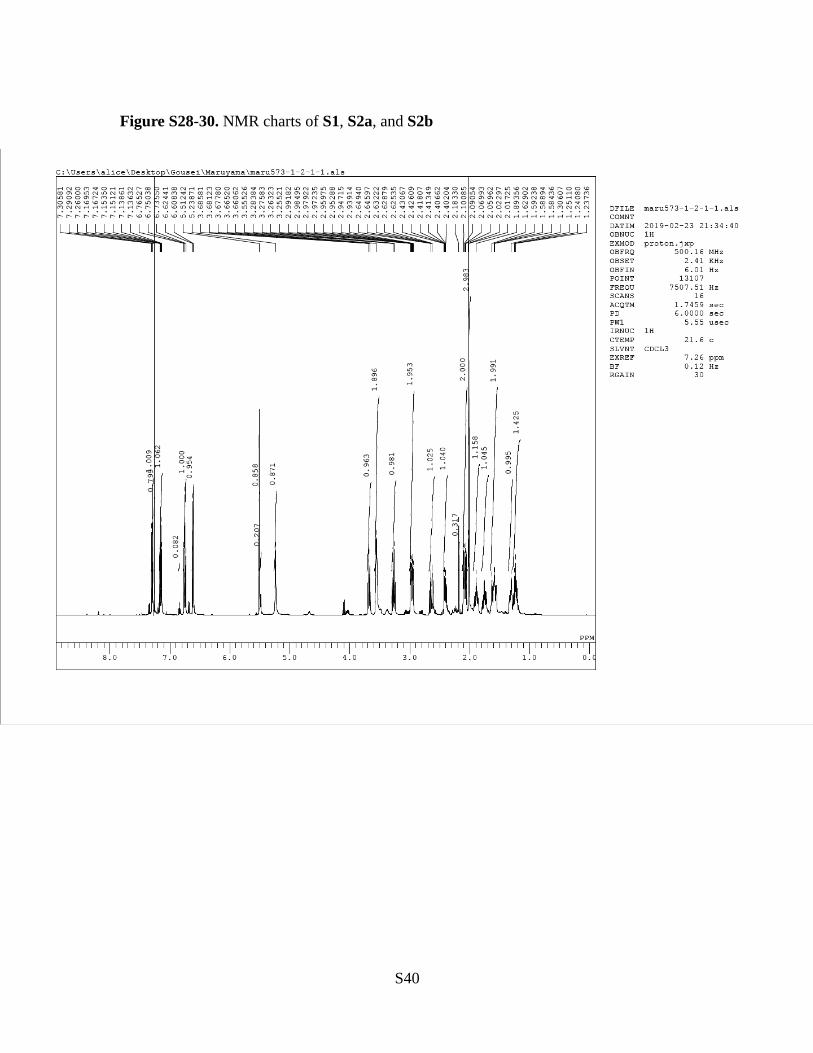
Figure S28-30. NMR charts of S1, S2a, and S2b
S40
S41

References
S42

1 T. Sonobe, K. Oisaki, M. Kanai, Chem. Sci. 2012, 3, 1572.2 S. Sato, K. Nakamura, H. Nakamura, ACS Chem. Biol. 2015, 10, 2633-2640.

download fileview on ChemRxivSI_toyama_V6499.docx (2.98 MiB)

Electrochemical Tryptophan-Selective Bioconjugation
Eisho Toyama,[a] Katsuya Maruyama,[a] Tomoya Sugai,[b] Mio Kondo,[c] Shigeyuki Masaoka,[c]
Tsuyoshi Saitoh,[b] Kounosuke Oisaki*[a] and Motomu Kanai*[a]
E-mail: [email protected]; [email protected]
[a] Graduate School of Pharmaceutical Sciences The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku,
Tokyo 113-0033, Japan
[b] International Institute for Integrative Sleep Medicine (WPI-IIIS), University of Tsukuba, 1-1-1
Tennodai, Tsukuba, Ibaraki 305-8575, Japan
[c] Institute for Molecular Science, National Institutes of Natural Sciences, Okazaki, 444-8787 Japan
Abstract
Bioconjugation reactions are a fundamental synthetic method for generating artificial
peptides and proteins. Despite the potentially superior properties of bioconjugates at hydrophobic
amino acid residues compared with those at hydrophilic amino acids, methods to target hydrophobic
amino acids with moderate reactivity under mild and metal-free conditions are limited. Here we report
the first electrochemically-promoted tryptophan (Trp)-selective bioconjugation of peptides and
proteins in neutral aqueous media. The unique electrochemical cooperation of two radicals, keto-
ABNO and 4-oxo-TEMPO, was critical to suppress both anodic overoxidation of the products and
cross reactivity. Systematic cyclic voltammetry analysis suggested that these two radicals, containing
similar redox potentials but contrasting steric demands, had distinct electrochemical roles (reactant
and electrochemical mediator). This new protocol will be an important advance toward clean and
scalable syntheses of chemically modified biologics.
Main Text
Bioconjugation reactions targeting proteinogenic amino acids are particularly important for
conferring supernatural functions to peptide/protein biomolecules, facilitating the development of
novel therapeutics, diagnostics, and biomaterials.[1] In recent years, bioconjugation methods targeting
poorly nucleophilic, less surface-exposed hydrophobic amino acid residues (e.g., tyrosine,[2-4]
tryptophan,[5,6] and methionine[7]) have attracted attention. Applying such synthetic methods to
bioconjugates will enhance their homogeneity and chemical space, thereby improving their
properties/efficacies and expanding their application scopes. The requirements for practical
bioconjugation reactions are as follows: (1) selectivity toward a targeted functional group of a specific

proteinogenic amino acid, (2) selective activation of the bioconjugation reagents over numerous
reactive functional groups in biomacromolecule substrates, (3) feasibility under physiologic
conditions (mild pH and temperature, aqueous media, and low concentration), and (4) avoidance of
excess reagents and toxic metallic reagents that would cause undesired cross reactivities and
cumbersome purifications. Satisfying these requirements is far more challenging when targeting less
reactive hydrophobic amino acid residues than when targeting nucleophilic amino acid residues such
as lysine and cysteine.
Merging electrochemical organic synthesis[8] and bioconjugation chemistry is an attractive
strategy toward achieving these aims. Traditionally, electrochemical protein modifications in
preparative scales have been limited to direct electrolysis for cysteine-cystine interconversions, site-
selective cleavage, and oxidative functionalizations.[9] In 2018, Gouin and co-workers reported the
first electrochemical protein bioconjugation by targeting tyrosine (Tyr) residues with a urazole
reagent.[3] The electrochemical conditions significantly improved the chemical yield and selectivity
of Barbas’ original conditions using a chemically preactivated reagent.[2] We report herein an
electrochemical tryptophan (Trp)-selective bioconjugation. The hybrid use of two N-oxyl radicals,
keto-ABNO and 4-oxo-TEMPO, was critical for the facile and high-yielding bioconjugation. In
contrast to our original report using chemical activation,[5] the electrochemical reaction developed
here proceeded under neutral pH with remarkably few side reactions.
We previously reported a transition metal-free, Trp-selective bioconjugation using a
sterically less-demanding organoradical, 9-azabicyclo[3.3.1]nonan-3-one-N-oxyl (keto-ABNO), in
the presence of substoichiometric NaNO2 in mildly acidic (pH ~ 3) aqueous media.[5] The active
species of this bioconjugation is oxoammonium cation A (see Scheme 3) generated in situ through
single-electron aerobic oxidation of keto-ABNO. Nitrogen oxide (NOx) produced from NaNO2 and
AcOH under air chemically activated keto-ABNO and promoted single-electron oxidation. Despite
the easy operation and broad substrate generality, the following features of the previous chemical
activation conditions are potentially problematic: (1) an acidic media that potentially hampered the
application to pH-sensitive proteins, and (2) NOx-induced S- and N-nitrosation side reactions.
Encouraged by the rich chemistry of N-oxyl radicals as electrochemical mediators,[10] we anticipated
that anodic electrochemical oxidation would promote the activation of keto-ABNO, leading to Trp-
selective bioconjugation under neutral pH without the use of any external oxidants (e.g., NOx) that
could cause undesired side reactions.
On the basis of this hypothesis, we initiated optimization studies of electrochemical
bioconjugation of Fmoc-protected peptide 1a as a model substrate using an ElectraSyn 2.0 kit (Table
1). Product 2a was produced in only 33% yield when a mixture of 1a (3 mM) and keto-ABNO (1
equiv) in an electrolyte solution (50 mM TBAP in CH3CN-H2O,1:1) was stirred for 1 h without electric
current (entry 1).[11] The application of constant voltage electrolysis (working electrode: graphite,

counter electrode: platinum) at 1.2 V vs Ag/AgCl to this solution for 1 F/mol (ca. 1 h), however,
markedly increased the yield of 2a to 75% with 18% of 1a recovered (entry 2). As the amount of
electric charge increased, 1a was completely consumed, but the amounts of overoxidation products
3a/3a’ increased (entries 3 and 4). The adduct of Trp‒keto-ABNO had a lower oxidation potential than
that of keto-ABNO.[12] Thus, electrolysis at a lower potential (1.0 V) successfully suppressed the
overoxidation, but the yield of 2a decreased (55%, entry 5). Application of 2.0 F/mol electric charge
increased the yield (78%, entry 6), but further application of electricity (>2 F/mol) at 1.0 V was not
possible due to increased resistance. When the amount of keto-ABNO was increased to 2 equiv, the
yield of 2a improved to 84%, but overoxidized products 3a/3a’ were still generated (entry 7).
For future application to electrochemical protein bioconjugation using precious payloads,
the stoichiometry of the reagent must be minimized. Therefore, we screened additives to facilitate the
electron-transfer processes. Interestingly, the addition of sterically-demanding TEMPO derivatives,
which themselves are inert in Trp bioconjugation under NOx conditions,[5] significantly increased the
yield of 2a (entries 8–12 vs entries 5 and 6). In all of these experiments, adducts of the TEMPO
derivatives with 1a were not detected at all. Among the conditions tested, the addition of 20 mol % 4-
oxo-TEMPO afforded the best result with complete consumption of 1a and minimum contamination
by overoxidized 3a/3a’ (97% yield of 2a, entry 12). The efficiency of the additives correlated with
their redox potentials, and TEMPO derivatives with higher oxidation potentials afforded higher yields
of 2a. The current efficiency (i.e., the slope of the graph depicting yield of 2a vs charge) was
approximately 1.5 times greater in the presence of 4-oxo-TEMPO than in its absence.[12]
Table 1. Optimization of Electrochemical Trp-Selective Bioconjugation
Entry
Substrate
(conc.)
Electrolyte solution Additive (equiv) Voltage (V vs
Ag/AgCl)
Charge (F/mol)
1
(%) 2
(%) 3
(%)
1 1a (3 mM)
50 mM TBAP/CH3CN-H2O (1:1) — — — 67 33 0
2 1a (3 mM)
50 mM TBAP/CH3CN-H2O (1:1) — 1.2 1.0 18 75 7
3 1a (3 mM)
50 mM TBAP/CH3CN-H2O (1:1) — 1.2 2.0 0 73 27
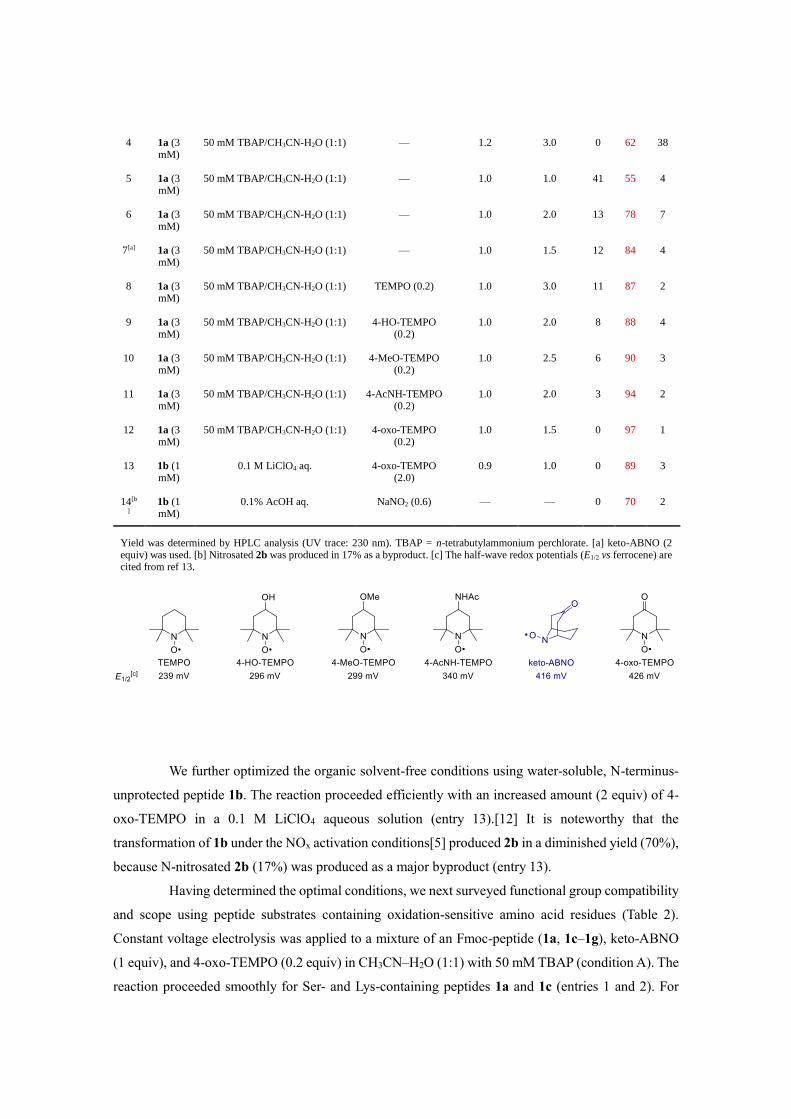
4 1a (3 mM)
50 mM TBAP/CH3CN-H2O (1:1) — 1.2 3.0 0 62 38
5 1a (3 mM)
50 mM TBAP/CH3CN-H2O (1:1) — 1.0 1.0 41 55 4
6 1a (3 mM)
50 mM TBAP/CH3CN-H2O (1:1) — 1.0 2.0 13 78 7
7[a] 1a (3 mM)
50 mM TBAP/CH3CN-H2O (1:1) — 1.0 1.5 12 84 4
8 1a (3 mM)
50 mM TBAP/CH3CN-H2O (1:1) TEMPO (0.2) 1.0 3.0 11 87 2
9 1a (3 mM)
50 mM TBAP/CH3CN-H2O (1:1) 4-HO-TEMPO (0.2)
1.0 2.0 8 88 4
10 1a (3 mM)
50 mM TBAP/CH3CN-H2O (1:1) 4-MeO-TEMPO (0.2)
1.0 2.5 6 90 3
11 1a (3 mM)
50 mM TBAP/CH3CN-H2O (1:1) 4-AcNH-TEMPO (0.2)
1.0 2.0 3 94 2
12 1a (3 mM)
50 mM TBAP/CH3CN-H2O (1:1) 4-oxo-TEMPO (0.2)
1.0 1.5 0 97 1
13 1b (1 mM)
0.1 M LiClO4 aq. 4-oxo-TEMPO (2.0)
0.9 1.0 0 89 3
14[b
] 1b (1 mM)
0.1% AcOH aq. NaNO2 (0.6) — — 0 70 2
Yield was determined by HPLC analysis (UV trace: 230 nm). TBAP = n-tetrabutylammonium perchlorate. [a] keto-ABNO (2 equiv) was used. [b] Nitrosated 2b was produced in 17% as a byproduct. [c] The half-wave redox potentials (E1/2 vs ferrocene) are cited from ref 13.
We further optimized the organic solvent-free conditions using water-soluble, N-terminus-
unprotected peptide 1b. The reaction proceeded efficiently with an increased amount (2 equiv) of 4-
oxo-TEMPO in a 0.1 M LiClO4 aqueous solution (entry 13).[12] It is noteworthy that the
transformation of 1b under the NOx activation conditions[5] produced 2b in a diminished yield (70%),
because N-nitrosated 2b (17%) was produced as a major byproduct (entry 13).
Having determined the optimal conditions, we next surveyed functional group compatibility
and scope using peptide substrates containing oxidation-sensitive amino acid residues (Table 2).
Constant voltage electrolysis was applied to a mixture of an Fmoc-peptide (1a, 1c–1g), keto-ABNO
(1 equiv), and 4-oxo-TEMPO (0.2 equiv) in CH3CN–H2O (1:1) with 50 mM TBAP (condition A). The
reaction proceeded smoothly for Ser- and Lys-containing peptides 1a and 1c (entries 1 and 2). For

His-, Tyr-, and Met-containing peptides 1d–1f, the reactions were slow under standard condition A.
By increasing the voltage and total charge (entry 3) and/or the amount of 4-oxo-TEMPO (entries 4
and 5), however, the reactions produced satisfactory yields. Although Cys-containing peptide 1g
produced the desired bioconjugation product in moderate yield (entry 6), the reaction proceeded in
high yield for disulfide bond-containing peptide 1h (entry 7). Moderate reactivity with free Cys-
containing peptides will not preclude the practical utility because surface-exposed Cys residues form
disulfide bonds in most proteins. Water-soluble peptides 1b, 1i, and 1j also produced satisfactory
yields without inducing notable side reactions (condition B, entries 8-10). Modification of the
commercially available Trp-containing drug leuprorelin acetate (1k) was also successful, affording the
product in 60% yield (entry 11).
Table 2. Substrate Scope of Short Peptides.
Entry Substrate Voltage (V)
Charge (F/mol)
Yield (%)
1 Fmoc-Gly-Ser-Asn-Trp-Gly-OH (1a) 1.0 1.5 97
2 Fmoc-Gly-Lys-Asn-Trp-Gly-OH (1c) 1.0 1.5 85
3 Fmoc-Gly-His-Asn-Trp-Gly-OH (1d) 1.2 3.0 86
4[a] Fmoc-Gly-Tyr-Asn-Trp-Gly-OH (1e) 1.0 3.0 98
5[b] Fmoc-Gly-Met-Asn-Trp-Gly-OH (1f) 1.0 2.5 85
6 Fmoc-Gly-Cys-Asn-Trp-Gly-OH (1g) 1.0 1.0 27
7
1.0 2.0 82
8 H-Gly-Ser-Asn-Trp-Gly-OH (1b) 0.9 1.0 89
9 H-Gly-Tyr-Asn-Trp-Gly-OH (1i) 0.9 1.5 78
10 H-Gly-Met-Asn-Trp-Gly-OH (1j) 0.9 1.5 80
11[c] Leuprorelin acetate: Pyr-His-Trp-Ser-Tyr-DLeu-Leu- Arg-Pro-NHEt・AcOH (1k)
0.9 2.0 60

Condition A (entries 1–7): 1 (3 mM) and 4-oxo-TEMPO (0.2 equiv) in 50 mM TBAP/CH3CN–H2O (1:1) unless otherwise noted. Condition B (entries 8-11): 1 (1 mM) and 4-oxo-TEMPO (2.0 equiv) in 0.1 M LiClO4 aq. (no organic solvent) unless otherwise noted. The reaction time was 1.0-3.0 h. Voltage was vs Ag/AgCl. Yield was determined by HPLC analysis (UV trace: 230 nm). [a] 4-oxo-TEMPO (2 equiv). [b] 4-oxo-TEMPO (1 equiv). [c] NaHCO3 (1 equiv) was added.
Another advantage of this electrochemical method is its facile applicability to one-pot,
orthogonal dual conjugation reactions by simply tuning the applied voltages.[14,15] We found that the
electrochemical method also promoted Tyr-selective oxidative bioconjugation with MeLum, originally
developed by Nakamura using hydrogen peroxide as a chemical oxidant for preactivation.[4g] Based
on this finding, the one-pot, electrochemical orthogonal double modifications of 1e bearing Tyr and
Trp residues were realized (Scheme 1). Thus, after the Tyr-selective bioconjugation of 1e with MeLum
at 1.2 V, the Trp-selective bioconjugation with keto-ABNO and 4-oxo-TEMPO was conducted at 1.0
V. Each bioconjugation reagent reacted one by one selectively with the target residues to produce
doubly modified peptide MeLum1-keto-ABNO-1e in 66% yield.
Scheme 1. One-pot, Orthogonal, Dual Electrochemical Modifications
Finally, the optimized reaction conditions for Trp-bioconjugation in an aqueous medium
without use of any organic solvents were applied to native proteins, lysozyme and bovine serum
albumin (Scheme 2). In the case of lysozyme, applying 1.0 F/mol electric charge at 0.9 V led to
complete consumption of the starting material. The corresponding lysozyme‒keto-ABNO conjugate
was obtained in nearly quantitative yield (MS detection). In the case of bovine serum albumin, 1.0

F/mol electric charge at 1.0 V cleanly led to the mono-adduct as the major product (~70% conversion
based on MS intensity), although some of the starting material remained. Therefore, the
electrochemical Trp-bioconjugation was generally applicable to peptides as well as protein substrates.
Scheme 2. Electrochemical Trp-Selective Bioconjugation of Proteins
To gain preliminary insight into the reaction mechanism, we conducted systematic cyclic
voltammetry (CV) measurements (Figure 1).[12] We used TEMPO as the additive for the CV studies,
because keto-ABNO and 4-oxo-TEMPO exhibited indistinguishable redox potentials.[12] First, we
assessed the interactions between keto-ABNO or TEMPO and a Trp residue using N-Ac-Trp-OEt as a
model substrate. A solution of keto-ABNO + N-Ac-Trp-OEt afforded new anodic peaks at ca. 0.75,
0.83, and 1.0 V (Figure 1a), suggesting an interaction between keto-ABNO and Trp (tentatively drawn
as structure C in Scheme 3). Notably, this interaction significantly decreased the oxidation potentials
of both keto-ABNO and Trp.[16] The cathodic peak currents disappeared because an irreversible
bioconjugation reaction proceeded between oxoammonium cation A derived from keto-ABNO and
Trp. In the case of TEMPO + N-Ac-Trp-OEt, however, the cyclic voltammogram was a simple
superposition of the two components (Figure 1b). Therefore, TEMPO and Trp neither interacted nor
reacted. The fact that no bioconjugation product was observed between TEMPO or its derivatives and
1a (Table 1) was consistent with this CV profile. Second, there was no significant electrochemical
interaction between two radicals in the absence of Trp; the cyclic voltammogram for keto-ABNO +
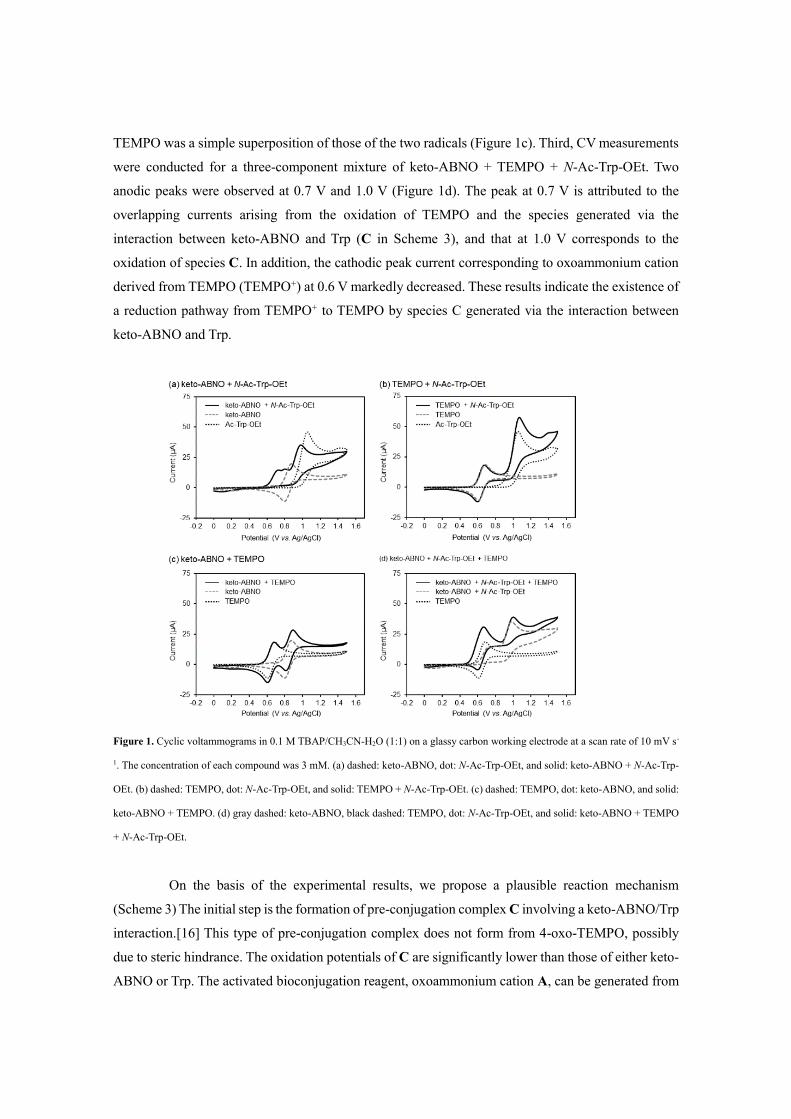
TEMPO was a simple superposition of those of the two radicals (Figure 1c). Third, CV measurements
were conducted for a three-component mixture of keto-ABNO + TEMPO + N-Ac-Trp-OEt. Two
anodic peaks were observed at 0.7 V and 1.0 V (Figure 1d). The peak at 0.7 V is attributed to the
overlapping currents arising from the oxidation of TEMPO and the species generated via the
interaction between keto-ABNO and Trp (C in Scheme 3), and that at 1.0 V corresponds to the
oxidation of species C. In addition, the cathodic peak current corresponding to oxoammonium cation
derived from TEMPO (TEMPO+) at 0.6 V markedly decreased. These results indicate the existence of
a reduction pathway from TEMPO+ to TEMPO by species C generated via the interaction between
keto-ABNO and Trp.
Figure 1. Cyclic voltammograms in 0.1 M TBAP/CH3CN-H2O (1:1) on a glassy carbon working electrode at a scan rate of 10 mV s-
1. The concentration of each compound was 3 mM. (a) dashed: keto-ABNO, dot: N-Ac-Trp-OEt, and solid: keto-ABNO + N-Ac-Trp-
OEt. (b) dashed: TEMPO, dot: N-Ac-Trp-OEt, and solid: TEMPO + N-Ac-Trp-OEt. (c) dashed: TEMPO, dot: keto-ABNO, and solid:
keto-ABNO + TEMPO. (d) gray dashed: keto-ABNO, black dashed: TEMPO, dot: N-Ac-Trp-OEt, and solid: keto-ABNO + TEMPO
+ N-Ac-Trp-OEt.
On the basis of the experimental results, we propose a plausible reaction mechanism
(Scheme 3) The initial step is the formation of pre-conjugation complex C involving a keto-ABNO/Trp
interaction.[16] This type of pre-conjugation complex does not form from 4-oxo-TEMPO, possibly
due to steric hindrance. The oxidation potentials of C are significantly lower than those of either keto-
ABNO or Trp. The activated bioconjugation reagent, oxoammonium cation A, can be generated from
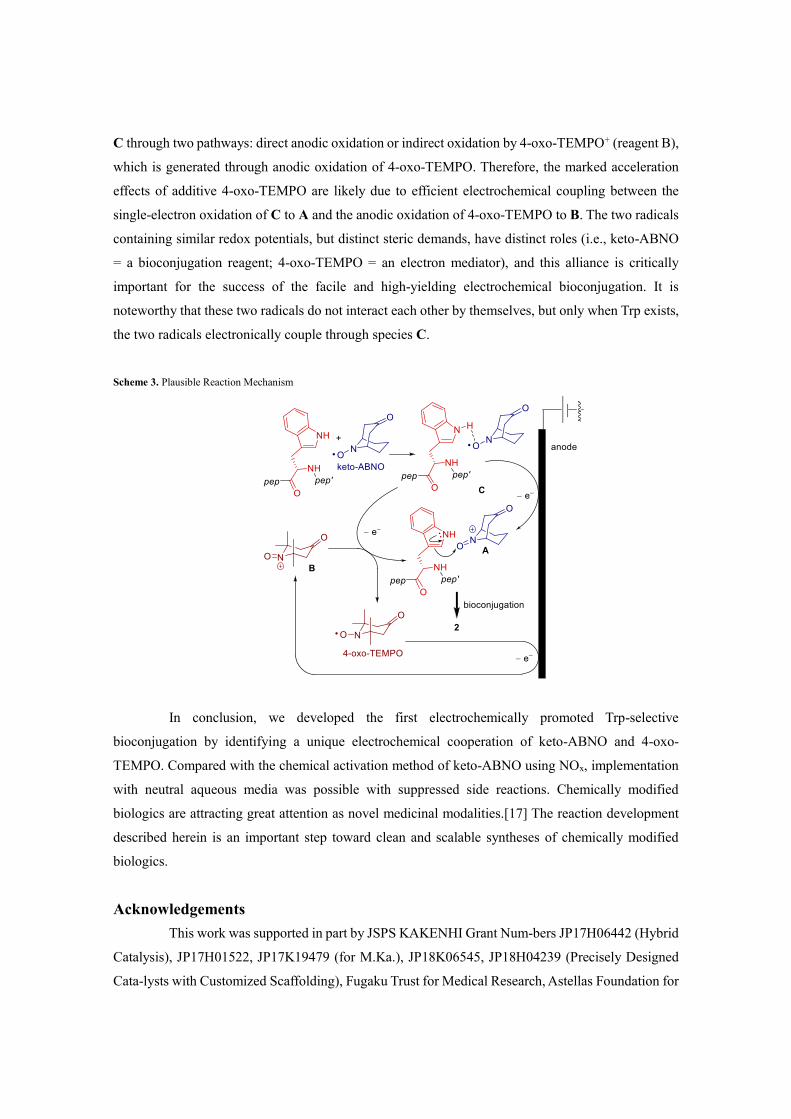
C through two pathways: direct anodic oxidation or indirect oxidation by 4-oxo-TEMPO+ (reagent B),
which is generated through anodic oxidation of 4-oxo-TEMPO. Therefore, the marked acceleration
effects of additive 4-oxo-TEMPO are likely due to efficient electrochemical coupling between the
single-electron oxidation of C to A and the anodic oxidation of 4-oxo-TEMPO to B. The two radicals
containing similar redox potentials, but distinct steric demands, have distinct roles (i.e., keto-ABNO
= a bioconjugation reagent; 4-oxo-TEMPO = an electron mediator), and this alliance is critically
important for the success of the facile and high-yielding electrochemical bioconjugation. It is
noteworthy that these two radicals do not interact each other by themselves, but only when Trp exists,
the two radicals electronically couple through species C.
Scheme 3. Plausible Reaction Mechanism
In conclusion, we developed the first electrochemically promoted Trp-selective
bioconjugation by identifying a unique electrochemical cooperation of keto-ABNO and 4-oxo-
TEMPO. Compared with the chemical activation method of keto-ABNO using NOx, implementation
with neutral aqueous media was possible with suppressed side reactions. Chemically modified
biologics are attracting great attention as novel medicinal modalities.[17] The reaction development
described herein is an important step toward clean and scalable syntheses of chemically modified
biologics.
Acknowledgements
This work was supported in part by JSPS KAKENHI Grant Num-bers JP17H06442 (Hybrid
Catalysis), JP17H01522, JP17K19479 (for M.Ka.), JP18K06545, JP18H04239 (Precisely Designed
Cata-lysts with Customized Scaffolding), Fugaku Trust for Medical Research, Astellas Foundation for

Research on Metabolic Disorders (for K.O.), JP16H04125, JP17H06444 (Hybrid Catalysis) (for S.M.),
JP15H05480, JP17K19185 (for M.Ko.).
Keywords
Electrochemistry • Bioconjugation • Protein • Organoradical • Tryptophan
References
[1] (a) N. Krall, F. P. da Cruz, O. Boutureira, G. J. L. Bernardes, Nat. Chem. 2016, 8, 103-113.
(b) J. N. deGruyter, L. R. Malins, P. S. Baran, Biochemistry 2017, 56, 3863-3873. (c) Q.-Y. Hu, F.
Berti, R. Adamo, Chem. Soc. Rev. 2016, 45, 1691−1719. (d) C. D. Spicer, B. G. Davis, Nat. Commun.
2014, 5, 4740. (e) Y. Wang, C. Wu, Biomacromolecules 2018, 19, 1804-1825.
[2] (a) H. Ban, J. Gavrilyuk, C. F. Barbas, III, J. Am. Chem. Soc. 2010, 132, 1523−1525. (b) H.
Ban, M. Nagano, J. Gavrilyuk, W. Hakamata, T. Inokuma, C. F. Barbas, III, Bioconjugate Chem. 2013,
24, 520−532.
[3] D. Alvarez-Dorta, C. Thobie-Gautier, M. Croyal, M. Bouzelha, M. Mével, D. Deniaud, M.
Boujtita, S. G. Gouin, J. Am. Chem. Soc. 2018, 140, 17120-17126.
[4] (a) N. S. Joshi, L. R. Whitaker, M. B. Francis, J. Am. Chem. Soc. 2004, 126, 15942−15943.
(b) S. D. Tilley, M. B. Francis, J. Am. Chem. Soc. 2006, 128, 1080−1081. (c) D. W. Romanini, M. B.
Francis, Bioconjugate Chem. 2008, 19, 153−157. (d) K. L. Seim, A. C. Obermeyer, M. B. Francis,
J. Am. Chem. Soc. 2011, 133, 16970−16976. (e) M. W. Jones, G. Mantovani, C. A. Blindauer, S. M.
Ryan, X. Wang, D. J. Brayden, D. M. Haddleton, J. Am. Chem. Soc. 2012, 134, 7406−7413. (f) S. Sato,
H. Nakamura, Angew. Chem. Int. Ed. 2013, 52, 8681-8684. (g) S. Sato, K. Nakamura, H. Nakamura,
ACS Chem. Biol. 2015, 10, 2633-2640. (h) E. J. Choi, D. Jung, J.-S. Kim, Y. Lee, M. Kim, Chem. Eur.
J. 2018, 24, 10948-10952.
[5] Y. Seki, T. Ishiyama, D. Sasaki, J. Abe, Y. Sohma, K. Oisaki, M. Kanai, J. Am. Chem. Soc.
2016, 138, 10798-10801.
[6] (a) J. M. Antos, J. M. McFarland, A. T. Iavarone, M. B. Francis, J. Am. Chem. Soc. 2009,
131, 6301−6308. (b) B. V. Popp, Z. T. Ball, J. Am. Chem. Soc. 2010, 132, 6660−6662. (c) W. Siti, A.
K. Khan, H. P. de Hoog, B. Liedberg, M. Nallani, Org. Biomol. Chem. 2015, 13, 3202−3206. (d) M.
B. Hansen, F. Hubalek, T. Skrydstrup, T. Hoeg-Jensen, Chem. Eur. J. 2016, 22, 1572−1576. (e) Y. Yu,
L.-K. Zhang, A. V. Buevich, G. Li, H. Tang, P. Vachal, S. L. Colletti, Z.-C. Shi, J. Am. Chem. Soc.
2018, 140, 6797–6800.
[7] (a) S. Lin, X. Yang, S. Jia, A. M. Weeks, M. Hornsby, P. S. Lee, R. V. Nichiporuk, A. T.
Iavarone, J. A. Wells, F. D. Toste, C. J. Chang, Science 2017, 355, 597−602. (b) M. T. Taylor, J. E.
Nelson, M.G. Suero, M. J. Gaunt, Nature 2018, 562, 563-568. (c) R. V. Maaskant, G. Roelfes,
ChemBioChem 2018, 20, 57-61.

[8] For recent comprehensive reviews in electrochemical organic synthesis: (a) M. Yan, Y.
Kawamata, P. S. Baran, Chem. Rev. 2017, 117, 13230-13319. (b) A. Wiebe, T. Gieshoff, S. Möhle, E.
Rodrigo, M. Zirbes, S. R. Waldvogel, Angew. Chem. Int. Ed. 2018, 57, 5594-5619. (c) S. Möhle, M.
Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe, S. R. Waldvogel, Angew. Chem. Int. Ed. 2018, 47, 6018-
6041.
[9] (a) D. J. Walton, J. Heptinstall, Prep. Biochem. Biotechnol. 2000, 30, 1-14. (b) M. Takahashi,
A. Handa, Y. Yamaguchi, R. Kodama, K. Chiba, J. Agric. Food Chem. 2016, 64, 6503-6507. (c) F. T.
G. van den Brink, T. Zhang, L. Ma, J. Bomer, M. Odijk, W. Olthuis, H. P. Permentier, R. Bischoff, A.
van den Berg, Anal. Chem. 2016, 88, 9190-9198.
[10] J. E. Nutting, M. R. Rafiee, S. S. Stahl, Chem. Rev. 2018, 118, 4834-4885.
[11] This background reaction might proceed through disproportionation of keto-ABNO to A
promoted by the C-terminal carboxylic acid of 1a. Under the same conditions, Ac-Trp-OEt did not
promote any background reaction. For disproportionation of N-oxyl radicals, see; (a) S.-y. Kishioka,
T. Ohsaka, K. Tokuda, Electrochim. Acta 2003, 48, 1589-1594. (b) V. D. Sen', V. A. Golubev, J. Phys.
Org. Chem. 2009, 22, 138-143. (c) V. D. Sen', I. V. Tikhonov, L. I. Borodin, E. M. Pliss, V. A. Golubev,
M. A. Syroeshkin, A. I. Rusakov, J. Phys. Org. Chem. 2015, 28, 17-24.
[12] See Supporting Information for details.
[13] M. B. Lauber, S. S. Stahl, ACS Catal. 2013, 3, 2612-2616.
[14] For consecutive electrochemical reactions in one pot in small molecule synthesis, see: (a) S.
Suga, M. Watanabe, J.-I. Yoshida, J. Am. Chem. Soc. 2002, 124, 14824−14825. (b) S. Suga, M.
Watanabe, C.-H. Song, J.-i. Yoshida, Electrochemistry 2006, 74, 672−679. (c) T. Nokami, R. Hayashi,
Y. Saigusa, A. Shimizu, C.-Y. Liu, K.-K. T. Mong, J.-i. Yoshida, Org. Lett. 2013, 15, 4520−4523. (d)
T. Nokami, Y. Isoda, N. Sasaki, A. Takaiso, S. Hayase, T. Itoh, R. Hayashi, A. Shimizu, J.-i. Yoshida,
Org. Lett. 2015, 17, 1525−1528. (e) T. Nokami, H. Tsuyama, A. Shibuya, T. Nakatsutsumi, Chem. Lett.
2008, 37, 942−943. (f) M. F. Hartmer, S. R. Waldvogel, Chem. Commun. 2015, 51, 16346−16348. (g)
Y. Kawabata, Y. Naito, T. Saitoh, K. Kawa, T. Fuchigami, S. Nishiyama, Eur. J. Org. Chem. 2014, 99-
104.
[15] For a recent review upon dual modification of biomolecules, see: A. Maruani, D. A. Richards,
V. Chudasama, Org. Biomol. Chem. 2016, 14, 6165-6178.
[16] Structural studies of how keto-ABNO interacts with Trp in aqueous solvent are difficult due
to the transient and radical characteristics. The following two types of interactions may be operative
based on previous reports; (1) For hydrogen-bonding interactions of N-oxyl radical and indole in
organic solvent, see: (a) Z. W. Qui, D. M. Grant, R. J. Pugmire, J. Am. Chem. Soc. 1984, 106, 557-
563. (2) For radical- interaction, see: (b) C. Estarellas, A. Frontera, D. Quiñonero, P. M. Deyà, Phys.
Chem. Chem. Phys., 2011, 13, 16698-16705.
[17] E. Valeur, S. M. Guéret, H. Adihou, R. Gopalakrishnan, M. Lemurerll, H. Waldmann, T. N.

Grossmann, A. T. Plowright, Angew. Chem. Int. Ed. 2017, 21, 10294-10323.
TOC
The first electrochemically-promoted tryptophan-selective bioconjugation in neutral aqueous media
was developed. Systematic CV analyses suggested that unique electrochemical alliance of two
structurally distinct N-oxyl radicals, keto-ABNO and 4-oxo-TEMPO, was critical for both high yield
and suppressed side reactions. This method was successfully applied to water-soluble peptides and
proteins, contributing to the progress of clean and scalable synthesis of chemically modified biologics.

download fileview on ChemRxivChemRxiv.pdf (658.94 KiB)


























![Radiosynthesis and Bioconjugation of [18 F]FPy5yne - Triumf](https://img.dokumen.tips/doc/110x75/6203aba2da24ad121e4c1a6b/radiosynthesis-and-bioconjugation-of-18-ffpy5yne-triumf.jpg)


