Embed Size (px)
Citation preview

Diagnosis and Treatment of KIT-MutantMetastatic MelanomaMegan Lyle, Melanoma Institute Australia, Sydney, New South Wales, AustraliaGeorgina V. Long, Melanoma Institute Australia; University of Sydney, Sydney; and Westmead Institute for Cancer Research,
Westmead, New South Wales, Australia
See accompanying artile on page 3182
The Oncology Grand Rounds series is designed to place original reports published in the Journal into clinical context. A casepresentation is followed by a description of diagnostic and management challenges, a review of the relevant literature, anda summary of the authors’ suggested management approaches. The goal of this series is to help readers better understand howto apply the results of key studies, including those published in Journal of Clinical Oncology, to patients seen in their ownclinical practice.
A 52-year-old man has unresectable locally recurrent melanoma of the left foot (Fig 1) and pulmonarymetastases. Nine months before this presentation, he underwent a wide local excision and sentinel nodebiopsy for an acral melanoma on his left heel. Pathology disclosed Breslow thickness of 4.8 mm, Clark levelIV, and tumor ulceration with a mitotic rate of 37 mitoses/mm2. Both sentinel nodes in the left groin werepositive for melanoma cells, which expressed S100, HMB45, and melan A. At subsequent left inguinaldissection, seven more nodes showed no additional nodal metastases. Within 3 months of his originalsurgery, the patient developed a local recurrence in the foot, and over the subsequent 6 months, he underwentserial local excisions and topical diphencyprone treatment. A recent staging scan showed at least 20 foci ofin-transit disease in the left lower leg and foot, as well as a solitary lung metastasis (12 mm). His EasternCooperative Oncology Group performance status is 1, with no significant comorbidities. High-resolutionmelt followed by sequencing of an in-transit metastasis showed there is no BRAF exon 15 mutation. How-ever, Sanger sequencing of KIT exons 9, 11, 13, and 17, performed as screening for a clinical trial enrollingpatients with metastatic acral and mucosal melanomas, showed an exon 13 K642E mutation.
CHALLENGES IN DIAGNOSIS AND MANAGEMENT
KIT aberrations in melanoma include KIT amplifications (increasedcopy number) and KIT mutations and do not necessarily occur to-gether (Table 1). Emerging evidence, including the phase II trial byHodi et al,6 reported in the article accompanying ours, suggests thatonly KIT mutations predict response to drug-targeted KIT inhibitionin the metastatic setting. The greatest diagnostic challenges are decid-ing which patients with metastatic disease should undergo KIT-mutation testing of their melanoma and identifying the full spectrumof KIT mutations. Additional challenges lie in the identification of thedrug-targetable mutations of the many KIT mutations in melanomaand determining which KIT inhibitor targets which mutation best.
KIT-mutant melanoma represents only approximately 3% ofmelanomas overall.7 KIT is one of many driver mutations identified inmelanoma so far,8 some of which are (almost absolutely) mutuallyexclusive. BRAF (approximately 45%)9-11 and NRAS (approximately20%)10-12 are the most frequently identified mutations and rarelycoexist. Others, such as GNAQ and GNA11, are much less common(1% to 2%) except in ocular melanoma, where these mutations are
found in 50% and 30% of patient cases, respectively.13,14 KIT muta-tions are most frequent in acral (arising on palms or soles or subun-gually) and mucosal (arising from mucosal epithelium of respiratory,alimentary, or genitourinary tract) melanomas, but they also occur inlow frequency in cutaneous melanomas associated with chronic sundamage (CSD; Table 1). KIT mutations in melanoma are dispersedacross the entire KIT gene in exons 9, 11, 13, 17, and 1815; thus, genetictesting requires sequencing of all these exons. Given the time, expense,and low yield of KIT mutations in an unselected population of patientswith melanoma, it is difficult to justify KIT testing for all patients asroutine practice.
SUMMARY OF THE RELEVANT LITERATURE
KIT is a receptor tyrosine kinase located on the cell surface and has arole in normal melanocyte development. Binding of the KIT receptorby its ligand, stem-cell factor, results in activation of downstreampathways, including the mitogen-activated protein kinase andphosphatidylinositol-3 kinase/AKT pathways, and is important in celldifferentiation, mobilization, proliferation, and survival.16
JOURNAL OF CLINICAL ONCOLOGY O N C O L O G Y G R A N D R O U N D S
VOLUME 31 � NUMBER 26 � SEPTEMBER 10 2013
3176 © 2013 by American Society of Clinical Oncology Journal of Clinical Oncology, Vol 31, No 26 (September 10), 2013: pp 3176-3181
2013 from 152.14.136.96Information downloaded from jco.ascopubs.org and provided by at North Carolina State University Libraries on November 7,
Copyright © 2013 American Society of Clinical Oncology. All rights reserved.2013 from 152.14.136.96Information downloaded from jco.ascopubs.org and provided by at North Carolina State University Libraries on November 7,
Copyright © 2013 American Society of Clinical Oncology. All rights reserved.2013 from 152.14.136.96Information downloaded from jco.ascopubs.org and provided by at North Carolina State University Libraries on November 7,
Copyright © 2013 American Society of Clinical Oncology. All rights reserved.2013 from 152.14.136.96Information downloaded from jco.ascopubs.org and provided by at North Carolina State University Libraries on November 7,
Copyright © 2013 American Society of Clinical Oncology. All rights reserved.2013 from 152.14.136.96Information downloaded from jco.ascopubs.org and provided by at North Carolina State University Libraries on November 7,
Copyright © 2013 American Society of Clinical Oncology. All rights reserved.2013 from 152.14.136.96Information downloaded from jco.ascopubs.org and provided by at North Carolina State University Libraries on November 7,
Copyright © 2013 American Society of Clinical Oncology. All rights reserved.2013 from 152.14.136.96Information downloaded from jco.ascopubs.org and provided by at North Carolina State University Libraries on November 7,
Copyright © 2013 American Society of Clinical Oncology. All rights reserved.

KIT aberrations, including somatic mutations and amplifica-tions, were first identified as arising frequently in specific histopatho-logic subtypes of melanoma, specifically mucosal, acral, andcutaneous melanomas associated with CSD.1 Subsequent studies havereported similar KIT mutation rates of 11% to 23% for acral and 9% to21% for mucosal melanomas; however, rates in cutaneous melanomawith evidence of CSD vary (Table 1).1-5 We found KIT mutations in3% of stage 3 or 4 cutaneous (nonacral, nonmucosal) melanoma(unpublished data). In 83 antecedent primary cutaneous melanomasscored for CSD using established criteria,17 the rate was 4% (two of 48)in patients with low CSD (score, 0 to 1) and 3% (one of 35) in thosewith high CSD (score, 2 to 3). In the subset of patients with acralmelanoma, the rate was 11% (three of 28), consistent with pub-lished data.
Testing for a KIT mutation is complex because testing for hotspotmutations will only detect mutations in predetermined locations; it is
not effective for screening nonrecurrent or multinucleotide substitu-tions, insertions, or deletions, genetic events often found in mutatedKIT.18 Therefore, sequencing of the relevant exons is the only reliablemethod to detect all KIT mutations. Expression of anti-CD117 anti-body as a marker of overexpression of KIT is not a reliable surrogatefor KIT aberrations in melanoma and should not be used as a screen-ing tool.2,4,19
Imatinib mesylate is a small-molecule inhibitor that selectivelybinds to the tyrosine kinases Abl and KIT as well as the platelet-derivedgrowth factor receptors alpha and beta.20 Several early trials of ima-tinib in metastatic melanoma conducted without consideration ofKIT mutation status showed no efficacy.21-23 After recognition of KITaberrations in melanoma subtypes, several case reports and seriessuggested responses with imatinib and other tyrosine kinase inhibitors(TKIs), such as sorafenib, sunitinib, and dasatinib, in patients withKIT mutations.24-30
BA
DC
2
1 6
7
14
Fig 1. Metastatic acral melanoma of theleft foot and leg.
Table 1. Summary of Published KIT Mutation and Amplification� Rates in Subtypes of Melanoma
Study
Acral Mucosal CSD Non-CSD (cutaneous)
Mutation Amplification Mutation Amplification Mutation Amplification Mutation Amplification
No. % No. % No. % No. % No. % No. % No. % No. %
Curtin et al1 3 of 28 11 7 of 28 25 8 of 38 21 10 of 38 26 3 of 18 17 1 of 18 5 0 of 18 0 0 of 18 0Beadling et al2 3 of 13 23 3 of 11 27 7 of 45 16 10 of 38 26 — — — — 1 of 58 1.7† 3 of 45 7†Handolias et al3 2 of 4 50 — — — — — — 2 of 98 2 — — 1 of 128 0.7 — —Kong et al4 23 of 193 12 14 of 193 7 16 of 167 10 17 of 167 10 6 of 29 21 1 of 29 3 5 of 62 8 2 of 62 3Carvajal et al5 18 of 84 21 8 of 83 10 17 of 93 18 14 of 91 15 5 of 32 16 3 of 30 10 — — — —
Abbreviation: CSD, chronic sun damage.�As determined by either polymerase chain reaction or fluorescence in situ hybridization.†Rate in cutaneous melanomas; not scored for CSD.
KIT-Mutant Melanoma
www.jco.org © 2013 by American Society of Clinical Oncology 3177
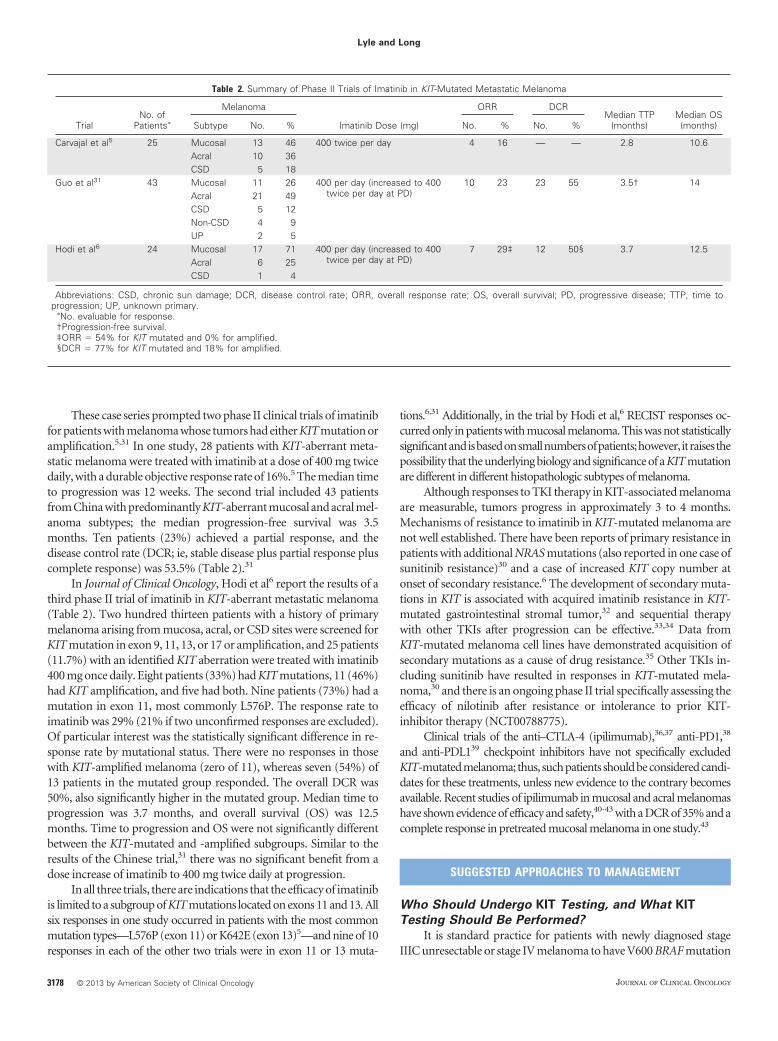
These case series prompted two phase II clinical trials of imatinibfor patients with melanoma whose tumors had either KIT mutation oramplification.5,31 In one study, 28 patients with KIT-aberrant meta-static melanoma were treated with imatinib at a dose of 400 mg twicedaily, with a durable objective response rate of 16%.5 The median timeto progression was 12 weeks. The second trial included 43 patientsfrom China with predominantly KIT-aberrant mucosal and acral mel-anoma subtypes; the median progression-free survival was 3.5months. Ten patients (23%) achieved a partial response, and thedisease control rate (DCR; ie, stable disease plus partial response pluscomplete response) was 53.5% (Table 2).31
In Journal of Clinical Oncology, Hodi et al6 report the results of athird phase II trial of imatinib in KIT-aberrant metastatic melanoma(Table 2). Two hundred thirteen patients with a history of primarymelanoma arising from mucosa, acral, or CSD sites were screened forKIT mutation in exon 9, 11, 13, or 17 or amplification, and 25 patients(11.7%) with an identified KIT aberration were treated with imatinib400 mg once daily. Eight patients (33%) had KIT mutations, 11 (46%)had KIT amplification, and five had both. Nine patients (73%) had amutation in exon 11, most commonly L576P. The response rate toimatinib was 29% (21% if two unconfirmed responses are excluded).Of particular interest was the statistically significant difference in re-sponse rate by mutational status. There were no responses in thosewith KIT-amplified melanoma (zero of 11), whereas seven (54%) of13 patients in the mutated group responded. The overall DCR was50%, also significantly higher in the mutated group. Median time toprogression was 3.7 months, and overall survival (OS) was 12.5months. Time to progression and OS were not significantly differentbetween the KIT-mutated and -amplified subgroups. Similar to theresults of the Chinese trial,31 there was no significant benefit from adose increase of imatinib to 400 mg twice daily at progression.
In all three trials, there are indications that the efficacy of imatinibis limited to a subgroup of KIT mutations located on exons 11 and 13. Allsix responses in one study occurred in patients with the most commonmutation types—L576P (exon 11) or K642E (exon 13)5—and nine of 10responses in each of the other two trials were in exon 11 or 13 muta-
tions.6,31 Additionally, in the trial by Hodi et al,6 RECIST responses oc-curred only in patients with mucosal melanoma. This was not statisticallysignificantandisbasedonsmallnumbersofpatients;however, itraisesthepossibility that the underlying biology and significance of a KIT mutationare different in different histopathologic subtypes of melanoma.
Although responses to TKI therapy in KIT-associated melanomaare measurable, tumors progress in approximately 3 to 4 months.Mechanisms of resistance to imatinib in KIT-mutated melanoma arenot well established. There have been reports of primary resistance inpatients with additional NRAS mutations (also reported in one case ofsunitinib resistance)30 and a case of increased KIT copy number atonset of secondary resistance.6 The development of secondary muta-tions in KIT is associated with acquired imatinib resistance in KIT-mutated gastrointestinal stromal tumor,32 and sequential therapywith other TKIs after progression can be effective.33,34 Data fromKIT-mutated melanoma cell lines have demonstrated acquisition ofsecondary mutations as a cause of drug resistance.35 Other TKIs in-cluding sunitinib have resulted in responses in KIT-mutated mela-noma,30 and there is an ongoing phase II trial specifically assessing theefficacy of nilotinib after resistance or intolerance to prior KIT-inhibitor therapy (NCT00788775).
Clinical trials of the anti–CTLA-4 (ipilimumab),36,37 anti-PD1,38
and anti-PDL139 checkpoint inhibitors have not specifically excludedKIT-mutatedmelanoma; thus, suchpatients shouldbeconsideredcandi-dates for these treatments, unless new evidence to the contrary becomesavailable. Recent studies of ipilimumab in mucosal and acral melanomashave shown evidence of efficacy and safety,40-43 with a DCR of 35% and acomplete response in pretreated mucosal melanoma in one study.43
SUGGESTED APPROACHES TO MANAGEMENT
Who Should Undergo KIT Testing, and What KIT
Testing Should Be Performed?
It is standard practice for patients with newly diagnosed stageIIIC unresectable or stage IV melanoma to have V600 BRAF mutation
Table 2. Summary of Phase II Trials of Imatinib in KIT-Mutated Metastatic Melanoma
TrialNo. of
Patients�
Melanoma
Imatinib Dose (mg)
ORR DCRMedian TTP
(months)Median OS(months)Subtype No. % No. % No. %
Carvajal et al5 25 Mucosal 13 46 400 twice per day 4 16 — — 2.8 10.6Acral 10 36CSD 5 18
Guo et al31 43 Mucosal 11 26 400 per day (increased to 400twice per day at PD)
10 23 23 55 3.5† 14Acral 21 49CSD 5 12Non-CSD 4 9UP 2 5
Hodi et al6 24 Mucosal 17 71 400 per day (increased to 400twice per day at PD)
7 29‡ 12 50§ 3.7 12.5Acral 6 25CSD 1 4
Abbreviations: CSD, chronic sun damage; DCR, disease control rate; ORR, overall response rate; OS, overall survival; PD, progressive disease; TTP, time toprogression; UP, unknown primary.
�No. evaluable for response.†Progression-free survival.‡ORR � 54% for KIT mutated and 0% for amplified.§DCR � 77% for KIT mutated and 18% for amplified.
Lyle and Long
3178 © 2013 by American Society of Clinical Oncology JOURNAL OF CLINICAL ONCOLOGY

testing performed on the most recently biopsied melanoma tissue ifavailable. If recent metastatic tissue is not available, other metastaticsites (eg, lymph nodes) or the primary tumor may be used becausethere is little discordance in BRAF and NRAS mutation profiles be-tween primary and metastatic sites.44,45 We are not aware of anyspecific evidence suggesting discordance of KIT mutation betweenmetastases and primary melanomas.
If the patient is BRAF mutation negative and has mucosal or acralmelanoma, he or she should undergo KIT mutation testing of exons 9,11, 13, 17, and 18 (Fig 2) because the pretest probability of a KITmutation is higher in these subgroups. In time, fewer KIT exons mayneed to be sequenced because evidence may show that some muta-tions are not TKI responsive. BRAF mutation–negative patientswith an unknown primary site should also undergo KIT mutationtesting, although recent evidence suggests that the molecular pro-file of these tumors is most consistent with cutaneous melanomarather then occult mucosal or ocular melanoma.46 Some centersroutinely test for KIT mutations in patients with BRAF mutation–negative cutaneous melanoma with evidence of CSD. In Australia,we do not because of our high prevalence of CSD and low pretestprobability. Our experience and published data3 suggest that in theAustralian population, KIT mutations are infrequent in cutaneousmelanoma irrespective of the degree of sun exposure. Testing forKIT amplification is not performed routinely, because it does notinfluence management.
At our institution and many other tertiary melanoma referralcenters, patients have their melanoma tissue tested for a panel ofcancer-related somatic mutations when first diagnosed with meta-static melanoma. Platforms using next-generation sequencing or Se-quenom (San Diego, CA)47 screen for a wider range of mutations,including but not limited to BRAF, NRAS, and KIT mutations (limitedhotspots in Sequenom). This decreases the number of patients requir-ing full KIT sequencing of exons 9, 11, 13, 17, and 18 because patientswith NRAS mutations or the most common KIT mutations havealready been identified. If the patient does not have a BRAF, NRAS,or common KIT mutation and has mucosal, acral, or an unknownprimary melanoma, full KIT sequencing is performed as previ-ously described.
Treatment of KIT Mutation–Positive
Metastatic Melanoma
The development of evidence-based guidelines for the manage-ment of KIT-mutated melanoma has been hampered by the smallpatient population and limited data available. However, the recentupdate of the National Comprehensive Cancer Network melanomaguidelines48 now references the use of imatinib in the setting of KIT-mutated metastatic melanoma.
We recommend that patients with KIT mutation–positive met-astatic melanoma be considered for clinical trials of targeted therapies(eg, KIT inhibitors) and/or immunotherapies (eg, PD1/PDL1 anti-bodies) if possible (Fig 2). If a clinical trial is not available, we wouldconsider off-label use of imatinib or use of ipilimumab. There is nowsome evidence for efficacy of ipilimumab in mucosal melanoma, asubtype enriched for KIT mutations,40-43 and there are no data tosuggest that a KIT-mutated tumor would not respond to immuneagents. In patients with a poor performance status resulting frommelanoma or a rapid rate of progression, our preference would be touse imatinib, with the expectation of a more rapid response to treat-ment. Alternatively, in the setting of patients with a good performancestatus and slowly progressing melanoma, we would choose ipili-mumab, which may result in a more durable response. Imatinib couldbe used second line after ipilimumab if needed. Given the short me-dian progression-free survival reported in the three phase II studies ofimatinib, if the performance status of a patient who had commencedimatinib treatment improved significantly, a strategy of imatinib in-terruption and treatment with ipilimumab could be considered, al-though there is no evidence supporting this.
In Australia, for example, imatinib is currently available if thepatient is able to pay for it or via compassionate access, so it is thereforenot always a reliable option. Unless an OS benefit is demonstrated, thisis unlikely to change. A meta-analysis of individual patient data fromphase II studies may provide more evidence for funding and regula-tory bodies to consider. Chemotherapy, such as with dacarbazine oroff-label use of nab-paclitaxel (Abraxane; Celgene, Summit, NJ), is anoption that we reserve for those patients who are unable to accessclinical trials, targeted therapies, or immunotherapies.
The patient described in the case presentation was enrolled ontoa phase II clinical trial of nilotinib in KIT-mutated metastatic acral or
Stage IIIcunresectable
or IV melanoma
BRAFmutation
test*
BRAFmutationpositive
BRAFmutationnegative
Acral/mucosal/unknown primary
KIT exon9,11,13,17,18sequencing†
KITmutationpositive
KITmutationnegative
Noteligiblefor trial
Imatinibor
Ipilimumab||
Eligiblefor clinical
trial‡
CutaneousCSD
Consider KITsequencing§
Fig 2. Algorithm for diagnosis and management of KIT-mutated metastatic melanoma. CSD, chronic sun damage. (*) Some centers perform somatic mutation panels,which may identify NRAS mutation or common KIT mutation and bypass the need for KIT sequencing. (†) Number of exons to sequence may be reduced as additionaldata become available on which mutations are responsive to KIT inhibitor therapy (eg, in phase II studies of imatinib, exons 11 and 13 mutations were most responsive).(‡) Patients should not be limited to trials for KIT-mutated melanoma only; they may be eligible for trials of immunotherapy (eg, PD-1 antibody) or combination therapy.(§) Not standard practice in Australia; more data are required on the significance of CSD. (�) Alternative options (in cases of disease progression or treatment intolerance)include other tyrosine kinase inhibitors (eg, sunitinib) or chemotherapy (eg, dacarbazine or nab-paclitaxel).
KIT-Mutant Melanoma
www.jco.org © 2013 by American Society of Clinical Oncology 3179

mucosal melanoma (NCT01028222). Although he did not have anobjective response to treatment, his lung metastasis remained stable,and he had local tumor progression on the foot that was resected. Hetolerated the nilotinib well and continued on study until RECISTtumor progression at 12 months. Second-line immunotherapy op-tions are under consideration.
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTSOF INTEREST
Although all authors completed the disclosure declaration, the followingauthor(s) and/or an author’s immediate family member(s) indicated afinancial or other interest that is relevant to the subject matter underconsideration in this article. Certain relationships marked with a “U” arethose for which no compensation was received; those relationships marked
with a “C” were compensated. For a detailed description of the disclosurecategories, or for more information about ASCO’s conflict of interest policy,please refer to the Author Disclosure Declaration and the Disclosures ofPotential Conflicts of Interest section in Information for Contributors.Employment or Leadership Position: None Consultant or AdvisoryRole: Georgina V. Long, Amgen (C), Bristol-Myers Squibb (C),GlaxoSmithKline (C), Novartis (C), Roche (C) Stock Ownership: NoneHonoraria: Georgina V. Long, Roche Research Funding: None ExpertTestimony: None Patents: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Financial support: Georgina V. LongProvision of study materials or patients: Georgina V. LongManuscript writing: All authorsFinal approval of manuscript: All authors
REFERENCES
1. Curtin JA, Busam K, Pinkel D, et al: Somaticactivation of KIT in distinct subtypes of melanoma.J Clin Oncol 24:4340-4346, 2006
2. Beadling C, Jacobson-Dunlop E, Hodi FS, etal: KIT gene mutations and copy number in mela-noma subtypes. Clin Cancer Res 14:6821-6828,2008
3. Handolias D, Salemi R, Murray W, et al:Mutations in KIT occur at low frequency in melano-mas arising from anatomical sites associated withchronic and intermittent sun exposure. Pigment CellMelanoma Res 23:210-215, 2010
4. Kong Y, Si L, Zhu Y, et al: Large-scale analysisof KIT aberrations in Chinese patients with mela-noma. Clin Cancer Res 17:1684-1691, 2011
5. Carvajal RD, Antonescu CR, Wolchock JD, etal: KIT as a therapeutic target in metastatic mela-noma. JAMA 305:2327, 2011
6. Hodi FS, Corless C, Giobbie-Hurder A, et al:Imatinib for melanomas harboring mutationally acti-vated or amplified KIT arising on mucosal, acral, andchronically sun damaged skin. J Clin Oncol 31:3182-3190, 2013
7. Postow MA, Carvajal RD: Therapeutic impli-cations of KIT in melanoma. Cancer J 18:137-141,2012
8. Hodis E, Watson IR, Kryukov GV, et al: Alandscape of driver mutations in melanoma. Cell150:251-263, 2012
9. Long GV, Menzies AM, Nagrial AM, et al:Prognostic and clinicopathologic associations of on-cogenic BRAF in metastatic melanoma. J Clin Oncol29:1239-1246, 2011
10. Lee JH, Choi JW, Kim YS: Frequencies ofBRAF and NRAS mutations are different in histolog-ical types and sites of origin of cutaneous mela-noma: A meta-analysis. Br J Dermatol 164:776-784,2011
11. Jakob JA, Bassett RL Jr, Ng CS, et al: NRASmutation status is an independent prognostic factorin metastatic melanoma. Cancer 118:4014-4023,2011
12. Devitt B, Liu W, Salemi R, et al: Clinicaloutcome and pathological features associated withNRAS mutation in cutaneous melanoma. PigmentCell Melanoma Res 24:666-672, 2011
13. Van Raamsdonk CD, Bezrookove V, Green G,et al: Frequent somatic mutations of GNAQ in uvealmelanoma and blue naevi. Nature 457:599-602,2008
14. Van Raamsdonk CD, Griewank KG, CrosbyMB, et al: Mutations in GNA11in uveal melanoma.N Engl J Med 363:2191-2199, 2010
15. COSMIC: Catalogue of somatic mutations incancer database. http://www.sanger.ac.uk/genetics/CGP/cosmic
16. Grichnik, J: Kit and melanocyte migration.J Invest Dermatol 126:945-947, 2006
17. Landi MT, Bauer J, Pfeiffer RM, et al: MC1Rgermline variants confer risk for BRAF-mutant mel-anoma. Science 313:521-522, 2006
18. Woodman SE, Lazar AJ, Aldape KD, et al:New strategies in melanoma: Molecular testing inadvanced disease. Clinical Cancer Res 18:1195-1200, 2012
19. Torres-Cabala CA, Wang WL, Trent J, et al:Correlation between KIT expression and KIT muta-tion in melanoma: A study of 173 cases with em-phasis on the acral-lentiginous/mucosal type. ModPathol 22:1446-1456, 2009
20. Heinrich MC, Griffith DJ, Druker BJ, et al:Inhibition of c-kit receptor tyrosine kinase activity bySTI 571, a selective tyrosine kinase inhibitor. Blood96:925-932, 2000
21. Kim KB, Eton O, Davis DW, et al: Phase II trialof imatinib mesylate in patients with metastaticmelanoma. Br J Cancer 99:734-740, 2008
22. Ugurel S, Hildenbrand R, Zimpfer A, et al: Lackof clinical efficacy of imatinib in metastatic mela-noma. Br J Cancer 92:1398-1405, 2005
23. Wyman K, Atkins MB, Prieto V, et al: Multi-center phase II trial of high-dose imatinib mesylatein metastatic melanoma. Cancer 106:2005-2011,2006
24. Handolias D, Hamilton AL, Salemi R, et al:Clinical responses observed with imatinib orsorafenib in melanoma patients expressing muta-tions in KIT. Br J Cancer 102:1219-1223, 2010
25. Hodi FS, Friedlander P, Corless CL, et al:Major response to imatinib mesylate in KIT-mutatedmelanoma. J Clin Oncol 26:2046-2051, 2008
26. Quintas-Cardama A, Lazar AJ, Woodman SE,et al: Complete response of stage IV anal mucosalmelanoma expressing KIT Val560Asp to the multiki-nase inhibitor sorafenib. Nat Clin Prac Oncol 5:737-740, 2008
27. Kalinsky K, Lee SJ, Lawrence DP, et al: Aphase II trial of dasatinib in patients with unresect-able locally advanced or stage IV mucosal, acral, andsolar melanomas: An Eastern Cooperative OncologyGroup study (E2607). J Clin Oncol 30:545s, 2012(suppl; abstr 8522)
28. Woodman SE, Trent JC, Stemke-Hale K, et al:Activity of dasatinib against L576P KIT mutant mel-anoma: Molecular, cellular, and clinical correlates.Mol Cancer Ther 8:2079-2085, 2009
29. Antonescu CR, Busam KJ, Francone TD, et al:L576P KIT mutation in anal melanomas correlateswith KIT protein expression and is sensitive tospecific kinase inhibition. Int J Cancer 121:257-264,2007
30. Minor DR, Kashani-Sabet M, Garrido M, et al:Sunitinib therapy for melanoma patients with KITmutations. Clin Cancer Res 18:1457-1463, 2012
31. Guo J, Si L, Kong Y, et al: Phase II, open-label,single-arm trial of imatinib mesylate in patients withmetastatic melanoma harboring c-KIT mutation oramplification J Clin Oncol 29:2904-2909, 2011
32. Antonescu CR, Besmer P, Guo T, et al: Ac-quired resistance to imatinib in gastrointestinal stromaltumor occurs through secondary gene mutation. ClinCancer Res 11:4182-4190, 2005
33. Prenen H, Cools J, Mentens N, et al: Efficacyof the kinase inhibitor SU11248 against gastrointes-tinal stromal tumor mutants refractory to imatinibmesylate. Clinical Cancer Res 12:2622-2627, 2006
34. Demetri GD, van Oosterom AT, Garrett CR, etal: Efficacy and safety of sunitinib in patients withadvanced gastrointestinal stromal tumour after fail-ure of imatinib: A randomised controlled trial. Lancet368:1329-1338, 2006
35. Todd JR, Becker TM, Kefford RF, et al: Sec-ondary c-Kit mutations confer acquired resistance toRTK inhibitors in c-Kit mutant melanoma cells. Pig-ment Cell Melanoma Res 26:518-526, 2013
36. Robert C, Thomas L, Bondarenko I, et al:Ipilimumab plus dacarbazine for previously un-treated metastatic melanoma. N Engl J Med 364:2517-2526, 2011
37. Hodi FS, O’Day SJ, McDermott DF, et al:Improved Survival with Ipilimumab in Patients withMetastatic Melanoma. N Engl J Med 363:711-723,2010
38. Topalian SL, Hodi FS, Brahmer JR, et al:Safety, activity, and immune correlates of anti-PD-1antibody in cancer. N Engl J Med 366:2443-2454,2012
39. Brahmer JR, Tykodi SS, Chow LQ, et al:Safety and activity of anti-PD-L1 antibody in patientswith advanced cancer. N Engl J Med 366:2455-2465, 2012
40. Lawrence D, McDermott DF, Hamid O, et al:Ipilimumab (Ipi) expanded access program (EAP) forpatients (pts) with stage III/IV melanoma: Safetydata by subgroups. Ann Oncol 23:368, (abstr)
Lyle and Long
3180 © 2013 by American Society of Clinical Oncology JOURNAL OF CLINICAL ONCOLOGY

41. Postow MA, Carvajal, R: The efficacy of ipili-mumab for patients with mucosal melanoma. Pig-ment Cell Melanoma Res 25:880, 2012 (abstr)
42. Shaw H, Larkin J, Corrie P, et al: Ipilimumabfor advanced melanoma in an expanded accessprogramme (EAP): Ocular, mucosal and acral sub-type UK experience. Ann Oncol 23:374, 2012 (abstr)
43. Del Vecchio M, Simeone E, Chiarion Seleni V:Efficacy and safety of ipilimumab in patients with pre-treated, mucosal melanoma: Experience from italian clin-ics participating in the European Expanded AccessProgramme (EAP). Ann Oncol 23:368, 2012 (abstr)
44. Omholt K, Platz A, Kanter L, et al: NRAS andBRAF mutations arise early during melanomapathogenesis and are preserved throughout tu-mor progression. Clin Cancer Res 9:6483-6488,2003
45. Colombino M, Capone M, Lissia A, et al:BRAF/NRAS mutation frequencies among primarytumors and metastases in patients with melanoma.J Clin Oncol 30:2522-2529, 2012
46. Dutton-Regester K, Kakavand H, Aoude,L,et al: Melanomas of unknown primary have amutation profile consistent with cutaneous sun
exposed melanoma. Pigment Cell Melanoma Res25:852, 2012 (abstr)
47. Sequenom: Genetic analysis. http://www.sequenom.com/genetic-analysis
48. Coit DG, Andtbacka R, Anker CJ, et al: Mela-noma, version 2.2013: Featured updates to theNCCN guidelines. J Natl Compr Canc Netw 11:395-407, 2013
DOI: 10.1200/JCO.2013.50.4662; publishedonline ahead of print at www.jco.org onAugust 12, 2013
■ ■ ■
Be the First to Hear When New Clinical Cancer Research Is Published Online
By signing up for JCO’s Early Release Notification, you will be alerted and have access to new articles posted online everyMonday, weeks before they appear in print. All Early Release articles are searchable and citable, and are posted on jco.orgin advance of print publication. Simply go to jco.org/earlyrelease, sign in, select “Early Release Notification,” and click theSUBMIT button. Stay informed—sign up today!
KIT-Mutant Melanoma
www.jco.org © 2013 by American Society of Clinical Oncology 3181

Acknowledgment
Supported by Melanoma Institute Australia.
Lyle and Long
© 2013 by American Society of Clinical Oncology JOURNAL OF CLINICAL ONCOLOGY





![Case Report A Rare Case of Metastatic Malignant Melanoma ...downloads.hindawi.com/journals/crigm/2014/312902.pdf · metastatic malignant melanoma of the GI tract [ ]. In fact, Wysocki](https://img.dokumen.tips/doc/110x75/5f9b841cf1457c0af634448c/case-report-a-rare-case-of-metastatic-malignant-melanoma-metastatic-malignant.jpg)























