Embed Size (px)
Citation preview
Det virtuelle kjemilaboratoriet
Trygve Helgaker
Centre for Theoretical and Computational Chemistry
Kjemisk institutt, Universitetet i Oslo
Etterutdanningskurs for lærere i Oslo kommune
Skolelaboratoriet, Kjemisk institutt, Universitetet i Oslo
Onsdag 10. oktober 2007
1

Eksperimentell kontra teoretisk kjemi
• Kjemi er en eksperimentell vitenskap!
• En teoretisk beregning (dvs. teoretisk kjemi) gir bare tall, ingen forstaelse!
“Every attempt to employ mathematical methods in the study of chemical
questions must be considered profoundly irrational. If mathematical analysis should
ever hold a prominent place in chemistry—an aberration which is happily
impossible—it would occasion a rapid and widespread degradation of that science.”
August Comte, 1748–1857
• Kvantekjemi er tuftet pa en dyp forstaelse av kjemiske systemer!
“The more progress sciences make, the more they tend to enter the domain of
mathematics, which is a kind of center to which they all converge. We may even
judge the degree of perfection to which a science has arrived by the facility with
which it may be submitted to calculation.”
Adolphe Quetelet, 1796–1874
2

Litt historikk
• partikler: atomteori (Dalton, 1808)
molekyler (Avogadro, 1811)
elektronet (Thomson, 1897)
fotoner (Einstein, 1905)
atomkjerner (Rutherford, 1911)
• kvantemekanikk: kvantisering (Planck, 1900)
Bohrs atommodell (1913)
partikkelbølger (de Broglie, 1924)
eksklusjonsprinsippet (Pauli, 1925)
bølgemekanikk: Schrodinger-ligningen (1926)
Heisenbergs usikkerhetsrelasjon (1927)
elektronspinn (Pauli, 1927)
relativistisk kvantemekanikk: Dirac-ligningen (1928)
• kvantekjemi: Lewis’ valensbindingsteori (1916)
kjerneseparasjon (Born–Oppenheimer, 1927)
hydrogenmolekylet (Heitler–London, 1927)
molekylorbitaler (Hund–Mulliken, 1927)
heliumatomet (Hylleraas, 1929)
Slater-determinanter (Slater, 1929)
3

Molekyler og kvantemekanikk
• Et molekyl er en samling N partikler med ulik masse og ladning holdt sammen av
elektrostatiske krefter.
• Partiklenes oppførsel (deres tilstand) er beskrevet av bølgefunksjonen—en en
matematisk funksjon av 4N romkoordinater og tiden:
Ψ = Ψ(x1, y1, z1, s1, x2, y2, z2, s2, . . . xN , yN , zN , sN , t)
• Bølgefunksjonen er en løsning av en differensialligning: Schrodinger-ligningen. I det
stasjonære tilfellet kan denne ligningen skrives pa formen
HΨ = EΨ
der den spinn- og feltfrie Hamilton-operatoren er gitt ved
H = −
X
i
h2
2mi
„
∂2
∂x2i
+∂2
∂y2i
+∂2
∂z2i
«
+e2
4πε0
X
i>j
ZiZj
rij
• Kvantisering: grensebetingelser (Ψ er kontinuerlig og gar mot null uendelig langt borte)
gir generelt kun løsninger for visse energier.
• Bølgefunksjonen inneholder all informasjon om systemet!
4
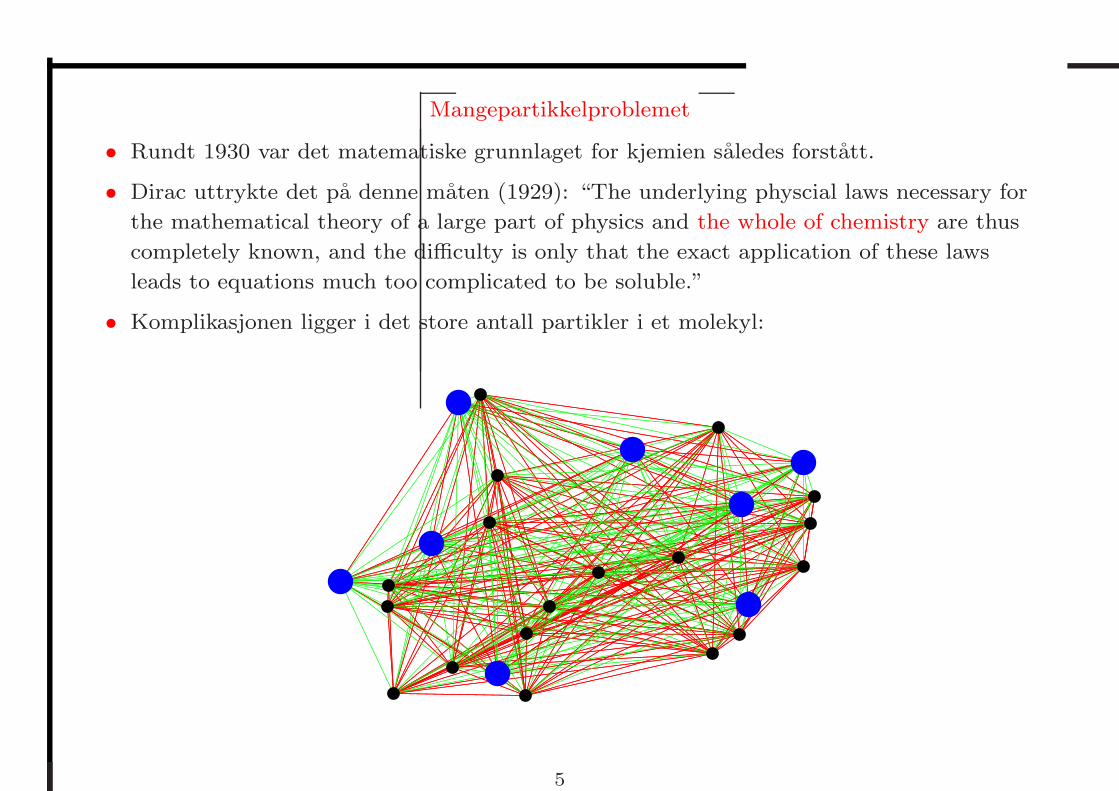
Mangepartikkelproblemet
• Rundt 1930 var det matematiske grunnlaget for kjemien saledes forstatt.
• Dirac uttrykte det pa denne maten (1929): “The underlying physcial laws necessary for
the mathematical theory of a large part of physics and the whole of chemistry are thus
completely known, and the difficulty is only that the exact application of these laws
leads to equations much too complicated to be soluble.”
• Komplikasjonen ligger i det store antall partikler i et molekyl:
5

Regnemaskinen—kvantekjemiens verktøy
• Hjelpen kom fra uventet hold, med utviklingen av den elektroniske regnemaskinen etter
annen verdenskrig:
ENIAC (Electronic Numerical Integrator and Computer) (1946)
• De siste 50 arene har datamaskinen gjennomgatt en eventyrlig utvikling, uttrykt i
Gordon Moore’s lov (1964):
Datamaskinens kapasitet dobles hver attende maned uten kostnadsøkning.
• En regnemaskin er i dag 10000 ganger raskere enn for 25 ar siden!
• Dette er en utvikling som ingen kunne forutse pa 30-tallet, og som har ført til at
kvantekjemiske beregninger i dag er blitt rutine.
• Mer generelt er Computational Science blitt et viktig fagfelt.
• I 1995 utgjorde beregninger ca. 15% av all kjemisk forskning. Denne andelen vokser med
ca. 1% i aret (H. Schaefer).
• Denne utviklingen skyldes at flere og flere ikke-teoretikere gjør teoretiske beregninger.
• Veksten vil sannsynligvis stagnere rundt 2035, nar halvparten av all kjemi vil være
eksperimentell og halvparten beregninger.
6

Moores lov
7

Eksakte og approksimative beregninger
• Selv om datamaskinene er blitt meget raske, kan vi i praksis ikke løse
Schrodinger-ligningen eksakt.
– Vi ma isteden gjøre forenklinger—dvs. lage gode beregningsmodeller
som tar hensyn til de viktigste effektene.
– Dette ma fortrinnsvis gjøres pa en ordnet mate, slik at beregningene
kan forbedres systematisk mot den eksakte løsningen.
• Slik etableres et hierarki av approksimasjoner—dvs. et system av stadig
mer nøyaktige og kostbare beregningsmodeller.
– Selv om vi saledes i prinsippet kan løse Schrodinger-ligningen eksakt,
løser vi den i praksis approksimativt, men pa en kontrollert mate.
– Da studentene er vant til eksakte (dvs. riktige eller gale) løsninger fra
skolen og studiene, finner de ofte denne bruken av modeller og
tilnærmede løsninger vanskelig i begynnelsen.
8

Born–Oppenheimer approksimasjonen: molekylets potensialflate
• Elektronene er lette, mens kjernene er tunge
– elektronene beveger seg raskt, kjerne sakte
• Løser Schrodinger-ligningen (eller Dirac-ligningen) for fastholdte kjerner:
H(R)Ψ(ri) = E(R)Ψ(ri)
– minima av E(R) svarer til stabile systemer, sadelpunkter til transition states, osv.
• Kjemien styres av elektronenes bevegelser eller “tilstand”
9

Born–Oppenheimer approksimasjonen: diatomiske potensialkurver
• Vi ma bestemme elektronenes energi som funksjon av kjernenes koordinater
H(R)Ψn(r1, r2; R) = En(R)Ψn(r1, r2; R)
2 4 6 8
-1.2
-1.0
-0.8
-0.6
-0.4
-0.2
0.2
– den elektroniske energi En(R) som funksjon av kjerneavstanden R i H2
• Noen tilstander er bindende, andre antibindende
– grunntilstanden har lavest energi og er den viktigste for kjemien
10

Elektroniske tilstander i H2
˛
˛
1Σ+g
¸
= 0.9939˛
˛1σ2g
¸
− 0.1106˛
˛1σ2u
¸
grunntilstanden˛
˛
1Σ+g
¸
= 0.1106˛
˛1σ2g
¸
+ 0.9939˛
˛1σ2u
¸
eksitert tilstand
-2 -1 0 1 2
0.5
-2-1 0 1 2
-2-1012
0.00
0.15
-2-1 0 1 2
-2 -1 0 1 2
-2
-1
0
1
2
-2 -1 0 1 2
0.5
-2-1 0 1 2
-2-1012
0.00
0.04
-2-1 0 1 2
-2 -1 0 1 2
-2
-1
0
1
2
11

Elektroniske tilstander i H2
• Eksiterte tilstander med apne skall 1σ1g1σ1
u:
–
˛
˛
˛
3Σ+u
E
(parallelle spinn, over) og˛
˛
˛
1Σ+u
E
(motsatt spinn, under)
-2 -1 0 1 2
0.5
-2-1 0 1 2
-2-1012
0.00
0.11
-2-1 0 1 2
-2 -1 0 1 2
-2
-1
0
1
2
-2 -1 0 1 2
0.5
-2-1 0 1 2
-2-1012
0.00
0.11
-2-1 0 1 2
-2 -1 0 1 2
-2
-1
0
1
2
12

Hartree–Fock-approksimasjonen
• Elektronenes bevegelse er meget komplisert
– hvert elektrons bevegelse er pavirket av alle andre elektroner
• I første omgang kan vi se bort ifra denne “elektronkorrelasjonen”
– for hvert elektron tar vi kun hensyn til den midlere effekten av alle
andre elektroner
– et elektron pavirkes saledes ikke av hvor de andre elektronene faktisk er,
men hvor de pleier a være
• Dette gir opphav til begrepet molekylorbitaler
– typisk feil pa 0.5% i totalenergien, 1% i struktur og 5% i andre
egenskaper som kraftkonstanter, skjermingskonstanter og
dipolmomenter.
– kan anvendes pa store systemer (mange hundre atomer)
13
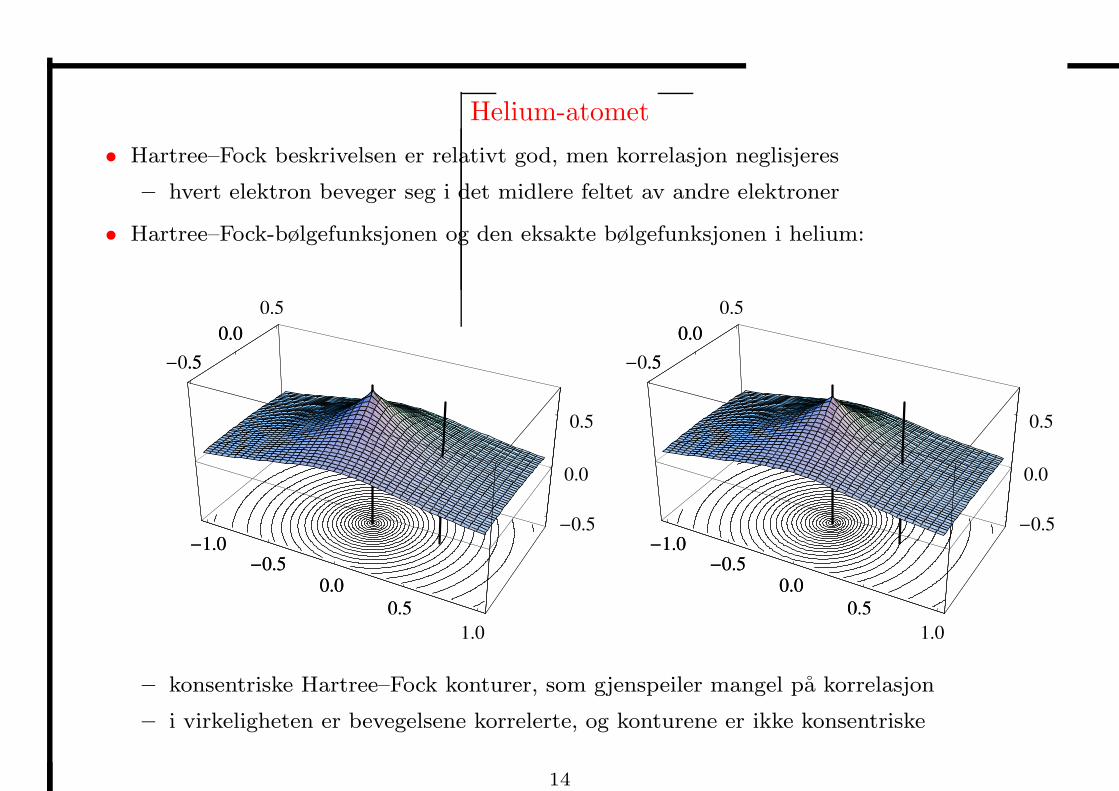
Helium-atomet
• Hartree–Fock beskrivelsen er relativt god, men korrelasjon neglisjeres
– hvert elektron beveger seg i det midlere feltet av andre elektroner
• Hartree–Fock-bølgefunksjonen og den eksakte bølgefunksjonen i helium:
-1.0-0.5
0.0
0.5
1.0
-0.5
0.0
0.5
-0.5
0.0
0.5
-1.0-0.5
0.0
0.5
-0.5
0.0
-
-1.0-0.5
0.0
0.5
1.0
-0.5
0.0
0.5
-0.5
0.0
0.5
-1.0-0.5
0.0
0.5
-0.5
0.0
-
– konsentriske Hartree–Fock konturer, som gjenspeiler mangel pa korrelasjon
– i virkeligheten er bevegelsene korrelerte, og konturene er ikke konsentriske
14
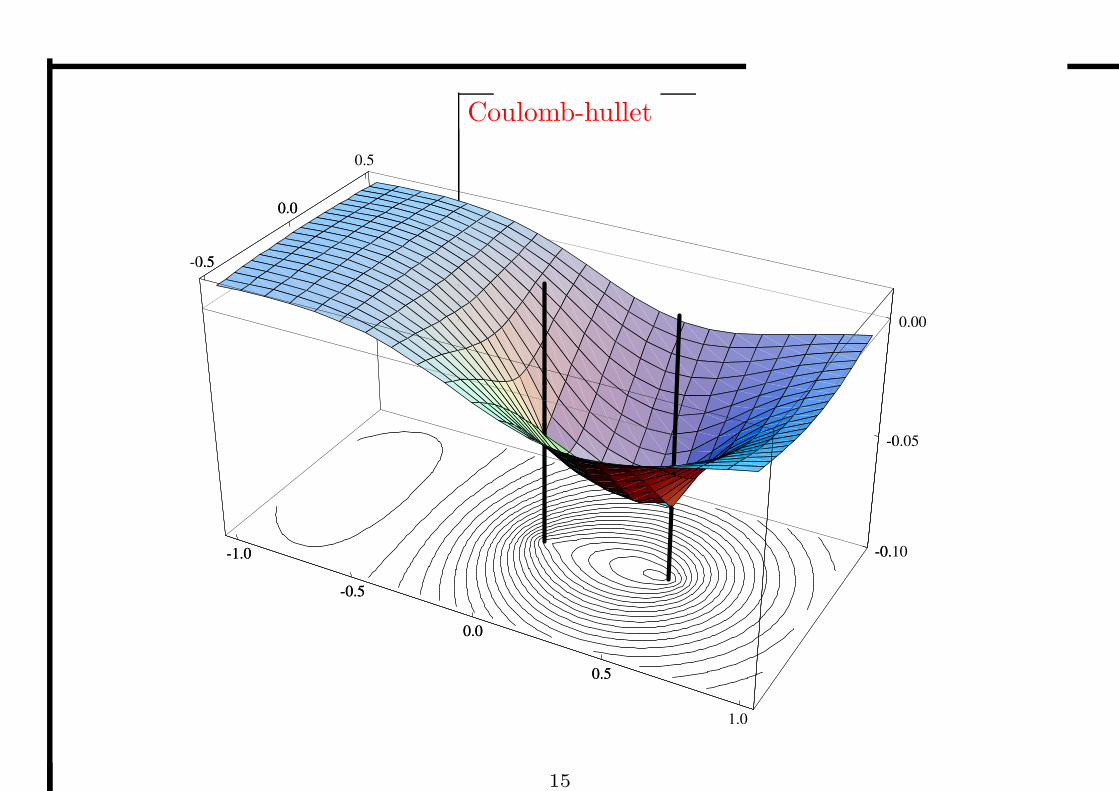
Coulomb-hullet
-1.0
-0.5
0.0
0.5
1.0
-0.5
0.0
0.5
-0.10
-0.05
0.00
-1.0
-0.5
0.0
0.5
-0.5
0.0
-0.10
-0.05
15

Elektronkorrelasjon og virtuelle eksitasjoner
• For a korrigere HF-modellen ma vi ta hensyn til de øyeblikkelige vekselvirkningene
mellom elektronene—elektronkorrelasjon:
– I rommet kan vi forestille oss at elektronene stadig kolliderer med hverandre og spres.
– I orbitalbildet manifesterer disse kollisjonene seg som eksitasjoner fra okkuperte til
virtuelle (ikke-okkuperte) spinnorbitaler.
• Dobbel-eksitasjoner:
– Den viktigste prosessen er kollisjoner mellom to elektroner.
– I orbitalbildet skjer dette ved at elektronene eksiteres fra to okkuperte til to virtuelle
spinnorbitaler. Dette kalles en par-eksitasjon eller dobbel-eksitasjon.
– Med hver slik eksitasjon assosierer vi en amplitude, som uttrykker sannsynligheten
for prosessen.
– Vi sier at beskrivelsen er korrelert.
• CCSD-modellen:
– Ved a la alle mulige enkel- og dobbel-eksitasjoner forega i alle mulige kombinasjoner
far vi coupled-cluster singles-and-doubles (CCSD) modellen.
– I CCSD-modellen er HF-feilen redusert typisk med en faktor tre eller fire.
– For høyere nøyaktighet ma trippel-eksitasjoner (CCSDT) tas med.
16
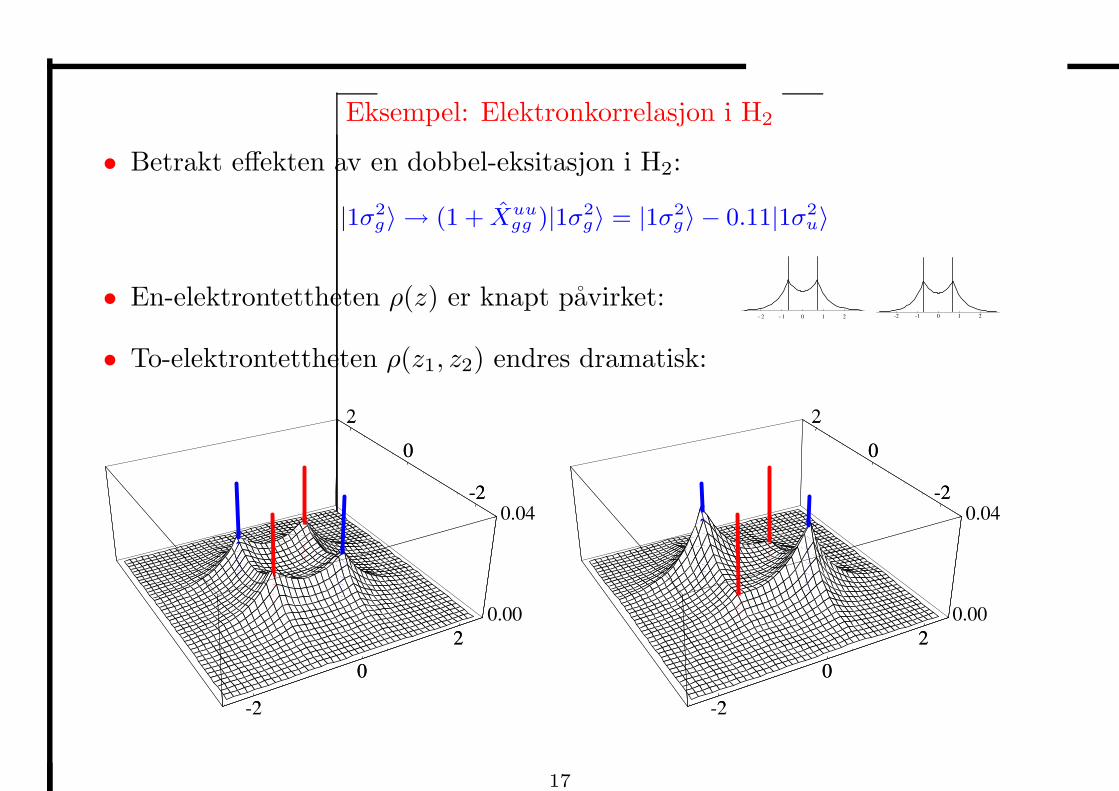
Eksempel: Elektronkorrelasjon i H2
• Betrakt effekten av en dobbel-eksitasjon i H2:
|1σ2g〉 → (1 + Xuu
gg )|1σ2g〉 = |1σ2
g〉 − 0.11|1σ2u〉
• En-elektrontettheten ρ(z) er knapt pavirket:-2 -1 0 1 2 -2 -1 0 1 2
• To-elektrontettheten ρ(z1, z2) endres dramatisk:
-2
0
2
-2
0
2
0.00
0.04
-2
0
2
-2
0
2
-2
0
2
-2
0
2
0.00
0.04
-2
0
2
-2
0
2
17

Korrelasjonskonvergens: bindingsavstander (pm)
HF 2-el. 3-el. 4-el. 5-el. rel. teori eksp. feil
HF 89.70 1.67 0.29 0.02 0.00 0.01 91.69 91.69 0.00
N2 106.54 2.40 0.67 0.14 0.03 0.00 109.78 109.77 0.01
F2 132.64 6.04 2.02 0.44 0.03 0.05 141.22 141.27 −0.05
CO 110.18 1.87 0.75 0.04 0.00 0.00 112.84 112.84 0.00
• Den ukorrelerte HF-modellen gir en god (kvalitativ) beskrivelse
– HF underestimerer bindingsavstanden med opp til 8.6 pm (for F2)
• Beskrivelse av elektronkorrelasjon forbedrer resultatene
– alle korrelasjonsbidrag er positive
– tilnærmet lineær konvergens, langsomst for F2
– overensstemmelse med eksperiment til 0.01 pm eller bedre unntatt for F2
– beregningenes kostnader øker hurtig med eksitasjonsniva:
HF: n4; CCSD: n6; CCSDT: n7; CCSDTQ: n8
• Relativistiske korreksjoner er sma unntatt for F2 (0.05 pm)
18
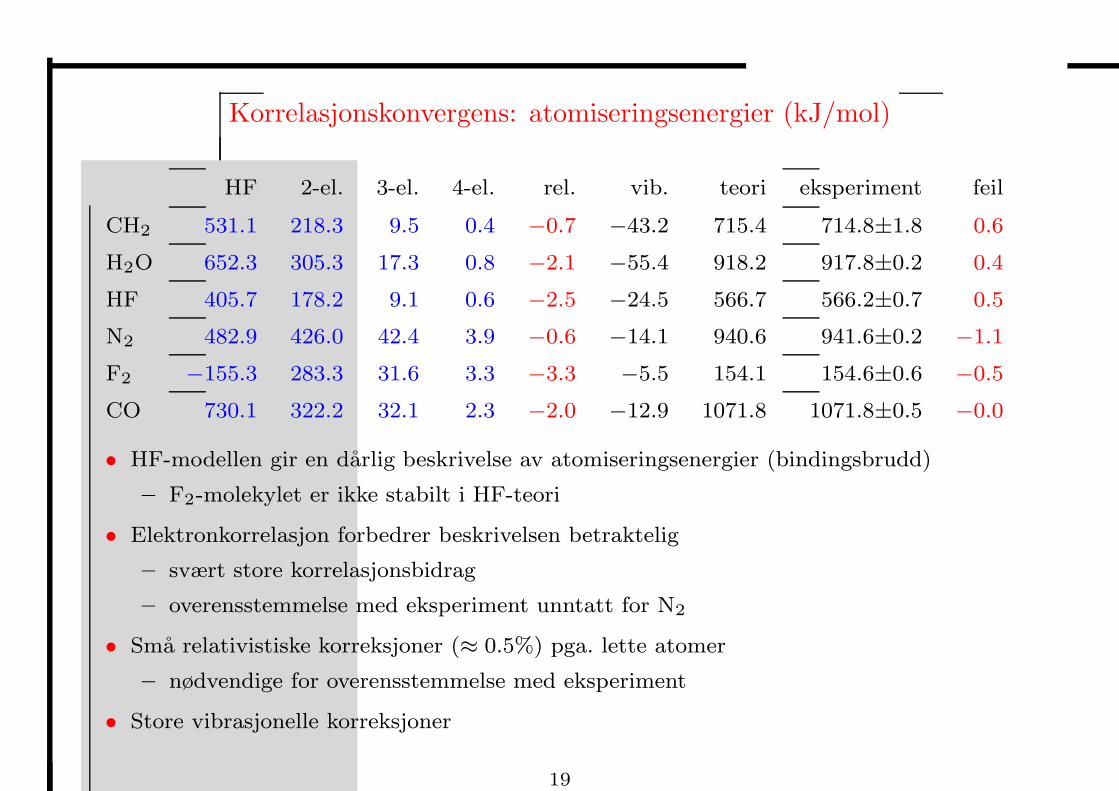
Korrelasjonskonvergens: atomiseringsenergier (kJ/mol)
HF 2-el. 3-el. 4-el. rel. vib. teori eksperiment feil
CH2 531.1 218.3 9.5 0.4 −0.7 −43.2 715.4 714.8±1.8 0.6
H2O 652.3 305.3 17.3 0.8 −2.1 −55.4 918.2 917.8±0.2 0.4
HF 405.7 178.2 9.1 0.6 −2.5 −24.5 566.7 566.2±0.7 0.5
N2 482.9 426.0 42.4 3.9 −0.6 −14.1 940.6 941.6±0.2 −1.1
F2 −155.3 283.3 31.6 3.3 −3.3 −5.5 154.1 154.6±0.6 −0.5
CO 730.1 322.2 32.1 2.3 −2.0 −12.9 1071.8 1071.8±0.5 −0.0
• HF-modellen gir en darlig beskrivelse av atomiseringsenergier (bindingsbrudd)
– F2-molekylet er ikke stabilt i HF-teori
• Elektronkorrelasjon forbedrer beskrivelsen betraktelig
– svært store korrelasjonsbidrag
– overensstemmelse med eksperiment unntatt for N2
• Sma relativistiske korreksjoner (≈ 0.5%) pga. lette atomer
– nødvendige for overensstemmelse med eksperiment
• Store vibrasjonelle korreksjoner
19

Korrelasjonskonvergens: (harmoniske) vibrasjonsfrekvenser ωe (cm−1)
RHF SD T Q 5 rel. theory exp. err.
HF 4473.8 −277.4 −50.2 −4.1 −0.1 −3.5 4138.5 4138.3 0.2
N2 2730.3 −275.8 −72.4 −18.8 −3.9 −1.4 2358.0 2358.6 −0.6
F2 1266.9 −236.1 −95.3 −15.3 −0.8 −0.5 918.9 916.6 2.3
CO 2426.7 −177.4 −71.7 −7.2 0.0 −1.3 2169.1 2169.8 0.7
• Vi ser den samme konvergens i beregninger av alle egenskaper
– for a fa overensstemmelse med eksperiment, ma vi ta hensyn til interaksjoner mellom
fire eller flere elektroner
– samtidig ma vi beregne relativistiske korreksjoner, selv for lette atomer
– for tunge atomer kan relativistiske effekter være større enn korrelasjonseffekter
20

Det virtuelle rom av orbitaler
• Vi beskriver kollisjoner som virtuelle eksitasjoner mellom elektroner:
φi(1)φj(2) → φa(1)φb(2) dobbel-eksitasjon
φi(1)φj(2)φk(3) → φa(1)φb(2)φc(3) trippel-eksitasjon
– resultatet er en endret romlig fordeling av elektronene
– for hver slik eksitasjon beregner vi en sannsynlighet for at den vil forega
– bølgefunksjonen fas ved a midle over alle mulige eksitasjone
• Vi ma ha mange virtuelle orbitaler for en nøyaktig beskrivelse
– beregningens kostnader skalerer typisk som n4, der n er antall virtuelle orbitaler
– en nøyaktig beskrivelse krever at vi har et stort rom av virtuelle orbitaler
21

Basis-sett av gaussfunksjoner
• I vare beregninger ekspanderer vi molekylorbitalene i gaussfunksjoner (Gaussian-type
functions eller GTOer) av formen
Gijk(rA, α) = xiAy
jAzk
A exp`
−αr2A
´
• minimal eller single-zeta (SZ) basis-sett:
– et sett av GTOer for hvert okkuperte atomskall (2s1p)
– rudimentær beskrivelse av elektronstrukturen
• dobbel-zeta (DZ) basis-sett:
– to sett av GTOer for hvert okkuperte atomskall (3s2p1d)
– tilstrekkelig for en kvalitativ beskrivelse
• trippel-zeta (TZ), kvadruppel-zeta (QZ) og større basis-sett:
– nødvendig for en kvantitativ beskrivelse
• Antall funksjoner pr. atom vokser hurtig:SZ DZ TZ QZ 5Z 6Z
5 14 30 55 91 140
22

Kvantekjemiens to hierarkier
• Kvaliteten av en ab-initio beregning er bestemt av:
1. bølgefunksjonsmodellen i N -elektronrommet—dvs. høyeste eksitasjonsprosess
2. basissettet i en-elektronrommet—dvs. størrelsen av det virtuelle rommet
• I hvert av disse to rommene har vi et hierarki av nivaer:
1. N -elektronhierarkiet:
coupled-cluster-eksitasjonsnivaer:
HF, CCSD, CCSDT, CCSDTQ, ...
2. en-elektronhierarkiet:
korrelasjonskonsistente basis-sett
SZ, DZ, TZ, QZ, ...
one -electron basis sets
wave-
functi
on
models
exac
tso
lutio
n
• Kvaliteten av beregningene øker systematisk nar vi beveger oss oppover i hierarkiene.
23

Atomiseringsenergier (kJ/mol)
-200 200
HFDZ
-200 200 -200 200
HFTZ
-200 200 -200 200
HFQZ
-200 200 -200 200
HF5Z
-200 200 -200 200
HF6Z
-200 200
-200 200
MP2DZ
-200 200 -200 200
MP2TZ
-200 200 -200 200
MP2QZ
-200 200 -200 200
MP25Z
-200 200 -200 200
MP26Z
-200 200
-200 200
CCSDDZ
-200 200 -200 200
CCSDTZ
-200 200 -200 200
CCSDQZ
-200 200 -200 200
CCSD5Z
-200 200 -200 200
CCSD6Z
-200 200
-200 200
CCSD(T)DZ
-200 200 -200 200
CCSD(T)TZ
-200 200 -200 200
CCSD(T)QZ
-200 200 -200 200
CCSD(T)5Z
-200 200 -200 200
CCSD(T)6Z
-200 200
24
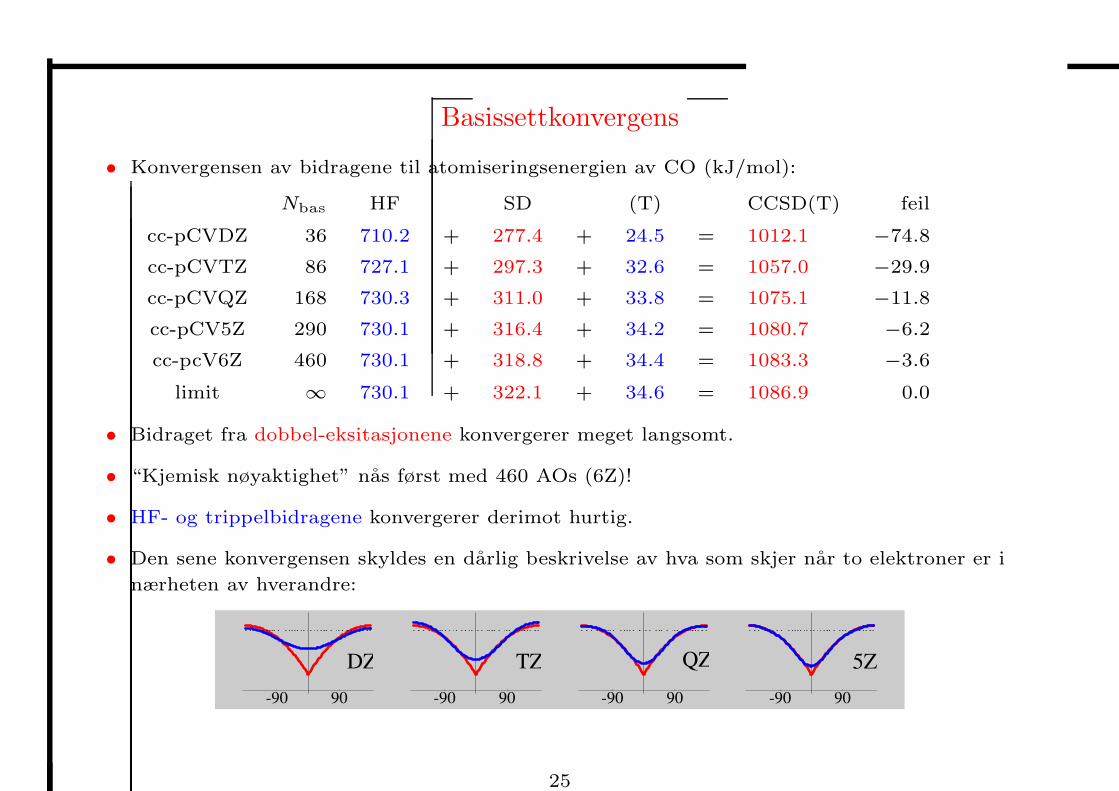
Basissettkonvergens
• Konvergensen av bidragene til atomiseringsenergien av CO (kJ/mol):
Nbas HF SD (T) CCSD(T) feil
cc-pCVDZ 36 710.2 + 277.4 + 24.5 = 1012.1 −74.8
cc-pCVTZ 86 727.1 + 297.3 + 32.6 = 1057.0 −29.9
cc-pCVQZ 168 730.3 + 311.0 + 33.8 = 1075.1 −11.8
cc-pCV5Z 290 730.1 + 316.4 + 34.2 = 1080.7 −6.2
cc-pcV6Z 460 730.1 + 318.8 + 34.4 = 1083.3 −3.6
limit ∞ 730.1 + 322.1 + 34.6 = 1086.9 0.0
• Bidraget fra dobbel-eksitasjonene konvergerer meget langsomt.
• “Kjemisk nøyaktighet” nas først med 460 AOs (6Z)!
• HF- og trippelbidragene konvergerer derimot hurtig.
• Den sene konvergensen skyldes en darlig beskrivelse av hva som skjer nar to elektroner er i
nærheten av hverandre:
-90 90
DZ
-90 90
TZ
-90 90
QZ
-90 90
5Z
25
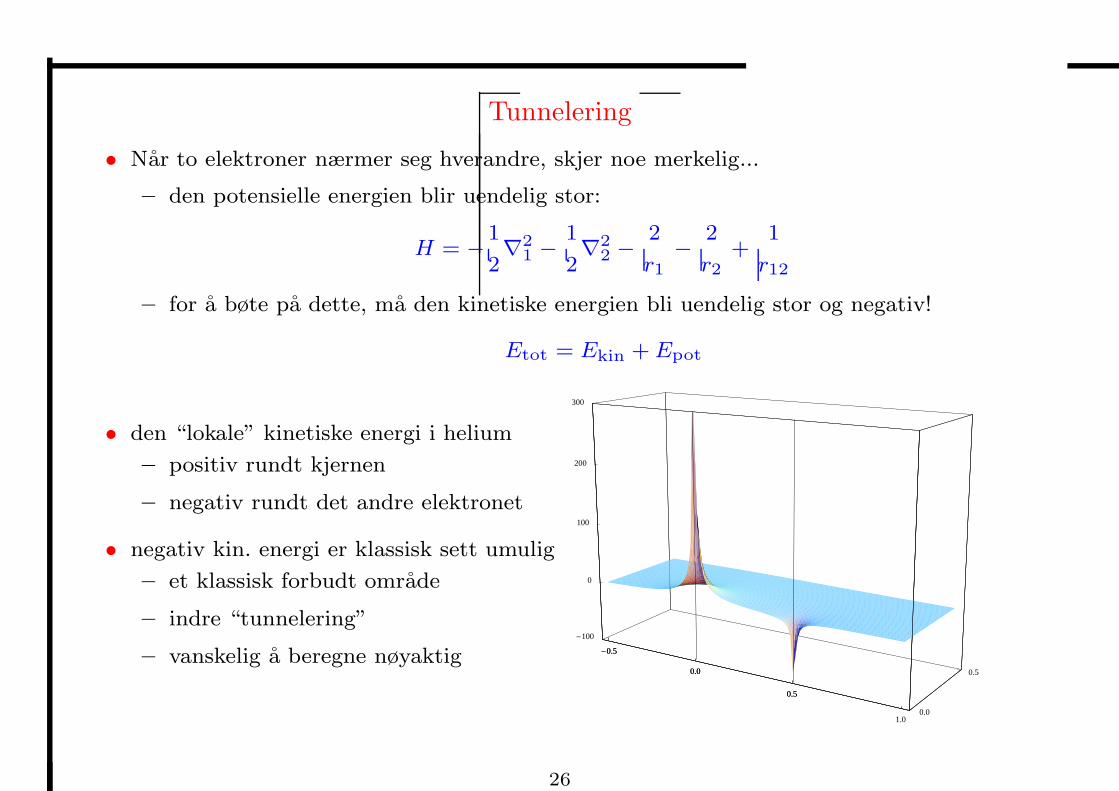
Tunnelering
• Nar to elektroner nærmer seg hverandre, skjer noe merkelig...
– den potensielle energien blir uendelig stor:
H = −1
2∇
21 −
1
2∇
22 −
2
r1
−2
r2
+1
r12
– for a bøte pa dette, ma den kinetiske energien bli uendelig stor og negativ!
Etot = Ekin + Epot
• den “lokale” kinetiske energi i helium
– positiv rundt kjernen
– negativ rundt det andre elektronet
• negativ kin. energi er klassisk sett umulig
– et klassisk forbudt omrade
– indre “tunnelering”
– vanskelig a beregne nøyaktig -0.5
0.0
0.5
1.00.0
0.5
-100
0
100
200
300
-0.5
0.0
0.5
26

Basissett-ekstrapolasjon
• Selv om konvergensen er sen, sa er den meget systematisk.
• Hvis basissettene er konstruert pa en optimal mate, sa er feilen i energien gitt ved
E∞ − EX = AX−3 + BX−4 + · · ·
der kardinaltallet X er 2 for DZ, 3 for TZ, 4 for QZ, etc.
• Dette betyr blant annet at feilen er proporsjonal med T−1/4, der T er CPU-tiden.
• Hvert nye siffer i svaret krever derfor 10000 ganger mer CPU tid!
1 minutt → 1 uke → 200 ar
• Imidlertid: fra to ulike energier EX og EY kan vi estimere basissettgrensen:
E∞ ≈ EX + AX−3
E∞ ≈ EY + AY −3
9
=
;
⇒ E∞ ≈X3EX − Y 3EY
X3 − Y 3
• Feilen i AE av CO reduseres kraftig ved ekstrapolasjon, og QZ gir kjemisk nøyaktighet:
kJ/mol DZ TZ QZ 5Z 6Z
uten ekstrapolasjon −73.5 −28.3 −11.4 −6.0 −3.5
med ekstrapolasjon −18.5 −0.7 0.0 0.0
27
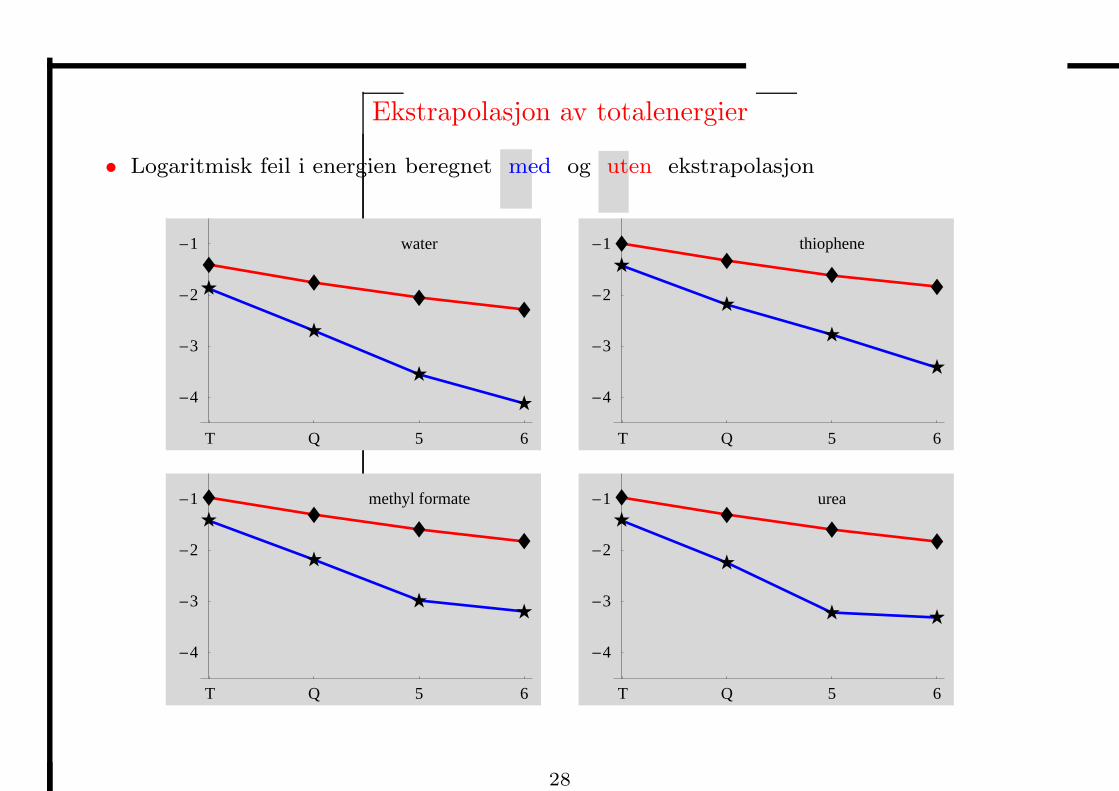
Ekstrapolasjon av totalenergier
• Logaritmisk feil i energien beregnet med og uten ekstrapolasjon
T Q 5 6
-1
-2
-3
-4
methyl formate
T Q 5 6
-1
-2
-3
-4
urea
T Q 5 6
-1
-2
-3
-4
water
T Q 5 6
-1
-2
-3
-4
thiophene
28

Sammenligning av CCSD(T) og eksperimentelle atomiseringsenergier
cc-pCVQZ cc-pCV(TQ)Z cc-pCV(56)Z eks.
HF 586.1 −7.0 592.8 −0.3 593.3 0.1 593.2(09)
O3 583.6 −32.6 600.8 −15.4 605.5 −10.7 616.2(17)
CH2 751.3 −5.7 758.7 1.6 757.9 0.9 757.1(22)
HNO 842.7 −18.8 858.5 −3.0 860.4 −1.1 861.5(03)
N2 936.3 −19.9 952.3 −4.0 954.9 −1.3 956.3(02)
H2O 963.5 −11.8 974.7 −0.5 975.5 0.2 975.3(01)
CO 1075.5 −11.2 1086.6 −0.1 1086.9 0.2 1086.7(05)
NH3 1232.7 −15.1 1247.3 −0.6 1247.4 −0.5 1247.9(04)
HCN 1294.1 −18.6 1311.3 −1.4 1311.0 −1.7 1312.8(26)
CH2O 1552.4 −14.2 1568.3 1.7 1568.0 1.4 1566.6(07)
CO2 1612.3 −20.1 1633.3 0.8 1633.2 0.7 1632.5(05)
C2H2 1681.0 −16.8 1698.8 1.0 1697.1 −0.8 1697.8(10)
CH4 1749.9 −9.4 1762.0 2.7 1759.4 0.1 1759.3(06)
C2H4 2343.6 −16.2 2363.2 3.4 2360.8 1.0 2359.8(10)
29

Entalpier for noen utvalgte reaksjoner (kJ/mol)
eks. CCSD(T)
C2H2 + H2 → C2H4 −203(2) −206
C2H2 + 3H2 → 2CH4 −446(2) −447
CO + H2 → CH2O −21(1) −23
N2 + 3H2 → 2NH2 −164(1) −165
F2 + H2 → 2HF −563(1) −564
O3 + 3H2 → 3H2O −933(2) −946
CH2O + 2H2 → CH4 + H2O −251(1) −250
H2O2 + H2 → 2H2O −365(2) −362
CO + 3H2 → CH4 + H2O −272(1) −273
HCN + 3H2 → CH4 + NH2 −320(3) −321
HNO + 2H2 → H2O + NH2 −444(1) −446
H2O + F2 → HOF + HF −129(4) −118
CO2 + 4H2 → CH4 + 2H2O −244(1) −244
2CH2 → C2H4 −844(3) −845
30

Bindingsavstander
HF MP2 CCSD CCSD(T) emp. exp.
H2O ROH 94.0 95.7 95.4 95.7 95.8 95.7
HNC RNH 98.2 99.5 99.3 99.5 99.5 99.4
NH3 RNH 99.8 100.8 100.9 101.1 101.1 101.1
N2H2 RNH 101.1 102.6 102.5 102.8 102.9 102.9
C2H2 RCH 105.4 106.0 106.0 106.2 106.2 106.2
C2H4 RCH 107.4 107.8 107.9 108.1 108.1 108.1
CH4 RCH 108.2 108.3 108.5 108.6 108.6 108.6
CH2O RCH 109.3 109.8 109.9 110.1 110.1 110.1
CH2 RCH 109.5 110.1 110.5 110.7 110.6 110.7
CO2 RCO 113.4 116.4 115.3 116.0 116.0 116.0
HNC RCN 114.4 117.0 116.2 116.9 116.9 116.9
C2H2 RCC 117.9 120.5 119.7 120.4 120.4 120.3
CH2O RCO 117.6 120.6 119.7 120.4 120.5 120.3
N2H2 RNN 120.8 124.9 123.6 124.7 124.6 124.7
31

Dalton
• Alle beregningene vi presenterer er gjort med vart eget program dalton.
• Arbeidet med dalton begynte for snart 25 ar siden, som et samarbeidsprosjekt med
Aarhus og Uppsala, senere ogsa med Odense, Stockholm og Tromsø.
• dalton 2.0 er gratis lisensiert til 862 grupper over hele verden (16% site-lisenser).
• daltons kildekode bestar av ca. 950 000 linjer.
• Funksjonalitet:
– Hartree–Fock, MCSCF, CC, MP2, CI, DFT
– energier
– strukturer
– eksitasjonsenergier og intensiteter
– vibrasjonsfrekvenser og intensiteter
– elektriske egenskaper (dipolmoment, polarisabiliteter, hyperpolarisabiliteter, osv)
– magnetiske egenskaper (skjermingskonstanter, spinn–spinnkoblingskonstanter osv.)
– dynamikk
• Hjemmeside: http://www.kjemi.uio.no/software/dalton/dalton.html
32

Dynamikk
• Vi har til na snakket om molekylers struktur og deres energetiske forhold.
• Kvantekjemiske metoder kan ogsa benyttes til studier av dynamikk og
reaksjonsmekanikk.
• Vi har utviklet metoder til a beregne atomenes bevegelser i reaksjoner
(deres trajektorier).
• Dette gjøres ved a beregne de kvantemekaniske krefter som beveger
kjernene.
• Dynamikken fremkommer ved a integrere Newtons ligninger:
mi
d2xi
dt2= −
dV (x1,x2, . . . ,xN )
xi
• Vi trenger ikke a konstruere potensialflaten V (x1,x2, . . . ,xN) pa forhand!
• Pa denne maten kan vi følge en kjemisk reaksjon i stor detalj i løpet av
noen femtosekunder.
33

Tetthetsfunksjonalteori
• I alle de metodene som vi har diskutert til na, beregner vi bølgefunksjonen—en meget
komplisert funksjon i 4N koordinater Φ(x1,x2, . . . ,xN ).
• For bestemmelsen av en bølgefunksjon benyttes ofte flere hundre millioner parameter,
noe som gjøre beregningene svært tunge.
• Imidlertid kan energien i prinsippet bestemmes som en funksjonal av tettheten E[ρ].
• Det er derfor mulig a omga bølgefunksjonen fullstendig og isteden beregne
elektrontettheten ρ(r)—en enkel funksjon i kun tre romlige koordinater r.
• Dette er grunnlaget for tetthetsfunksjonalteori: density functional theory (DFT).
• Problemet er at den eksplisitte formen av E[ρ] er ikke kjent—ikke en gang i prinsippet.
• En rekke approksimative tetthetsfunksjonaler er utviklet og gir gode resultater.
• Dessverre er disse funksjonalene konstruert pa et empirisk grunnlag og kan ikke
forbedres pa en systematisk mate.
• Det finnes derfor ikke i dag noe hierarki av nivaer i DFT.
• DFT beregningene kan derfor ikke forfines mot den eksakte grensen!
• DFT er likevel av umatelig stor praktisk nytte, da den lettere kan anvendes pa store
systemer.
34

NMR spinn–spinnkoblingskonstanter
• Som et eksempel pa anvendelse av DFT skal vi se pa beregningen av NMR
spinn–spinnkoblingskonstanter.
• Kjernene i et molekyl er utstyrt med magnetiske momenter MP :
– kjernene vil orientere seg i forhold til hverandre
– den direkte koblingen forsvinner pga molekylrotasjon
– kjernene kobler isteden indirekte, via elektronene
• Hvert kjernepar P og Q er karakterisert ved en koblingskonstant JP Q
– disse konstantene bestemmer oppsplittingen av linjene i et NMR-spektrum
– spinn-spinnkoblingskonstantene kan beregnes fra elektronenes bølgefunksjon
– HF-modellen gir av forskjellige grunner darlige resultater
– med DFT er slike beregninger blitt rutine (etter 2000)
• Hvis vi i tillegger beregner kjernenes skjermingskonstanter, sa kan hele NMR-spekteret
av et molekyl simuleres
• Review-artikkel: Helgaker, Jaszunski og Ruud, Chem. Rev. 99, 293 (1999)
35

Simulerte NMR-spektra av vinyllitium (200 MHz)
0 100 200
MCSCF
0 100 200 0 100 200
B3LYP
0 100 200
0 100 200
experiment
0 100 200 0 100 200
RHF
0 100 200
36
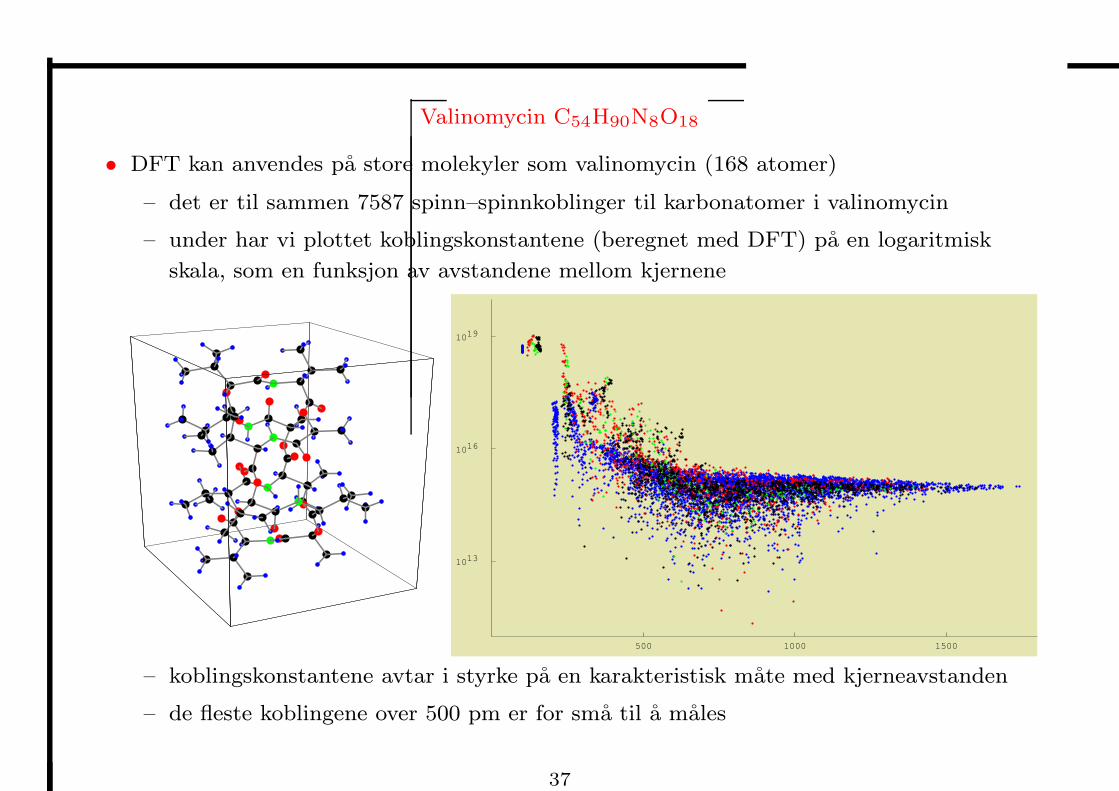
Valinomycin C54H90N8O18
• DFT kan anvendes pa store molekyler som valinomycin (168 atomer)
– det er til sammen 7587 spinn–spinnkoblinger til karbonatomer i valinomycin
– under har vi plottet koblingskonstantene (beregnet med DFT) pa en logaritmisk
skala, som en funksjon av avstandene mellom kjernene
500 1000 1500
1019
1016
1013
– koblingskonstantene avtar i styrke pa en karakteristisk mate med kjerneavstanden
– de fleste koblingene over 500 pm er for sma til a males
37
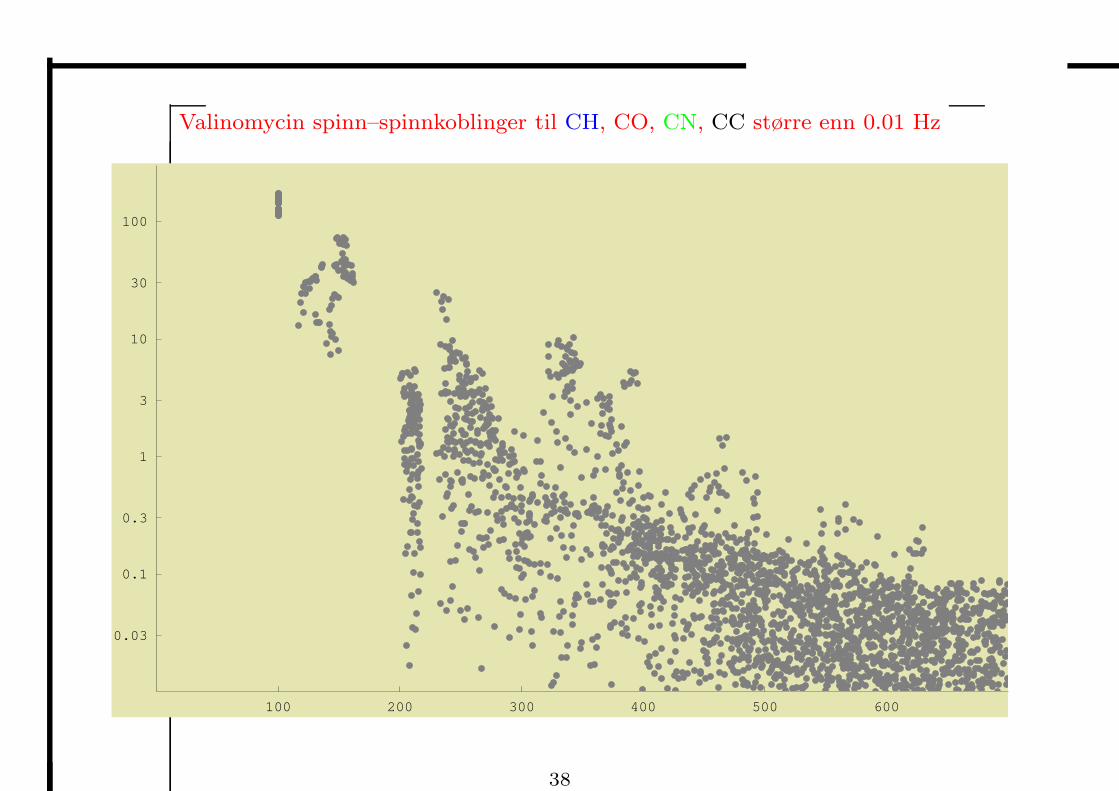
Valinomycin spinn–spinnkoblinger til CH, CO, CN, CC større enn 0.01 Hz
100 200 300 400 500 600
100
30
10
3
1
0.3
0.1
0.03
38
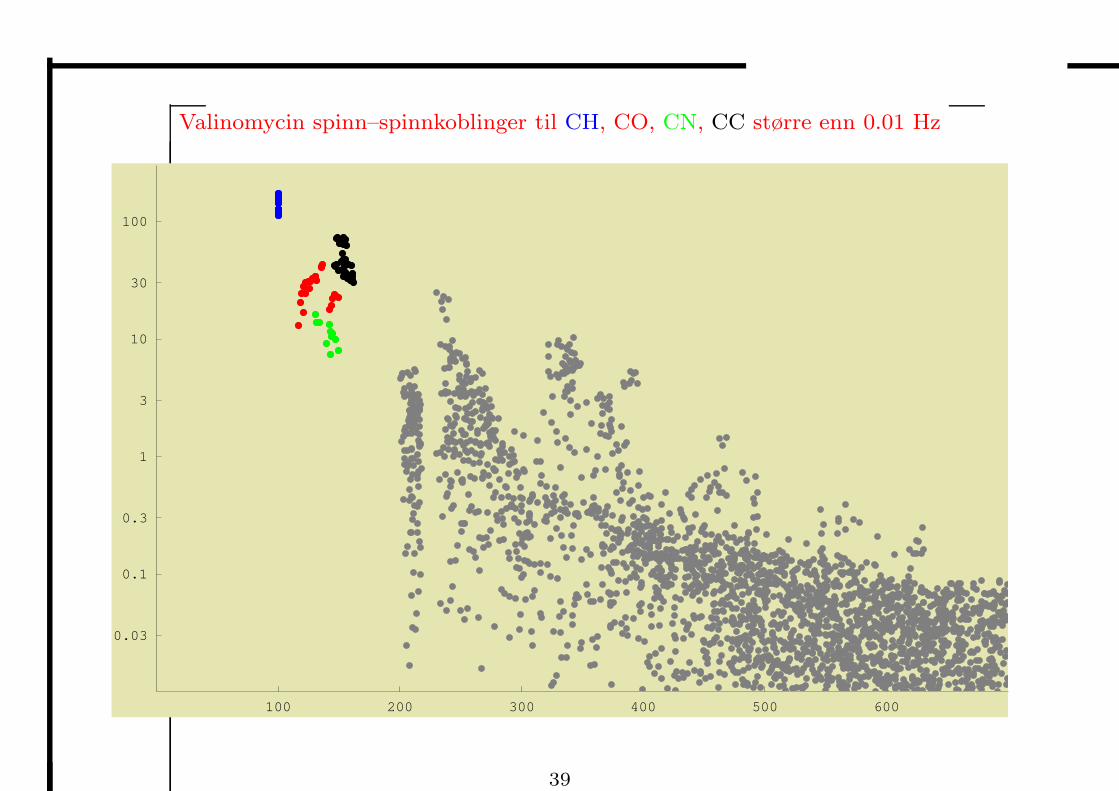
Valinomycin spinn–spinnkoblinger til CH, CO, CN, CC større enn 0.01 Hz
100 200 300 400 500 600
100
30
10
3
1
0.3
0.1
0.03
39
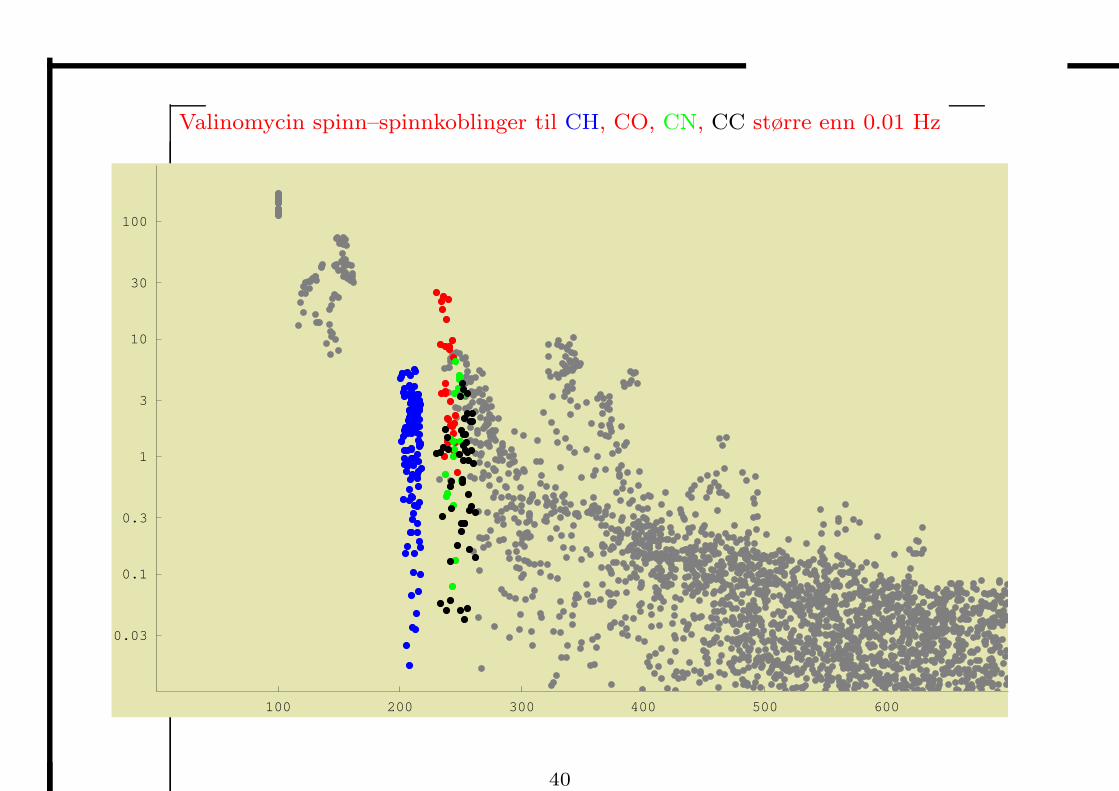
Valinomycin spinn–spinnkoblinger til CH, CO, CN, CC større enn 0.01 Hz
100 200 300 400 500 600
100
30
10
3
1
0.3
0.1
0.03
40

Valinomycin spinn–spinnkoblinger til CH, CO, CN, CC større enn 0.01 Hz
100 200 300 400 500 600
100
30
10
3
1
0.3
0.1
0.03
41

Valinomycin spinn–spinnkoblinger til CH, CO, CN, CC større enn 0.01 Hz
100 200 300 400 500 600
100
30
10
3
1
0.3
0.1
0.03
42

Valinomycin LDA/6-31G spin–spin couplings to CH, CO, CN, CC
500 1000 1500
100
1
0.01
0.0001
43

Oppsummering
• Kvantekjemien er et meget allsidig verktøy.
• Vi sitter med nøkkelen til molekylene: bølgefunksjonen. Fra denne kan vi i prinsippet
trekke ut all informasjon om kjemiske systemer.
• Vi har demonstrert dette for en del viktige molekylære egenskaper.
• Vi har lagt vekt pa presisjon og nøyaktighet for a belyse de ulike modellene.
• I praktiske anvendelser av kvantekjemiske metoder gjøres beregningene pa en mer
pragmatisk mate, da interessen er knyttet mer til det kjemiske problemet enn til
metoden.
• Det finnes mange kjemiske problemer der de kvantekjemiske metoder fortsatt ikke
strekker til!
44




























