Embed Size (px)
Citation preview

Density Functional Theory and
Exchange-Correlation Functionals
Weitao Yang, Duke University
Durham, North Carolina
2H
Funding
NSF
NIH
DOE
Theory
Biological
Nano
Material
Isfahan
May 2016 1

Outline
1. Introduction to the Fundamental Principles of DFT
2. Kohn-Sham Equations and Density Functional
Approximations
3. Fractional Perspectives of DFT
R. G. Parr and W. Yang, “Density functional theory of atoms
and molecules”, Oxford Univ. Press, 1989
A. J. Cohen, P Mori-Sanchez, and W. Yang, Chem. Rev, 2012
2

The Schrodinger Equation for N electrons
Hª(x1;x2;:::xN) = Eª(x1;x2;:::xN)
x= r; s
H = T + Vee +
NX
i
vext(ri)
T =
NX
i
¡1
2r2
Vee =
NX
i<j
1
rij
vext(r) = ¡X
A
¡ZA
rAi
3

Wavefunction--the curse of dimensionality
Hª=Eª# of electrons wavefunction amount of data
1
2
… … …
N
ª(r1)
ª(r1; r2) 106103
•The amount of data contained in wavefunction grows exponentially
with the number of electrons!
•The problem is caused by the exponential increase in volume
associated with adding extra dimensions to a (mathematical) space.
ª(r1;r2; :::rN) 103N
N » 1¡10000
4

The curse of dimensionality in QM
The problem is caused by the exponential increase in
volume associated with adding extra dimensions to a
(mathematical) space.
5

Exponential growth of information
N, positions and types of atoms
H
103N in ª(r1;r2; :::rN)
+
+, N, v(r)
v(r) is the electrostatic potential from the nuclei
m
v(r) =
AtomsX
A
¡ ZA
jr¡RAj
1.
2.
?
6
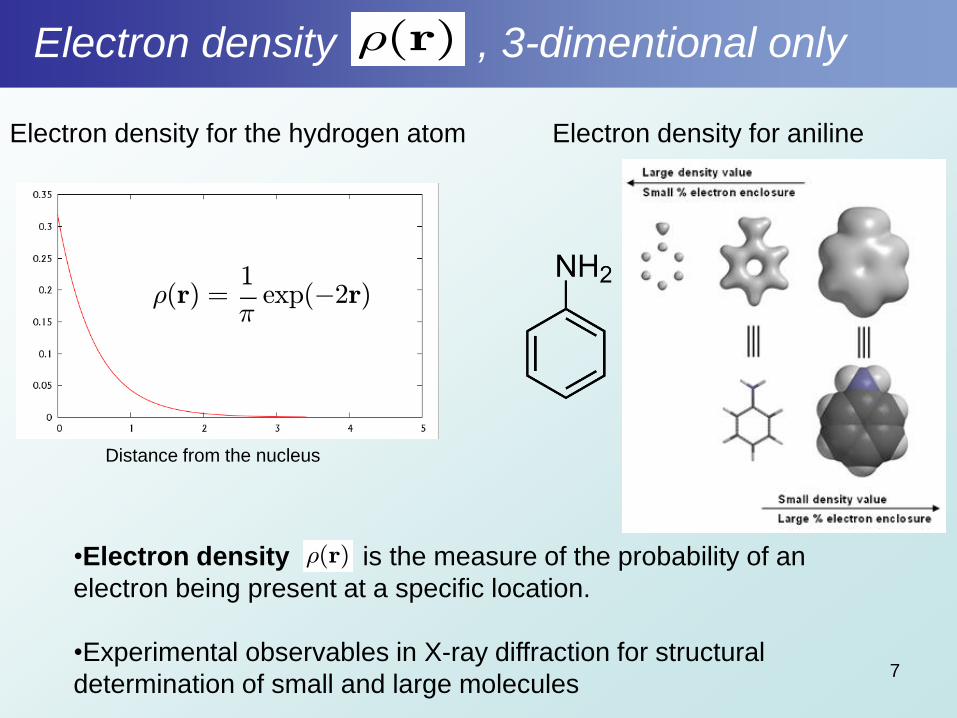
Electron density , 3-dimentional only
7
•Electron density is the measure of the probability of an
electron being present at a specific location.
•Experimental observables in X-ray diffraction for structural
determination of small and large molecules
½(r)
Electron density for anilineElectron density for the hydrogen atom
Distance from the nucleus
½(r) =1
¼exp(¡2r)
½(r)

Density Functional Theory
Density functional theory (DFT) (1964, 1965)
1
2.
8
½(r)()N;v(r)()ª
The Nobel Prize in Chemistry 1998
Walter Kohn Pierre C. Hohenberg Lu J. Sham
½(r) =X
i
jÁi(r)j2 Just the sum of molecular orbital densities

DFT: Exchange-correlation energy
9
E = E[½(r)]
= Kinetic energy + potential energy
+ Coulomb interaction energy
+ Exc[½(r)]
E
Exc[½(r)]
•Exchange-correlation energy
•The only unknown piece in the energy
•About 10% of E

Approximations in exchange-correlation energy
10
Exc[½(r)]
• 1965: Kohn and Sham, Local Density Approximation (LDA)
• 1980s-1990s: Axel Becke, Robert Parr, John Perdew
•Generalized Gradient Approximation (GGA)
•Hybrid Functionals (B3LYP, PBE0)

Density Functional Theory
• Structure of matter: atom, molecule, nano,
condensed matter
• Chemical and biological functions
• Electronic
• Vibrational
• Magnetic
• Optical (TD-DFT)
11
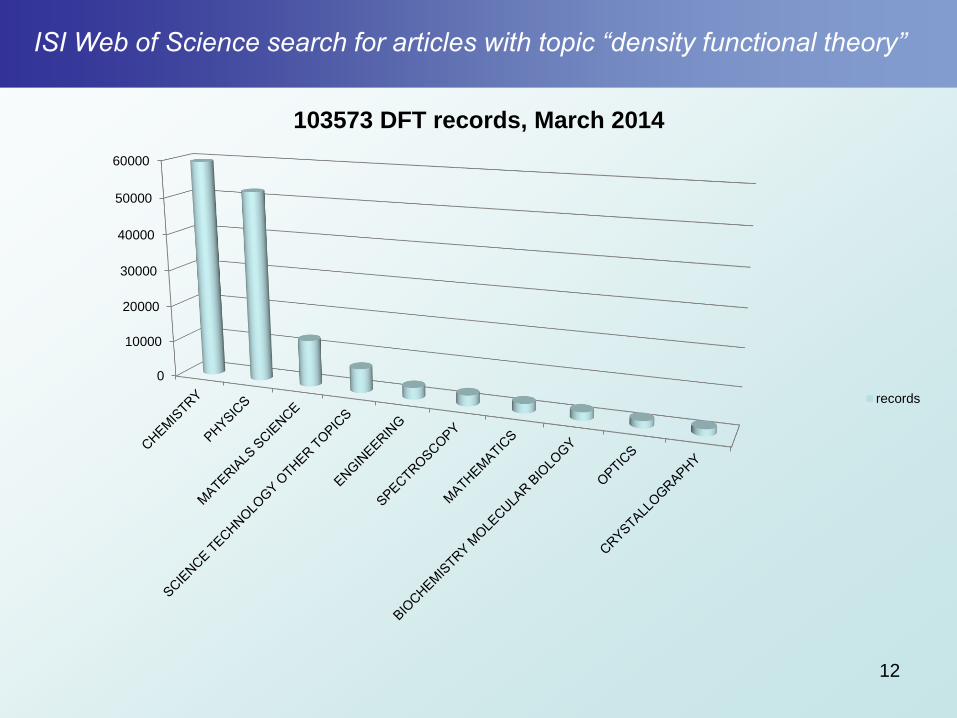
ISI Web of Science search for articles with topic “density functional theory”
12
0
10000
20000
30000
40000
50000
60000
103573 DFT records, March 2014
records

A deeper look at the Hamiltonian
PN
i vext(ri) =Rdrvext(r)
PN
i ±(r¡ ri)
H = T + Vee +
NX
i
vext(ri)
PN
i vext(ri)Only depends on atoms and molecules
Rdrf(r)±(r¡ r0) = f(r0)
13

Introducing the electron density
hªjNX
i
vext(ri) jªi =
ZdxN jª(x1;x2;:::xN )j2
NX
i
vext(ri)
=
ZdxN jª(x1;x2;:::xN )j2
Zdrvext(r)
NX
i
±(r¡ ri)
=
Zdrvext(r)
ZdxN jª(x1;x2;:::xN )j2
NX
i
±(r¡ ri)
=
Zdrvext(r)½(r)
14

½(r) =
ZdxN jª(x1;x2;:::xN )j2
NX
i
±(r¡ ri)
= hªjNX
i
±(r¡ ri) jªi
= N hªj ±(r¡ r1) jªi
= N
ZdxN jª(x1;x2;:::xN )j2 ±(r¡ r1)
= N
Zds1dx2:::dxN jª(rs1;x2;:::xN )j2
Introducing the electron density
15

½(r)the electron density
½(r)
1. The electron density at r in physical space
2. The probability of finding an electron at r
3. A function of three variables
½(r) ¸ 0
Zdr½(r) = N
Zdr jr½(r)j2 <1
16

For a determinant wave function
j©0i = jÂ1Â2:::ÂiÂj:::ÂNi½(r) = h©0j
NX
i
±(r¡ ri) j©0i
=
NX
i
h©0j ±(r¡ ri) j©0i
=
NX
i
Zdxi jÂi(xi)j2 ±(r¡ ri)
=
NX
i
Zdxi jÂi(xi)j2 ±(r¡ ri)
=
NX
i
ZdriX
si
jÁi(ri)¾(si)j2 ±(r¡ ri)
=
NX
i
jÁi(r)j2 17

The variational principle for ground states
Most QM methods use the wavefunction as the
computational variable and work on its optimization.
18

An alternative route
Goal: Looking for the max height, and the tallest kid in a school.
1) We can first look for the tallest kid in each class
2) then compare the result from each class
19

An alternative route
Goal: Looking for the max height, and the tallest kid in a school.
1) We can first look for the tallest kid in each class
2) then compare the result from each class
h(Ki) is the height of kid Ki
H(J) = maxKi2class(J) fh(Ki)g, the max height of class J20

An alternative route
is the original variable
H(J) = maxKi2class(J) fh(Ki)g is a constrained search
i
is the reduced variableJ
21

The constrained-search formulation of DFT
Levy, 197922

The constrained search
F [½(r)] = minª!½(r)
hªjT + Vee jªi1. A functional of density; given a density, it gives a
number.
2. A universal functional of density, independent of
atoms, or molecules.
B. The ground state energy is the minimum
E0 = min½(r)
Ev[½(r)]
= min½(r)
½F [½(r)] +
Zdrvext(r)½(r)
¾
A. We have a new
C. is the new reduced variable ½(r)23

The condition on the density
For what density the energy functional is defined?
-- It has to come from some wavefunciton, N-representable
-- the N-representability is insured if
F [½(r)] = minª!½(r)
hªjT + Vee jªi
½(r) ¸ 0
Zdr½(r) = N
Zdr jr½(r)j2 <1 24

The Thomas Fermi Approximation for T
1. Divide the space into many small cubes
2. Approximate the electrons in each cubes as independent
particles in a box (Fermions)
3. T= Sum of the contributions from all the cubes
² =h2
8ml2(n2x + n2y + n2z) =
h2
8ml2R2²
Eigenstate energies for particle in a cubic box
With quantum numbers nx; ny; nz = 1;2;3; ::::
Each state can have two electrons (spin up and spin down).
25

The number of states (with energy < ²) ©(²)
©(²) =1
8
4¼R3²
3=
¼
6(8ml2
h2²)
32
Density of states
g(²) =d©(²)
d²=
¼
4(8ml2
h2)32 ²
Two electrons occupy each state. The kinetic energy is
t = 2
Z ²F
0
²g(²)d² =8¼
5(2m
h2)32 l3²
52
F
N = 2
Z ²F
0
g(²)d² =8¼
3(2m
h2)32 l3²
32
F
The Thomas Fermi Approximation for T
26

The Thomas Fermi Approximation for T
To express t as a function of density
t =3
10(3¼2)
23 l3(
N
l3)53
N
l3! ½
l3 ! dr
TTF [½] =X
t = CF
Zdr½(r)
53
CF =3
10(3¼2)
23
---Local density approximation for T 27

The Thomas Fermi energy functional
J[½] = 12
Rdrdr0 ½(r)½(r
0)jr¡r0j
For the Vee functional, use the classical electron-electron interaction
energy
ETF [½] = CF
Zdr½(r)
53 +
1
2
Zdrdr0
½(r)½(r0)
jr¡ r0j +
Zdrvext(r)½(r)
The Thomas-Fermi total energy functional is
The minimization of ETF [½] leads to the Euler-Lagrange equation, with ¹
for the constraint ofRdr½(r) = N .
¹ =±Ev[½]
±½(r)=
±TTF [½]
±½(r)+ vJ(r) + vext(r)
or
5
3CF½(r)
23 +
Zdr0
½(r0)
jr¡ r0j + vext(r) = ¹28

The Thomas Fermi Dirac energy functional
Dirac exchange energy functional
ELDAx [½] = ¡CD
Z½(r)
43 dr
Slater, Gaspar, the X® approximation
EX®x [½] = ¡3
2®CD
Z½(r)
43 dr
CD = 34( 3¼)13
Adding the local approximation to the exchange energy
29

1 Question to the students
What is the Thomas –Fermi kinetic energy functional in
2 dimensional space?
In 3dTTF [½] = CF
Zdr½(r)
53
CF =3
10(3¼2)
23
30

Comments on TF theory
•Beautiful and simple theory, with density as
the basic variable
BUT,
•the energy is NOT a bound to the exact
energy
•no shell structure for atoms, no quantum
oscillation
•no bounding for molecules
•Main problem is in the local
approximation for T
31

2. Kohn-Sham Theory and Density Functional
Approximations
•Kohn-Sham equations
•Generalized Kohn-Sham equations
•Exchange and correlation energy functional
•Adiabatic Connections
•Density Functional Approximations
40

Kohn-Sham theory, going beyond the Thomas-Fermi
Approximation
Use a non-interacting electron system, —Kohn-Sham reference system,
to calculate the electron density and kinetic energy
j©si = jÂ1Â2:::ÂiÂj:::ÂNi
½(r) =
NX
i
jÁi(r)j2
Ts[½] = h©sjNX
i
¡1
2r2i j©si
=
NX
i
ZdriÁ
¤i (ri)
µ¡1
2r2i
¶Ái(ri)
=
NX
i
hÁij ¡1
2r2 jÁii
41

Kohn-Sham theory
½(r) =
NX
i
jÁi(r)j2 Ts[½] =
NX
i
hÁij ¡1
2r2 jÁii
² ½(r) =Pi jÁi(r)j2 is the true electron den-
sity, but Ts[½] is not T [½]: However, Ts[½] is
a very good approximation to Ts[½]
² The essence of KS theory is to solve Ts[½]
exactly in terms of orbitals
42

Kohn-Sham theory
½(r) =
NX
i
jÁi(r)j2 Ts[½] =
NX
i
hÁij ¡1
2r2 jÁii
Introduce the exchange correlation energy functional
F [½] = T [½] + Vee[½]
= Ts[½] + J [½] +Exc[½]
Exc[½] = T [½]¡ Ts[½] + Vee[½]¡ J [½]
J[½] = 12
Rdrdr0 ½(r)½(r
0)jr¡r0j
43

Kohn-Sham Equations
½(r) =
NX
i
jÁi(r)j2 Ts[½] =
NX
i
hÁij ¡1
2r2 jÁii
In terms of the orbitals, the KS total energy functional is now
Ev[½] =
NX
i
hÁij ¡1
2r2 jÁii+ J[½] +Exc[½] +
Zdrvext(r)½(r)
The g.s. energy is the minimum of the functional w.r.t. all
possible densities. The minimum can be attained by
searching all possible orbitals
W [Ái] = Ev[Ái]¡NX
i
"i f< ÁijÁi > ¡1g
±W [Ái]
±Ái(r)= 0 44

W [Ái] = Ev[Ái]¡NX
i
"i f< ÁijÁi > ¡1g±W [Ái]
±Ái(r)= 0
The orbitals fjÁiig are the eigenstates of an
one-electron local potential vs(r) if we have
explicit density functional for Exc[½] (KS equa-
tions)
µ¡12r2+ vs(r)
¶jÁii = "i jÁii ;
or a nonlocal potential vNLs (r,r0), if we have
orbital functionals for Exc[Ái] (generalized KS
equations)
µ¡12r2+ vNLs (r,r0)
¶jÁii = "GKSi jÁii : 45

vs(r) =±Exc[½]
±½(r)+ vJ(r) + vext(r)
vNLs (r,r0) =±Exc[±½s(r
0; r)]
±½s(r0; r)+ [vJ(r) + vext(r)] ±(r
0 ¡ r)
µ¡12r2+ vs(r)
¶jÁii= "i jÁii ;
µ¡12r2+ vNLs (r,r0)
¶jÁii= "GKSi jÁii :
Kohn-Sham (KS)
Generalized Kohn-Sham (GKS)
One electron Equations
46

Feastures of Kohn-Sham theory
1. With N orbitals, the kinetic energy is treated rigorously. In
comparison with the Thomas-Fermi theory in terms of
density, it is a trade of computational difficulty for
accuracy.
2. The KS or GKS equations are in the similar form as the
Hartree-Fock equations and can be solved with similar
efforts.
3. KS or GKS changes a N interacting electron problem into
an N non-interacting electrons in an effective potential.
4. The E_xc is not known, explicitly. It is about 10% of the
energy for atoms. 47

Adiabatic connection: from Kohn-Sham reference system to
the true physical system
½(r) =
NX
i
jÁi(r)j2
So far, the E_xc is expressed in a form that is not appealing, not
revealing, not inspiring…
What is the relation of the Kohn-Sham reference system to the true
physical system?
• The same electron density
• Anything else?48

Adiabatic connection: from Kohn-Sham reference system to
the true physical system
½(r) =
NX
i
jÁi(r)j2
49

Adiabatic connection: from Kohn-Sham reference system to
the true physical system
• Exc in terms of wavefunction!
• Compared with
• What is the integrand at ?
• Starting point to make approximations –
DFA (Density functional approximation)
52
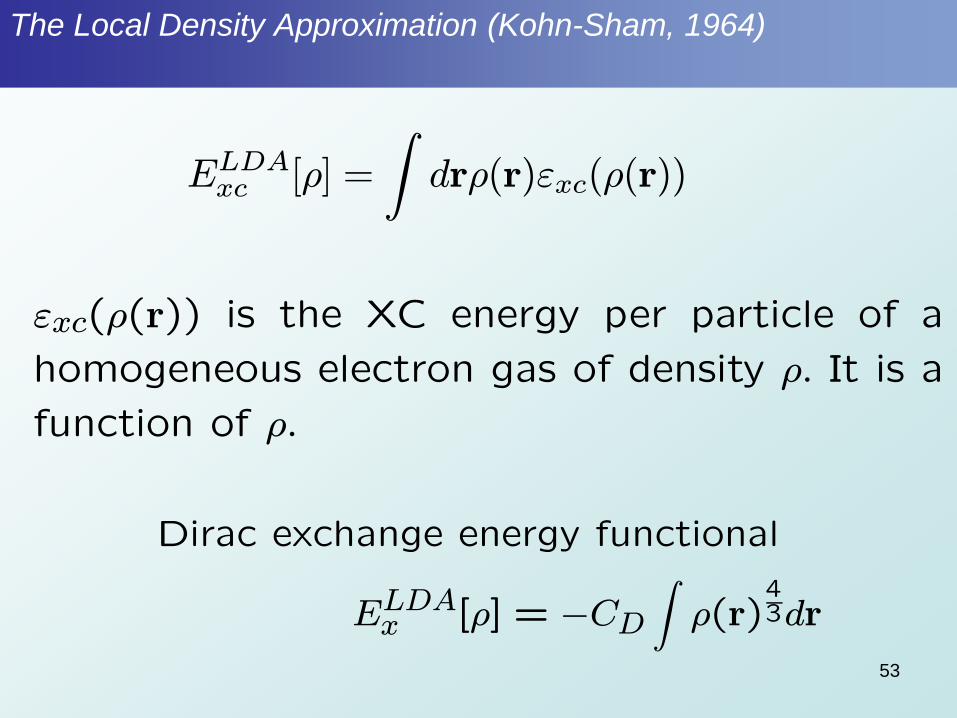
The Local Density Approximation (Kohn-Sham, 1964)
ELDAxc [½] =
Zdr½(r)"xc(½(r))
"xc(½(r)) is the XC energy per particle of a
homogeneous electron gas of density ½: It is a
function of ½:
Dirac exchange energy functional
ELDAx [½] = ¡CD
Z½(r)
43dr
53
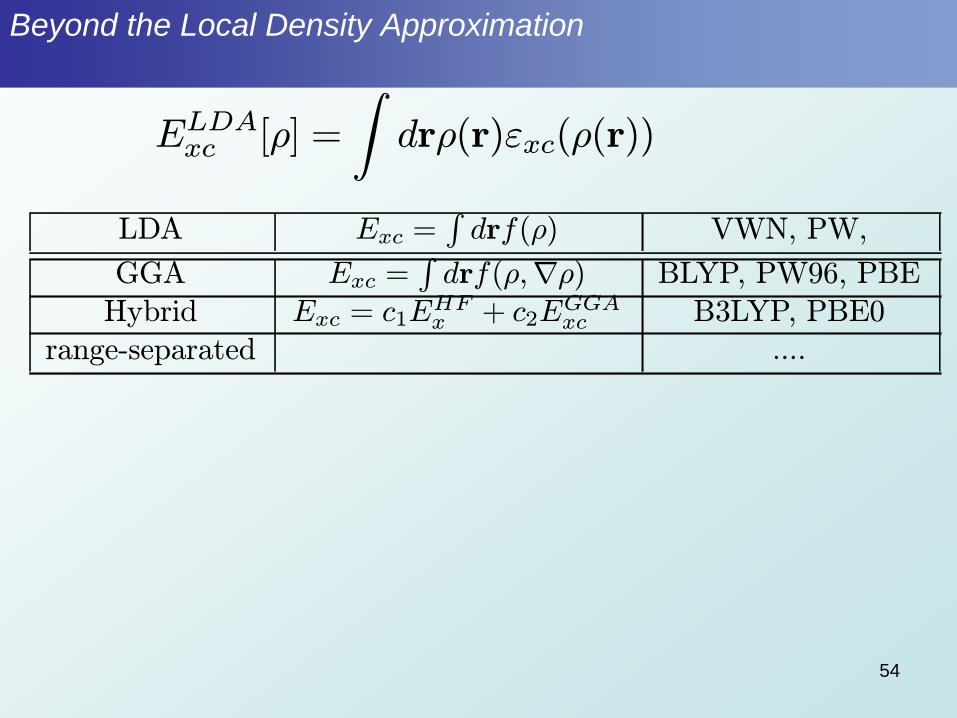
Beyond the Local Density Approximation
ELDAxc [½] =
Zdr½(r)"xc(½(r))
LDA Exc =Rdrf(½) VWN, PW,
GGA Exc =Rdrf(½;r½) BLYP, PW96, PBE
Hybrid Exc = c1EHFx + c2E
GGAxc B3LYP, PBE0
range-separated ....
54
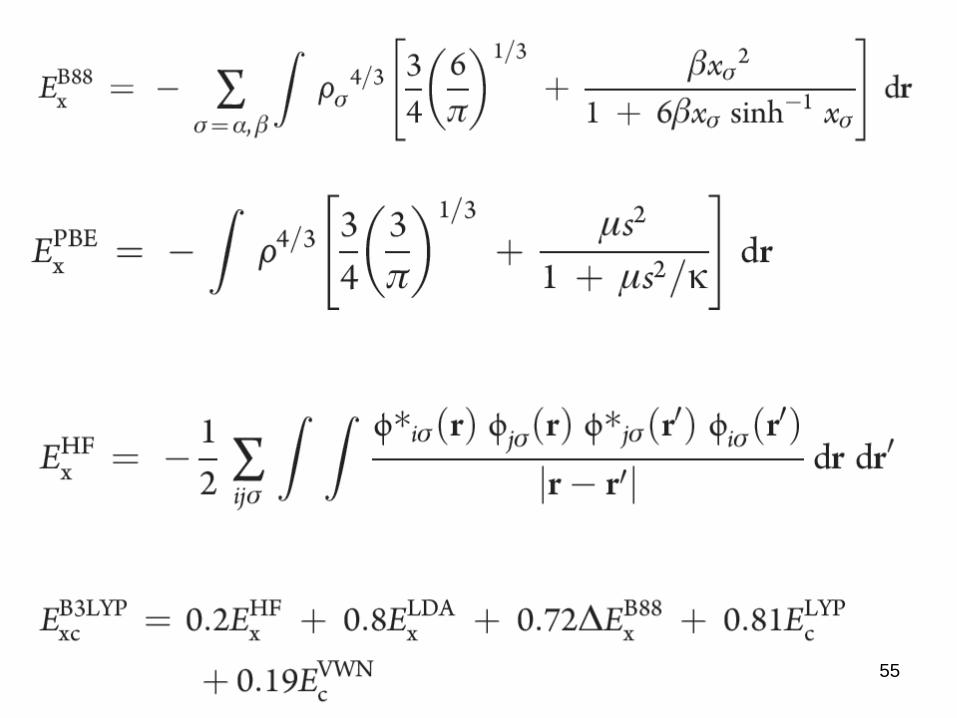
55
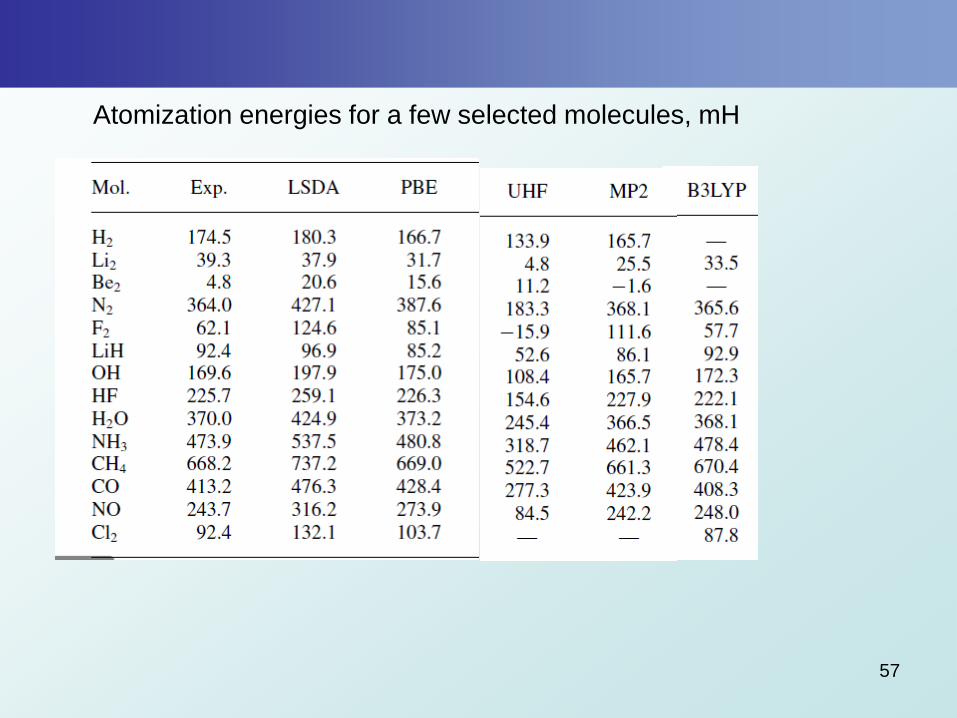
Atomization energies for a few selected molecules, mH
57
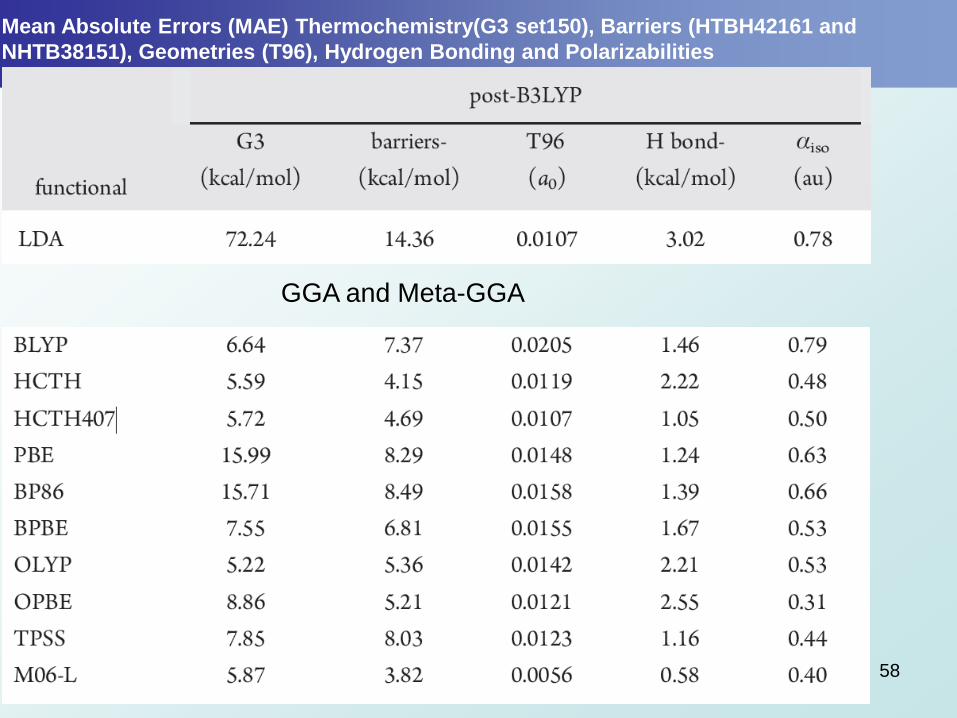
Mean Absolute Errors (MAE) Thermochemistry(G3 set150), Barriers (HTBH42161 and
NHTB38151), Geometries (T96), Hydrogen Bonding and Polarizabilities
GGA and Meta-GGA
58
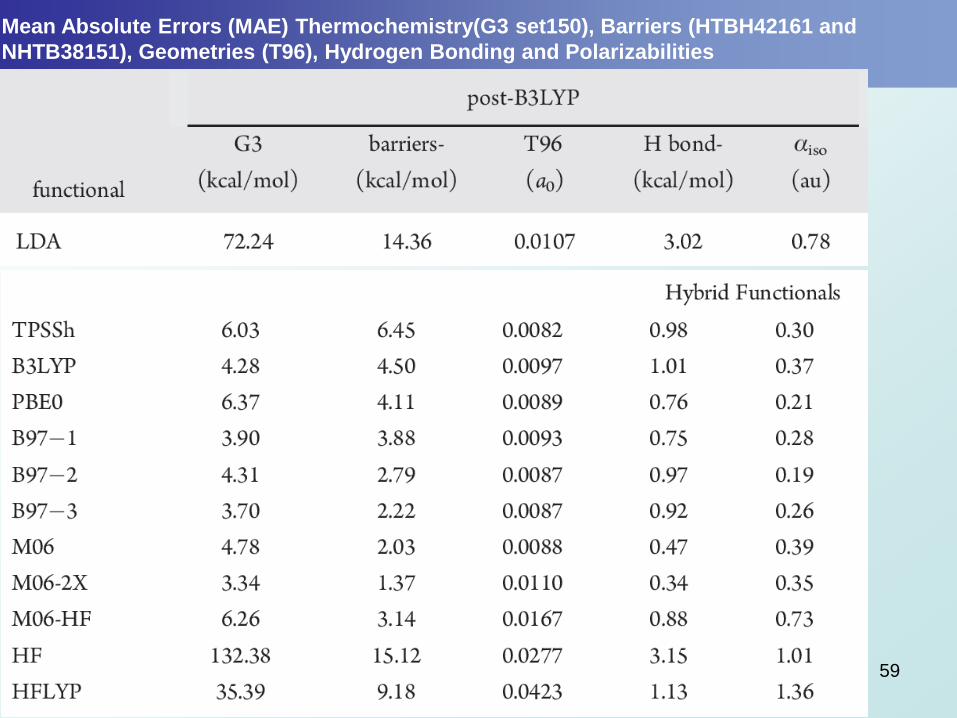
Mean Absolute Errors (MAE) Thermochemistry(G3 set150), Barriers (HTBH42161 and
NHTB38151), Geometries (T96), Hydrogen Bonding and Polarizabilities
59

Mean Absolute Errors (MAE) Thermochemistry(G3 set150), Barriers (HTBH42161 and
NHTB38151), Geometries (T96), Hydrogen Bonding and Polarizabilities
60

A large class of problems
• Wrong dissociation limit for molecules and ions
• Over-binding of charge transfer complex
• too low reaction barriers
• Overestimation of polarizabilities and hyperpolarizabilities
• Overestimation of molecular conductance in molecular electronics
• Incorrect long-range behavior of the exchange-correlation potential
• Charge-transfer excited states
• Band gaps too small
• Diels-Alder reactions, highly branched alkanes, dimerization of aluminum complexes
Fractional Charges
Delocalization Error61
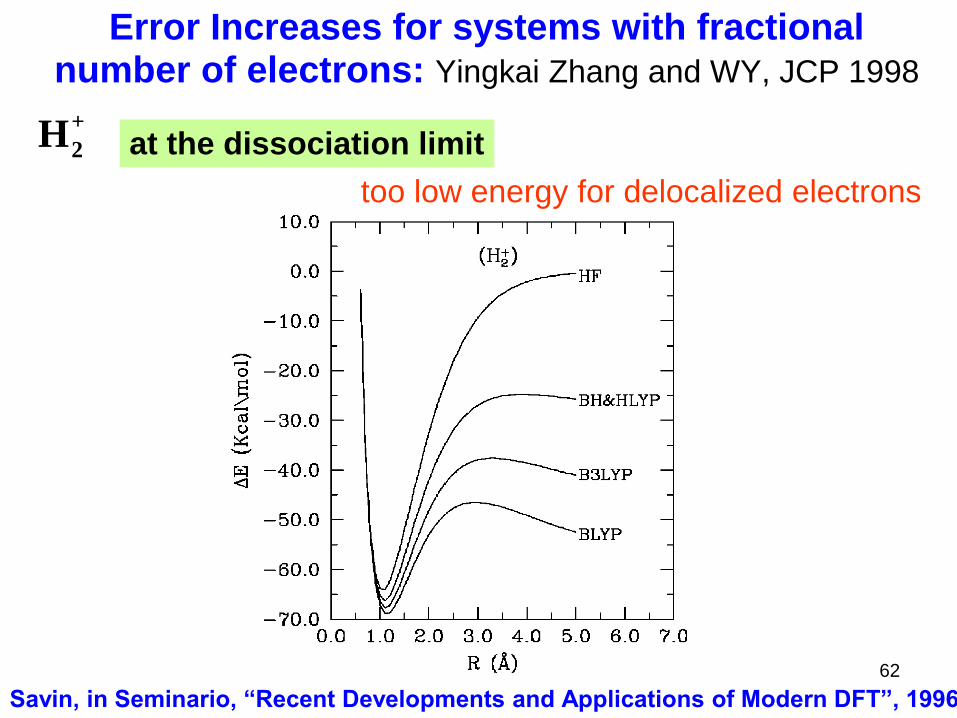
Error Increases for systems with fractional number of electrons: Yingkai Zhang and WY, JCP 1998
too low energy for delocalized electrons
Savin, in Seminario, “Recent Developments and Applications of Modern DFT”, 1996
+
2H at the dissociation limit
62

DFT for fractional number of electronsfrom grand ensembles,
Perdew, Parr, Levy, and Balduz, PRL. 1982
63

Where can you find fractional charges?
WY, Yingkai Zhang and Paul Ayers, PRL, 2000 – pure states
+
2H
(0) (1)E E E
1
2
1 1 12 2 2
( ) ( ) 2 ( )E E E E
ProtonA
ProtonB
e
ProtonB
eProtonA
at the dissociation limit
e/2Proton
A
ProtonB
e/2
(1) (0)E E E
1 1 1 12 2 2 2
( ) : ( ) (0) (1) (1)E N E E E E 64
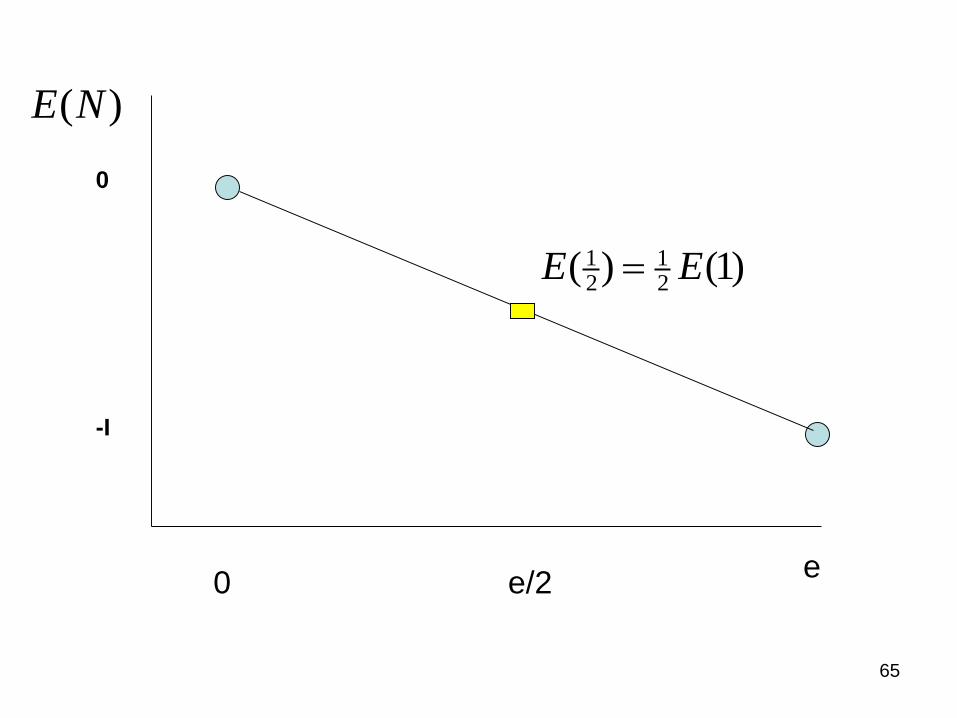
The linearity condition in fractional charges: The energy of e/2
ee/2
1 12 2
( ) (1)E E
0
0
-I
( )E N
65
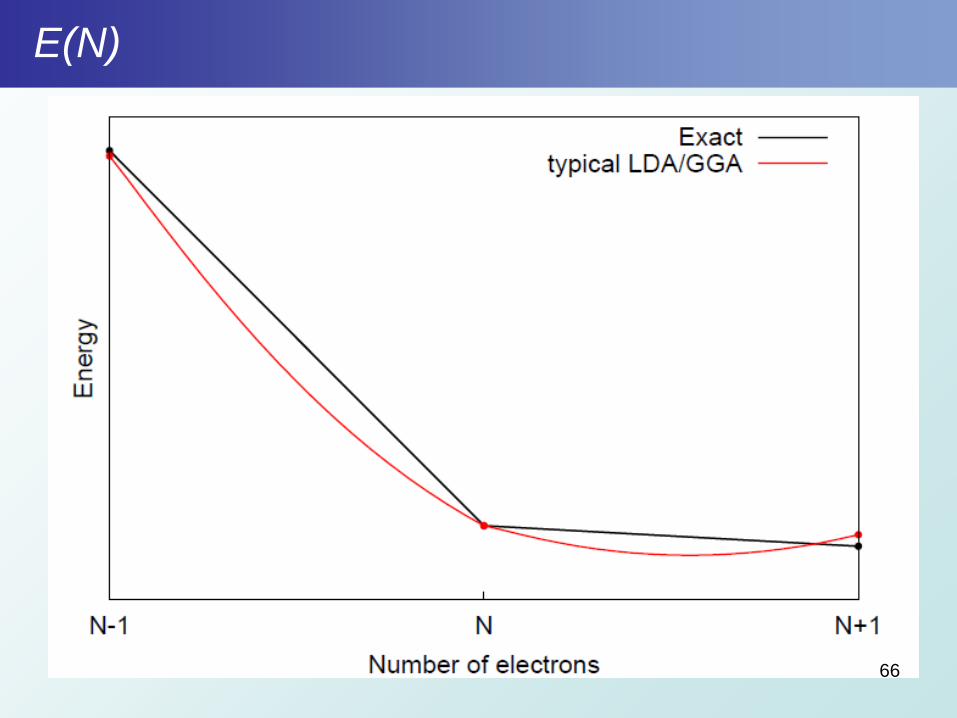
E(N)
66

12 ( ) ( ) ( 1),
2For -convex, < delocalizedN E N E N E N
( )A dimer, with separation: each monomer has E N
12 ( ) ( ) ( 1),
2For -concave, > localizedN E N E N E N
2 x + e
67

Localization Error
Delocalization Error
Delocalization and Localization ErrorPaula Mori-Sanchez, Aron Cohen and WY, PRL 2008
Consequence of Delocalization Error1. predicts too low energy for delocalized distributions
2. gives too delocalized charge distributions 68

Delocalization Error
Define the Delocalization Error as the violation of the
linearity condition for fractional charges
Cohen, Mori-Sanchez and Yang, 2008 Science69

Delocalization Error vs. Self-Interaction Error (SIE)
Delocalization Error (Mori-Sanchez, Cohen and Yang, PRL 2008)• Two SIE-free functionals: Becke06, and MCY2 (2006) did not solve the problems (Self-
interaction-free exchange-correlation functional for thermochemistry and kinetics, Mori-Sanchez, Cohen and Yang, JCP 2006)
• Many-electron SIE were used in 2006 (Mori-Sanchez, Cohen and Yang, JCP 2006, A. Ruzsinszky, J. P. Perdew, G. I. Csonka, O. A. Vydrov, and G. E. Scuseria, JCP 2007)
• Delocalization Error agrees with SIE for one electron systems. For general systems, it reveals the true relevant mathematical error of approximate functionals, and captures the physical nature of the error—delocalization.
SIE (Perdew-Zunger 1982)• Exc error for one-electron systems• Self-Interaction Correction (SIC) forces the correction for
every one-electron orbital, improves atomic systems.• SIC does not improve molecular systems in general (kind
over corrections). • SIC fractional extension (0<N<1, Zhang and Yang, JCP 1998)
explained problem) +
2H
70

Delocalization Error in GGA, LDA, B3LYP
• Energy of dissociation of molecular ion: too low
• Charge transfer complex energy: too low
• Transition state energy: too low
• Charge transfer excitation energy: too low
• Band gap: too low
• Molecular conductance: too high
• (Hyper)polarizability for long molecules: too high
• Diels-Alder reaction products, highly branched alkanes, dimerization of aluminum complexes: too high
Too low energy for fractional charge systems
Cohen, Mori-Sanchez and Yang, 2008 Science71

Seeing the delocalization error 2 7( )Cl H O
Where is the negative charge ?
72

Another large class of problems
• huge error dissociation of chemical bonds
• transition metal dimmers
• some magnetic properties
• strongly correlated systems
• Mott insulators, high superconductors
• degeneracy and near degeneracyc
T
Fractional Spins
Static Correlation Error
Aron Cohen, Paula Mori-Sanchez and WY, JCP, 2008
73
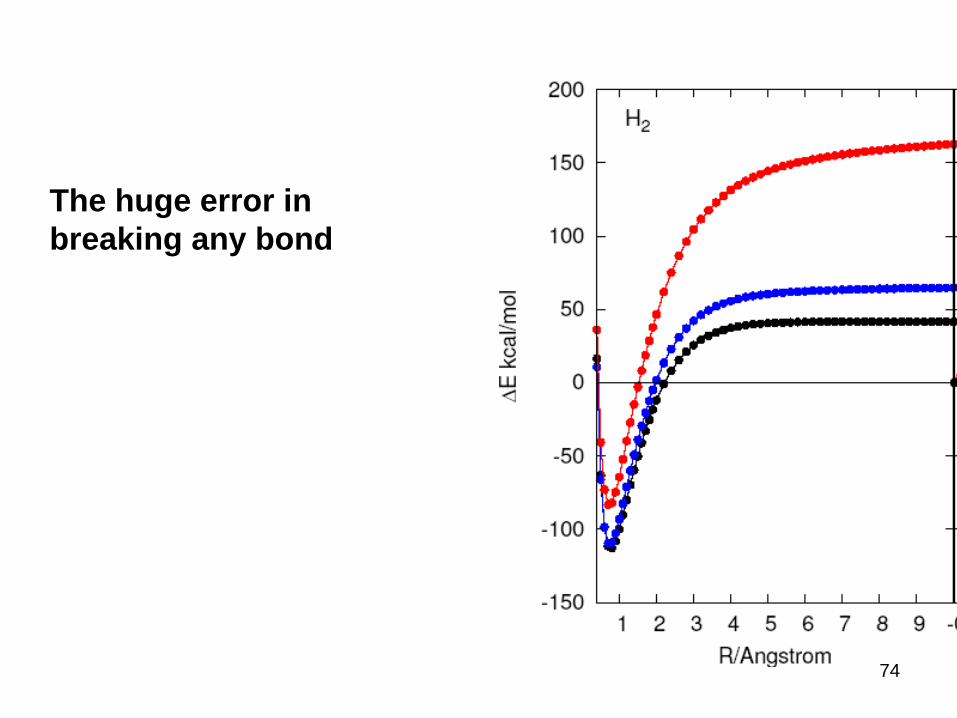
The huge error in
breaking any bond
74

Where can you find fractional spins?
Aron Cohen, Paula Mori-Sanchez and Yang, 2008, JCP
Yang, Ayers and Zhang, PRL 2000
2H
( ) ( )E E E
1
2
2 ( ) 2 ( )E E E
Proton
A
Proton
B↑
Proton
B
Proton
A
at the dissociation limit
Proton
A
Proton
B
( ) ( )E E E
2( ) ( ) ( )E E E
↓
↑ ↓
↑↓↑↓
Fractional Spin H Atom: half spin up electron and half spin down electron75
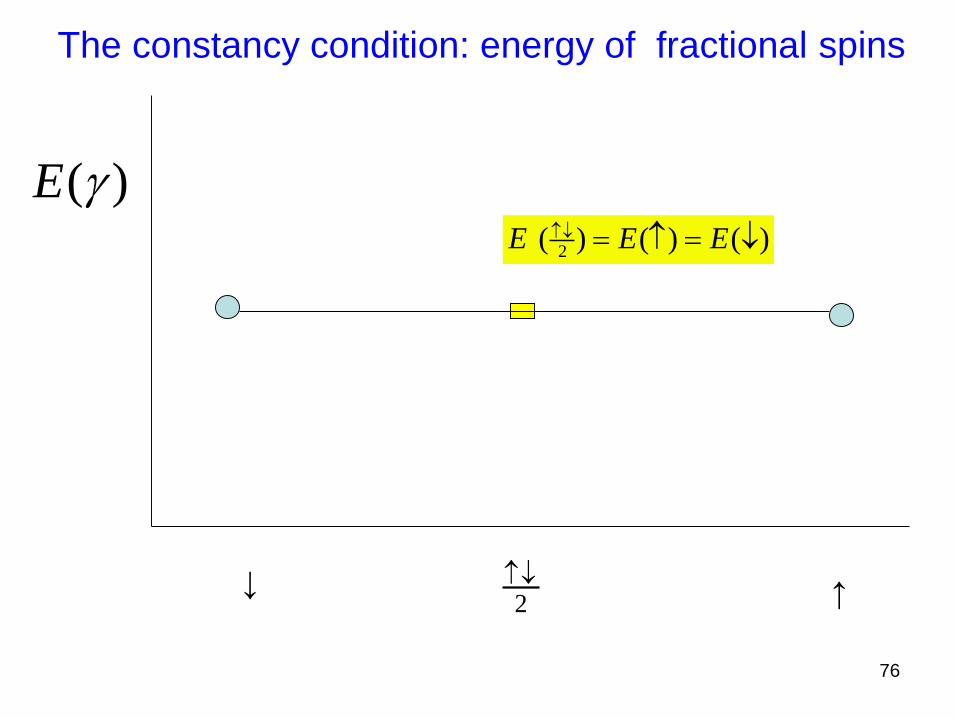
The constancy condition: energy of fractional spins
( )E 2
( ) ( ) ( )E E E
↑
↑2
76
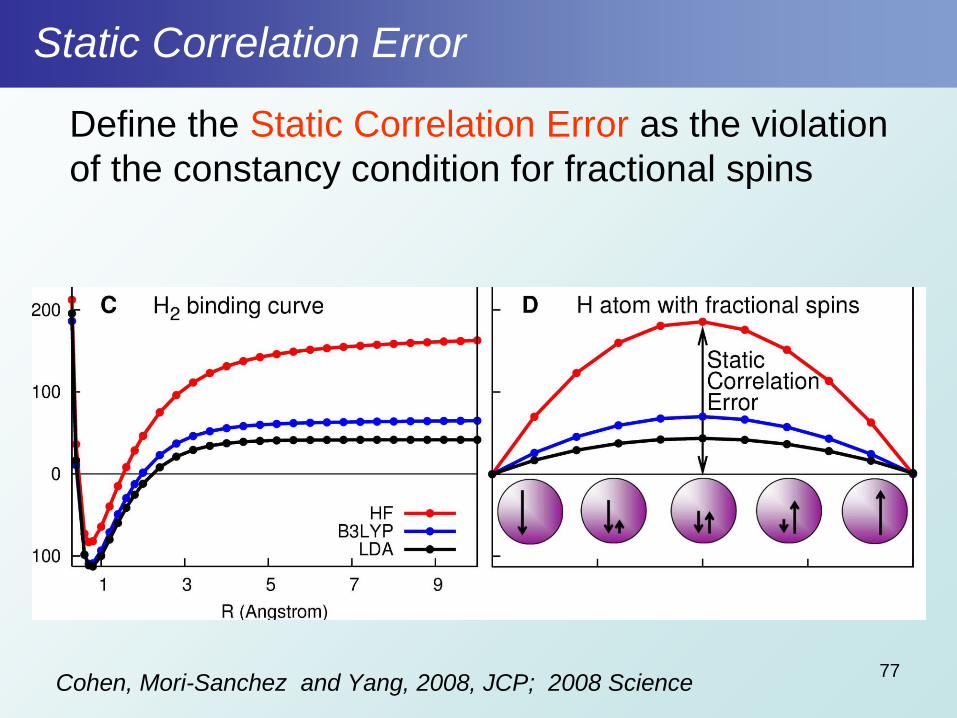
Static Correlation Error
Define the Static Correlation Error as the violation
of the constancy condition for fractional spins
Cohen, Mori-Sanchez and Yang, 2008, JCP; 2008 Science77

78
Why we have to deal with fractional number of electrons?
½(r)()ª(r1; r2; :::rN)
E = hªjH jªiE = E[½(r)]
Wavefunction TheoryDensity Functional Theory
Fractional charge can occur Integers, always!
Many-electron theories based on
•Green function
•Density matrix

Exact conditions on DFT
, ,[ ]i N i N i NiE c E E
1 1(1 ) (1 )N N N NE E E
, 1, 1(1 ) (1 )i N i j N j N Ni jE c d E E
•!! The exact XC functional cannot be an explicit and differentiable functional of the electron density/density matrix, either local or nonlocal.
•Valid for density functionals, and also for 1-body density matrix functionals, 2-RDM theory, and other many-body theories.
Fractional Charge: 1982: Perdew, Levy, Parr and Baldus
Fractional Spins: 2000, PRL, WY, Zhang and Ayers;2008, JCP, Cohen, Moris-Sanchez, and WY
Fractional Charges and Spins: 2009: PRL, Moris-Sanchez, Cohen and WY
79
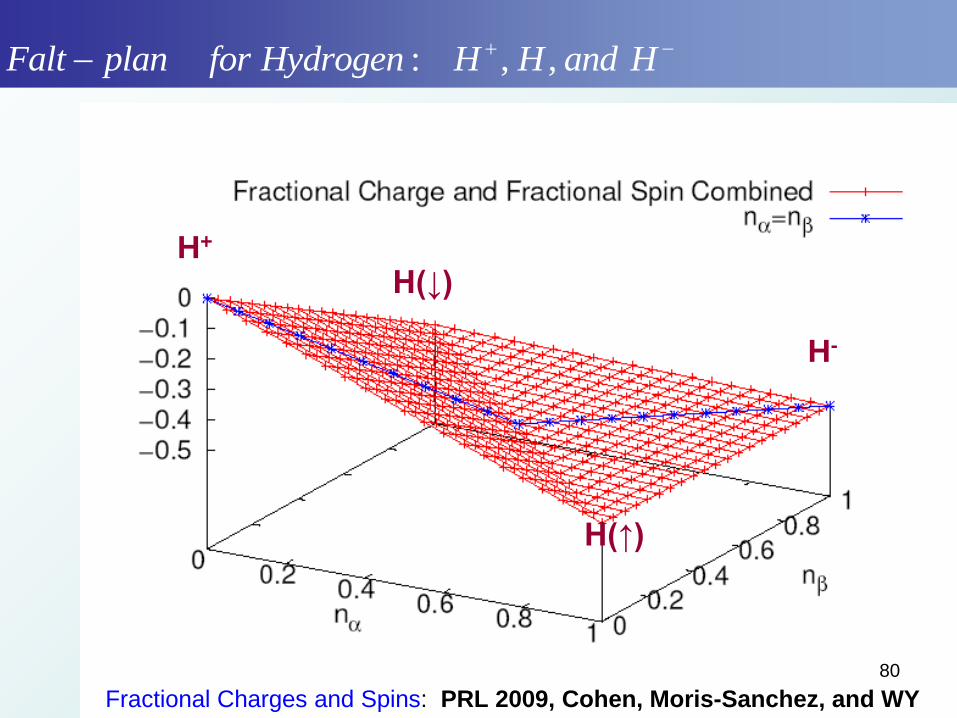
: , ,Falt plan for Hydrogen H H and H
H+
H(↓)
H-
H(↑)
Fractional Charges and Spins: PRL 2009, Cohen, Moris-Sanchez, and WY
80

81

Band GapDefinition of fundamental gap
derivative
Only if is linear.
82

How can fundamental gap be predicted in DFT
• LUMO energy is the chemical potential for electron addition
• HOMO is the chemical potential for electron removal
• Fundamental gaps predicted from DFT with KS, or GKS
calculations, as the KS gap or the GKS gap
• For orbital functionals, the LUMO of the KS (OEP) eigenvalue
is NOT the chemical potential of electron addition.
Thus the KS gap is not the fundamental gap predicted by the
functional.
@Ev(N)
@N= hÁf jHe® jÁfi
WY, Mori-Sanchez and Cohen, PRB 2008, JCP 201283
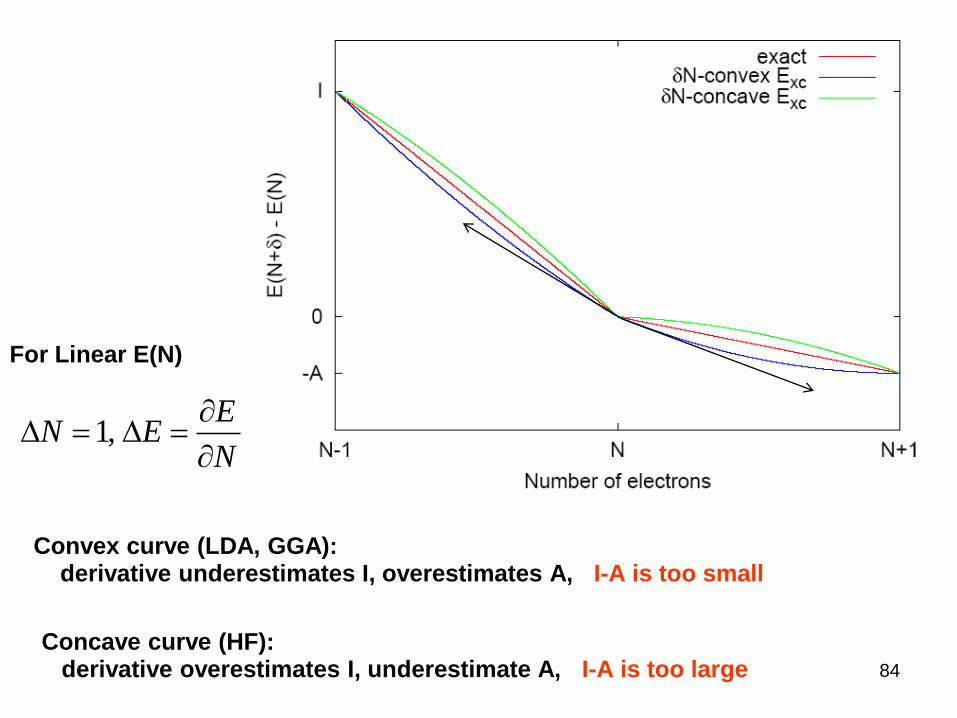
Convex curve (LDA, GGA):derivative underestimates I, overestimates A, I-A is too small
Concave curve (HF):derivative overestimates I, underestimate A, I-A is too large
1,E
N EN
For Linear E(N)
84
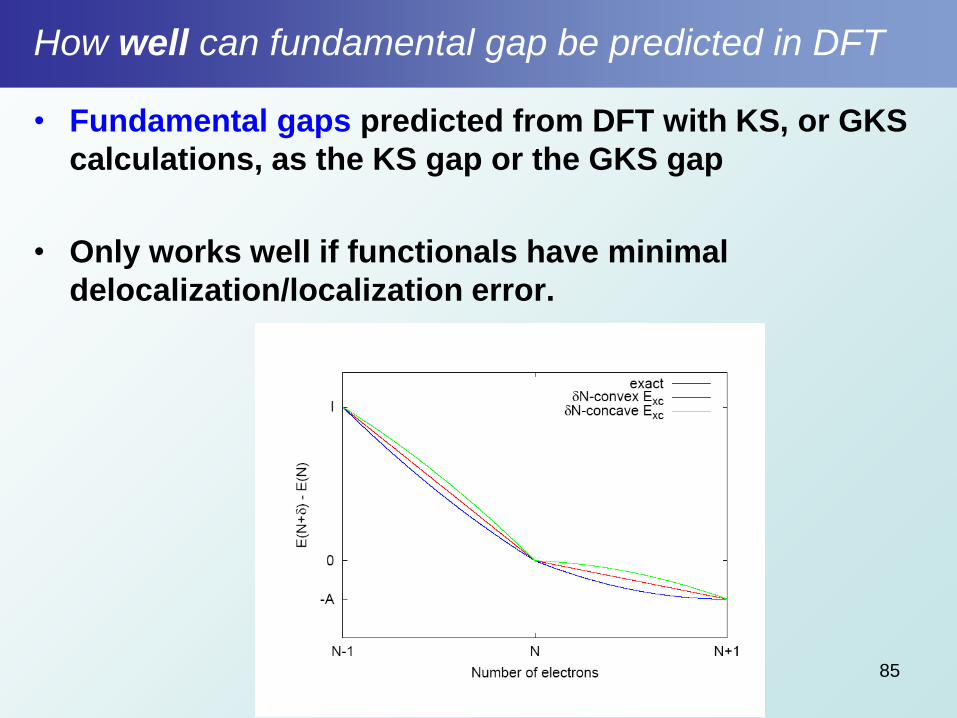
How well can fundamental gap be predicted in DFT
• Fundamental gaps predicted from DFT with KS, or GKS
calculations, as the KS gap or the GKS gap
• Only works well if functionals have minimal
delocalization/localization error.
85

2012
86

Improving band gap prediction in density functional theory from
molecules to solidsPRL, 2011, Xiao Zheng, Aron J. Cohen, Paula Mori-
Sanchez, Xiangqian Hu, and Weitao Yang
Paula Mori-Sanchez(Univ. Autonoma Madrid)
Xiao Zheng(USTC)
Aron J. Cohen(Cambridge) Xiangqiang Hu
87

88
Nonempirical Scaling correction
To linearize an energy component, Ecomp, for a system of N + n (0 < n < 1) electrons, we construct
1 and 0 at E reproduces and , with linearly scales )(E compcomp nnnnN~
The SC to Ecomp is
It is then important to cast ΔEcomp into a functional form, so that it depends explicitly on ρ(r), or on Kohn-Sham first-order reduced density matrix ρs(r,r’)
88

89
Energy versus fractional electron number
The SC significantly restores linearity condition for energy89

90
Band gaps by various DFA and S-DFASummary of band-gap prediction for a variety of systems
The SC preserves the accuracy of I, A, and integer gaps, while it improves significantlyon HOMO and LUMO energies, and derivative gaps
S-MLDA predicts reasonable band gaps with consistent accuracy for systems of allsizes, ranging from atoms and molecules to solids
90

91
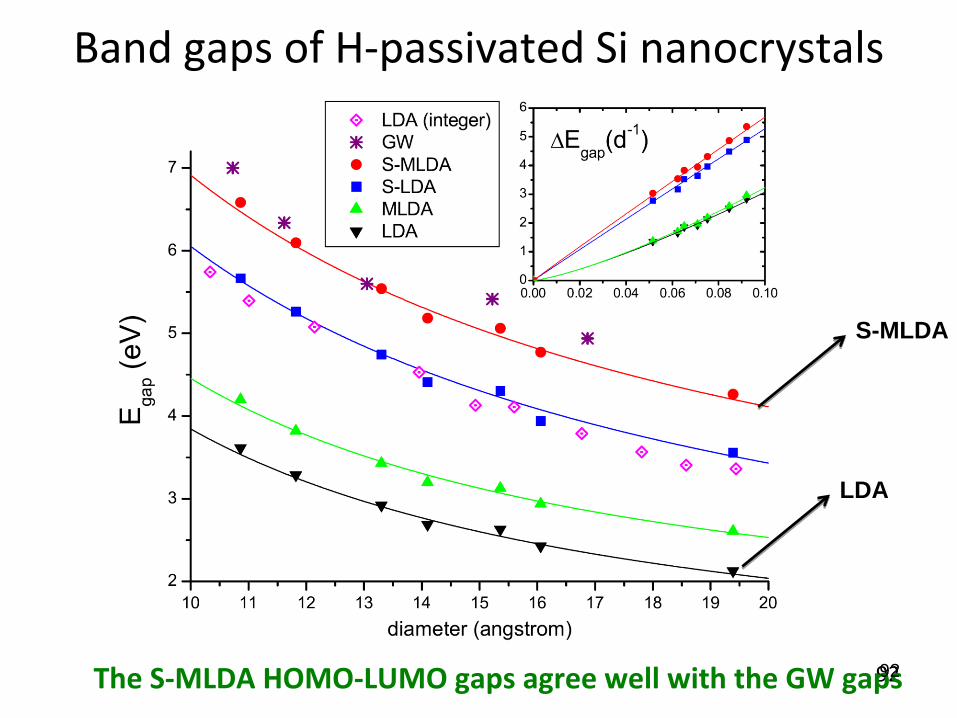
Band gaps of H-passivated Si nanocrystalsComputational details
Systems under study: H-passivated spherical Si nanocrystals. The largest system isSi191H148 with a diameter of 20 angstrom
Geometries optimized by B3LYP with Lanl2dz ECP basis set, except that the largestsystem Si191H148 is optimized with semiempirical PM3 method
Diffusive basis functions are important to obtain accurate HOMO-LUMO gaps: 6-31G for H atoms; an sp-shell with an exponent of 0.0237 au and a d-shell with anexponent of 0.296 au added to Lanl2dz basis for Si atoms
Si191H148
91
92
Band gaps of H-passivated Si nanocrystals
The S-MLDA HOMO-LUMO gaps agree well with the GW gaps
S-MLDA
LDA
92

Local Scaling Correction, PRL, 2015
Paula Mori-Sanchez
(Univ. Autonoma Madrid)
Xiao Zheng
(USTC)
Aron J. Cohen
(Cambridge))
Chen Li
(Duke)
93
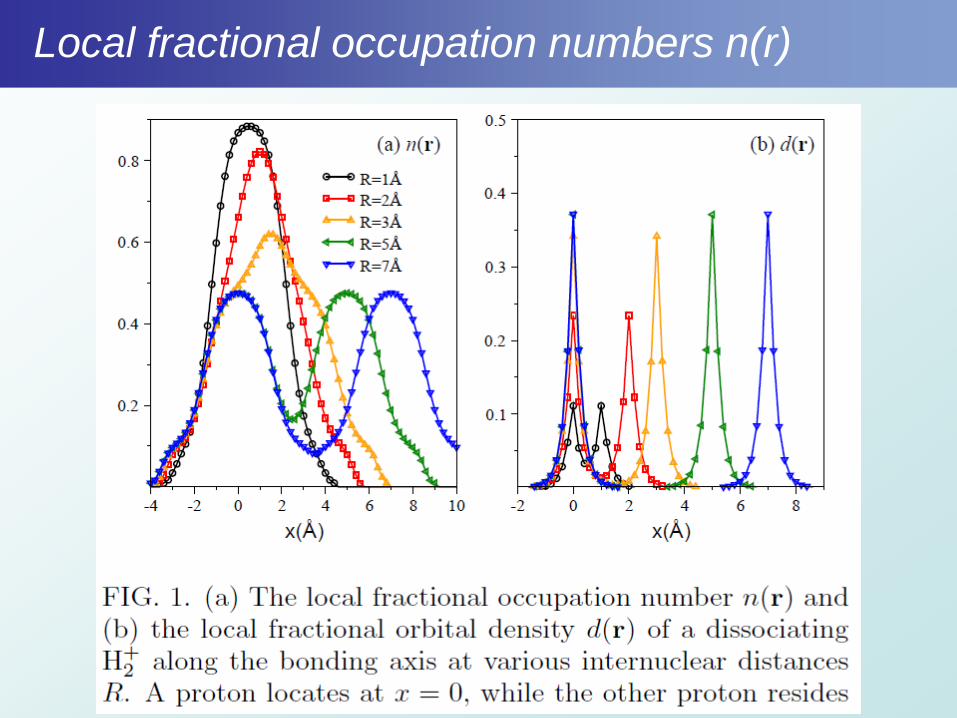
Local fractional occupation numbers n(r)

Local Scaling Correction
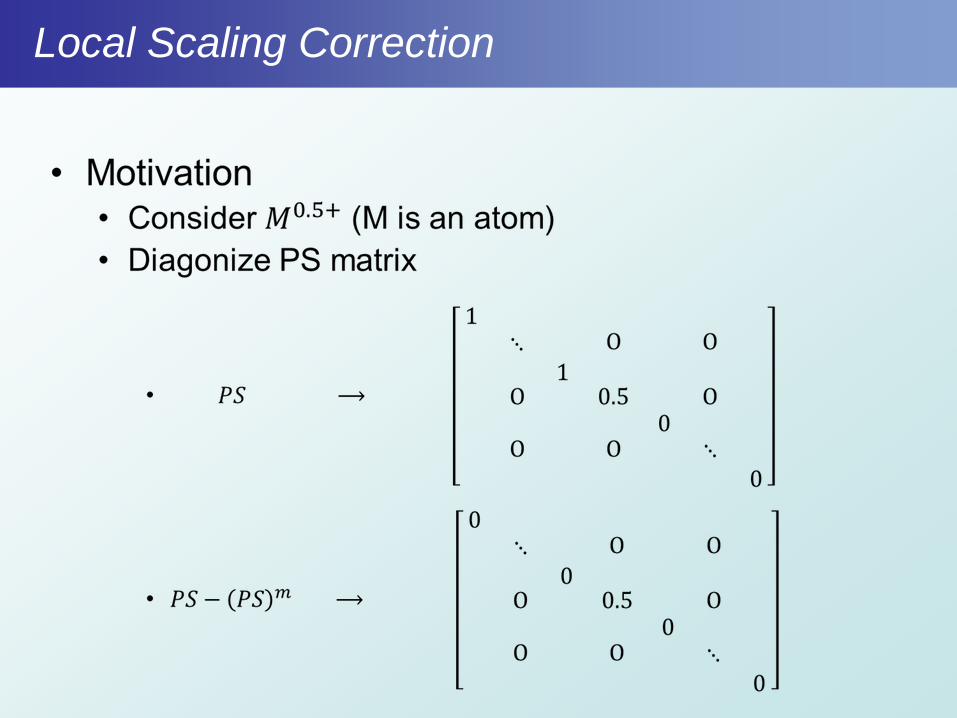
Local Scaling Correction

Local Scaling Correction

Local Scaling Correction
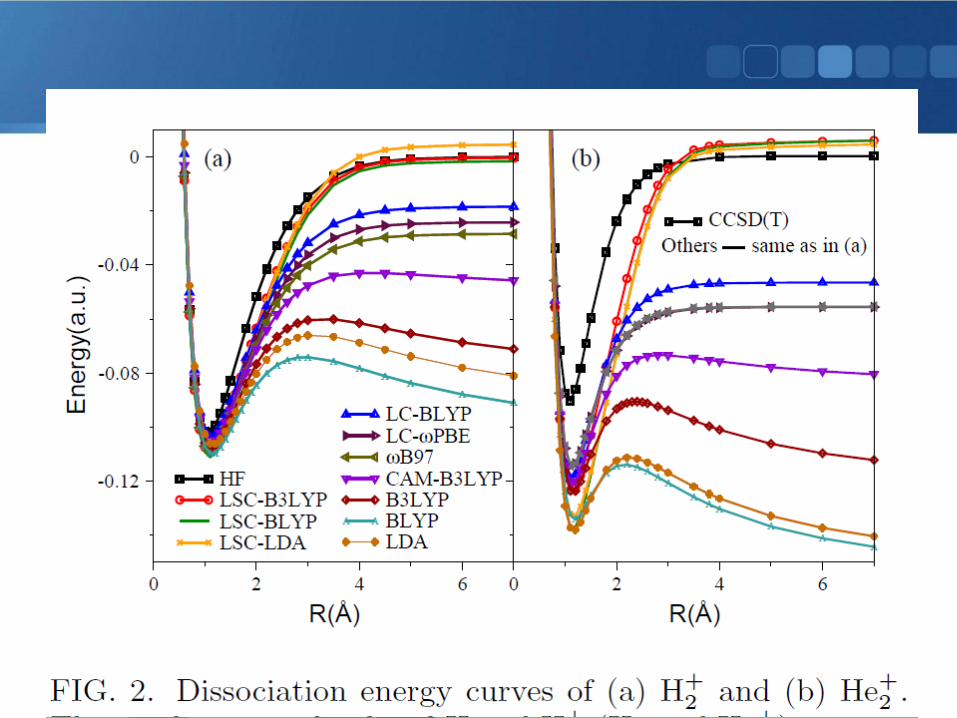
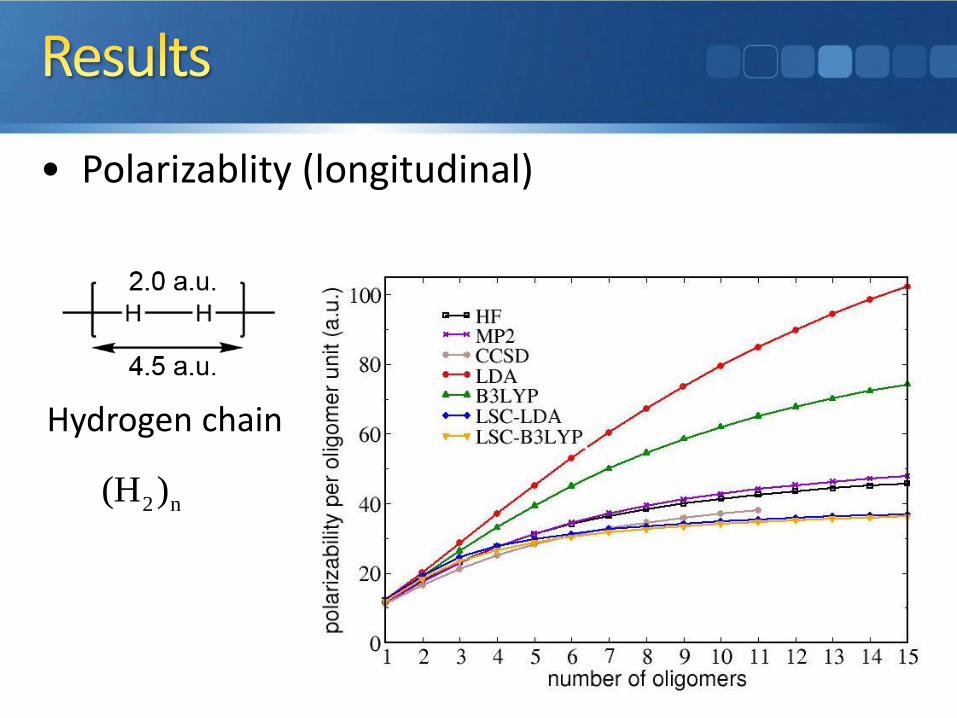
• Polarizablity (longitudinal)
Hydrogen chain
2 n(H )

• Polarizablity (longitudinal)
Polyethilene (PY)
2 nH(C ) H
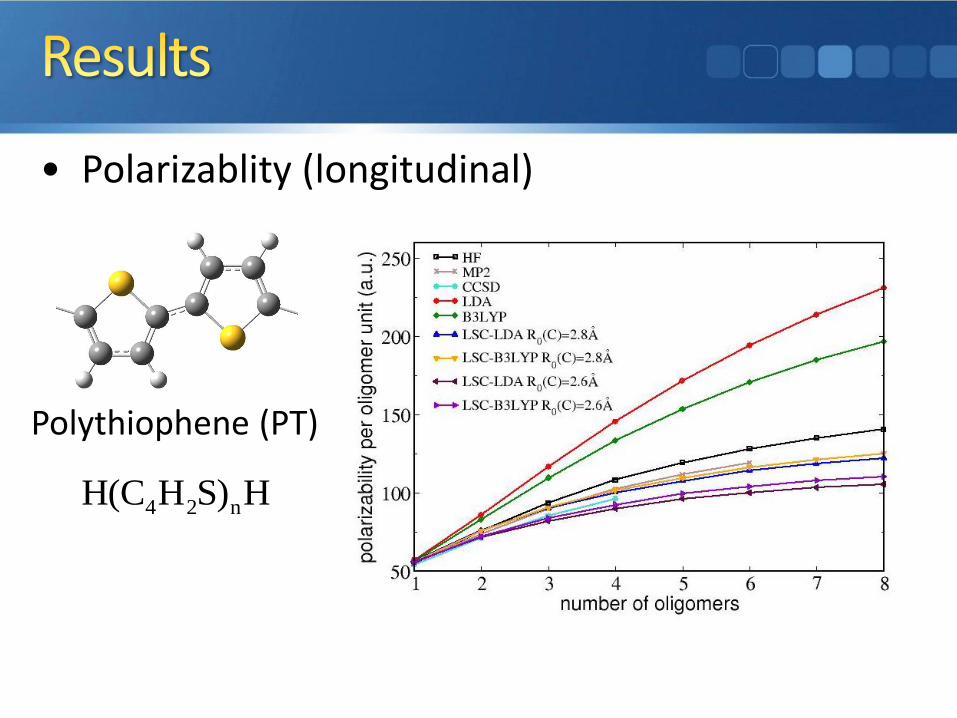
• Polarizablity (longitudinal)
Polythiophene (PT)
4 2 nH(C H S) H
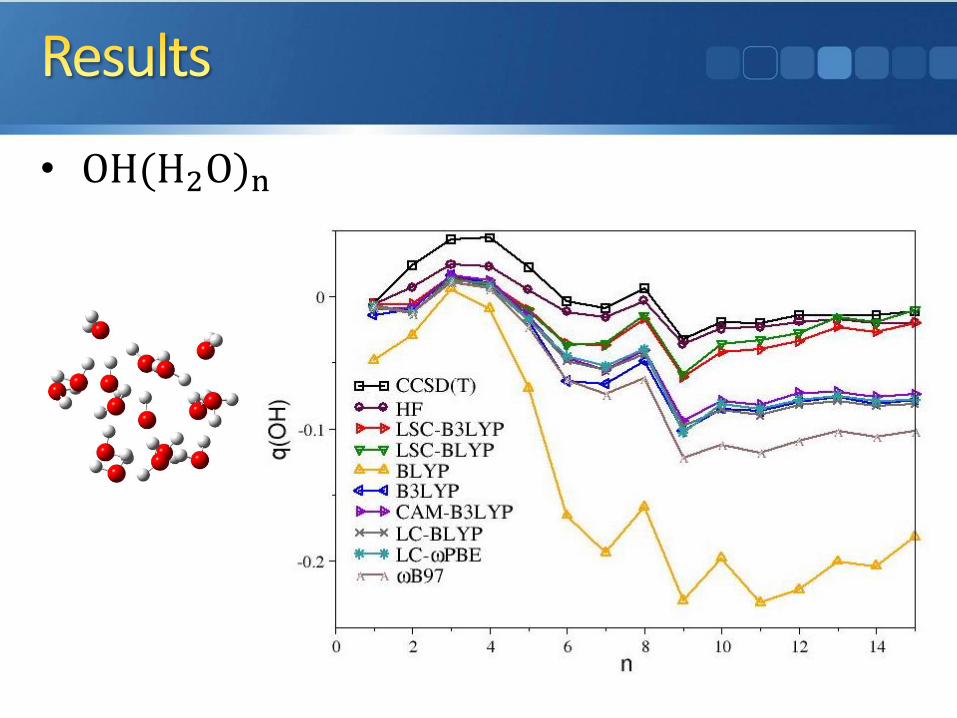
• OH(H2O)n





























