Embed Size (px)
Citation preview

DE NOVO DESIGN OF A THYMIDYLATE KINASE
INHIBITOR

Notes• dTMP is one of the building blocks for DNA synthesis• Enzyme inhibition inhibits DNA synthesis and cell division• Enzyme inhibitors are potential anticancer agents• Inhibitors can be modelled on the substrate or cofactor
Thymidylate Synthase Thymidylate Synthase
HN
N
O
O
O
HOH
OP
Thymidylatesynthase
5,10-Methylene-tetrahydrofolate
dUMP
HN
N
O
O
O
HOH
OP
CH3
dTMP
Dihydrofolicacid
EnzymeEnzyme ReactionReaction EnzymeEnzyme ReactionReaction

Notes• Provides a 1-C unit for biosynthetic pathways• Inhibitors can be based on the cofactor structure• Difficult to gain selectivity between enzymes using the same
cofactor
HN
N NH
N
N
O
N
H CO2H
CO2H
O
H2N
5,10-Methylenetetrahydrofolate
Dihydrofolic acid
HN
N NH
HN
HN
O
N
H CO2H
CO2H
O
H2N
CofactorCofactor CofactorCofactor
Thymidylate Synthase Thymidylate Synthase

Notes• The design of a novel inhibitor based on the structure of the
binding site • Crystallize target enzyme with known inhibitors• Establish structure by X-ray crystallography• Molecular modelling studies to carry out following• Identify binding site and binding regions• Design structure to fit the binding site• Incorporate functional groups to make binding interactions• Possibility of better selectivity between different targets
De Novo DesignDe Novo Design

Notes• 5-Fluorodeoxyuridylate binds to the substrate binding site• CB 3717 binds to the cofactor binding site
Enzyme inhibitorsEnzyme inhibitors
HN
N
O
O
O
HOH
OH
F
HN
N
N
O
N
H CO2H
CO2H
O
H2N
H
Pteridine
CB 37175-Fluorodeoxyuridylate
De Novo DesignDe Novo Design

Notes• Identifies binding interactions of pteridine ring• Identifies available amino acids and bridging water molecule
Binding interactions for CB 3717Binding interactions for CB 3717
HO
H
O
Arg-21
NH
N
Ala-263
Asp-169O
O
N
N
O
N
RH
H
H
De Novo DesignDe Novo Design
Notes• Remove CB 3717 in silico• Set up a grid in the empty binding site• Place an aromatic CH probe at each grid point• Measure hydrophobic interactions• The binding pocket for the pteridine ring is hydrophobic• Identify a hydrophobic scaffold to fit the pocket• Scaffold must be smaller than binding pocket to allow
introduction of functional groups• Add functional groups to make additional binding interactions
De Novo Design De Novo Design

Notes• Hydrophobic naphthalene group forms van der Waals
interactions with the binding site• Room for additional binding groups
Naphthalene
De Novo Design De Novo Design

Notes• Lactam introduced to allow additional hydrogen bond
interactions with the binding site• Naphthostyryl scaffold
Naphthalene
HN
O 1
85
Naphthostyrylscaffold
De Novo Design De Novo Design

Notes• Amino substituent added to gain access to vacant region• Placed at 5-position for synthetic feasibility• Can vary N-alkyl groups without introducing an asymmetric
centre
Naphthalene
HN
O 1
85
Naphthostyrylscaffold
HN
O
NR2
De Novo DesignDe Novo Design

NotesThe benzyl group mimics the benzene ring of the cofactor
Naphthalene
HN
O 1
85
Naphthostyrylscaffold
HN
O
NR2 HN
O
N
Me
Substitution
De Novo DesignDe Novo Design

Notes• Phenylsulfonylpiperazine group is added for water solubility• Positioned to protrude from binding site• Makes contact with surrounding water• No desolvation penalty
Naphthalene
HN
O 1
85
Naphthostyrylscaffold
HN
O
NR2 HN
O
N
Me
Substitution
HN
O
N
Me
S
N
O
O
NH
De Novo DesignDe Novo Design

Notes• De novo designed inhibitor is now synthesized and tested • Active inhibitor• Co-crystallized with enzyme• Crystal structure determined
Naphthalene
HN
O 1
85
Naphthostyrylscaffold
HN
O
NR2 HN
O
N
Me
Substitution
HN
O
N
Me
S
N
O
O
NH
De Novo DesignDe Novo Design
N
O
NR2
H
N
O
NR2
H
ShiftShift
Water shifted Water shifted
Binding Interactions
IntendedIntended ActualActual
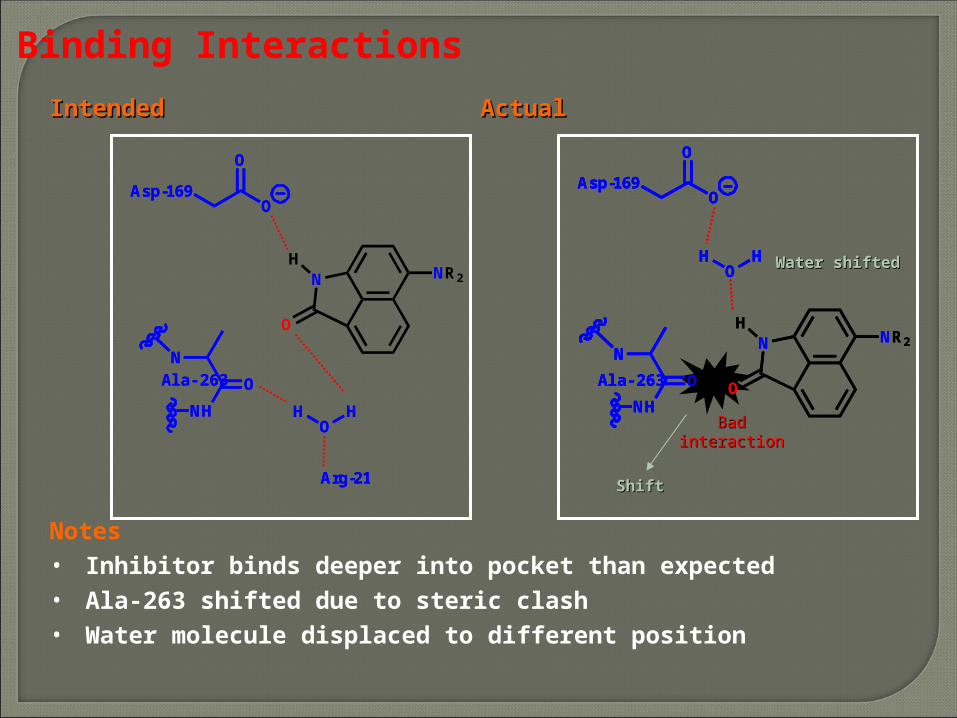
Notes• Inhibitor binds deeper into pocket than expected• Ala-263 shifted due to steric clash• Water molecule displaced to different position
HO
H
O
Arg-21
NH
N
Ala-263
Asp-169O
O
O
NH
N
Ala-263
HO
H
Asp-169O
O
HO
H
O
Arg-21
NH
N
Ala-263
Asp-169O
O
O
NH
N
Ala-263
HO
H
Asp-169O
O
BadBadinteractioninteraction
N
O
NR2
H
O
NH
N
Ala-263
HO
H
Asp-169O
O
Notes• Molecular modelling studies of actual binding interactions• Identifies 4 regions for modification• Possible analogues are overlaid with lead compound to test
whether they fit the binding site • Synthesis of analogues
HN
R2
N
R1
S
R4
O
O
R3
Structure-based Drug DesignStructure-based Drug Design

Region R1
• Substituent fits hydrophobic pocket• Pocket becomes hydrophilic with depth• Polar functional group at the end of the alkyl chain may be
beneficial
• CH2CH2OH has greater binding affinity
• Methyl better than ethyl
HN
R2
N
R1
S
R4
O
O
R3
Structure-based Drug Design

Region R2
• Carbonyl oxygen replaced with amidine group• Capable of binding to Ala-263 instead of repelling it• More basic and protonated - allows ionic interaction and
stronger hydrogen bonding interaction• Increased inhibition• Binding interactions identified from crystal structure
HN
R2
N
R1
S
R4
O
O
R3
Structure-based Drug Design

Binding interactionsBinding interactions
Structure-based Drug DesignStructure-based Drug Design
HO
H
O
Arg-21
NH
N
Ala-263
Asp-169O
O
N
N
HNR2
H
H
Ionic and stronger H-bonding interactions
H-Bonding
Notes• Binding interactions as expected• Ala-263 not displaced• Bridging water molecule not displaced

Region R3
• Small hydrophobic pocket available in the region• Methyl or chloro-substituent both beneficial for activity
HN
R2
N
R1
S
R4
O
O
R3
Structure-based Drug Design

Region R4
Morpholine ring found to be beneficial for selectivity andpharmacokinetics
Structure-based Drug Design
HN
R2
N
R1
S
R4
O
O
R3
Notes• Structures are synthesized containing combinations of the
optimum groups at each position• Amidine is the most important group for enhanced activity• Inclusion of all the optimum groups is not beneficial
HN
R2
N
R1
S
R4
O
O
R3
Structure-based Drug Design

Notes• Amidine, morpholine and methyl group are introduced• No change at R3
• Potent inhibitor (500 x more active)• Structure taken forward for clinical trials
Structure-based Drug Design
N N
CH3
S
N
O
OH2N
O
HN
O
N
Me
S
N
O
O
NH

Principles• De novo design is useful in designing lead compounds for
structure-based drug design• Designed structure should be ‘loose fitting’ and flexible• Allows possibility of different binding modes if binding does
not take place as predicted• Allows scope for further modification and drug optimization• Compounds should be synthetically feasible• Compounds should be in a stable conformation• Desolvation penalties need to be considered
De Novo Design De Novo Design





























