Embed Size (px)
Citation preview

Journal of Food Biochemistry
29
(2005) 1–12.
All Rights Reserved.
©
Copyright 2005, Blackwell Publishing
1
COVALENT IMMOBILIZATION OF BOVINE PHOSPHOLIPASE A
2
SEUNG-HEE NAM and MARIE K. WALSH
1
Department of Nutrition and Food SciencesUtah State University
Logan, UT 84322-8700
Received for Publication October 16, 2003 Accepted for Publication February 2, 2004
ABSTRACT
Bovine phospholipase A
2
(PPLA
2
) was immobilized onto controlled poreglass (CPG) beads using four different immobilization methods. PPLA
2
wasimmobilized directly to CPG using glutaraldehyde without and with reductionby sodium borohydride (PP1 and PP2). Whey protein isolate was directlyimmobilized to CPG as a spacer followed by immobilization of PPLA
2
withoutand with reduction (PP3 and PP4). Immobilized enzyme samples were char-acterized with respect to total amount of protein immobilized, activity with afluorescent substrate and stability over 3 weeks. Among the methods, PP2 andPP4 showed the highest enzyme activity. All methods but PP2 showed asignificant decrease in enzyme activity over 3 weeks. Enzyme immobilized bytwo methods (PP2 and PP4) were compared with soluble enzyme for thehydrolysis of egg phospholipids. Soluble and immobilized enzyme (PP4)resulted in similar free fatty acid values.
INTRODUCTION
Immobilized enzymes are widely used in the food industry and offerseveral advantages over bulk or free enzyme preparations. Advantages includegreater productivity of the enzyme, automation and continuous processing,precise control of the extent of reaction, easy product recovery and the enzymedoes not contaminate the final product (Swaisgood and Horton 1989).
Examples of immobilized enzymes for food applications include glucoseisomerase, beta-galactosidase, proteases and lipases. Immobilized glucoseisomerase is used in the production of high fructose corn syrup because of thehigh costs associated with purification of this enzyme from yeast sources.Immobilized beta-galactosidase is employed for the hydrolysis of lactose insweetened dairy products (Heng and Glatz 1994). Proteases such as trypsin
Blackwell Science, LtdOxford, UKJFBCJournal of Food Biochemistry0145-8884Copyright 2005 by Food & Nutrition Press, Inc., Trumbull, Connecticut.2005291112Short Note
IMMOBILIZATION OF PHOSPHOLIPASE A
2
S.-H. NAM and
M.K. WALSH
1
Corresponding author. TEL: 1-435-797-2177; FAX: 1-435-797-2379; EMAIL: [email protected]

2 S.-H. NAM and M.K. WALSH
(Huang
et al
. 1999; Wang
et al
. 1999) and pepsin (Katchalski-Katzir 1993)are currently being used in an immobilized form for the limited hydrolysis ofmilk and wheat proteins. Improving the characteristics of complex fats can bedone with phospholipase A
2
(PPLA
2
), which selectively hydrolyzes the esterlinkage at the sn-2 position of phospholipids forming lyso-compounds. Thisbioconversion is important in food science because lysophospholipids arestrong bioemulsifiers, enhancing emulsion stability by improving solubility ofhydrophobic and/or hydrophilic compounds in oil and water phases (Madoery
et al
. 1995).The two methods currently used to immobilize phospholipase A
2
arenoncovalent adsorption and covalent cross-linking. Noncovalent adsorption isthe simplest and most economical method, but usually results in high enzymeleaching from the support matrix (Madoery
et al
. 1995, 1999). The result isa loss of enzyme productivity and contamination of the final product. Covalentimmobilization does not involve enzyme leaching from the matrix, but thismethod is more complex and expensive (Ferreira
et al
. 1993, 1994; Shen andCho 1995; Chen and Chen 1998).
Lipase, covalently immobilized using glutaraldehyde, retained a signifi-cant amount of activity with minimal leaching of the enzyme into the finalproduct (Rucka and Turkiewica 1989; Rucka
et al
. 1990; Cho and Rhee 1993).Immobilization techniques incorporating a linker molecule have previouslybeen more successful than direct coupling to the support, and treatment withsodium borohydride has been shown to increase stability in glutaraldehyde-immobilized proteases (Li and Walsh 2000).
Immobilization methods suitable for food applications are limited tothose employing techniques with GRAS (generally recognized as safe) status.Covalent attachment of PPLA
2
to solid supports has involved the use of toxicchemicals such as carbodiimide and hexane (Ferreira
et al
. 1993, 1994; Chenand Chen 1998). Here, we produced immobilized PPLA
2
matrices usingglutaraldehyde, which does have GRAS status (Cho and Rhee 1993).
The objectives of the present study were (1) to develop a covalentlyimmobilized PPLA
2
using glutaraldehyde with or without a linker protein(whey protein isolate) and a reducing agent (sodium borohydride) and (2) tocompare soluble with immobilized enzyme activity using a fluorescent sub-strate and liquid eggs.
MATERIALS AND METHODS
Materials and Chemicals
PPLA
2
(Bovine Pancreas, EC 3.1.1.4) was obtained from M.G. Wald-baum (Gaylor, MN). Other reagents included 3-aminopropyltriethoxylsilane

IMMOBILIZATION OF PHOSPHOLIPASE A
2
3
(Acros, Geel, Belgium); glutaraldehyde, 25%, Grade II, and methyl sulfoxide(Sigma Chem. Co., St. Louis, MO); sodium borohydride (Fisher Scientific,Pittsburgh, PA); and whey protein isolate (WPI) containing 90% protein(Glanbia, Twin Falls, ID). Support material was controlled pore glass (CPG),180–840 microns, 2191 Å average pore diameter (CPG, Inc., Lincoln Park,NJ). Fluorescent substrate specific for PPLA
2
activity [2-(6-(7-nitrobenz-2-oxa-1, 3-diazol-4-yl) amino) hexanoyl-1-hexadecanoly sn-glycero-3-phos-phocholine (NBD C
6
-HPC)] was purchased from Molecular Probes (Eugene,OR).
Enzyme Immobilization
Acid-cleaned CPG-beads were silanized with 3-aminopropyltriethoxysi-lane according to Walsh and Swaisgood (1993) and activated using 2% (v/v)glutaraldehyde in 50 mM sodium phosphate buffer, pH 6.5 for 1 h at roomtemperature. Excess reagent was washed off with distilled water as describedby Li and Walsh (2000). PPLA
2
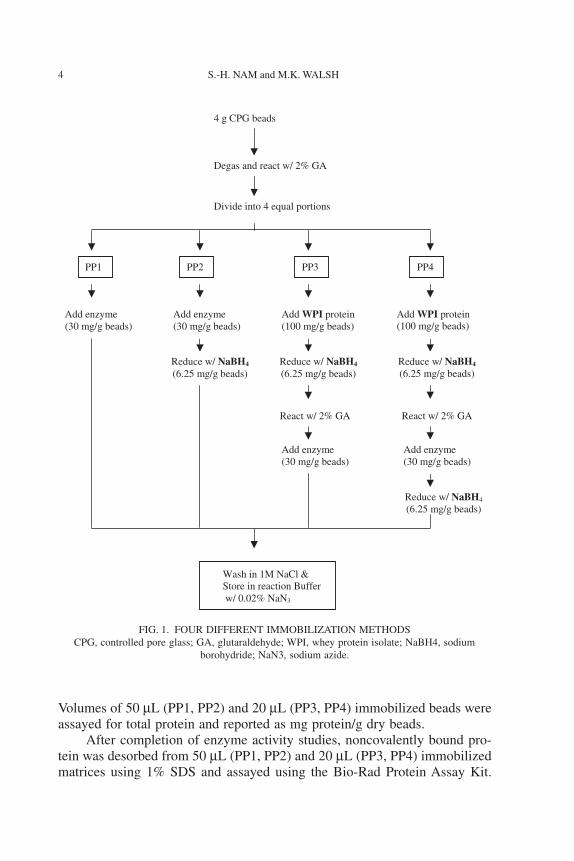
was coupled to the activated beads by directcovalent attachment or using cross-linker proteins (WPI) as described by Liand Walsh (2000). Aliquots of both samples were further reacted with sodiumborohydride to compare the influence of reducing on enzyme activities(Fig. 1).
For direct immobilization, 10 mL enzyme solution (3 mg protein/mL)was added to 1 g beads and allowed to react at room temperature for 4 h onan orbital shaker (PP1). Enzyme-coupled beads were reduced with 6.25 mgsodium borohydride in 10 mL water for 30 min (PP2). Beads were washedwith 10 vol of 1 M NaCl in 50 mM sodium phosphate buffer, pH 6.5 to removefree enzyme (Li and Walsh 2000). For immobilization involving a cross-linker,a solution of 100 mg WPI in 25 mL 50 mM sodium phosphate buffer, pH 6.5,was mixed with the glutaraldehyde activated beads for 1 h at room tempera-ture. Excess protein was removed with distilled water and the WPI-coatedbeads were reduced by the addition of 10 mL water containing 6.25 mgsodium borohydride. The reduced, WPI-coated beads were activated with 2%glutaraldehyde as described above. After completely washing off excess glu-taradehyde with 10 vol of 50 mM sodium phosphate buffer, pH 6.5, 10 mL ofenzyme solution (3 mg protein/mL) was added to 1 g beads and allowed toreact at room temperature for 4 h (PP3). Sodium borohydride (6.25 mg) in10 mL of 50 mM phosphate buffer, pH 6.5, was added slowly to the enzyme-coupled beads before washing with 10 vol of 1 M NaCl in 50 mM sodiumphosphate buffer, pH 6.5, to remove free enzyme (PP4).
The beads were assayed for immobilized protein after immobilizationwas complete. Total amount of protein immobilized onto the CPG was deter-mined directly by a dye-binding assay as described in Bond
et al
. (1992).
4 S.-H. NAM and M.K. WALSH
Volumes of 50
m
L (PP1, PP2) and 20
m
L (PP3, PP4) immobilized beads wereassayed for total protein and reported as mg protein/g dry beads.
After completion of enzyme activity studies, noncovalently bound pro-tein was desorbed from 50
m
L (PP1, PP2) and 20
m
L (PP3, PP4) immobilizedmatrices using 1% SDS and assayed using the Bio-Rad Protein Assay Kit.
FIG. 1. FOUR DIFFERENT IMMOBILIZATION METHODSCPG, controlled pore glass; GA, glutaraldehyde; WPI, whey protein isolate; NaBH4, sodium
borohydride; NaN3, sodium azide.
React w/ 2% GA
Add enzyme(30 mg/g beads)
4 g CPG beads
Degas and react w/ 2% GA
Divide into 4 equal portions
PP1 PP2 PP3 PP4
Add enzyme(30 mg/g beads)
Add WPI protein (100 mg/g beads)
Reduce w/ NaBH4
(6.25 mg/g beads)Reduce w/ NaBH4
(6.25 mg/g beads)
Add WPI protein (100 mg/g beads)
Reduce w/ NaBH4
(6.25 mg/g beads)
Wash in 1M NaCl & Store in reaction Buffer w/ 0.02% NaN3
Add enzyme(30 mg/g beads)
Reduce w/ NaBH4
(6.25 mg/g beads)
React w/ 2% GA
Add enzyme(30 mg/g beads)

IMMOBILIZATION OF PHOSPHOLIPASE A
2
5
Proteins desorbed from the matrices are classified as noncovalently boundenzyme.
Enzyme Activity
Immobilized and soluble enzyme activities were determined using afluorescent substrate (NBD C
6
-HPC) or liquid egg yolks. The fluorescentsubstrate (NBD C
6
-HPC) in 50 mM HEPES buffer (pH 7) was added to UV-pass fluorescent cuvettes containing phospholipase-immobilized glass beadsin 10 mM Tris buffer, pH 8, 100 mM KCl, 4 mM CaCl
2
(with modificationsdescribed by Moreau 1989). The absorbance maximum was 466 nm with anemission of 533 nm. The final concentration of substrate was 10
m
M. Thechange in fluorescence was measured every 5 min for 60 min and the slopeof this plot resulted in an activity unit of
D
RFU/min. For each aliquot of beads(50
m
L of PP1, PP2 and 20
m
L of PP3, PP4) assayed for activity, the dryweight of the beads was also determined to obtain an activity value per grambeads. Activity is expressed as relative fluorescence units
D
RFU/min/g beads.After each 5 min activity measurement, the cuvettes containing the beads wereinverted twice prior to the next measurement. Soluble and immobilizedenzymes were stored for 3 weeks in 10 m
M
Tris (pH 8), 100 KCl, 4 m
M
CaCl
2
buffer containing 0.02% NaN
3
at 4C.Enzyme activity with liquid egg yolks was determined using Easy Eggs,
pasteurized liquid egg product (M.G. Waldbaum, Gaylor, MN). Immobilizedor soluble enzyme was added to 5 g of liquid eggs in 150 mL, Teflon-lined,screw-capped tube and incubated at room temperature with shaking (40 rpm)using a single-platform shaker (Reliable Scientific, Inc., Hernando, MS) for30 min or 120 min. PPLA
2
activity was obtained by manual titration ofreleased fatty acids as described by Helrich (1990) with modifications. Eggfat was first extracted using anhydrous MgSO
4
(5 g) and ethyl ether (60 mL)at 30C for 30 min with occasional shaking. After evaporating ether undervacuum, samples were dried for 1 h at 105C. A phenolphthalein and toluene-methanol mixture (30 mL, 2:1, v/v) was added dropwise to samples. Manualtitration was accomplished potentiometrically with 0.02 N methanolic-potas-sium hydroxide. Activity values are given in free fatty acid (FFA) (mL KOH/g sample).
Immobilized Enzyme Stability
To monitor the stability of the immobilized enzyme preparations, aliquotsof the matrices were assayed once a week for 3 weeks with the fluorescentassay. Beads were washed in 50 mM Tris (pH 8) prior to enzyme analysis.This gives the stability of the enzyme under controlled conditions and pro-vides an indication of stability over storage.

6 S.-H. NAM and M.K. WALSH
Statistical Analysis
A 4
¥
4 factorial design was used to determine the influence of theimmobilization method (PP1, PP2, PP3 and PP4) and time (week 0, 1, 2 and3) on the immobilized enzyme activity. A 3
¥
2 factorial design was used todetermine the influence of enzyme type (soluble, PP2 and PP4) and incubationtime (30 and 120 min) on the enzyme activity on liquid eggs. Samples wereprepared and assayed in triplicate for each factorial experiment. The data wereanalyzed by
MANOVA
(STATISTICA/Mac program; Stat Soft Inc., Tulsa, OK)and results reported to be statistically significant have
P
£
0.05.
RESULTS AND DISCUSSION
Enzyme Immobilization
Four different methods were performed to determine the influence of alinker protein (WPI) and a reducing agent (sodium borohydride) on the immo-bilization efficiency and activity of PPLA
2.
The four immobilization methodswere (1) direct enzyme immobilization (PP1), direct enzyme immobilizationplus reduction (PP2), enzyme immobilization using a linker protein (PP3) andenzyme immobilization using a linker protein plus reduction (PP4). High ionicstrength solution (1 M NaCl) was applied to remove noncovalently attachedenzyme after immobilization. CPG beads were used as the support matrix inthis study because this material possesses a greater surface area per unitvolume than nonporous beads, thus allowing preparation of an immobilizedbiocatalyst with high enzyme activity (Li and Walsh 2000).
We used 10 mM Tris buffer, pH 8.0, for the enzyme reaction as thehighest reported enzyme activity was achieved between pH 7.4 and 8.0(Thuren
et al
. 1987, 1988). It was also reported that the pH optimum is similarfor both soluble (pH 8.3) and immobilized enzyme (pH 8.2) and the immobi-lized enzyme rate is less sensitive to pH changes (Thuren
et al
. 1988). In thisstudy, 4 mM CaCl
2
was added into enzyme–substrate reaction because cal-cium is essential for achieving the maximum activity for both free and immo-bilized PPLA
2
(Kim
et al
. 2001). Madoery
et al
. (1995) also found that 5 mMCaCl
2
in the reaction leads to a 61 to 100% increase in PPLA
2
activity. Thespecific affinity of Ca
2
+
for soluble or immobilized PPLA
2
is in the mM rangeand the values are about 10
2
-
10
3
times higher than the specific affinity of Ca
2
+
for phospholipid (Madoery
et al
. 1999).
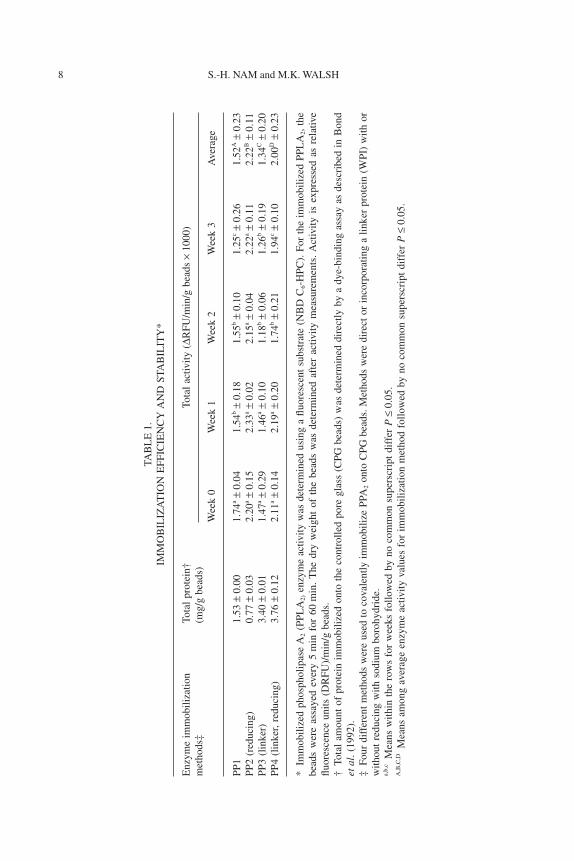
Immobilization Efficiency and Stability
Immobilization efficiency and stability for four immobilization methodsare shown in Table 1. For immobilization efficiency, the total amount of

IMMOBILIZATION OF PHOSPHOLIPASE A
2
7
immobilized protein and the total activity per gram beads were measured atweeks 0, 1, 2 and 3. The total amount of protein immobilized varies primarilywith respect to the use of a linker protein. PP1 and PP2 did not employ alinker protein and show a lower total amount of immobilized protein than PP3and PP4 (Table 1).
Enzyme activity was determined using a fluorometric assay and reportedas
D
RFU/min/g beads. The main effects of immobilization method(
P
= 0.001), time (P = 0.003), and their interaction (P = 0.009) were signifi-cantly different. The average activity for each immobilization method wasdifferent from each other although the average activities of PP2 and PP4approached nonsignificance (P = 0.04). These two methods incorporated areducing agent that reduces the labile imino bonds, resulting in a strongcovalent bond between the enzyme and the matrix. This reducing step mayalso reduce disulfide bonds present in enzymes, resulting in lower activity,although this was not observed with this enzyme (Fleer et al. 1981; Jeyaseelanet al. 2000).
Soluble enzyme activity was 4.98 DRFU/min per milligram protein withno change over a 3-week period. PP2 showed a relatively high activity (2.22DRFU/min/g beads) compared with PP1 (1.52 DRFU/min/g beads). This maybe a result of the amount of enzyme immobilized because immobilized phos-pholipase specific activity decreased sharply with increasing protein loading(Chen and Chen 1998). Enzyme molecules with less steric restrictions maylead to a more favorable spatial arrangement for the enzyme-substrate com-plex formation (Ferreira et al. 1993; Madoery et al. 1995).
Our immobilization method showed about 50% activity retention com-pared with soluble enzyme, which is higher than the previously reportedvalues of 20% (Ferreira et al. 1994) and 20–50% (Ferreira et al. 1993; Chenand Chen 1998), but lower than those of other studies showing 60–70% (Shenand Cho 1995) and 82% activity (Madoery et al. 1995) compared with solubleenzyme.
Higher immobilization efficiency has been obtained with noncovalentadsorption (Madoery et al. 1995, 1999) compared with covalent cross-linking(Ferreira et al. 1993, 1994; Chen and Chen 1998) and with snake venomPPLA2 than pancreatic PPLA2. Covalent cross-linking methods can lead tomore conformational changes than noncovalent adsorption because of themultiple and complex reactions.
We obtained a low coupling yield for which there are at least two possiblereasons. First, the amount of enzyme offered for immobilization to the solidsupport was in excess or second, the immobilization process led to undesirablemodifications of catalytically important residues including lysine, asparticacid and glutamic acid residues in PPLA2 (Chen and Chen 1998). PPLA2
coupled through e-amino groups showed a decrease in the coupling yield
8 S.-H. NAM and M.K. WALSH
TAB
LE
1.
IMM
OB
ILIZ
AT
ION
EFF
ICIE
NC
Y A
ND
STA
BIL
ITY
*
Enz
yme
imm
obili
zatio
nm
etho
ds‡
Tota
l pr
otei
n†(m
g/g
bead
s)To
tal
activ
ity (
DRFU
/min
/g b
eads
¥ 1
000)
Wee
k 0
Wee
k 1
Wee
k 2
Wee
k 3
Ave
rage
PP1
1.53
± 0
.00
1.74
a ± 0
.04
1.54
b ± 0
.18
1.55
b ± 0
.10
1.25
c ± 0
.26
1.52
A ±
0.2
3PP
2 (r
educ
ing)
0.77
± 0
.03
2.20
a ± 0
.15
2.33
a ± 0
.02
2.15
a ± 0
.04
2.22
a ± 0
.11
2.22
B ±
0.1
1PP
3 (l
inke
r)3.
40 ±
0.0
11.
47a ±
0.2
91.
46a ±
0.1
01.
18b ±
0.0
61.
26b ±
0.1
91.
34C ±
0.2
0PP
4 (l
inke
r, re
duci
ng)
3.76
± 0
.12
2.11
a ± 0
.14
2.19
a ± 0
.20
1.74
b ± 0
.21
1.94
c ± 0
.10
2.00
D ±
0.2
3
*Im
mob
ilize
d ph
osph
olip
ase
A2
(PPL
A2)
enz
yme
activ
ity w
as d
eter
min
ed u
sing
a fl
uore
scen
t su
bstr
ate
(NB
D C
6-H
PC).
For
the
im
mob
ilize
d PP
LA
2, th
ebe
ads
wer
e as
saye
d ev
ery
5 m
in f
or 6
0 m
in.
The
dry
wei
ght
of t
he b
eads
was
det
erm
ined
aft
er a
ctiv
ity m
easu
rem
ents
. A
ctiv
ity i
s ex
pres
sed
as r
elat
ive
fluor
esce
nce
units
(D
RFU
)/m
in/g
bea
ds.
†To
tal
amou
nt o
f pr
otei
n im
mob
ilize
d on
to t
he c
ontr
olle
d po
re g
lass
(C
PG b
eads
) w
as d
eter
min
ed d
irec
tly b
y a
dye-
bind
ing
assa
y as
des
crib
ed i
n B
ond
et a
l. (1
992)
.‡
Four
dif
fere
nt m
etho
ds w
ere
used
to
cova
lent
ly i
mm
obili
ze P
PA2
onto
CPG
bea
ds. M
etho
ds w
ere
dire
ct o
r in
corp
orat
ing
a lin
ker
prot
ein
(WPI
) w
ith o
rw
ithou
t re
duci
ng w
ith s
odiu
m b
oroh
ydri
de.
a,b,
cM
eans
with
in t
he r
ows
for
wee
ks f
ollo
wed
by
no c
omm
on s
uper
scri
pt d
iffe
r P
£ 0
.05.
A,B
,C,D
Mea
ns a
mon
g av
erag
e en
zym
e ac
tivity
val
ues
for
imm
obili
zatio
n m
etho
d fo
llow
ed b
y no
com
mon
sup
ersc
ript
dif
fer
P £
0.0
5.

IMMOBILIZATION OF PHOSPHOLIPASE A2 9
because of the affected lysine residues, which played an important role in thecatalysis and/or maintaining enzyme structure (Shen and Cho 1995). Interest-ingly, our immobilization result is inconsistent with those of previous studies,which showed that when the Œ-amino groups are used to couple to a support,the coupling yield is high but the activity of immobilized enzyme towardsubstrates decreases drastically. We show a low coupling yield with a 50%activity level based on bead protein content. Other studies have shown thatthe coupling of lipase carboxylic groups to the solid support improves theactivity retention up to 50% of original activity with low coupling yield (Fleeret al. 1981; Ferreira et al. 1993; Shen and Cho 1995).
For immobilization stability, PP1, PP3 and PP4 showed decreasedenzyme activity over the 3-week testing. The amount of noncovalently boundprotein was determined by treating the matrices with 1% SDS after completionof the activity studies. No noncovalently bound protein was detected, whichmay be a result of the detection limit (minimum 2.5 mg/mL) of the proteinassay
Enzyme Activity on Liquid Eggs
Activity of soluble versus immobilized enzyme is shown in Table 2.Immobilized enzyme preparations that showed the highest activity (PP2 andPP4) were used in this assay. Each enzyme format (soluble and immobilized)was tested at room temperature at two different times, 30 and 120 min.Approximately the same units of enzyme activity (600 units based on thefluorometric assay) were used in each format.
TABLE 2.ENZYME ACTIVITY ON LIQUID EGGS*
Samples Free fatty acids (FFA)†(mL KOH/g sample) 30 min
120 min
No enzyme 14.76a ± 0.89 14.86a ± 1.19Soluble enzyme* 25.85b ± 2.72 42.22c ± 2.43Immobilized enzyme*
PP2 (reducing) 21.40d ± 1.39 36.05e ± 0.65PP4 (reducing, linker) 25.36b ± 1.31 41.86c ± 2.44
* Approximately 600 units of enzyme (soluble or immobilized) were added to 5 g of liquid eggs andincubated for 30 min or 120 min at room temperature.† The FFA value after incubation was determined with the manual titration method using methanolic-potassium hydroxide. Activity values are given in FFA (mL KOH/g sample). Values are the means ofthree experiments ± the standard deviation.a, b, c, d, e Fatty acid values with no common superscript differ P £ 0.05.

10 S.-H. NAM and M.K. WALSH
As shown in Table 2, the FFA value of no enzyme remained constant ateach time point. For both the soluble and immobilized enzymes, the FFA valueincreased with an increase in time. Soluble and immobilized enzyme (PP4)exhibited similar activities; therefore, the immobilized enzyme preparationswere at least as active as the soluble. Enzyme activity on liquid eggs wassignificantly affected by enzyme type (P = 0) and incubation time (P = 0) butnot affected by their interaction (P = 0.09).
CONCLUSIONS
Significant differences were observed among the four immobilizationmethods with respect to total activity. These differences most likely resultedfrom the use of a reducing agent to form a more stable imino bond betweenthe enzyme and the matrix. The use of a cross-linker, in this case WPI, didnot lead to higher enzyme activity using the fluorescent substrate. Theinfluence of the reducing agent did lead to a more stable and highly activematrix.
Activity of each enzyme format (immobilized and soluble) on liquid eggyolks was similar, demonstrating that immobilization of PPLA2 does lead toa matrix that can be used on liquid eggs.
ACKNOWLEDGMENTS
This research was supported by the Western Dairy Center and UtahAgricultural Experiment Station, Utah State University, Logan, UT 84322-4810. Approved as journal paper number 7562.
REFERENCES
BOND, M., PONTOPPIDAN, H. and PEPPER, D.S. 1992. Direct-dye binding– a quantitative assay for solid phase immobilized protein. Anal.Biochem. 200, 195–198.
CHEN, J.P. and CHEN, J.Y. 1998. Preparation and characterization ofimmobilized phospholipase A2 on chitosan beads for lowering serumcholesterol concentration. J. Molec. Catal. B 5, 483–490.
CHO, S. and RHEE, J.S. 1993. Immobilization of lipases for effective inter-estification of fats and oils in organic solvent. Biotechnol. Bioeng. 41,204–210.

IMMOBILIZATION OF PHOSPHOLIPASE A2 11
FERREIRA, J.P.M., SASISEKHARAN, R., LOUIE, O. and LANGER, R.1993. Influence of chemistry in immobilizaton of cobra venom phospho-lipase A2: implications as to mechanism. Biochemistry 32, 8098–8102.
FERREIRA, J.P.M., SASISEKHARAN, R., LOUIE, O. and LANGER, R.1994. Study on the functional subunits of phospholipase A2 by enzymeimmobilization. Biochem. J. 303, 527–530.
FLEER, E.A.M., VERHEIJ, H.M. and HAAS, G.H. 1981. Modification ofcarboxylate groups in bovine pancreatic phospholipase A2. Eur. J. Bio-chem. 113, 283–288.
HELRICH, K. 1990. Acid value of butter fat. In Official Methods of Analysis,5th Ed., pp. 837–838, AOAC Inc., Arlington, VA.
HENG, M.H. and GLATZ, C.E. 1994. Ion exchange immobilization ofcharged beta-galactosidase fusions for lactose hydrolysis. Biotechnol.Bioeng. 44, 745–752.
HUANG, X.L., CATIGNANI, G.L. and SWAISGOOD, H.E. 1999. Modifica-tion of rheological properties of whey protein isolates by limited proteol-ysis. Nahrung 43, 79–85.
JEYASEELAN, K., ARMUGAM, A., DONGHUI, M. and TAN, N.H. 2000.Structure and phylogeny of the venom group I phospholipase A2 gene.Mol. Biol. Evol. 17, 1010–1021.
KATCHALSKI-KATZIR, E. 1993. Immobilized enzymes—learning frompast successes and failures. TIBTECH. 11, 64–69.
KIM, J., LEE, C., OH, J. and B. 2001. Production of egg yolk lysolecithinwith immobilized phospholipase A2 Enzyme Microbial. Technol. 29,587–592.
LI, X. and WALSH, M.K. 2000. Influence of limited proteolysis with immo-bilized or soluble enzymes on the whiteness of skim milk. J. FoodBiochem. 24, 265–274.
MADOERY, R., GATTONE, C.G. and FIDELIO, G. 1995. Bioconversion ofphospholipids by immobilized phospholipase A2. J. Biotechnol. 40, 145–153.
MADOERY, R., GATTONE, C.G. and FIDELIO, G. 1999. The effect ofphospholipase A2 immobilization upon calcium interaction: a kineticstudy. J. Biochem. 126, 1060–1066.
MOREAU, R.A. 1989. An evaluation of NBD-phospholipids as substrates forthe measurement of phospholipase and lipase activities. Lipids 24, 691–699.
RUCKA, M. and TURKIEWICA, B. 1989. Hydrolysis of sunflower oil bymeans of hydrophobic membrane with lipolytic activity. Biotechnol.Letts. 11, 167–172.
RUCKA, M., TURKIEWICA, B. and ZUK, J.S. 1990. Polymeric membranesfor lipase immobilization. JAOCS. 76, 887–889.

12 S.-H. NAM and M.K. WALSH
SHEN, Z. and CHO, W. 1995. Highly efficient immobilization of phospholi-pase A2 and its biomedical applications. J. Lipid Res. 36, 1147–1151.
SWAISGOOD, H.E. and HORTON, H.R. 1989. Immobilized enzymes asprocessing aids or analytical tools. In Biocatalyst in Agricultural Bio-technology, (J.R. Whitaker and P.E. Sonnet, eds.) pp. 242–261, ACSSymposium Series 389, American Chemical Society, Washington, DC.
THUREN, T., VIRTANEN, J.A., VERGER, R. and KINNUNEN, P.K.J.1987. Hydrolysis of 1-palmitoyl-2-[6-(pyren-1-yl) ]hexanoyl-sn-glycero-3-phospholipids by phospholipase A2: effect of the polar head-group.Biochem. Biophys. Acta. 917, 411–417.
THUREN, T., VIRTANEN, J.A., SOMERHARJU, P.J. and KINNUNEN,P.K.J. 1988. Phopholipase A2 assay using an intramolecularly quenchedpyrene-labeled phospholipid analog as a substrate. Anal. Biochem. 170,248–255.
WALSH, M.K. and SWAISGOOD, H.E. 1993. Characterization of a chemi-cally conjugated b-galactosidase bioreactor. J. Food Biochem. 17, 283–292.
WANG, Q., ALLEN, J.C. and SWAISGOOD, H.E. 1999. Binding of lipo-philic nutrients to beta-lactoglobulin prepared by bioselective adsorption.J. Dairy Sci. 82, 257–264.






![Review Article: Immobilized Molecules Using Biomaterials ... · Cao L, 2005 [5]. 1.4.2. Covalent Binding . This method of immobilization involves formation of a covalent bond between](https://img.dokumen.tips/doc/110x75/5c024fdd09d3f2fa038de969/review-article-immobilized-molecules-using-biomaterials-cao-l-2005-5.jpg)






















