Embed Size (px)
Citation preview

ANALYTICAL BIOCHEMISTRY 237, 109–114 (1996)ARTICLE NO. 0207
Cloning Differentially Expressed Genesby Linker Capture Subtraction
Meiheng Yang and Arthur J. SytkowskiLaboratory for Cell and Molecular Biology, Division of Hematology and Oncology, New England Deaconess Hospital,Department of Medicine, Harvard Medical School, Boston, Massachusetts 02215
Received December 8, 1995
probes, subtractive hybridization utilizing DNA/DNAWe have developed a simple and effective method, hybrids or DNA/RNA hybrids, RNA fingerprint, and
designated linker capture subtraction (LCS), for clon- differential display (1–5). Recently, PCR-coupled sub-ing differentially expressed genes between two cell tractive processes have been reported (6–12). Each oftypes or between cells treated in two different ways. these methods has achieved some success and each hasIn the first step of the method, two mRNA populations some inherent limitations. Differential display (5) hasare converted to double-stranded cDNAs, fragmented, problems of ‘‘false positives,’’ redundancy, and under-and ligated to linkers for PCR amplification. In the representation of certain mRNA species. cDNA–RDAsecond step, the linkered DNA (tester) from one mRNA (12) is a labor-intensive process, and its efficiency re-population is hybridized to an excess of the unlinkered mains to be evaluated.DNA (driver) from the other mRNA population, fol- We have sought to overcome some of these problemslowed by incubation with mung bean nuclease which
and have now developed a method designated as linkerdigests single-stranded DNA specifically. This leavescapture subtraction (LCS).1 This method is related op-only tester–tester homohybrids to be amplified byerationally to RDA (10), that is, subtraction coupled toPCR in the following step, so as to achieve an enrich-PCR amplification. However, it does not rely on a ki-ment of tester-specific sequences. The amplified PCRnetic mechanism of enrichment as does RDA. Rather,products are then used as tester for another round ofit achieves enrichment by specifically preserving PCR-subtraction. The process of subtraction is carried outpriming sites of target sequences, using mung beanthree times, and the final PCR products are insertednuclease as the mediator. Also, it is a much less labor-into a vector for clonal analysis. We have used theintensive process. We have applied LCS to the humanstrategy to begin to clone and identify the genes ex-prostate cancer cell lines LNCaP and PC-3, which havepressed differentially between the human prostate
cancer cell lines LNCaP and PC-3, which have differ- different tumorigenic and metastatic potentials. It hasent tumorigenic and metastatic potentials. We demon- resulted in the rapid and effective isolation of genesstrated strong enrichment of target sequences. We also expressed differentially between the two cell lines.report the identities of two of the genes expressed dif-ferentially in these cell lines. One is prostate-specific MATERIALS AND METHODSantigen (PSA) which is known to be expressed in
Cell Culture and cDNA PreparationLNCaP but not in PC-3. The other is vimentin, the dif-ferential expression of which has not been reported Human prostate cancer lines LNCaP and PC-3 cellspreviously in these prostate cancer cells. q 1996 Academic (American Type Culture Collection, Rockville, MD) werePress, Inc. cultured in RPMI 1640 medium with 10% fetal bovine
serum, 95% air/5% CO2 at 377C. Total RNA was isolatedby a guanidinium thiocyanate/phenol method (13).Poly(A)/ RNA was selected through oligo(dT)25–Dyna-The isolation and identification of differentially ex-beads (Dynal Inc., Lake Success, NY). cDNA was synthe-pressed genes is of great importance in the study ofsized from 2 mg of poly(A)/ RNA using a SuperScriptembryogenesis, cell growth and differentiation, and
neoplastic transformation. A variety of methods havebeen employed to achieve this end. They include differ- 1 Abbreviations used: LCS, linker capture subtraction; AP, ampli-
fication primer; PSA, prostate-specific antigen.ential screening of cDNA libraries with selective
1090003-2697/96 $18.00Copyright q 1996 by Academic Press, Inc.All rights of reproduction in any form reserved.
AID AB 9555 / 6m15$$$201 04-24-96 22:37:21 aba AP: Anal Bio

YANG AND SYTKOWSKI110
Choice System (GIBCO, Gaithersburg, MD) according 20 ml of pH-shift buffer A (1 mM ZnCl2, 10 mM Naacetate, pH 5.0) was added and the solution was di-to the manufacturer’s instruction. Oligo(dT)12–18 was
used to prime the first strand of cDNA synthesis. vided into five aliquots. They were incubated with 0,0.85, 1.75, 3.5, or 7 U of mung bean nuclease (Promega),respectively, 377C, 30 min. To each sample 80 ml of pH-Restriction Enzyme Digestion, Linker Ligation, andshift buffer B (10 mM Tris–HCl, pH 8.9, 50 mM KCl,PCR Amplificationand 0.1% Triton X-100) was added. They were heatedThe double-stranded cDNA was digested with AluI (957C, 5 min) to inactive the mung bean nuclease. Thenand RsaI and then ligated with a double-stranded oligo- 20 ml of enzyme solution (10 mM Tris–HCl, pH 8.9, 50deoxynucleotide linker, which had a blunt end and a mM KCl, and 0.1% Triton X-100, 1 mM dNTPs, 5 mM2-base 3 * protruding end: AP, 10 mM MgCl2 and 5 U Taq polymerase (Promega))was added. The PCR reaction was run under the same
ACTCTTGCTTGGACGAGCTCT conditions as above. Each sample was electrophoresedon 2% agarose gel. The sample with the most abundantACTGAGAACGAACCTGCTCGAGA-pproducts of 0.1–1.0 kb was selected as tester for an-other round of subtraction. The above process was re-The linker contained an AluI/SacI site near the bluntpeated twice with 2.5 mg of driver DNA and 0.025 mg ofend as indicated. The top strand was designated thetester DNA. To test for enrichment of target sequences,amplification primer (AP). The bottom strand wasPCR products derived from subtraction cycles 0–3 werephosphorylated at the 5* end. The linker was preparedelectrophoresed on 4% NuSieve agarose (FMC, Rock-by annealing the two strands. An equal mass of eachland, ME), transferred to GeneScreen Plus membraneof the two oligodeoxynucleotides was combined. The(Dupont/NEN, Boston, MA), and probed with the ran-mixture was heated to 907C for 2 min and then alloweddom-labeled PCR products (with linkers removed) ofto cool to room temperature. The ligation was carriedthe third round of subtraction (see Results).out by mixing 1 mg of cut cDNA, 5 mg of linker, 11
ligation buffer (Stratagene, La Jolla, CA) and 4 WeissConstruction of Subtractive Library and Clonalunits of T4 DNA Ligase (Stratagene) in a volume of 10
Analysisml, 87C, 20 h. The reaction mixture was electrophoresedthrough a 2% low-melt agarose gel to remove the unli- After three rounds of subtraction, the PCR-amplifiedgated linkers. The linker-ligated cDNA fragments in products were purified (Gene Clean), digested withthe size range of 0.1–1.0 kb were collected. SacI, inserted into dephosphorylated pGEM-7Zf(/)
Linker-ligated cDNA fragments in agarose were am- (Promega) at the SacI site, and transformed into com-plified directly by PCR using AP as primer. The reac- petent Escherichia coli JM109 cells. We prepared twotion (100 ml) contained 10 mM Tris–HCl, pH 8.9, 50 subtractive libraries:mM KCl, 0.1% Triton X-100, 200 mM dNTPs, 1 mM AP,2 mM MgCl2, 1 ml of melted agarose, and 5 U Taq
LNCaP (tester)/PC-3 (driver) Å ‘‘L-P,’’ andpolymerase (Promega), running for 30 cycles (947C, 1min; 557C, 1 min; 727C, 1 min). The amplified cDNA PC-3 (tester)/LNCaP (driver) Å ‘‘P-L’’ in this way.fragments were purified using a Gene-Clean kit(Bio101, Vista, CA) and were used as the initial mate-
Forty-eight white colonies from each library wererial for subtractive hybridization.picked randomly and inoculated into LB / Amp me-dium in individual wells of a 96-well plate. Two replica
Subtractive Hybridization DNA dot-blots were prepared on GeneScreen Plus fil-ters using 25 ml of bacterial cells per well. The replicaTwenty micrograms of PCR-amplified driver DNA
was digested with AluI (50 U), 377C, 2 h, followed by dot-blots were processed according to Brown and Knud-son (14) and probed with random-labeled driver DNAsSacI (50 U), 1 h to cleave the linker so that driver
DNA could not be amplified later. After digestion, the from LNCaP and PC-3, respectively.Candidate-positive colonies were boiled for 5 min inproducts were purified using Gene-Clean.
The digested driver DNA (2.5 mg) and nondigested 20 ml H2O and centrifuged. DNA in the supernatantwas amplified by PCR using universal vector primertester DNA (0.1 mg) were mixed, vacuum-dried, and
redissolved in 4 ml of a buffer containing 15 mM N-(2- T7 and SP6 for 20 cycles of 947C, 1 min; 557C, 1 min;727C, 1 min. The PCR products were electrophoresedhydroxyethyl)piperazine-N *-(3-propane sulfonic acid)
(EPPS), pH 8.0, with 1.5 mM EDTA, overlaid with min- on 2% agarose. The desired bands were excised andpurified (Gene Clean). The products were subjected toeral oil, and denatured by heating for 5 min at 1007C.
One microliter of 5 M NaCl was added, and the DNA direct DNA sequencing (15), and were employed to pre-pare probes for Northern blot analyses.was hybridized for 20 h at 677C. After hybridization,
AID AB 9555 / 6m15$$$202 04-24-96 22:37:21 aba AP: Anal Bio

LINKER CAPTURE SUBTRACTION 111
of subtraction. The process of subtractive hybridiza-tion, mung bean nuclease digestion, and PCR amplifi-cation is carried out three times. Finally, the PCR prod-ucts of the third round of subtraction are used toprepare a subtraction library by inserting them into avector.
Cloning and Analysis of Differentially ExpressedGenes between the Human Prostate Cancer CellLine LNCaP and PC-3
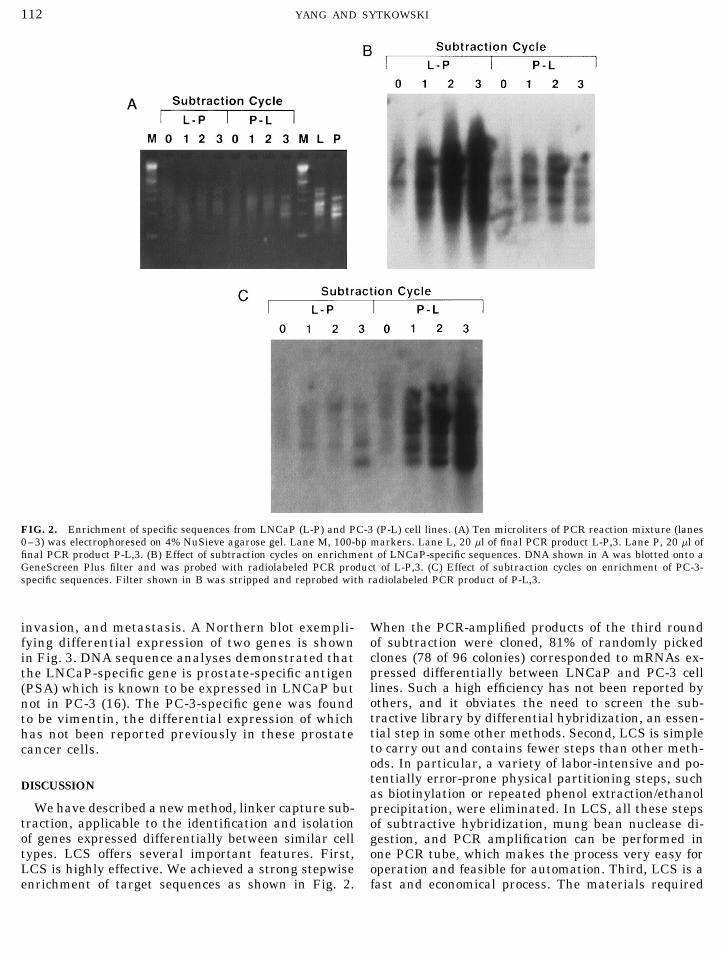
We have used the strategy to begin to clone and iden-tify the genes expressed differentially between the hu-man prostate cancer cell lines LNCaP and PC-3, whichhave different tumorigenic and metastatic potentials.After three cycles of subtraction, the PCR productswere cleaved with SacI, inserted into pGEM-7Zf(/),and transformed into E. coli JM109 cells. Figure 2Ashows the electrophoretic analysis of the PCR-ampli-fied DNA derived from subtraction cycles 0–3. Theoriginal unsubtracted DNAs from LNCaP (lane L-P, 0)and PC-3 (lane P-L, 0) moved as a smear between 0.1and 1.0 Kb. As subtraction rounds were performed, dis-tinct bands were seen (lanes L-P, 1–3; P-L, 1–3). Theintensity and resolution of these bands increased pro-gressively with successive subtraction. When labeledPCR products of the third round of subtraction wereelectrophoresed on a 6% sequencing gel, 50–60 bandscould be seen (not shown). DNA of the agarose gel ofFig. 2A was transferred to Gene Screen Plus mem-brane, and probed with the labeled PCR products ofthe third round of subtraction L-P, 3 (Fig. 2B) or P-L,FIG. 1. Schematic illustration of linker capture subtraction.3 (Fig. 2C). The results indicate strong enrichment ofdifferentially expressed sequences.
After three rounds of subtraction, the PCR-ampli-RESULTSfied products were inserted into pGEM-7Zf(/) and
Experimental Strategy transformed into E. coli JM109 cells. We randomlypicked 48 white colonies from each of the librariesThis method is designed to isolate genes expressed
differentially between two cell types or between cells and grew them in LB medium in individual wellsof a 96-well plate. Two replica DNA dot-blots weretreated in two different ways (Fig. 1). In the first step,
both tester DNA and driver DNA are prepared. This is prepared and probed with the labeled driver DNAsfrom LNCaP (lane P-L, 0) and PC-3 (lane L-P, 0),accomplished by digesting the double-stranded cDNA
with restriction enzymes of choice, ligating the frag- respectively. A comparison of the hybridization in-tensity of a clone in two replica membranes revealedments to linkers, and carrying out a PCR reaction with
linker sequence as primer. The driver DNA is digested the relative abundance of the transcript in the twocell types. Over two-thirds of the selected cloneswith restriction enzymes to remove the linker se-
quence. In the second step, the linkered tester DNA is demonstrated significant differences in abundance.We tested clones further with Northern blot and se-hybridized to an excess of driver DNA (with linkers
removed) followed by incubation with mung bean quence analyses. From 78 colonies, 15 distinct cloneswere identified which correspond to mRNAs ex-nuclease which digests single-stranded DNA specifi-
cally. This leaves only linkered tester–tester homohy- pressed differentially between LNCaP and PC-3 celllines. The extent of differential expression rangedbrids and unlinkered homo- and heterohybrids. In the
following step, the linkered tester–tester homohybrids from severalfold to ú100-fold. In addition to fivenovel genes, the identified genes included some veryare amplified by PCR with linker sequence as primer
to fulfill the first round of enrichment. The amplified interesting known genes which are or may be in-volved in signal transduction, tumor growth, tumorPCR products are then used as tester for another round
AID AB 9555 / 6m15$$$202 04-24-96 22:37:21 aba AP: Anal Bio
YANG AND SYTKOWSKI112
FIG. 2. Enrichment of specific sequences from LNCaP (L-P) and PC-3 (P-L) cell lines. (A) Ten microliters of PCR reaction mixture (lanes0–3) was electrophoresed on 4% NuSieve agarose gel. Lane M, 100-bp markers. Lane L, 20 ml of final PCR product L-P,3. Lane P, 20 ml offinal PCR product P-L,3. (B) Effect of subtraction cycles on enrichment of LNCaP-specific sequences. DNA shown in A was blotted onto aGeneScreen Plus filter and was probed with radiolabeled PCR product of L-P,3. (C) Effect of subtraction cycles on enrichment of PC-3-specific sequences. Filter shown in B was stripped and reprobed with radiolabeled PCR product of P-L,3.
invasion, and metastasis. A Northern blot exempli- When the PCR-amplified products of the third roundof subtraction were cloned, 81% of randomly pickedfying differential expression of two genes is shownclones (78 of 96 colonies) corresponded to mRNAs ex-in Fig. 3. DNA sequence analyses demonstrated thatpressed differentially between LNCaP and PC-3 cellthe LNCaP-specific gene is prostate-specific antigenlines. Such a high efficiency has not been reported by(PSA) which is known to be expressed in LNCaP butothers, and it obviates the need to screen the sub-not in PC-3 (16). The PC-3-specific gene was foundtractive library by differential hybridization, an essen-to be vimentin, the differential expression of whichtial step in some other methods. Second, LCS is simplehas not been reported previously in these prostateto carry out and contains fewer steps than other meth-cancer cells.ods. In particular, a variety of labor-intensive and po-tentially error-prone physical partitioning steps, suchDISCUSSIONas biotinylation or repeated phenol extraction/ethanol
We have described a new method, linker capture sub- precipitation, were eliminated. In LCS, all these stepstraction, applicable to the identification and isolation of subtractive hybridization, mung bean nuclease di-of genes expressed differentially between similar cell gestion, and PCR amplification can be performed intypes. LCS offers several important features. First, one PCR tube, which makes the process very easy forLCS is highly effective. We achieved a strong stepwise operation and feasible for automation. Third, LCS is a
fast and economical process. The materials requiredenrichment of target sequences as shown in Fig. 2.
AID AB 9555 / 6m15$$$202 04-24-96 22:37:21 aba AP: Anal Bio

LINKER CAPTURE SUBTRACTION 113
such as DMSO, formamide, or glycerol would achieveother representations of the mRNA population of thecells under study. Moreover, thermostable DNA poly-merases from different vendors may also give differentrepresentations because these enzymes appear to havedifferent efficiencies in amplifying large-size frag-ments.
We added the same linker to the tester and driverin our experiment. Previously, Balzer and Baumlein(17) reported the addition of different linkers to thetester and driver in order to avoid the necessity of re-striction enzyme digestion of driver and to eliminatecontamination of residual linkered driver. We foundthat the addition of different linkers gave an unequiva-lent representation of starting mRNAs for tester anddriver, probably due to sequence-contexting of primersor so-called PCR ‘‘bias,’’ a tendency to amplify somesequences preferentially. Therefore, we found it advan-FIG. 3. Linker capture subtraction reveals differential expression
of prostate-specific antigen (PSA) and vimentin in LNCaP (L) and tageous to use the same linker for both tester andPC-3 (P) cells. Northern analysis. Total RNA from either cell was driver rather than different linkers. While it is trueelectrophoresed and probed with radiolabeled cDNA of two differen- that the driver linkers cannot be completely removedtially expressed clones isolated by LCS. Top, autoradiograms; bot-
by a restriction enzyme, we believe that our protocoltom, ethidium bromide-stained gels.itself has a mechanism to eliminate the contaminationproblem of the residual linkered driver. Since the un-linkered driver is present at high excess in the reaction,are kept to a minimum. The procedure, from isolation
of mRNA to construction of the subtractive library, can the residual linkered driver should be driven out bythe unlinkered driver. Of course, too high a level ofbe completed within 1 week.
Unlike RDA (10) which uses a kinetic mechanism of linkered driver would still pose a problem for efficiencyof enrichment. Therefore, we designed a linker withenrichment, LCS achieves enrichment by specifically
preserving PCR-priming sites (linkers) of target se- both AluI and SacI sites included to ensure maximumremoval of linker sequences from driver. Incubationquences. Mung bean nuclease plays a central role in
the process. It removes linkers of all other linkered with AluI first and then SacI was used to avoid incorpo-ration of driver DNA into the library, since the SacIsequences except for tester–tester homohybrids. The
nuclease also digests single-stranded DNA in the hy- site was used for library construction.How to achieve an enrichment for both abundant andbridization solution which is an abundant species that
might otherwise cause high background or even failure rare target genes is always an issue for the methodsof cloning differentially expressed genes. For LCS, weof enrichment. The use of the enzyme appears to be
more reliable and efficient than other physical parti- think that the hybridization time might be the de-termining factor. Kinetically, short hybridization timestioning methods as indicated by the high enrichment
of target sequences and efficient isolation of numerous favor enrichment of more abundant sequences, whilelonger times allow rare sequences to bind. As long asdifferentially expressed genes in our experiments. Exo-
nuclease VII, also specific for ssDNA, could be em- enough time is given for hybridization, rare target se-quences can remain in the reaction. Indeed, moderateployed in the LCS protocol. It has the added advantage
of a pH optimum near to those of the subtractive hy- to rare genes are included in the genes we identified(data not shown). Moreover, as noted by Hubank andbridization and PCR reaction, thus eliminating the
need for pH-shift buffers in the process. Schatz (12), unwanted dominant sequences (i.e., al-ready identified sequences in the reaction) can beThe digestion of double-stranded cDNA with differ-
ent restriction enzymes gives a representation of the driven out by supplementing driver with unlinkeredcorresponding sequences. This would allow less domi-mRNA population. In the example of linker capture
subtraction presented here, we employed AluI and nant species to be isolated. One may also try to normal-ize tester and driver before subtraction. Here we sug-RsaI. Obviously, the use of other enzymes would give
different representations and may result in the isola- gest a procedure based on reassociation kinetics. First,both tester and driver are denatured and hybridizedtion of genes different from those achievable using AluI
and RsaI. Also, the use of different PCR conditions such for a short time (e.g., 1 h). Then, the linkers of hybrids(presumably more abundant sequences) are removedas additional concentrations of magnesium, different
annealing temperatures, and the addition of reagents by restriction enzymes. Finally, the remaining single-
AID AB 9555 / 6m15$$$202 04-24-96 22:37:21 aba AP: Anal Bio

YANG AND SYTKOWSKI114
stranded fraction of DNA is amplified by PCR. Proper ACKNOWLEDGMENTSsampling of target genes that differ in abundance by This work was supported by a NIH NRSA 1 F32 DK 09364 to M.Y.
and U.S. Navy Grant N00014-93-1-0776, N.I.H. Grant DK38841, andonly a few-fold is another issue. By adjustment of theNATO Grant 890509 to A.J.S.tester/driver ratios and by more cycles of subtraction,
these genes should be isolated. REFERENCESWe suggest that linker capture subtraction will be 1. Mather, E. L., Alt, F. W., Bothwell, A. L. M., Baltimore, D., and
Koshland, M. E. (1981) Cell 23, 369–378.generally applicable to experiments such as those re-2. Hedrick, S. M., Cohen, D. I., Nielsen, E. A., and Davis, M. M.ported here as well as to studies of differential gene
(1984) Nature 308, 149–153.expression in cells incubated in the absence or presence3. Davis, R. L., Weintraub, H., and Lassar, A. (1987) Cell 51, 987–of cytokines, growth factors or other biologically active 1000.
molecules. Moreover, the method should also prove use- 4. Welsh, J., Chada, K., Dalal, S. S., Cheng, R., Ralph, D., andful in finding differences between genomic DNAs. Obvi- McColelland, M. (1992) Nucleic Acids Res. 20, 4965–4970.
5. Liang, P., and Pardee, A. B. (1992) Science 257, 967–971.ously, this method will not detect important genes criti-6. Straus, D., and Ausubel, F. M. (1990) Proc. Natl. Acad. Sci. USAcal for biological events whose mRNAs are not
87, 1889–1893.expressed differentially. One should also not expect7. Sive, H. L., and John, T. S. (1988) Nucleic Acids Res. 16, 10937.that in one experiment this method will provide the 8. Wieland, I., Bolger, G., Asouline, G., and Wigler, M. (1990) Proc.
entire array of differentially expressed genes between Natl. Acad. Sci. USA 87, 2720–2724.cell types, although by modification of conditions de- 9. Wang, Z., and Brown, D. D. (1991) Proc. Natl. Acad. Sci. USA
88, 11505–11509.scribed above, it may be possible to achieve this goal.10. Lisitsyn, N., Lisitsyn, N., and Wigler, M. (1993) Science 259,While this article was in preparation, two different
946–951.methods which may reach the same endpoint as LCS11. Zeng, J., Gorski, R. A., and Hamer, D. (1994) Nucleic Acids Res.
were reported. Schena et al. (18) developed a robotic 22, 4381–4385.system for generating high-density microarrays of com- 12. Hubank, M., and Schatz, D. G. (1994) Nucleic Acids Res. 22,
5640–5648.plementary DNA clones. By labeling samples with dif-13. Xie, W. Q., and Rothblum, L. I. (1991) BioTechniques 11, 325–ferent dyes, differences in gene expression could be
327.quantified. Velculescu et al. (19) developed another14. Brown, S. E., and Knudson, D. L. (1991) BioTechniques 10, 719–techniques called serial analysis of gene expression, or 722.
SAGE. It relies on the fact that a sequence as short as 15. Winship, P. R. (1989) Nucleic Acids Res. 17, 1266.nine base pairs is sufficient to identify 95% of human 16. Blok, L. J., Kumar, M. V., and Tindall, D. J. (1995) Prostate 26,
213–224.genes, provided that the sequence is picked from the17. Balzer, H. J., and Baumlein (1994) Nucleic Acids Res. 14, 2853–same place in all the genes surveyed. Both approaches
2854.may allow a broad view of patterns of gene expression18. Schena, M., Shalon, D., Davis, R. W., and Brown, P. O. (1995)simultaneously. In our experience, LCS is at least as Science 270, 467–470.
effective as these new methods, and it affords the ad- 19. Velculescu, V. E., Zhang, L., Vogelstein, B., and Kinzler, K. W.(1995) Science 270, 484–487.vantages of efficiency, simplicity, and practicality.
AID AB 9555 / 6m15$$$202 04-24-96 22:37:21 aba AP: Anal Bio
![WhiB5, a Transcriptional Regulator That Contributes to ... · .bioinfo.cnio.es/]) (41), using the print-tip lowess method after back-ground subtraction. Genes significantly differentially](https://img.dokumen.tips/doc/110x75/608836f405d0b82a904408c1/whib5-a-transcriptional-regulator-that-contributes-to-41-using-the-print-tip.jpg)




























