Embed Size (px)
Citation preview

Journal of Neurology, Neurosurgery, and Psychiatry 1985;48:145-149
Cerebellar degeneration in dominantly inheritedspastic paraplegiaCL SCHOLTZ, M SWASH
From the Departments ofNeuropathology and Neurology, The London Hospital and The London HospitalMedical College, London, UK
SUMMARY The clinical features of five affected members in three generations of a family withdominantly inherited Strumpell's spastic paraplegia are described, together with the pathologicalfindings in two cases. The late presentation and slow progression of the disease encompassfeatures of the types I and II of other authors illustrating the heterogeneous expression of thedisorder. Cerebellar involvement was evident clinically and pathologically.
Hereditary spastic paraplegia is a disorder character-ised by slowly increasing spasticity and weakness ofthe lower limbs.' Since Seeligmuller s original study2and the reports of Strumpell34 and Lorrain5 approx-imately 200 families with the disorder have beendescribed.6" Pathologically it is characterised bydegeneration of the corticospinal tracts and, to alesser extent, of the posterior columns.' 67 It maypresent as a pure form, "Strumpell-Lorrain Dis-ease" , in which degeneration of the corticospi-nal tracts occurs below the decussation of thepyramids,7 or as a complicated form in which there isextension of the disease process into other neuronalsystems.8 In the latter, optic nerve involvement andretinal degeneration have been described andextrapyramidal features, distal atrophy of the upperlimbs, and dementia may also occur.9 Mild cerebel-lar ataxia may occur in the arms in some cases, butataxia of gait is uncommon.'0 Hereditary spasticparaplegia is of adult onset and progression is slow;longevity is usually unimpaired.Two forms of pure dominantly inherited spastic
paraplegia have been recognised.78 In the Type 1form, spasticity of the lower limbs is more markedthan weakness. In this group of patients there is usu-ally a history of delay in developmental milestones,particularly in walking, and the disorder usually pre-sents before the age of 35 years. Progression is slowand disability variable. In the Type 2 form muscleweakness, distal sensory loss and urinary urgency oreven incontinence may be evident. This form usually
Address for reprint requests: Dr M Swash, The London Hospital,Whitechapel, London El 1BB, UK.Received 3 April 1984 and in revised form 19 July 1984.Accepted 21 July 1984
presents after the age of 35 years but progressesmore rapidly resulting in more disability than in theType 1 form. Families affected by a similar disorder,but inherited as an autosomal recessive trait havealso been reported, but this mode of inheritance isuncommon.8 " Many of these latter families showother features, in addition to corticospinal tractinvolvement.8
In this report we describe the clinical andpathological features of. a late-onset, dominantlyinherited form of the disease in which ataxia accom-panied severe spastic paraplegia.
Clinical features
The family tree is illustrated in fig 1.Generation II (six members; one affected)I.1. female, I1.2. female, I1.3. female. Died in infancycause unknown.II.4. female. Died aged 83 yr. She was bedridden for manyyears before her death. Postmortem examination carriedout 40 years ago at another hospital showed degenerationof the lateral columns and spinocerebellar tracts. Nofurther details are known'11.5. female. Died in childhood.11.6. male. Died aged 36 yr of pulmonary tuberculosis.
Generation III (six members; two affected)111.1. female. Died aged 60 yr of pneumonia and renaldisease.111.2. female. Presented aged 50 yr with gradually progres-sive spastic paraplegia. She required a wheelchair by theage of about 70 years. No cerebellar ataxia was observed inthe arms. Eye movements and vision were normal. Shedied at the age of 84 years. Postmortem examination wasperformed.111.3. female. Died aged 5 yr of meningitis.
145
Protected by copyright.
on 30 May 2018 by guest.
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp.48.2.145 on 1 February 1985. D
ownloaded from
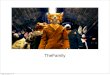
T CLINICAL FEATURES NOT KNOWN
1 2 3 4 5 6
0 0 N 0
1 2 3 4 5
L Li
IV 1 2 3lE3s 0 &
6
.0 0 0 Ts .0
4 5 6 7 8 9
0 0
Male OI) U)riUa fe ct d0A f ct ed JDiabeticFemale Unaffected Affected
Fig 1 Genealogical table offamily.
111.4. male. Died aged 2 yr of meningitis.111.5. male. Died aged 11 yr of diabetes mellitus.111.6. male. Died aged 76 yr. In the 5th decade he com-plained of unsteadiness of gait and weakness, and thisprogressed slowly. He was completely disabled by the ageof 63 yr and died suddenly of myocardial infarction. Post-mortem examination was not carried out.
Scholtz, SwashGeneration IV (nine members; two affected)In this generation no neurological disease has developed inthe offspring of Case 111. 1, but two have diabetes mellitus.IV.1. male. Died aged 70 yr of diabetes mellitus.IV.2. female. Died aged 51 yr of myocardial infarction.IV.3. female. Died aged 63 yr of diabetes mellitus.There are four offspring of case 111.2. of whom one has
neurological disease.IV.4. female. Died aged 71 yr. No neurological symptoms.IV.5. male. Now aged 69 yr. This patient, a physician,presented at the age of 47 yr because he was less coordi-nated and because he noted that his toes were catching onthe ground. Examination showed little abnormality at thistime but in the next ten years he slowly developed cerebel-lar ataxia in the legs, and to a lesser extent in the arms,accompanied by extensor plantar responses, mild spastic-ity, and increased reflexes in the legs. Strength in the legswas normal and sensation was intact. At the age of 59 yrthere was mild cerebellar ataxia in the arms but in the legsthere was marked spasticity with ataxia of gait, withoutweakness; the knee jerks were present and the plantarresponses were extensor. Slight urgency of defaecation andmicturition with occasional incontinence of urine werenoted. There was distal sensory impairment in the legs, andthe ankle jerks were absent. The fundi were normal. Theglucose tolerance test was normal. Although the disorderhas slowly progressed his disability has remained moderateand he is mobile with a walking frame. There is no intellec-tual impairment.
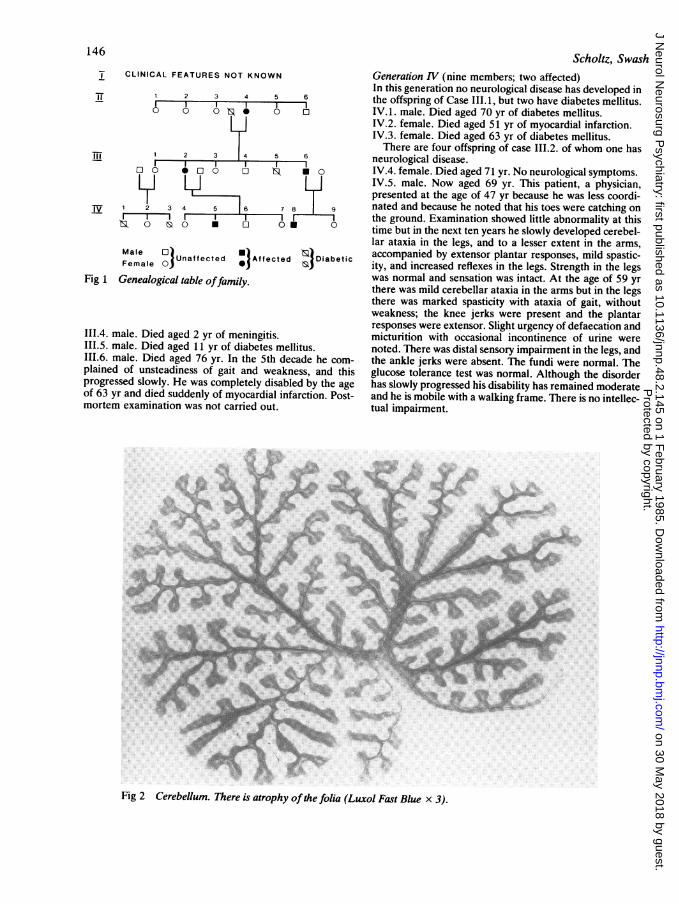
Fig 2 Cerebellum. There is atrophy of the folia (Luxol Fast Blue x 3).
146
--gi,10.
Protected by copyright.
on 30 May 2018 by guest.
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp.48.2.145 on 1 February 1985. D
ownloaded from
Cerebellar degeneration in dominantly inherited spastic paraplegia 147
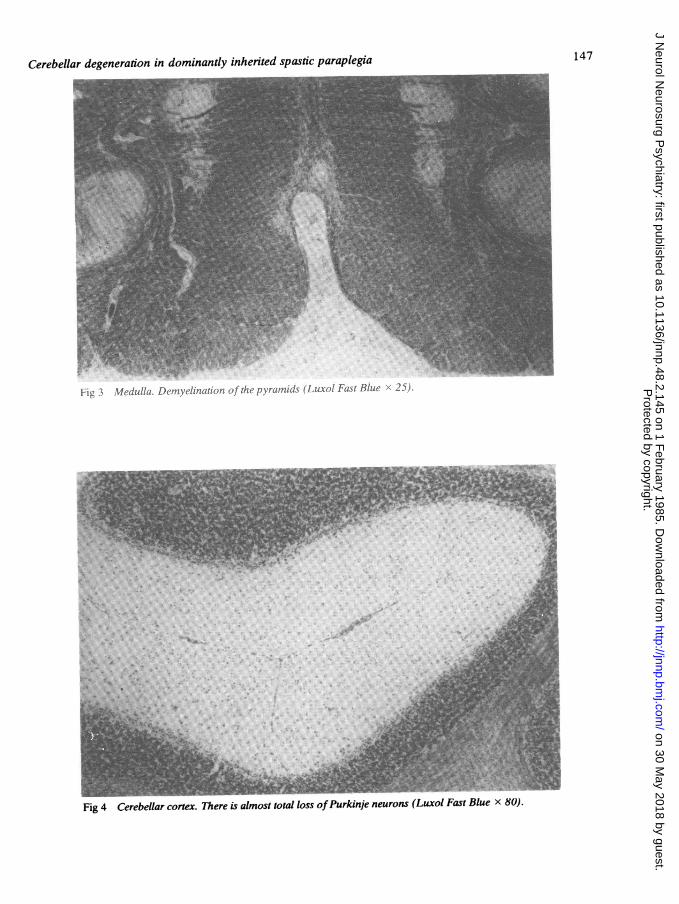
f:jL I %lecdulla. L)cat'litioOf c1.7xratniids (Luxol Fast Blue 22i).
't
Fig 4 Cerebellar cortex. There is almost total loss ofPurkinje neurons (Luxol Fast Blue x 80).
Protected by copyright.
on 30 May 2018 by guest.
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp.48.2.145 on 1 February 1985. D
ownloaded from
148
C,.
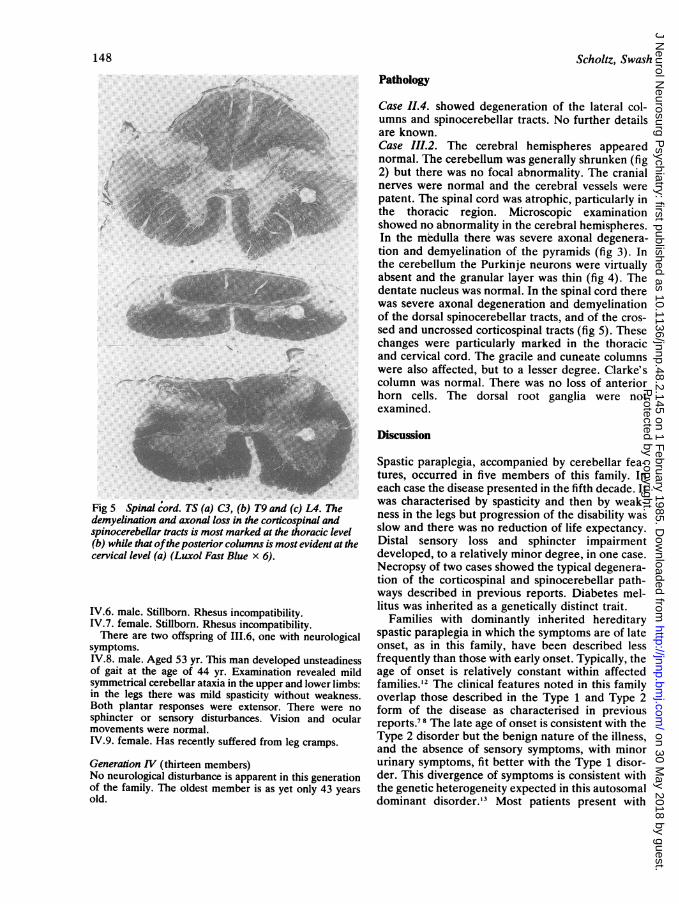
Fig 5 Spinal cord. TS (a) C3, (b) T9 and (c) L4. Thedemyelination and axonal loss in the corticospinal andspinocerebellar tracts is most marked at the thoracic level(b) while that ofthe posterior columns is most evident at thecervical level (a) (Luxol Fast Blue x 6).
IV.6. male. Stillborn. Rhesus incompatibility.IV.7. female. Stillborn. Rhesus incompatibility.There are two offspring of 111.6, one with neurological
symptoms.IV.8. male. Aged 53 yr. This man developed unsteadinessof gait at the age of 44 yr. Examination revealed mildsymmetrical cerebellar ataxia in the upper and lower limbs:in the legs there was mild spasticity without weakness.Both plantar responses were extensor. There were no
sphincter or sensory disturbances. Vision and ocularmovements were normal.IV.9. female. Has recently suffered from leg cramps.
Generation IV (thirteen members)No neurological disturbance is apparent in this generationof the family. The oldest member is as yet only 43 yearsold.
Scholtz, Swash
Pathology
Case 11.4. showed degeneration of the lateral col-umns and spinocerebellar tracts. No further detailsare known.Case III.2. The cerebral hemispheres appearednormal. The cerebellum was generally shrunken (fig2) but there was no focal abnormality. The cranialnerves were normal and the cerebral vessels werepatent. The spinal cord was atrophic, particularly inthe thoracic region. Microscopic examinationshowed no abnormality in the cerebral hemispheres.In the medulla there was severe axonal degenera-tion and demyelination of the pyramids (fig 3). Inthe cerebellum the Purkinje neurons were virtuallyabsent and the granular layer was thin (fig 4). Thedentate nucleus was normal. In the spinal cord therewas severe axonal degeneration and demyelinationof the dorsal spinocerebellar tracts, and of the cros-sed and uncrossed corticospinal tracts (fig 5). Thesechanges were particularly marked in the thoracicand cervical cord. The gracile and cuneate columnswere also affected, but to a lesser degree. Clarke'scolumn was normal. There was no loss of anteriorhorn cells. The dorsal root ganglia were notexamined.
Discussion
Spastic paraplegia, accompanied by cerebellar fea-tures, occurred in five members of this family. Ineach case the disease presented in the fifth decade. Itwas characterised by spasticity and then by weak-ness in the legs but progression of the disability wasslow and there was no reduction of life expectancy.Distal sensory loss and sphincter impairmentdeveloped, to a relatively minor degree, in one case.Necropsy of two cases showed the typical degenera-tion of the corticospinal and spinocerebellar path-ways described in previous reports. Diabetes mel-litus was inherited as a genetically distinct trait.
Families with dominantly inherited hereditaryspastic paraplegia in which the symptoms are of lateonset, as in this family, have been described lessfrequently than those with early onset. Typically, theage of onset is relatively constant within affectedfamilies.'2 The clinical features noted in this familyoverlap those described in the Type 1 and Type 2form of the disease as characterised in previousreports.78 The late age of onset is consistent with theType 2 disorder but the benign nature of the illness,and the absence of sensory symptoms, with minorurinary symptoms, fit better with the Type 1 disor-der. This divergence of symptoms is consistent withthe genetic heterogeneity expected in this autosomaldominant disorder.'3 Most patients present with
Protected by copyright.
on 30 May 2018 by guest.
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp.48.2.145 on 1 February 1985. D
ownloaded from

Cerebellar degeneration in dominantly inherited spastic paraplegiaspastic paraplegia alone, but pathological studieshave shown involvement of the posterior columnsand of the ascending spinocerebellar pathways. Inthe Type 2 form of familial spastic paraplegia, ataxiaof gait may be present often in association withslight distal sensory impairment, but pure cerebellarataxia of gait appears to be very uncommon."4 15
Indeed, most reports do not comment on thepresence or absence of cerebellar signs, or of thehistological features in the cerebellum. It is con-ceivable that this absence of reports of cerebellarinvolvement reflects the overwhelming predomi-nance of the spastic paraplegia. In this Type 2 form,however, the degeneration may extend into otherneuronal groups.
In our case III.2. necropsy showed a virtuallycomplete loss of cerebellar Purkinje cells. Thenumber of Purkinje cells decreases with advancingage so that in the ninth decade about 75% of thesecells remain;'6 the changes in Case I11.2 could notthus be explained by ageing. The dentate nucleuswas spared. In the spinal cord there was degenera-tion not only of the corticospinal tracts but also ofspinocerebellar tracts and posterior columns. Thispatient's son (case IV.5) also has mild cerebellarataxia in addition to spastic paraplegia.
Greenfield'7 classified dominantly inherited spas-tic paraplegia with the hereditary spinocerebellardegenerations. He argued that several dominantlyinherited examples of the latter, described ashereditary spastic ataxia were shown at necropsy tohave lesions involving the spinal cord. In theSchut-Haymaker family,'8 for example the diseasedifferentiated in the third generation into severaltypes, resembling respectively Friedreich's ataxia,hereditary cerebellar ataxia and hereditary spasticparaplegia with minimal ataxia. Similarly in theMenzel form of hereditary cerebellar ataxia'9 thereis spinal cord degeneration, resembling that found inour cases, with cerebellar degeneration, and atrophyof the olives and of the pons.
We thank Case IV.5, a physician, who recognisedthe unusual nature of his familial disorder, and DrCharles Simpson of Vancouver, Canada and DrRonald Henson, London, for providing us withdetails of the clinical features.
References
'Schwartz GA. Hereditary (familial) spastic Paraplegia.Arch Neurol Psychiat 1952;68:655-82.
2 Seeligmuller A. Skelerose der seitenstrange des Ruck-enmarks bei 4 Kindern derselben Familie, DeutscheMed. Wochnschr 1876;2:185-6.
3 Strumpell A. Beitrage zur Pathologie des Ruckenmarks,Arch Psychiat Nervenkr 1880; 10: 676-717.
4 Strumpell A. Die primare Seitenstrangsklerose (Spas-tische Spinalparalyse) Deutsch Nervenheilkunde1904;27:291-339.
Lorrain M. Contributon a l'etude de la Paraplegie Spas-modique Familiale. Paris. G Steinheil 1898.
6 Schwartz GA, Liu C-N. Hereditary (familial) spasticparaplegia. Arch Neurol 1965; 75: 144-62.
Behan WMH, Maia M. Strumpell's familial spastic para-plegia: genetics and neuropathology. J NeurolNeurosurg Psychiatry 1974;37:8-20.
8 Harding AE. Hereditary "pure" spastic paraplegia: aclinical and genetic study of 22 families. J NeurolNeurosurg Psychiatry 1981;44:871-83.
Price GE. Familial lateral sclerosis (spastic paralysis). JNerv Ment Dis 1939;90:51-3.
'° Skre H. Hereditary spastic paraplegia in Western Nor-way. Clin Genet 1974;6:165-83.
"Harding AE. Classification of the hereditary ataxias andparaplegias. Lancet 1983;ii: 1151-5.
12 Holmes GL, Shaywitz BA. Strumpell's pure familialspastic paraplegia: case study and review of the litera-ture. J Neurol Neurosurg Psychiatry 1977;40: 1003-8.
13 Harris H, Smith CAB. The sib-sib age of onset correla-tion among individuals suffering from a hereditarysyndrome produced by more than one gene. AnnEugenics 1947/9; 14:309-18.
Landau WM, Gitt JJ. Hereditary spastic paraplegia andhereditary ataxia: a family demonstrating a variety ofphenotypic manifestations. Arch Neurol Psychiat1951;66:346-54.
Schmit RW. Hereditary ataxia; a clinical study throughsix generations. Arch Neurol Psychiat 1950;63:535-68.
16 Corsellis JAN. Some observations on the Purkinje cellspopulation and on brain volume in humansageing. In:Neurobiology of Aging Vol 3. Terry D, Gershon S.eds. Raven Press New York 1976:205.
Greenfield JG. The Spino-Cerebellar Degenerations.Oxford Blackwell.
18 Schut JW, Haymaker W. Hereditary ataxia: pathologicstudy of five cases of common ancestry. J NeuropathClin Neurol 1951;1: 189-213.
Menzel P. Beitrage zur kenntnis der hereditaren ataxieund Kleinhirnatrophie. Arch Psychiat u Nervenkr,1891:22:160.
149
Protected by copyright.
on 30 May 2018 by guest.
http://jnnp.bmj.com
/J N
eurol Neurosurg P
sychiatry: first published as 10.1136/jnnp.48.2.145 on 1 February 1985. D
ownloaded from