Embed Size (px)
Citation preview

Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin,a nuclear-encoded mitochondrial metalloprotease. Cell 1998; 3: 973–983.
6. Vazza G, Zoreta M, Boaretto F, Micaglio GF, Sartori V, Mostacciuolo ML. Anew locus for autosomal recessive spastic paraplegia associated with mentalretardation and distal motor neuropathy, SPG14, maps to chromosome3q27-q28. Am J Hum Genet 2000; 67: 504–509.
7. Hughes CA, Byrne PC, Webb S, McMonagle P, Patterson V, Hutchison M,Parfery NA. SPG15, a new locus for autosomal recessive complicated HSP onchromosome 14q. Neurology 2001; 56: 1230–1233.
8. Shibasaki Y, Tanaka H, Iwabuchi K, Kawasaki S, Kondo H, Uekawa K, UedaM, Kamiya T, Katayama Y, Nakamura A, Takashima H, Nakagawa M, MasudaM, Utsumi H, Nakamuro T, Tada K, Kurohara K, Inoue K, Koike F, Sakai T,Tsuji S, Kobayashi H. Linkage of autosomal recessive hereditary spasticparaplegia with mental impairment and thin corpus callosum to chromosome15A13-15. Ann Neurol 2000; 48: 108–112.
9. Nakamura A, Izumi K, Umehara F, Kuriyama M, Hokezu Y, Nakagawa M,Shimmyozu K, Izumo S, Osame M. Familial spastic paraplegia with mentalimpairment and thin corpus callosum. J Neurol Sci 1995; 131:35–42.
10. Ueda M, Katayama Y, Kamiya T, Mishina M, Igarashi H, Okubo S, Senda M,Iwabuchi K, Terashi A. Hereditary spastic paraplegia with a thin corpuscallosum and thalamic involvement in Japan. Neurology 1998; 51(6):1751–1754.
11. Katayama T, Sakamoto N, Kuroda K, Yahara O, Ugawa Y. A case of spasticparaparesis with mental deterioration and markedly thin corpus callosum-callosal dysfunction demonstrated by magnetic stimulation. Rinsho Shinkeigaku1998; 38(5): 418–422.
12. Wakabayashi K, Koyobashi H, Kawakai S, Kondo H, Takahashi H. Autosomalrecessive spastic paraplegia with hypoplastic corpus callosum, multisystemdegeneration and ubiquitinated eosinophilic granules. Acta Neuropathol (Berl)2001; 101(1): 69–73.
13. Casaubon LK, Melanson M, Lopes-Cendes I, Marineau C, Andermann E,Andermann F, Weissenbach J, Prevost C, Bouchard J-P, Mathieu J, RouleauGA. The gene responsible for a severe form of peripheral neuropathy andagenesis of the corpus callosum maps to chromosome 15q. Am J Hum Genet1996; 58: 28–34.
14. Howard HC, Mount DB, Rochefort D, Byun N, Dupre N, Lu J, Fan X, Song L,Riviere J-B, Prevost C, Horst J, Simonati A, et al. The K–Cl cotransporter KCC3is mutant in a severe peripheral neuropathy associated with agenesis of thecorpus callosum. Nat Genet 2002; 32: 384–392.
15. Naiman JL, Fraser FC. Agenesis of the corpus callosum. A report of two cases insiblings. Arch Neurol Psychiat 1955; 74: 182–185.
16. Andermann F, Andermann E, Joubert M, Karpati G, Carpenter S, Melancon D.Familial agenesis of the corpus callosum with anterior horn cell disease. Asyndrome of mental retardation, areflexia, and paraplegia. Trans Am NeurolAssoc 1972; 97: 242–244.
17. Fransen E, Vits L, Van Camp G, Willems PJ. The clinical spectrum of mutationsin L1, a neuronal cell adhesion molecule. Am J Med Genet 1996; 64(1): 73–77.
18. Martinez Murillo F, Kobayashi H, Pegoraro E, Galluzzi G, Creel G, Mariani C,Farina E, Ricci E, Alfonso G, Pauli RM, Hoffman EP. Genetic localization of anew locus for recessive familial spastic paraparesis to 15q13-15. Neurology1999; 53(1): 50–56.
19. Teive HA, Iwamoto FM, Della Coletta MV, Camargo CH, Bezerra RD,Minguetti G, Werneck LC. Hereditary spastic paraplegia associated with thincorpus callosum. Arq Neuropsiquiatr 2001; 59: 784–789.
Cavernous angiomapresenting as epilepsy 13years after initial diagnosis
Kensuke Murakami1 MDMD,, Kunihiko Umezawa1 MDMD,,
Mitsuomi Kaimori2 MDMD,, Michiharu Nishijima1 MDMD
1Department of Neurosurgery and 2Department of Pathology, Aomori
Prefectural Central Hospital, Aomori, Japan
Summary A 22-year-old man presented with tonic-clonic seizure
and was admitted to our hospital. He had suffered from frequent
headaches, and had been diagnosed with a brain tumour on MRI 13
years ago. However, neither further examination nor follow-up neu-
roimaging study have been performed. Computed tomography and
magnetic resonance imaging demonstrated an intraaxial tumor with
granular calcification in the right frontal lobe, attached to the adjacent
dura mater, which was enlarged compared with the lesion on CT
13 years before. The lesion was surgically excised through right
frontal craniotomy. Histopathological analysis indicated cavernous
angioma.
In cavernous angioma in younger children, more aggressive
surgical indications than in adults may be favorable both to prevent
hemorrhagic complications and to confirm pathologic diagnosis.
ª 2004 Elsevier Ltd. All rights reserved.
Journal of Clinical Neuroscience (2004) 11(4), 430–432
0967-5868/$ - see front matter ª 2004 Elsevier Ltd. All rights reserved.
doi:10.1016/j.jocn.2003.05.010
Keywords: Cavernous angioma, Epilepsy, Surgery
Received 11 December 2002
Accepted 17 May 2003
Correspondence to: Dr. Kensuke Murakami, Department of Neurosurgery,
Aomori Prefectural Central Hospital, 2-2-1 Higahshi-tsukurimichi, Aomori
030-8553, Japan. Tel.: +81-17-726-8111; Fax: +81-17-726-1885;
E-mail: [email protected]
INTRODUCTION
Cavernous angioma are intracranial lesions observed in 0.5–0.7%of the population. It has been reported that the initial presentingsymptom is epileptic seizure in 38% and intracranial haemorrhagein 24%.1 Although asymptomatic cavernous angiomas have beenincidentally diagnosed since computed tomography (CT) andmagnetic resonance imaging (MRI) were introduced into clinicalmedicine,2 no clear management strategy for asymptomatic le-sions has been established.
We describe a case of a frontal cavernous angioma presentingwith epilepsy following an asymptomatic period of 13 years afterinitial diagnosis.
CASE REPORT
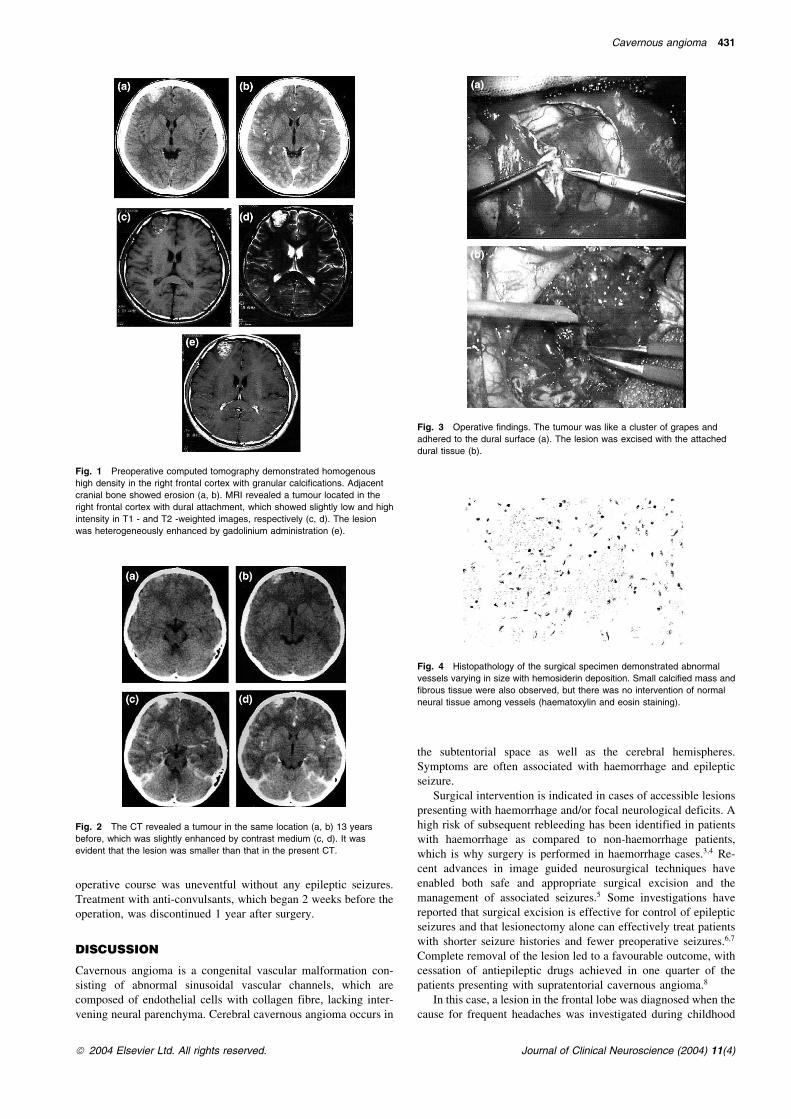
A 22 year old man was admitted to our hospital with a tonic-clonicseizure. Epileptic attacks had occurred 3 times during the 2months before presentation. On admission the patient was alertwith no neurological deficits. He had a past history of frequentheadaches 13 years ago and at that time an intracranial lesion wasdiscovered on head CT in another hospital. The headaches im-proved immediately afterwards and no further examinations wereperformed. An anti-convulsant was given on this admission. CTdemonstrated a highly dense lesion in the right frontal lobe,25� 20� 20 mm in size. There were high density granules indi-cating calcification in the lesion (Fig. 1(a) and (b)). MRI dem-onstrated hypointensity and hyperintensity on T1 and T2 weightedimages, respectively (Fig. 1(c) and (d)). The lesion was15� 12� 18 mm in size on the CT performed 13 years before(Fig. 2(a) and (b)) and had definitely enlarged. The pre-operativeneuroimaging diagnosis was a calcified intraaxial tumour,including cavernous angioma and low grade glioma. The tumourwas surgically excised to clarify the pathological diagnosis. Totalremoval was performed through a right frontal craniotomy(Fig. 3). Since the tumour was adhered to the dura mater, it wasexcised en bloc. Post-operative histopathologic analysis demon-strated sinusoidal vessels without the interposition of normalneural tissue (Fig. 4), indicating cavernous angioma. The post-
430 Murakami et al.
Journal of Clinical Neuroscience (2004) 11(4) ª 2004 Elsevier Ltd. All rights reserved.
operative course was uneventful without any epileptic seizures.Treatment with anti-convulsants, which began 2 weeks before theoperation, was discontinued 1 year after surgery.
DISCUSSION
Cavernous angioma is a congenital vascular malformation con-sisting of abnormal sinusoidal vascular channels, which arecomposed of endothelial cells with collagen fibre, lacking inter-vening neural parenchyma. Cerebral cavernous angioma occurs in
the subtentorial space as well as the cerebral hemispheres.Symptoms are often associated with haemorrhage and epilepticseizure.
Surgical intervention is indicated in cases of accessible lesionspresenting with haemorrhage and/or focal neurological deficits. Ahigh risk of subsequent rebleeding has been identified in patientswith haemorrhage as compared to non-haemorrhage patients,which is why surgery is performed in haemorrhage cases.3;4 Re-cent advances in image guided neurosurgical techniques haveenabled both safe and appropriate surgical excision and themanagement of associated seizures.5 Some investigations havereported that surgical excision is effective for control of epilepticseizures and that lesionectomy alone can effectively treat patientswith shorter seizure histories and fewer preoperative seizures.6;7
Complete removal of the lesion led to a favourable outcome, withcessation of antiepileptic drugs achieved in one quarter of thepatients presenting with supratentorial cavernous angioma.8
In this case, a lesion in the frontal lobe was diagnosed when thecause for frequent headaches was investigated during childhood
Cavernous angioma 431
ª 2004 Elsevier Ltd. All rights reserved. Journal of Clinical Neuroscience (2004) 11(4)
Fig. 1 Preoperative computed tomography demonstrated homogenous
high density in the right frontal cortex with granular calcifications. Adjacent
cranial bone showed erosion (a, b). MRI revealed a tumour located in the
right frontal cortex with dural attachment, which showed slightly low and high
intensity in T1 - and T2 -weighted images, respectively (c, d). The lesion
was heterogeneously enhanced by gadolinium administration (e).
Fig. 2 The CT revealed a tumour in the same location (a, b) 13 years
before, which was slightly enhanced by contrast medium (c, d). It was
evident that the lesion was smaller than that in the present CT.
Fig. 3 Operative findings. The tumour was like a cluster of grapes and
adhered to the dural surface (a). The lesion was excised with the attached
dural tissue (b).
Fig. 4 Histopathology of the surgical specimen demonstrated abnormal
vessels varying in size with hemosiderin deposition. Small calcified mass and
fibrous tissue were also observed, but there was no intervention of normal
neural tissue among vessels (haematoxylin and eosin staining).

13 years ago. The lesion had been asymptomatic since diagnosisand the presenting seizures was the initial symptom. The clinicalcourse of this case seems to be uncommon, but suggests that anasymptomatic course as long as 13 years does not necessarilyindicate an actual asymptomatic lesion. The lesion had definitelyenlarged when compared to its size 13 years ago. Pre-operativeMRI demonstrated no evidence of repeated haemorrhage such ashaemosiderin deposition. Intratumoural calcification on the pre-operative CT is sometimes observed in low grade glioma as wellas in cavernous angioma. It was therefore difficult to diagnosepreoperatively as a cavernous angioma with only these findings.Histological analysis of the pathology was a primary goal of theplanned surgery in this case.
Histopathology demonstrated characteristic findings of cav-ernous angioma, sinusoidal structures without intervening braintissue. Cavernous angioma has been, in general, assumed to be acongenital lesion and the expansion of the lesion is usually sub-sequent to haemorrhage.7;9 On the other hand, however, the denovo formation of cavernous angioma is recognised in the patientsaffected by the familial form and undergoing radiotherapy.10–12
Cavernous angioma shows expansile growth by enlargement ofcavernous matrix or cyst like a neoplastic lesion.13;14 It is con-troversial whether the lesion is congenital or neoplastic. In thepresent case, the lesion had grown for 13 years since the initialdiagnosis without evidence of intralesional or perilesional haem-orrhage on neuroimaging studies. However, histopathologic ex-amination revealed that the intravascular space of sinusoidalvessels was partly expanded and was thrombosed and organisedwith and without calcification. These histological alterationsmight be implicated in the growth of the lesion (see Fig. 3).
Recently, detection of asymptomatic cavernous angioma hasbeen increasing since the advent of CT and MRI.2 The manage-ment of symptomatic cavernous angioma, however, is still beingdebated, because its natural history is still not clearly understood,especially in paediatric cases. It has been reported that the rate ofepilepsy is 1.51% per year and that the rate of haemorrhage is0.25–2.3% per year.4;7 Increased risk of rebleeding after the initialhaemorrhage and the decreased effectiveness of lesionectomy inpatients with a long seizure history could justify early surgicalintervention for cavernous angioma.15 It is documented that trueepilepsy is less common and may be related to haemorrhagiccomplications such as chronic or recurrent microbleeding.16
Haemorrhagic lesions seem to be an indiction for surgical exci-sion, although the potential risks and benefits of surgery due toaccessibility and eloquence of the lesions should be carefullydetermined17 (see Fig. 4).
In paediatric cases, the choice of management for asymptom-atic lesions is more difficult than in adults, because the naturalhistory is not clearly known. However, some groups have dem-onstrated a high risk of haemorrhagic complications in children ascompared to adults and advocate surgery.16;18;19 Considering thelonger life span anticipated in paediatric patients, an aggressivesurgical approach is favoured even for asymptomatic lesions lo-cated in non-eloquent areas as well as symptomatic lesions.1
Furthermore, surgical intervention is reasonable for the purpose ofdifferential diagnosis of pathology to rule out other malignanttumours when the neuroimaging study is not typical of cavernousangioma.
We have presented a case of a frontal cavernous angioma pre-sentingwith epilepsy following an asymptomatic period of 13 years.Surgical intervention is necessary in cases with repeat haemor-rhages indicated by neuroimaging studies. An aggressive surgicalapproach is indicated in paediatric cases as opposed to adult cases.Surgical excision is essential for lesions located in non-eloquentareas for the purpose of precise pathologic diagnosis.
REFERENCES
1. Giombini S, Morello G. Cavernous angiomas of the brain. Account of fourteenpersonal cases and reviewof the literature.ActaNeurochir (Wien) 1978; 40: 61–82.
2. Requena I, AriasM, Lopez-Ibor L, Pereiro I, Barba A, AlonsoA et al. Cavernomasof the central nervous system: clinical and neuroimaging manifestations in 47patients. J Neurol Neurosurg Psychiatry 1991; 54: 590–594.
3. Aiba T, Tanaka R, Koike T, Kameyama S, TakedaN, Komata T. Natural hisotyr ofintracranial cavernous malformation. J Neurosurg 1995; 83: 56–59.
4. Kim DS, Park YG, Choi JU, Chung SS, Lee KC. An analysis of the naturalhistory of cavernous malformation. Surg Neurol 1997; 48: 9–17.
5. Buckingham MJ, Crone KR, Ball WS, Berger TS. Management of cerebralcavernous angiomas in children presenting with seizures. Child Nerv Syst 1989;5: 347–349.
6. Cohen DS, Zubay GP, Goodman RR. Seizure outcome after lesionectomy forcavernous malformations. J Neurosurg 1995; 83: 237–242.
7. Del Curling Jr O, Kelly Jr DL, Elster AD, Craven TE. An analysis of the naturalhistory of cavernous angiomas. J Neurosurg 1991; 75: 702–708.
8. Casazza M, Broggi G, Franzini A, Avanzini G, Spreafico R, Brcchi M et al.Supratentorial cavernous angiomas and epileptic seizures: preoperative courseand post operative outcome. Neurosurgery 1996; 39: 26–32.
9. Kondziolka D, Lunsford LD, Kestle JRW. The natural history of cerebralcavernous malformation. J Neurosurg 1995; 83: 820–824.
10. Detwiler PW, Porter RW, Zabramski JM, Spetzler RF. De novo formation of acentral nervous system cavernous malformation: implication for predicting riskof haemorrhage. J Neurosurg 1997; 87: 629–632.
11. Pozzati E, Acciarri N, Tognetti F, Marliani F, Giangaspero F. Grown,subsequent bleeding and do novo appearance of cerebral cavernous angiomas.Neurosurgery 1996; 38: 662–669.
12. Zabramski JM, Wascher TM, Spetzler RF et al. The natural history of familialcavernous malformation: results of an ongoing study. J Neurosurg 1994; 80:422–432.
13. Houtteville JP. Brain cavernoma: a dynamic lesion. Surg Neurol 1997; 48: 610–614.
14. Siddiqui AA, Jooma R. Neoplastic growth of cerebral cavernous malformationpresenting with impending cerebral herniation: a case report and review of theliterature on de novo growth of cavernomas. Surg Neurol 2001; 56: 42–45.
15. Maggi G, Aliberti F, Ruggiero C, Pittore L. Cerebral cavernous angiomas incritical areas. Reports of three cases in children. J Neurosurg Sci 1997; 41:353–357.
16. Mottolese C, Hermier M, Stan H, Jouvet A, Saint-Pierre G, Froment JC et al.Central nervous system cavernomas in the pediatric age group. Neurosurg Rev2001; 24: 55–71.
17. Smit LM, Halbertsma FJ. Cerebral cavernous hemangioma in childhood. Clinicalpresentation and therapeutic considerations. Childs Nerv Syst 1997; 13: 522–525.
18. Di Rocco C, Iannelli A, Tamburrini G. Cavernomas of the central nervoussystem in children. A report of 22 cases. Acta Neurochir (Wien) 1996; 138:1267–1274.
19. Di Rocco C, Iannelli A, Tamburrini G. Surgical management of paediatriccerebral cavernomas. H Neurosurg Sci 1997; 41: 343–347.
Lumbosacral clear-cellmeningioma treated withsubtotal resection andradiotherapy
Ronald Boet1, Ho-Keung Ng2, Sekhar Kumta3,
Leung-Cho Chan4, Kwok-Wing Chiu4, Wai-Sang Poon
1Division of Neurosurgery, Department of Surgery, Chinese University of
Hong Kong, Hong Kong SAR, People’s Republic of China, 2Department of
Anatomical and Cellular Pathology, Chinese University of Hong Kong,
Hong Kong SAR, People’s Republic of China, 3Department of Orthopedics and
Traumatology, Chinese University of Hong Kong, Hong Kong SAR, People’s
Republic of China, 4Department of Clinical Oncology, Chinese University of
Hong Kong, Hong Kong SAR, People’s Republic of China
Summary We wish to report a rare case of clear-cell meningioma in
the lumbosacral region in a 34-year-old male patient who presented
to us with lower back pain and leg pain. The management of the
432 Boet et al.
Journal of Clinical Neuroscience (2004) 11(4) ª 2003 Elsevier Ltd. All rights reserved.





























