Embed Size (px)
Citation preview

BLEEDING DISORDERS
Dr. MUBARAK ABDELRAHMAN
MD PEDIATRICS AND CHILD HEALTH
Assistant Professor
FACULTY OF MEDICINE -JAZAN

Objectives:
By the end of this lecture, the student should be able to:
• Describe the mechanism of homeostasis.
• Identify causes, presentation and management of the common bleeding and clotting disorders.
• Evaluate a patient with bleeding and/or clotting disorder.

Haemostatic Mechanism
The classic haemostatic mechanism include:
-vascular response
-platelets adhesion
-coagulation factor

Vascular Factor
• The vessel wall is first line of defense against blood loss.
• Local vasoconstriction is induced by a neurogenic reflex and is maintained by vaso constrictor substances released by platelets.

Platelet haemostatic activity
1. Stimulate vasoconstriction of injured vessels.
2. They form haemostatic plug to seal small vessel wall.
3. Play a role in fibrin clot formation.

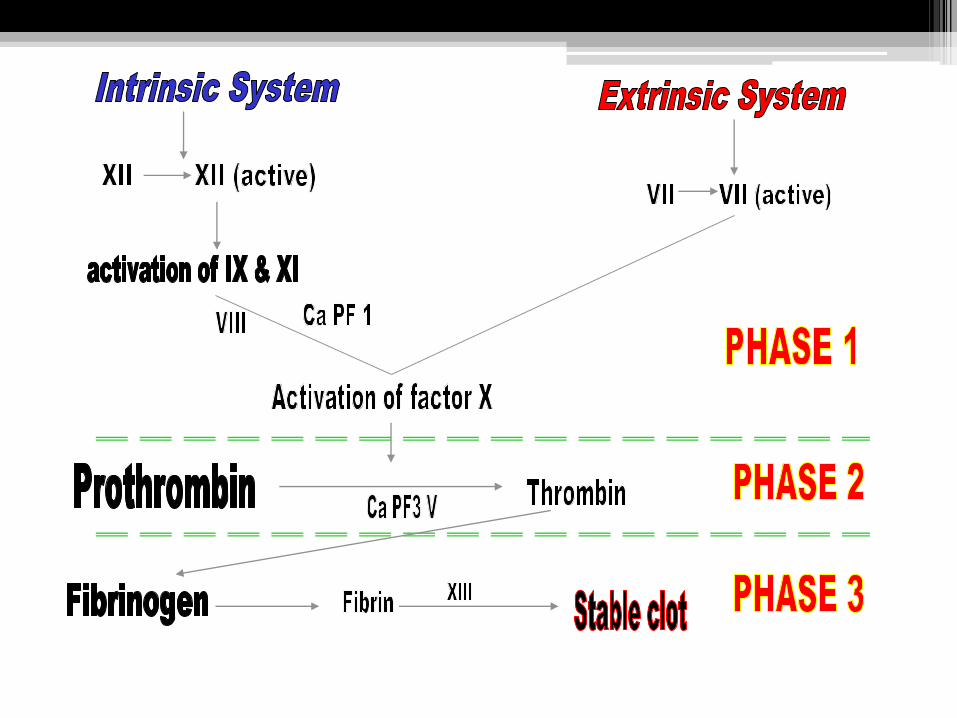
The fibrin clot formation
• The clot formation depends on the presence of:
( ionized calcium, platelets and the coagulation factor).
• Clot formation pass through 3 phases:
PHASE 1: Activation of factor X mediated by intrinsic or extrinsic system.
PHASE 2: Conversion of pro-thrombin to active thrombin.
PHASE 3: Formation of fibrin clot from fibrinogen

Bleeding disorders
1- Platelet disorder:A-↓ number (thrombocytopenia) due to:-
#Decreased platelet production
-Bone marrow depression
Congenital
Fanconi`s anemia
Acquired
Drugs(chemotherapy)
infection e.g. hepatitis
-Bone marrow infiltration
Leukemia
Neuroblastoma

B-Increased platelet destruction#immune mechanism
-ITP-Collagen vascular disorder e.g. SLE.
#non immune mechanism-↑ platelet consumption e.g. DIC
-hypersplenism
C-Abnormal platelet function
1-hereditary : -Glanzmann Thrombasthenia
-Bernard Soulier Disorder
-von Willebrand Disorder
2-acquired: -drugs e.g. Aspirin
-toxic metabolic product (e.g. in uremia)
- autoantibodies,immune complex.

2- Vascular disordera- Hereditary e.g. hemorrhagic , telangiectasisb-Acquired:
1-Henoch-Schonlein purpura.2-Septicemia.3-collagen disorder e.g. SLE
3- Coagulation DisorderCongenital
-Hemophilia A, B, C-von Willebrand-Afibrinogenemia
Acquired-DIC-Deficient Vit. K-Hemorrhagic Disorder of the newborn-Liver Disease

Hereditary Clotting Factor Deficiency
Factor VIII or factor IX deficiency
(Hemophilia A or B):
• The most common inherited clotting disease.
• They has similar clinical pictures and similar patterns of inheritance. (considered one disease)

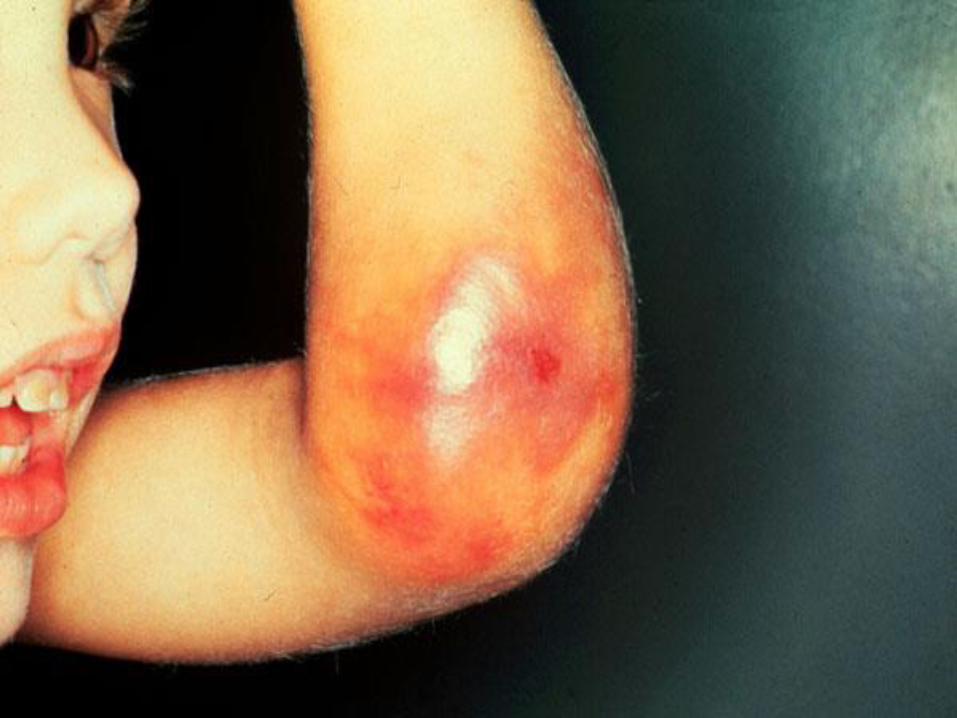
Clinical Manifestation:• According to severity.
• Bleeding may be spontaneous, after minor, moderate or significant injury.
Severe If factor VIII less than 1%
Moderate If factor VIII less than 1-5%
Mild If factor VIII less than 6-30%

Clinical Manifestation:
• Bleeding symptoms may present from birth or occur in fetus. (factor VIII and factor IX do not cross the placenta)
• Occasionally , neonate with hemophilia may sustain intracranial hemorrhage.
• Only 30% of affected male infant with hemophilia bleed with circumcision.
• Many affected children bleed during teeth eruption.

Laboratory Findings:-
1. Reduced level of factor VIII or factor IX.
2. PTT is prolonged.
3. Platelet count , bleeding time, PT are normal.
4. Prolonged clotting time.
5. ↑ thromoplastin generation time.

Treatment :
General Treatment:
1. Prevention of trauma.
2. Careful dental hygiene & regular dental examination.
3. Avoid aspirin.
4. Vaccinate against HEPATITIS B.
5. If surgery needed it should be carried out under cover of replacement therapy.

Replacement therapy:
• Mild hemophilia may be treated with infusion of cryoprecipitate or desmopressin (DDAVP).
• Moderate or severe type, the store amount of factor VIII is inadequate, so give:-
*Fresh Frozen Plasma
*Factor VIII
Factor Half-Life, h
VIII 8-12
IX 24

Complications of Hemophilia
1. Chronic joint destruction secondary to haemoarthrosis.
2. Transfusion-transmitted infectious diseases e.g. HIV, Hepatitis, ...
3. Development of inhibitor to factor XIII & IX (in 10% of all hemophilia leading to poor clinical response to therapy).

Von Willebrand Disease
• It is the most common hereditary bleeding disorder.
• Inherited as autosomal dominant.

Pathophysiology:
von Willebrand factor has 2 major functions:
1- VWF acts as a bridge between subendothelial matrix & platelets.
2- VWF also serves as the carrier protein for plasma factor VIII .

VON WILLEBRAND'S DISEASE has three main subtypes:
Type 1 is a simple deficiency of von Willebrand's factor but the protein is otherwise normal.
Type 2 there is a molecular defect in the structure of the protein.
Type 3 there is a complete lack of von Willebrand's factor.

Lab. Findings:
1-↑ bleeding time.
2-↑ clotting time.
3-↑ PTT.
4-↓ factor VIII & VWF.
5- defective platelet adhesion but platelet number are normal.

Treatment:
Minor bleeding problems, may not require specific treatment.
For more serious bleeding, desmopressin (DDAVP).
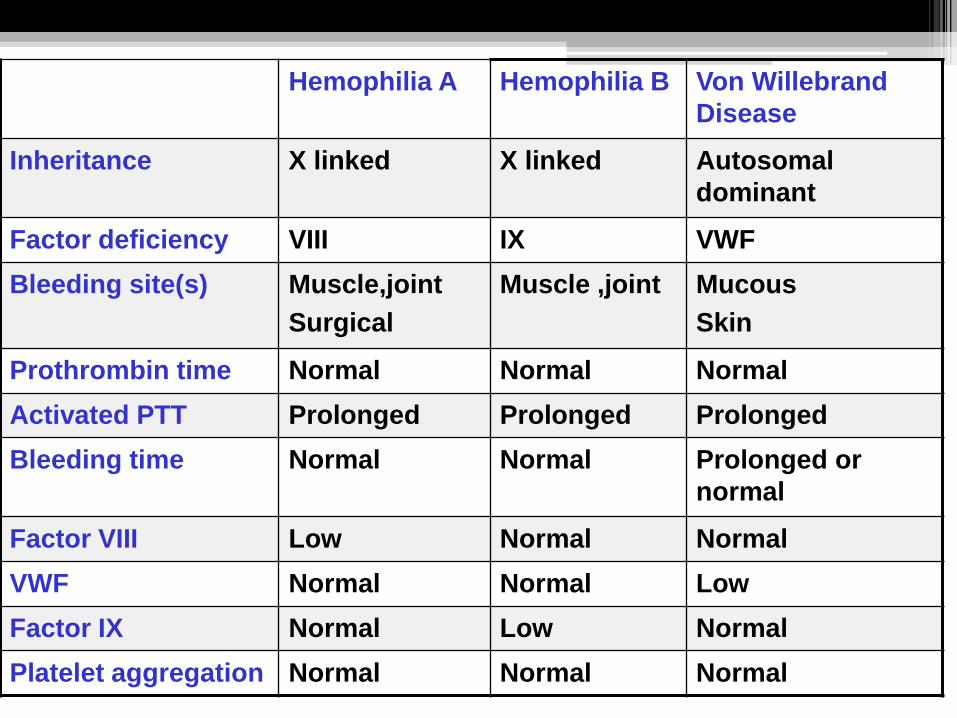
Hemophilia A Hemophilia B Von Willebrand
Disease
Inheritance X linked X linked Autosomal
dominant
Factor deficiency VIII IX VWF
Bleeding site(s) Muscle,joint
Surgical
Muscle ,joint Mucous
Skin
Prothrombin time Normal Normal Normal
Activated PTT Prolonged Prolonged Prolonged
Bleeding time Normal Normal Prolonged or
normal
Factor VIII Low Normal Normal
VWF Normal Normal Low
Factor IX Normal Low Normal
Platelet aggregation Normal Normal Normal

Hemorrhagic Disease of Newborn
A moderate decrease of vitamin K dependent coagulation factor (II,VII,IX,X) normally occur in all new born infants by 2nd – 3rd day after delivery with gradual return to birth level by 7th –10th day of age.

Causes:
1-Small amount of vit. K in the breast milk.
2-Imaturity of the infant liver.
3-Absence of bacterial flora in the intestine which is also responsible for synthesis of vit. K.

Clinical manifestations:
• Bleeding from umbilicus, mucous membranes, GI tract.
• Bleeding following circumcision and venipuncture.
• Hematomas at sites of trauma e.g. cephalohematomas and bruising.
• Intracranial bleeding can occur (the main cause of mortality and long-term morbidity).

Investigations:
1- Coagulation + PT + PTT = prolonged.
2- Bleeding time + platelet = normal.
3- Vitamin K direct assay is not useful because levels normally are low in newborns.
4- The diagnosis is confirmed if administration of vitamin K stopped the bleeding and reduced the PT value.

Treatment:* IV vit.K, plasma or blood transfusion.
Prevention:
*1 mg of vit. K is given I.M. after delivery for every newborn infant.

There are 2 abnormalities in the platelet:
1- function 2-number
Thrombathenia Thrombocytopenia
Acquired congenital acquired congenital
Platelet Disorders

Immune Thrombocytopenic Purpura (ITP)
• Thrombocytopenia with normal bone marrow and the absence of toxic exposure or a disease associated with a low platelet count.
• Manifest as an acute condition (in children) and a chronic condition (in adults).

Etiology:
Autoimmune thrombocytopenia 1- 4 weeks following exposure to a common viral infection.
(commonly with EBV,HIV)

Clinical Manifestation:
1- The classic presentation of ITP from 1-4 years old who has sudden onset of generalized petechiae and purpura.
2- Often there is bleeding gums & mucous membrane.
3- History of preceding viral infection 1-4 weeks.

Differential Diagnosis:
• Splenic sequestration.
• Aplastic process such as Fanconi anemia.
• Megakaryocytic thrombocytopenia.
• Hypersplenism.
• Leukemia.

Laboratory Finding:
1-Hb/TWBC & differential should be normal; but HB may be decreased if profuse nose bleeding or menorrhagia.
2- Severe thrombocytopenia (platelet count < 20X10*9) is common .
3- Antinuclear antibody test is more often positive in
adolescents with chronic ITP.
4- PT & PTT are normal.

Treatment:
• Life-threatening bleeding requires critical care interventions.
• Platelet transfusion is indicated for controlling severe hemorrhage.
• In the patient with known ITP, high-dose steroids or IV immunoglobulin (IVIG).
• Splenectomy.

Henoch-Schőnlein Purpura
(Vaculitis)
Definition:-
It is a hypersensitivity vasculitis involving the small blood vessels of skin, joints, gut and kidneys.

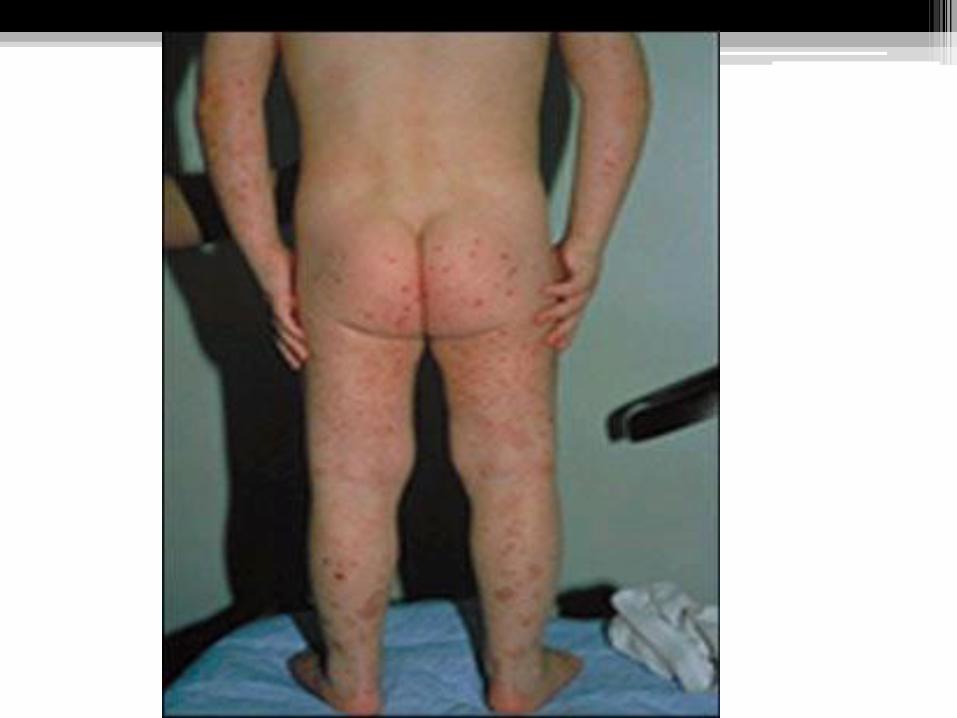
Clinical Manifestation:
1. Skin rash (100% of cases).
2. Arthritis(65% of cases).
3. Abdominal Manifestation(65% of cases).
4. Renal Manifestation(20% of cases).
5. C.N.S. (not common. Presented with convulsion, paresis and coma).

The evaluation of a child with BleedingHistory:1. Sites of bleeding.2. Severity and duration. 3. Age of symptom onset.4. Was the bleeding spontaneous or after trauma?5. If there is similar problem in the family or past history of same
personal condition ?6. If symptom correlate with degree of injury or trauma?7. Does bruising occur spontaneously?8. Are there lump with bruises or when there is minimal
trauma?9. History of previous surgery, dental procedures or
tonsillectomy?10. In past pubertal female it is important to take carefully
menstrual history.

Physical Examination:
The examination should focus on whether symptoms are primarily associated with
• mucus membrane or skin (muco-cutaneous bleeding) or
• muscle and joint (deep bleeding).

Laboratory test:1-Test for vascular & platelet disorder.
*Bleeding time: the bleeding time assesses the function of platelets & their interaction with vascular wall.Normal range:4-8 minutes
*platelet count: normal (150,000-400,000)2-Test for coagulation factor.
*Activated PTT: intrinsic factor VII,IX,XI normally 25-40 seconds.
*Prothrombin time (PT): extrinsic clotting system, it measures II,V,VII,X.
Normal range: 12-14 seconds.*Thrombin time:-Normal thrombin time 11-15 second.
Prolonged thrombin time occur in fibrinogen deficiency factor 2. It measures final step of clotting cascade.




























