Embed Size (px)
Citation preview

Autosomal dominant vererbte Erkrankungen
Prof. Dr. med. Jürgen KohlhasePraxis für Humangenetik
Freiburgwww.humangenetik-freiburg.de

Mutationstypen
• Genommutationen: numerische Änderungen des Chromosomensatzes (Polyploidie, Aneuploidie)
• Genmutationen (ein Gen betroffen):DeletionenInsertionenPunktmutationen
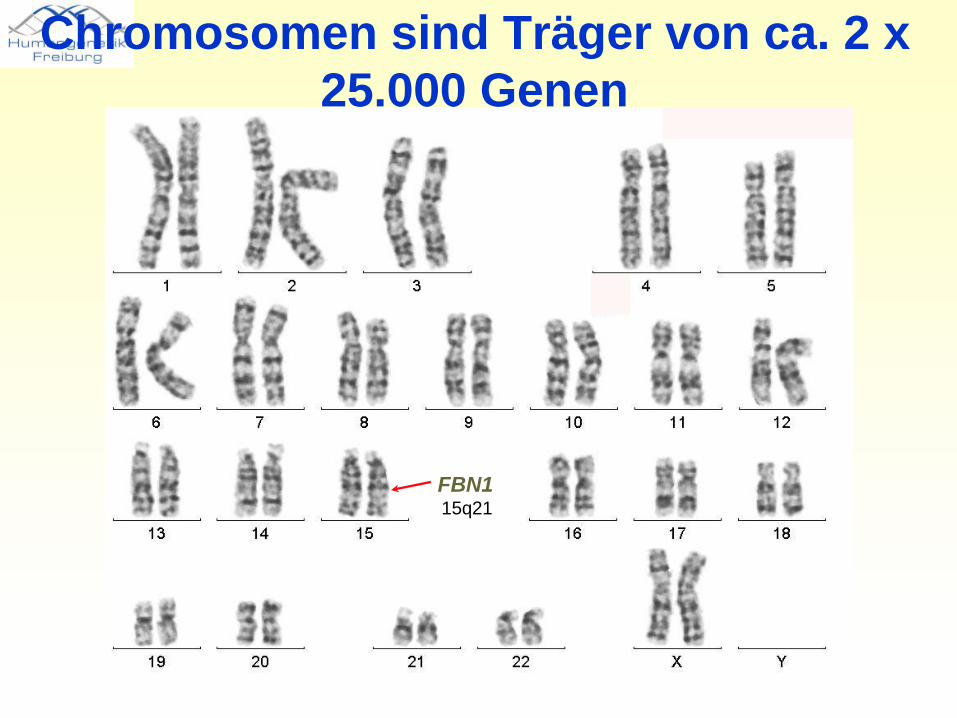
FBN115q21
Chromosomen sind Träger von ca. 2 x 25.000 Genen

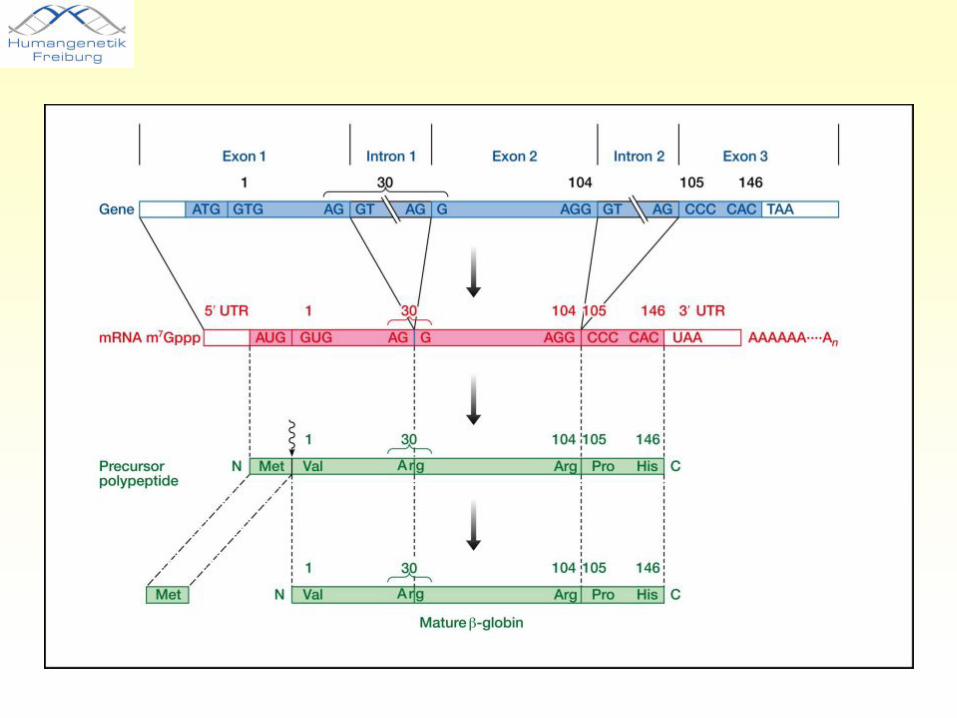
Gen
• Einheit, die für ein Protein kodiert und/ oder in eine mRNA transkribiert wird
• Besteht aus Exons (Bestandteile der reifen mRNA), Introns und regulatorischen Regionen
• Mutationen sind meist in kodierenden Exons zu finden, gelegentlich aber auch in anderen Regionen

Deletionen
• Verlust von mindestens einem Basenpaar (keine feste Grenze), auch ganzer Gene. Effekt: Verlust/ Verkürzung wichtiger Proteinabschnitte, Frameshift(Leserasterverschiebung), Gendosisverminderung.
T I L G G VNormal ACG ATC CTG GGG GGT GTGMut delG ACG ATC CTG GGG GTG TGA
T I L G V STOP

Insertionen/ Duplikationen
• Einbau mindestens einem Basenpaar (keine feste Grenze). Effekt: Verlängerung des Proteins, Einbau falscher Aminosäuren, Frameshift(Leserasterverschiebung), Erhöhung der Gendosis.
T I L G G VNormal ACG ATC CTG GGG GGT GTGMut insA ACG ATA CCT GGG GGG TGT
T I P G G C

Punktmutationen• Veränderung eines Basenpaares.
Effekt: Verkürzung des Proteins (Stopp- oder „Nonsense“-Mutation), Einbau falscher Aminosäuren („missense“-Mutation).
T I L G G V QNormal ACG ATC CTG GGG GGT GTG CAGMut T>C ACG ATC CCG GGG GGT GTG CAG
T I P G G V Q
Mut C>T ACG ATC CTG GGG GGT GTG TAGT I L G G V X

Transition versus Transversion
• Transition: Purin zu Purin (A>G), Pyrimidin zu Pyrimidin (C>T)
• Transversion: Purin zu Pyrimidin (A>C,T), (G>C,T) Pyrimidin zu Purin (C>A,G), (T>A,G)
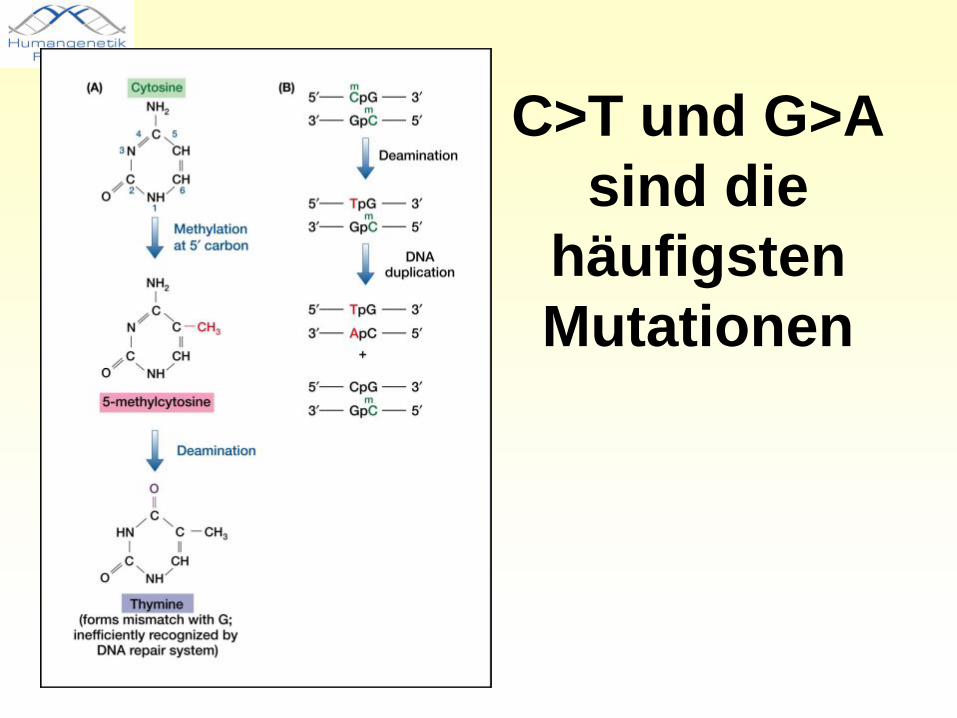
C>T und G>A sind die
häufigsten Mutationen
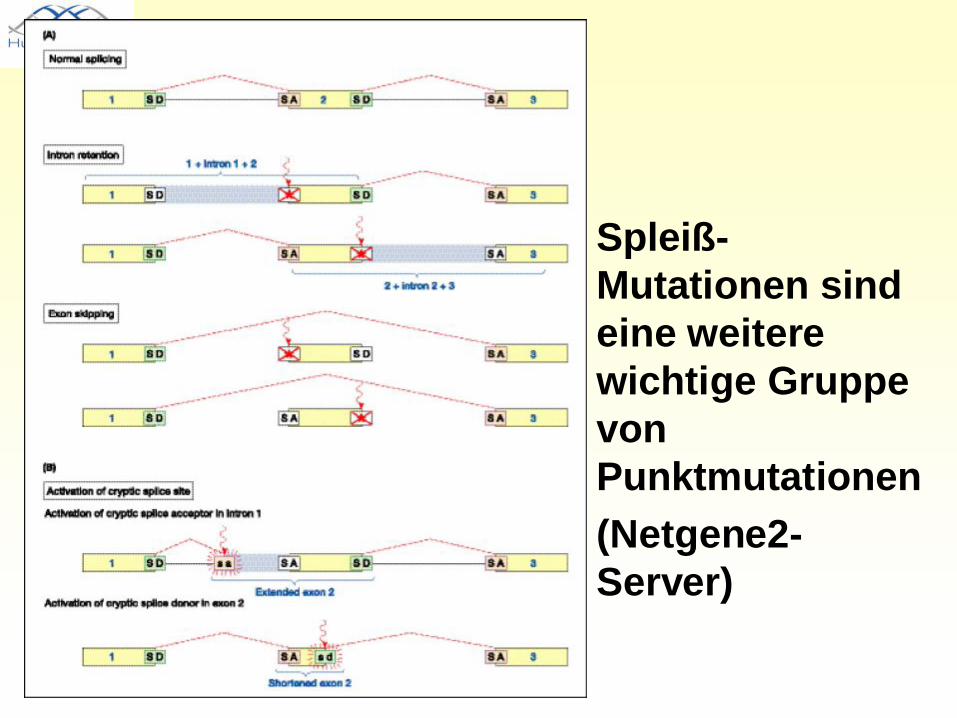
• Spleiß-Mutationen sind eine weitere wichtige Gruppe von Punktmutationen
• (Netgene2-Server)

Was ist autosomal-dominant?
• Die Vererbung erfolgt über ein Nicht-Geschlechtschromosom (Autosom)
• Jeder gesunde Mensch hat i.d.R. von allen autosomalen Genen zwei Kopien, eine vom Vater, eine von der Mutter
• Ist eine davon defekt, kommt es zur Krankheit, d.h. die zweite Kopie kann die Mutation nicht ausgleichen

I1 2
I I 1 2 3
I I I
I V
1 2 3
1 2 3
Was ist autosomal-dominant?• Suggestiv: Vererbung über drei
konsekutive Generationen• Beweisend: Mutter-Sohn- und
Vater-Sohn-Vererbung schließen geschlechtschromosomalen Erbgang aus
• Beispiel PKD

Polyzystische Nierenerkrankung
• Zunehmende Nierengröße
• Verschlechterung der Nierenfunktion
• Endstadium Nierenversagen
• Gene: PKD1(Chromosom 16), PKD2(Chromosom 4)
I1 2
I I 1 2 3
I I I
I V
1 2 3
1 2 3
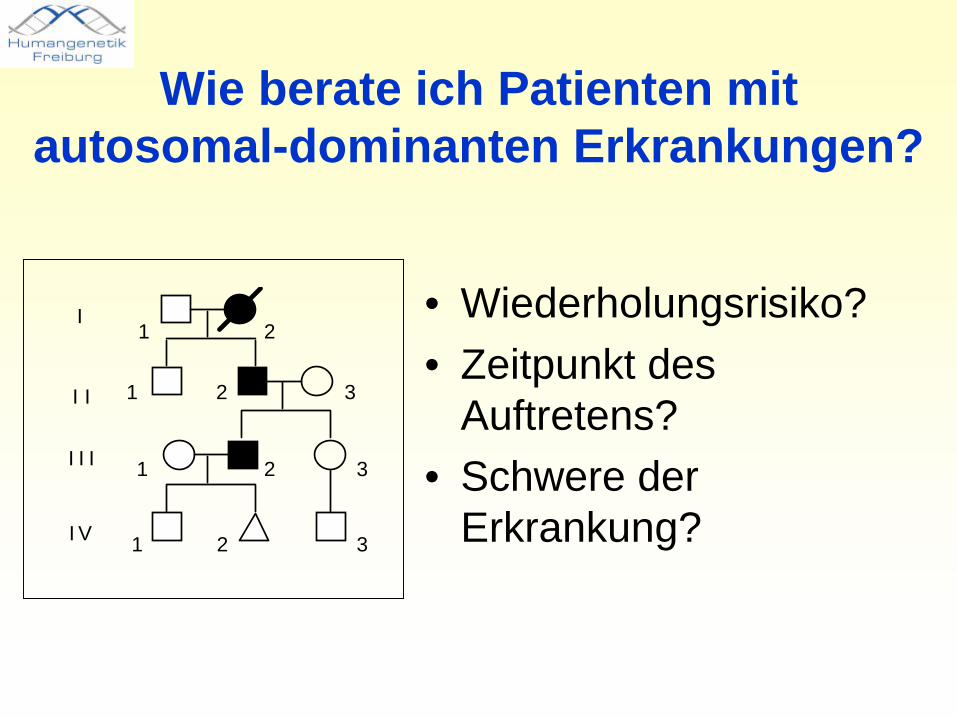
Wie berate ich Patienten mit autosomal-dominanten Erkrankungen?
• Wiederholungsrisiko?• Zeitpunkt des
Auftretens?• Schwere der
Erkrankung?
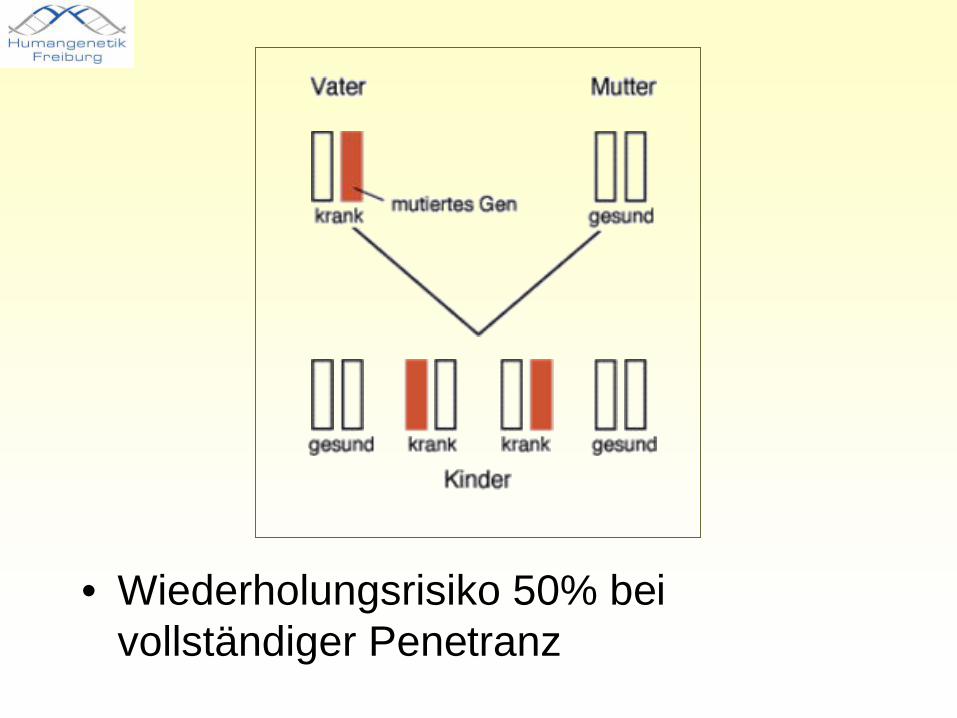
• Wiederholungsrisiko 50% bei vollständiger Penetranz

Neurofibromatose 1• Gen auf Chromosom 17q11.2• Protein: Neurofibrin• Inzidenz: 1/1000 (Israel) - 1/ 3500 (SF)• Café au lait-Flecke• Neurofibrome• Seltener Skoliosen, mentale Retardierung,
Pseudarthrose der Tibia, Phäochromozytom, Meningiom, Gliom, Akusticusneurinom, Opticusneurinom, Hypertension
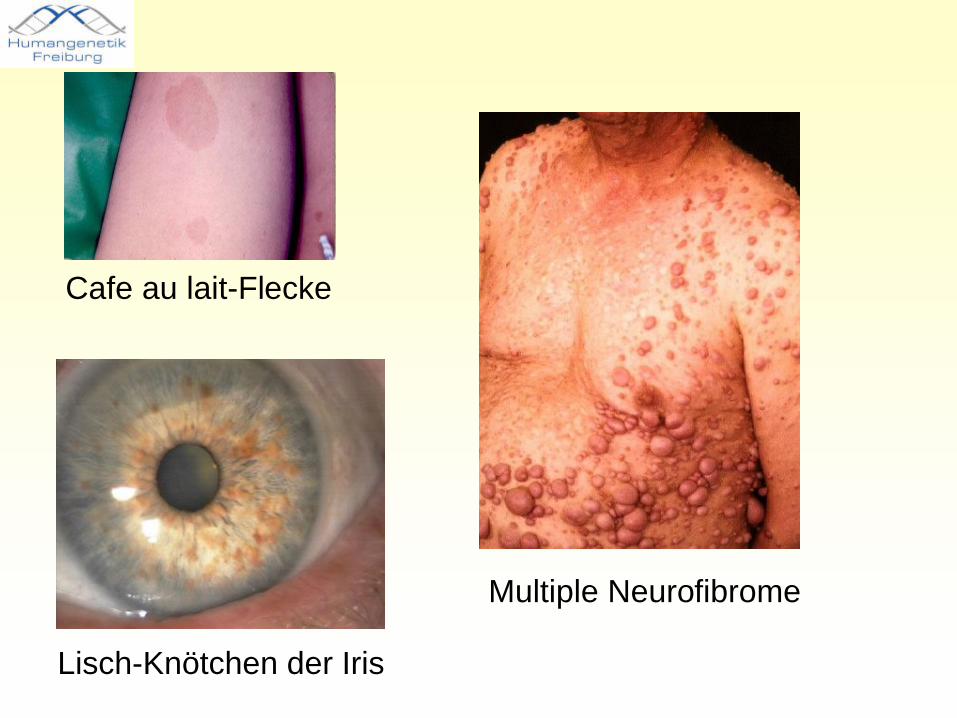
Cafe au lait-Flecke
Lisch-Knötchen der Iris
Multiple Neurofibrome

Marfan-Syndrom• Bindegewebserkrankung, Häufigkeit 1-2/10000• Meist verursacht durch Mutationen von FBN1 auf Chromosom
15q21• Betrifft verschiedene Systeme:
– Kardiovaskuläres System: Aortendilatation, Risiko dissezierender Aortenaneurysmen
– Okuläres System: Linsenluxation, abgeflachte Kornea– Skelettsystem: Kielbrust, Trichterbrust,
Armspanne/Körperlänge >1.05, positives Handgelenk- und Daumenzeichen, Skoliose >20% oder Wirbelgleiten, eingeschränkte Ellbogenstreckung, Plattfuß
– Lunge: Spontanpneumothorax– Dura: Ektasie– Haut: Striae
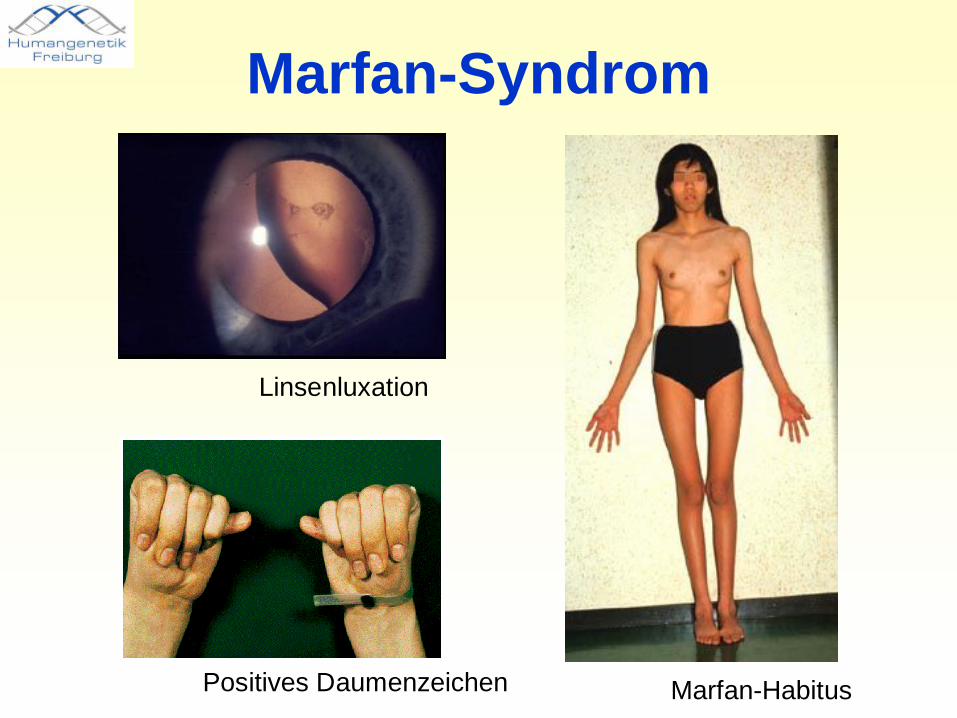
Marfan-Syndrom
Linsenluxation
Marfan-HabitusPositives Daumenzeichen

Marfan-Syndrom• Problematik: rechtzeitige Diagnosestellung zur Prophylaxe von
lebensbedrohlichen Komplikationen• Vorgehen:
– klinische Diagnostik bei einem Indexpatienten, dabei Überprüfung aller betroffenen Systeme nach den sog. Gent-Kriterien
– Molekulargenetische Untersuchung des FBN1-Gens: Mutation wird in max. 90% der „Gent-positiven“ Personen gefunden
– Molekulargenetische Untersuchung von Verwandten: Identifizierung von Mutationsträgern und Angebot eines besonderen Vorsorgeprogramms insbesondere zur Früherkennung von relevanten Gefäßkomplikationen
• Personen ohne FBN1-Mutation mit starkem klinischen Verdacht können unerkannte Mutationen in FBN1 oder Mutationen in anderen Genen (TGFBR2 und TGFBR1) tragen.

Der „sporadische Fall“• Sporadischer Fall:
Keimzellmutation (dominante Neumutation) oder Keimbahnmosaik
• Erbgang nur bei Kenntnis der Erkrankung erkenntlich

CHARGE-Syndrom• 1 auf 10000 bis 1 auf 8500 Neugeborene• Fast ausschliesslich sporadisch, wenige familiäre Fälle • Wiederholungsrisiko von 1-2% (empirisch)• Lange Zeit als Assoziation bezeichnet, Erbgang oft
verkannt.• CHARGE: C = coloboma, H = Herzfehler, A = Atresia
der Choanae, R = Retardation (Wachstum und Entwicklung), G = (Uro)Genital-Anomalien, E = Ear Anomalien und/oder Hörstörungen.
• LKG-Spalte, Tracheo-Ösophageale Fistel oder Atresie, Gesichtslähmung, Schluckprobleme, zerebrale Anfälle, Mikrozephalie, Anomalien der Hypophyse, Schwäche des Immunsystems.

CHARGE-Syndrom
• Ursache: heterozygote CHD7-Mutationen• Also: autosomal-dominante Erkrankung mit fast
ausschliesslich sporadischen Fällen• 50% Risiko für Kinder Betroffener (selten)
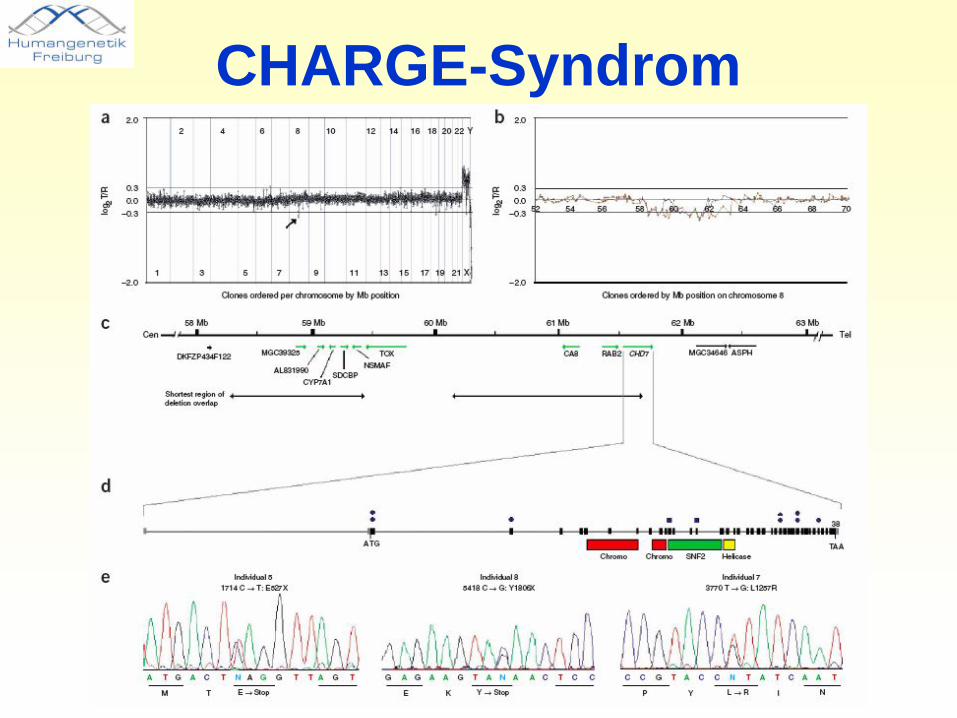
CHARGE-Syndrom
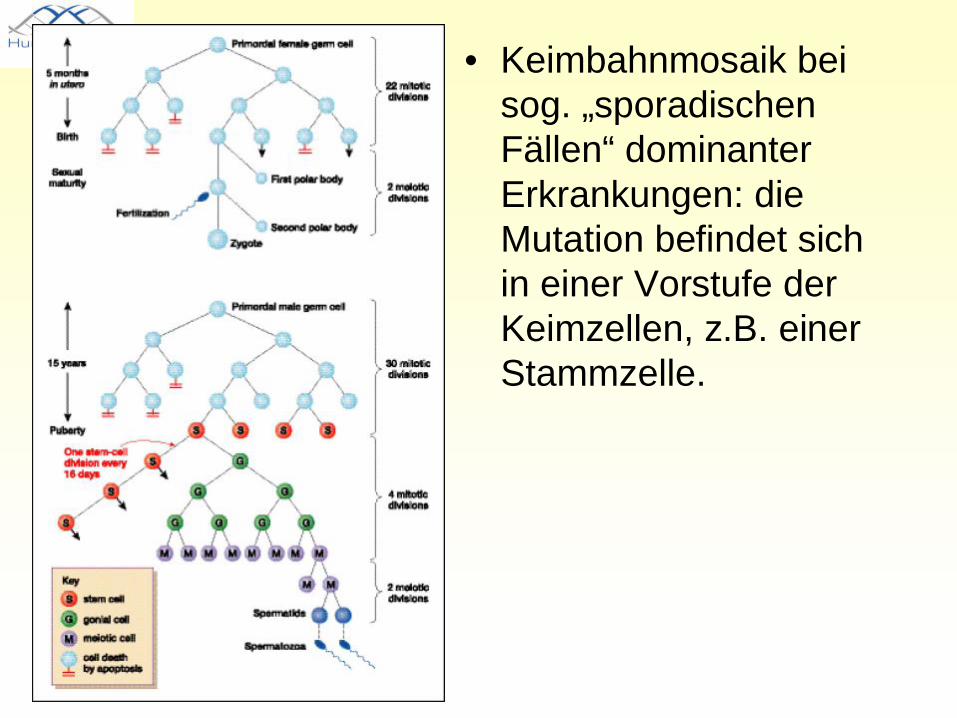
• Keimbahnmosaik bei sog. „sporadischen Fällen“ dominanter Erkrankungen: die Mutation befindet sich in einer Vorstufe der Keimzellen, z.B. einer Stammzelle.

Beratung bei sporadischen Fällen
• Mutationsanalyse wenn möglich beim Kind durchführen (EDTA-Blut). Wenn eine Mutation gefunden wird, Eltern untersuchen.
• Bei Nachweis der Mutation bei den Eltern in Lymphozyten-DNA (EDTA-Blutprobe): 50% Risiko für weitere Kinder
• Kein Mutationsnachweis in elterlicher Lymphozyten-DNA: meist ca. 2-5% Wiederholungsrisiko

Penetranz
• Anteil der betroffenen Mutationsträger an der Gesamtheit der Mutationsträger, z.B. 70% (reduzierte Penetranz) oder 100% (vollständige Penetranz)
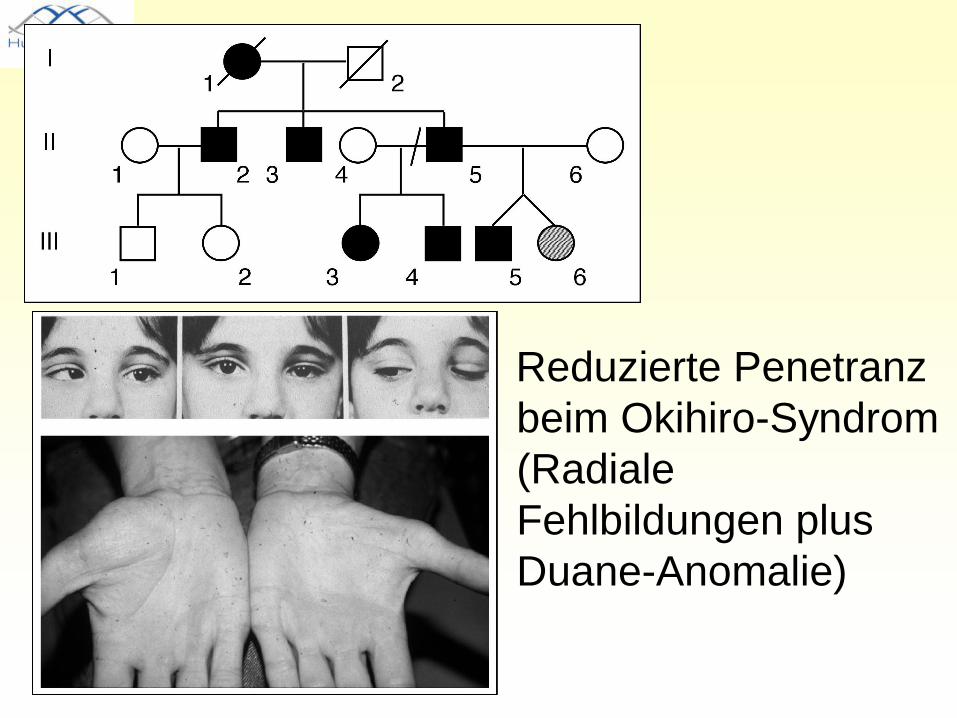
Reduzierte Penetranz beim Okihiro-Syndrom (Radiale Fehlbildungen plus Duane-Anomalie)

Variable Expressivität
• Erkrankungen manifestieren sich unterschiedlich schwer bei Betroffenen derselben Familie oder verschiedener Familien
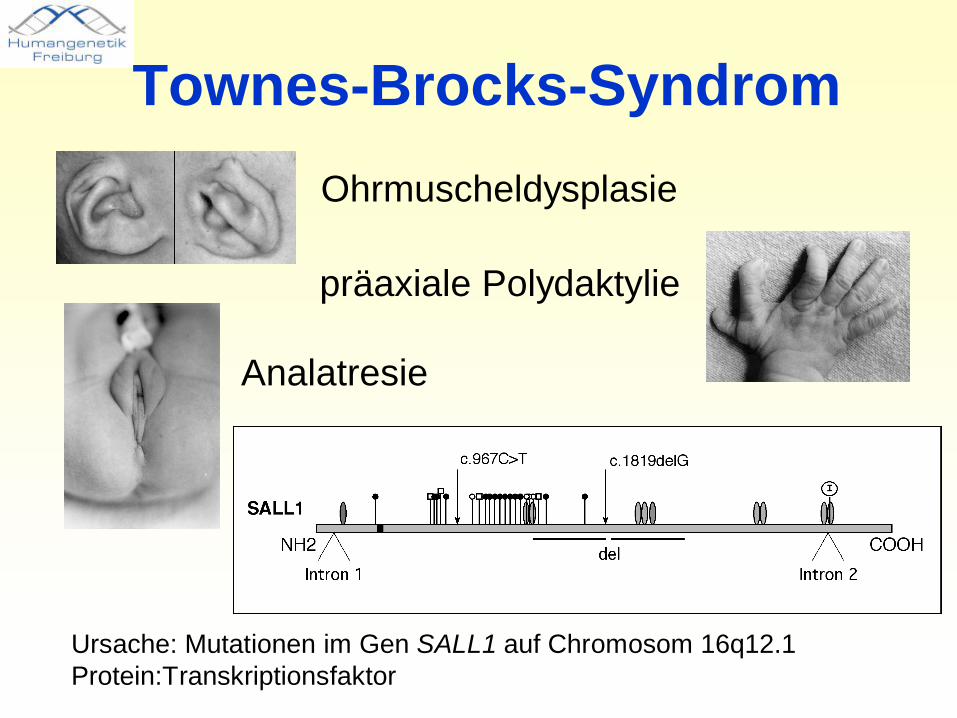
Townes-Brocks-SyndromOhrmuscheldysplasie
präaxiale Polydaktylie
Analatresie
Ursache: Mutationen im Gen SALL1 auf Chromosom 16q12.1 Protein:Transkriptionsfaktor
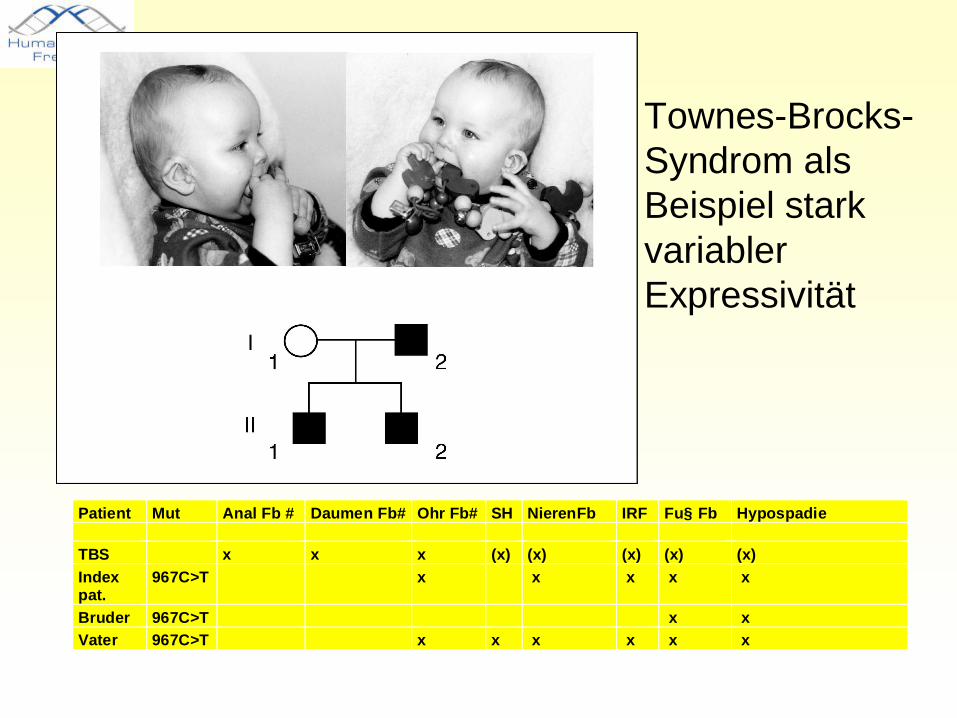
• Townes-Brocks-Syndrom als Beispiel stark variabler Expressivität
Patient Mut Anal Fb # Daumen Fb# Ohr Fb# SH NierenFb IRF Fu§ Fb Hypospadie
TBS x x x (x) (x) (x) (x) (x)Indexpat.
967C>T x x x x x
Bruder 967C>T x xVater 967C>T x x x x x x
4
2 31
21 3
1 23
5 6 7 8 9
41 2 3 5 6 7 8
4 56 7
4 5
I
I I
I I I
I V
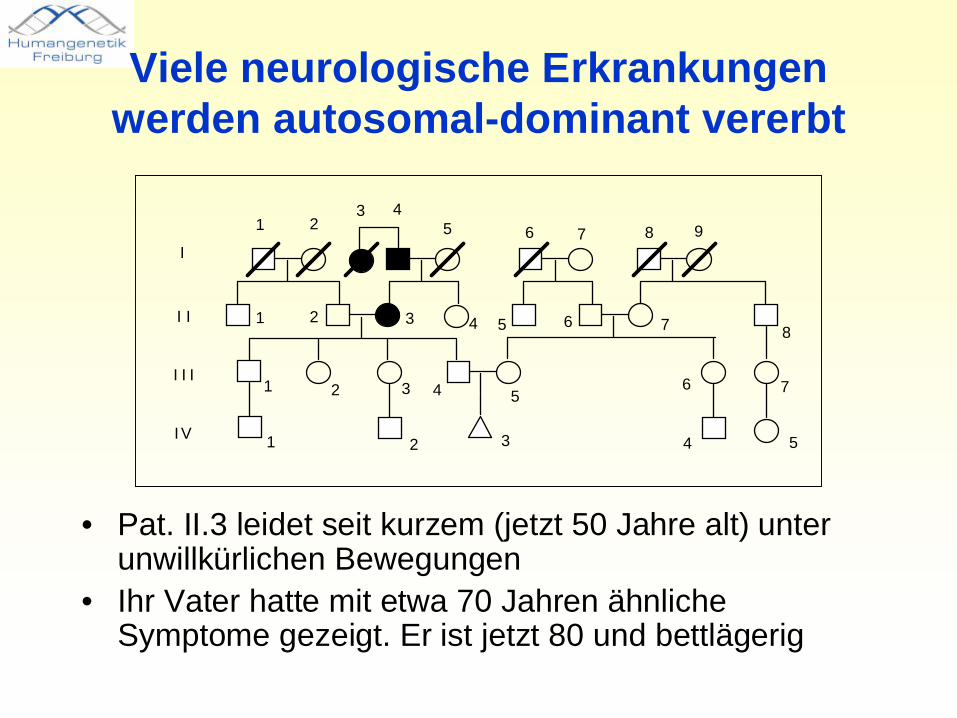
Viele neurologische Erkrankungen werden autosomal-dominant vererbt
• Pat. II.3 leidet seit kurzem (jetzt 50 Jahre alt) unter unwillkürlichen Bewegungen
• Ihr Vater hatte mit etwa 70 Jahren ähnliche Symptome gezeigt. Er ist jetzt 80 und bettlägerig

Chorea Huntington
• Erste Symptome im Allgemeinen zwischen dem 35. und dem 45. Lebensjahr (also selten vor dem Heiratsalter!) auf. Krankheitsbeginn vor dem 10. und nach dem 60. Lebensjahr selten.
• Anfangs nicht selten psychische Auffälligkeiten: Betroffene sind vermehrt reizbar, aggressiv oder enthemmt, verlieren ihre Spontanität oder zeigen zunehmende Ängstlichkeit (im Spätstadium Demenz).
• Bewegungsunruhe der Extremitäten, plötzlich auftretende unwillkürliche Bewegungen von Armen, Beinen, Kopf oder Rumpf.
• Grimassenschneiden, in Extremfällen tänzelnder Gang, der den Namen "Veitstanz" (griech. Choreia = Tanz) prägte.
• abgehackte Sprache, Schluckstörungen.• Muskelsteifheit mit Bewegungsverminderung im Spätstadium• Dauer bis zum Tod ca. 5-20 Jahren (abhängig von der Schwere der
Erkrankung).

Mutationswirkungen• Stoppmutationen, Deletionen: Menge des
Genprodukts halbiert, was zur normalen Funktion nicht ausreicht (Haploinsuffizienz).
• Andere Möglichkeit: Stoppmutationen führen zu kürzeren Proteinen, die mit dem Normalprotein interferieren (dominant-negative Wirkung)
• Missense-Mutationen, CAG-Repeatveränderungen: veränderte Proteine haben veränderte Eigenschaften, echte dominante (gain of function) oder auch dominant-negative Wirkung

Dominant negative Mutationen in Kollagen-Genen sind schwer-wiegender als Null-Mutationen
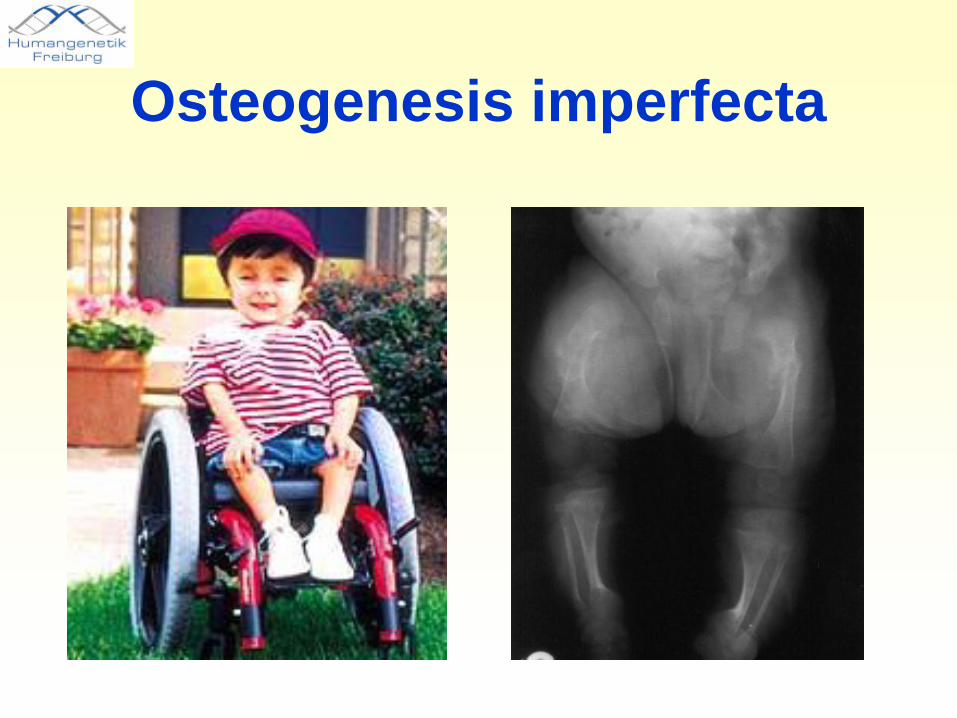
Osteogenesis imperfecta

Genomische Erkrankungen
Prof. Dr. med. Jürgen KohlhasePraxis für Humangenetik Freiburg
www.humangenetik-freiburg.de

Was sind genomischeErkrankungen?
• Krankheiten, die aufgrund wiederkehrender DNA-Rearrangements in „instabilen genomischen Regionen“ entstehen
• der klinische Phänotyp ist eine Konsequenz einer abnormalen Gendosis eines oder mehrerer Gene
• inter- und intrachromosomale Rearrangements werden begünstigt durch regionsspezifische „low-copy repeats“ (LCRs) und resultieren aus nicht-allelischen homologen Rekombinationen (NAHR)
• LCRs umfassen ca. ~10–400 kb genomischer DNA und zeigen ≥97% Sequenzübereinstimmung.
• NAHR-verursachte Erkrankungen zeigen übereinstimmende Bruchpunkte. Eine zweite Form genomischer Erkrankungen ist durch wiederkehrende Deletionen unterschiedlicher Größe gekennzeichnet, der Mechanismus ist nicht NAHR.

Was sind genomischeErkrankungen?
• Unterschied zu nicht-genomischen erblichen Erkrankungen: solche entstehen durch Punktmutationen oder kleinere Deletionen/ Insertionen und mangelhafte DNA-Replikation oder -Reparatur
• Sind in der Regel häufiger als monogeneErkrankungen durch Punktmutationen
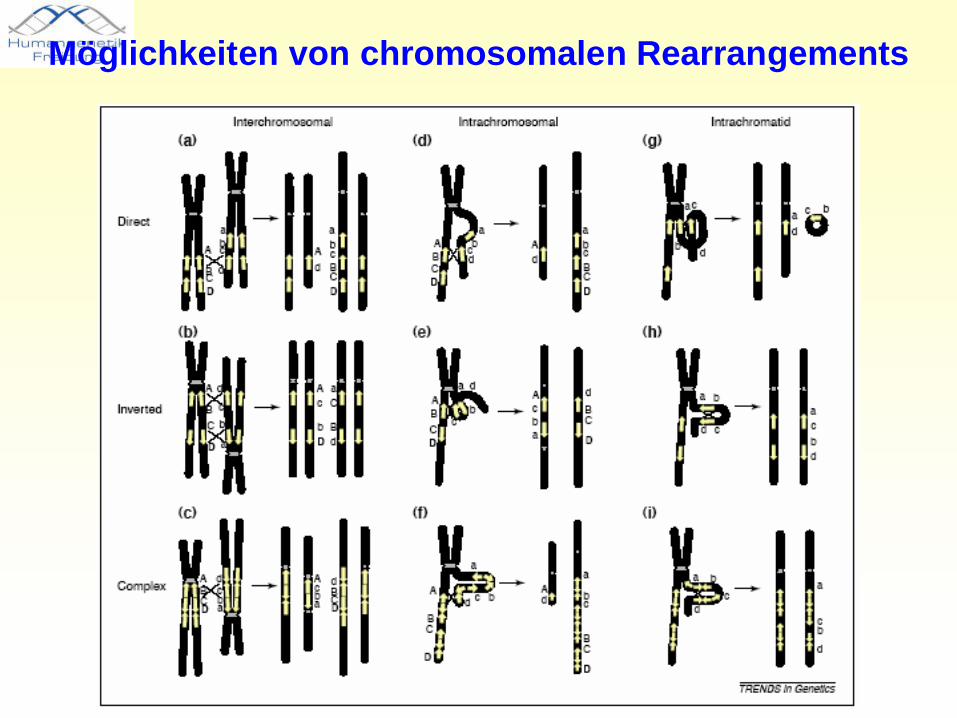
Möglichkeiten von chromosomalen Rearrangements
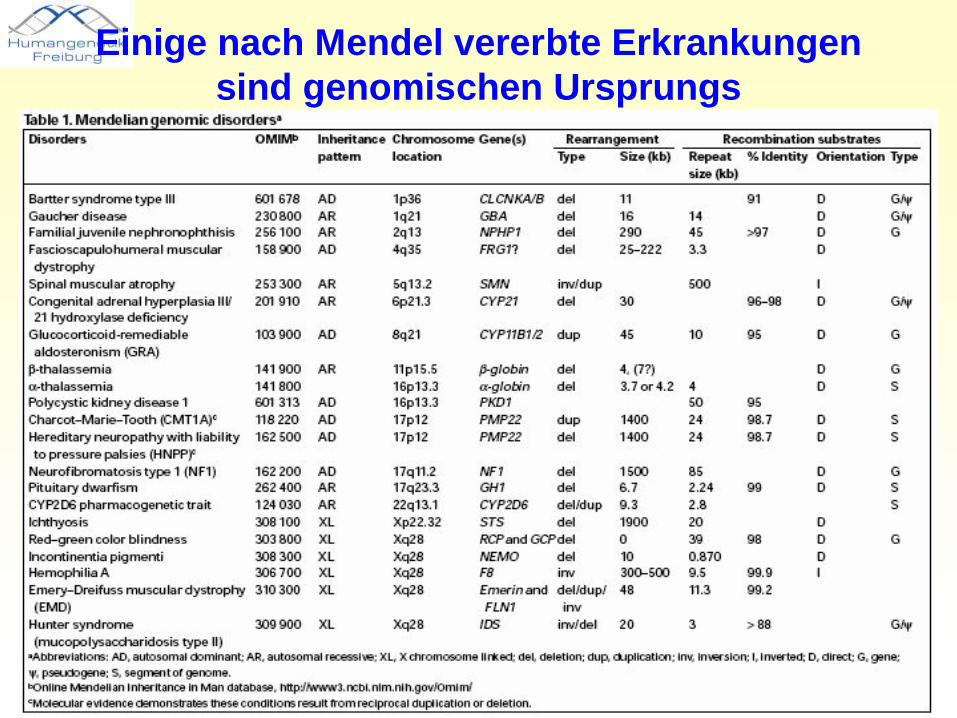
Einige nach Mendel vererbte Erkrankungen sind genomischen Ursprungs

„Contiguous gene syndromes“genomischen Ursprungs

Genomische Erkrankungen
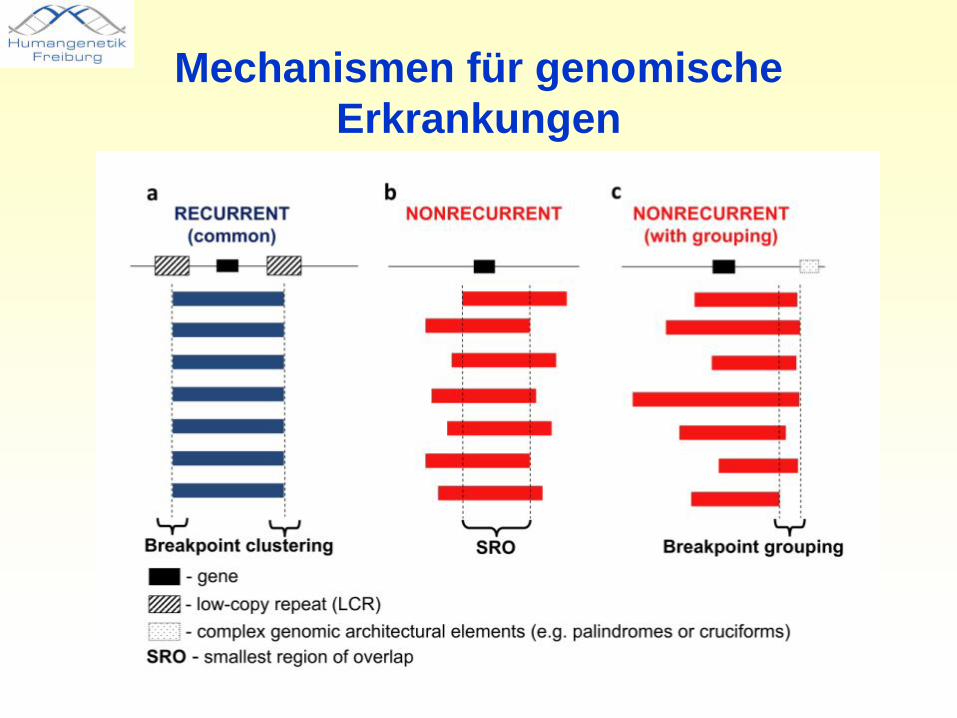
Mechanismen für genomischeErkrankungen
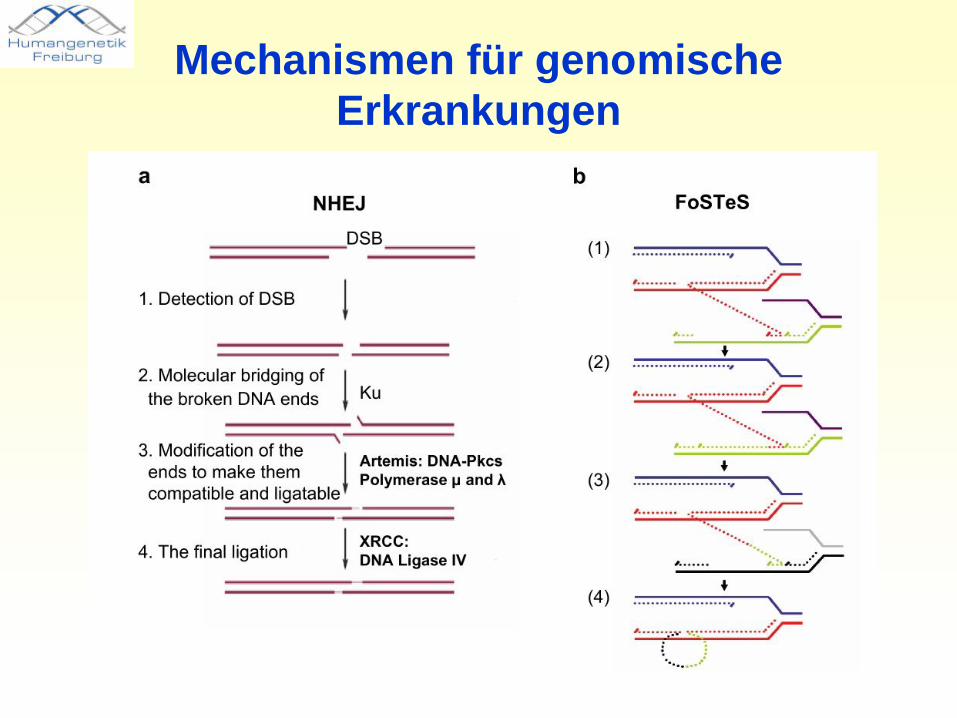
Mechanismen für genomischeErkrankungen

Genomische Erkrankungen
Beispiele:• HNPP/ CMT1A• WBS• DGS/VCFS• Sotos-Syndrom

Charcot-Marie-Tooth-Erkrankung• Synonym: Hereditäre Motorisch-Sensible Neuropathie• langsam progrediente Degeneration der peripheren Nerven• motorische Nervenfasern meist wesentlich stärker betroffen
als sensorische• Sekundär Untergang einzelner Muskelfasern und damit
neurale Muskelatrophie, meist zuerst an Fuß- und Unterschenkelmuskulatur. Später auch obere Extremitäten betroffen. Spätfolgen: schwere Gangstörung oder zu einer Beeinträchtigung der Feinbeweglichkeit der Hände.
• Ausprägung intra- und interefamiliär sehr variabel.• verschiedene Typen der HMSN werden unterschieden.
Meist autosomal-dominant vererbt, aber auch X-chromosomal- und autosomal-rezessive Vererbung.
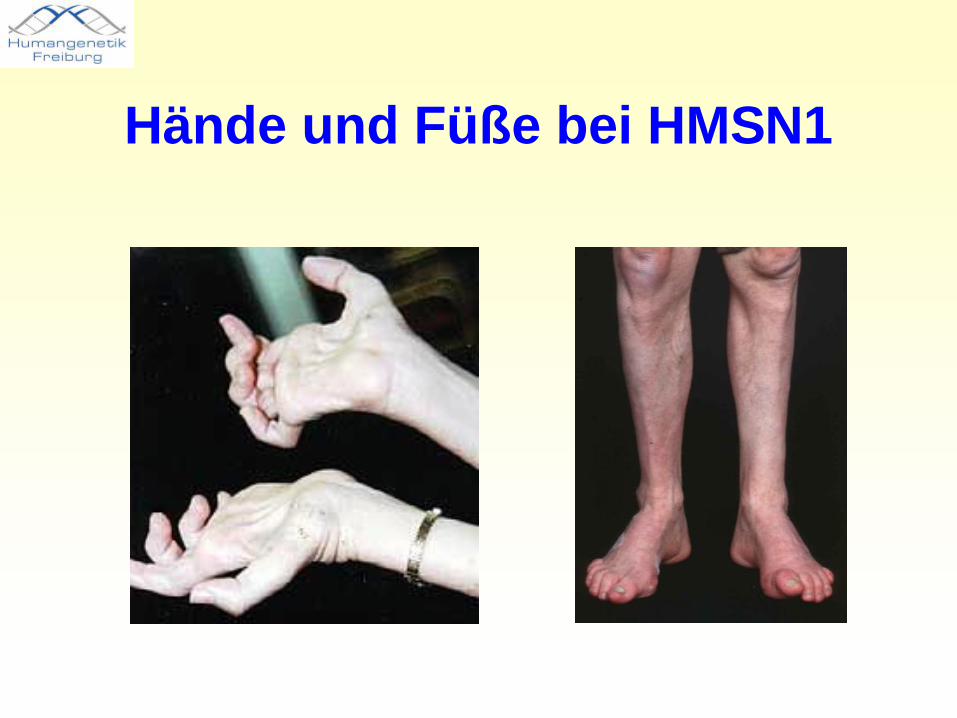
Hände und Füße bei HMSN1

Hereditäre Neuropathie mit Neigung zu Druckläsionen (tomakulöse Neuropathie, HNPP)
• Erkrankung der peripheren Nerven: Verdickung der Markscheide und später segmentale Entmarkung der Nerven.
• polyneuropathische Beschwerden mit Neigung zu Druckläsionen
• Sensibilitätsstörungen und/ oder Lähmungen bevorzugt an den Extremitäten.
• Erste Symptome zwischen dem ersten und dritten Lebensjahrzehnt.
• Auslöser: längerdauernder Druck, kleinere Traumata, Kälte. • Prophylaxe: Vermeidung auslösender Faktoren
(insbesondere bei Operationen!)• Autosomal-dominante Vererbung
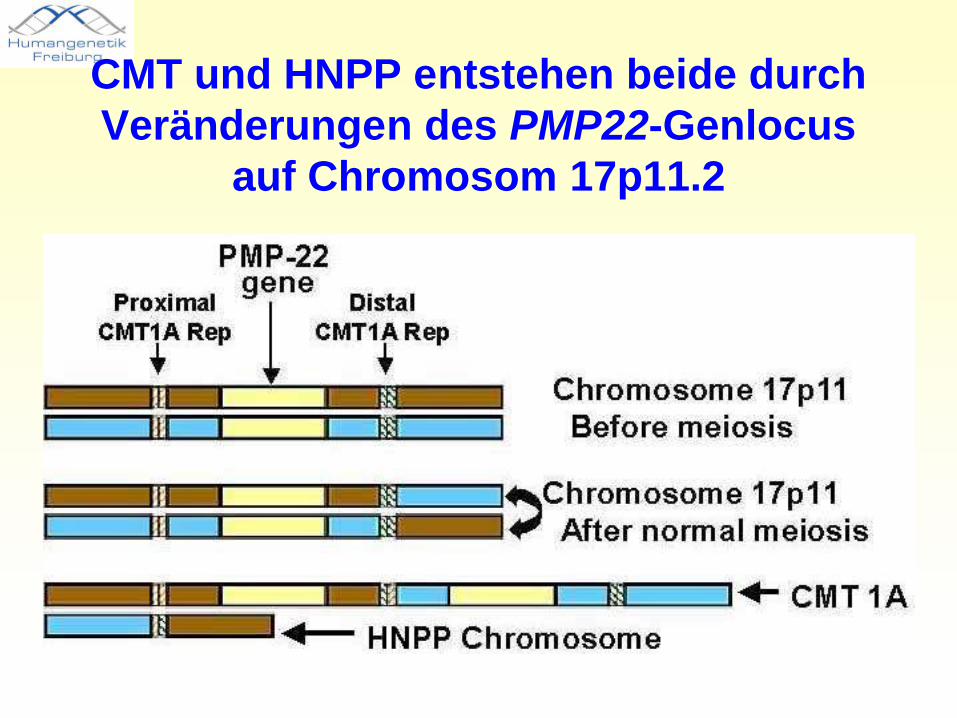
CMT und HNPP entstehen beide durch Veränderungen des PMP22-Genlocus
auf Chromosom 17p11.2
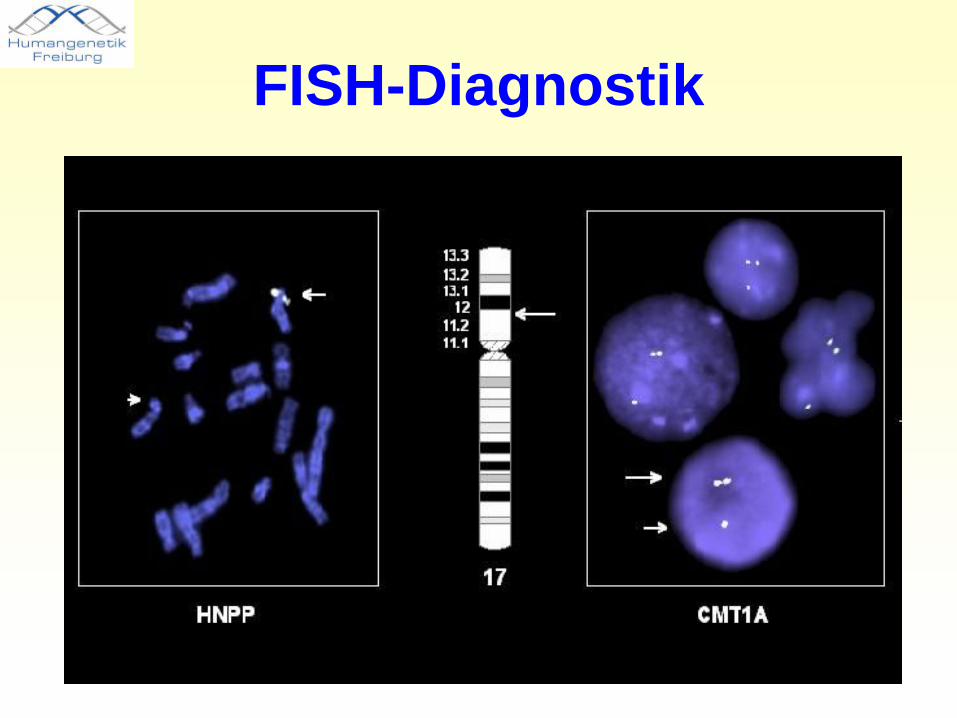
FISH-Diagnostik

Williams-Beuren-Syndrom• breite Stirn• Mikrozephalie• abgerundete Nasenspitze,
antevertierte Nasenlöcher• volle Wangen• großer Mund mit evertierter
Unterlippe• kleines Kinn (Mikrogenie) • Strabismus• Hypoplastische Zähne• Kleinwuchs• IQ meist zwischen 35 und 70• Gefäßstenosen, besonders
supravalvuläre Aortenstenose• Ca. 1/10000 Neugeborene
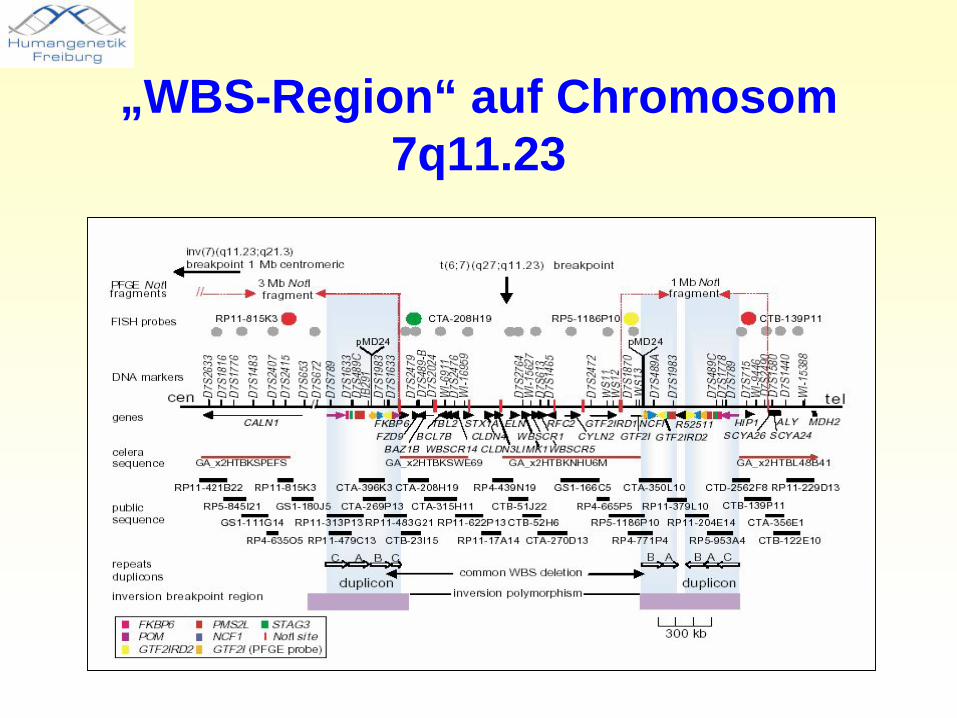
„WBS-Region“ auf Chromosom 7q11.23

Ursache
• „Contiguous gene syndrome“ durch ungleiche meiotische Rekombination
• in 95% der Fälle: definierte 1,5 Mb-Deletion (heterozygot)
• Deletion umfaßt 17 Gene• restliche 5% der Fälle: kein
nachweisbares Chromosomen-Rearrangement

Inversion zwischen Duplikons• Bei ca. 1/3 der Eltern von
Patienten findet man eine Inversion der WBS-Region, die sonst bei ca. 5% der Bevölkerung zu finden ist
• Die Inversion liegt auf dem Chromosom, welches beim Kind die Deletion trägt
• Prädisposition zur Deletion!
Duplikon

Zusammenfassung WBS• Das Williams-Beuren-Syndrom entsteht durch
eine heterozygote Deletion mehrerer Gene auf Chromosom 7q11.23
• Die WBS-Region zeigt häufig eine Inversion bei Eltern von Betroffenen
• Inversion und Deletion werden durch Duplikons(sog. Low-Copy-Repeats) vermittelt
• Die typische Inversion führt nicht zu phänotypischen Ausprägungen, aber scheint für eine Deletion zu prädisponieren.
• Bei Eltern mit Inversion ist daher ein leicht erhöhtes Wiederholungsrisiko anzunehmen

Das 22q11.2 Deletions-Syndrom
Synonyme:
Di-George-Syndrom (DGS):Thymus- und Nebenschilddrüsenhypoplasie oder –aplasie. Ausfall der zellulären Immunität, Herzfehler, Hypokalzämie, Gesichtsdysmorphien
Velo-Cardio-Faciales Syndrom (VCFS):Gaumenspalte, Herzfehler (rechtsseitiger Aortenbogen), Gesichtsdysmorphien, kleine Körpergröße, Hypotonie, dünne Hände und Zehen, Lernschwäche und geistige Retardierung
Conotruncal anomaly face syndrome (CAFS/CAS):Klinische Übereinstimmung mit DGS, in Japan beschriebenKardiovaskuläre Ausprägung

• Meist de novo Deletionen (90 %), weniger als 10 % vererbt
• 80-90% der Patienten haben die gleiche 3Mb-Deletion (ca. 30 Gene), Phänotypen variieren stark
• Ca. 10% zeigen eine kleinere 1,5Mb-Deletion, die aber nicht zwingend zu einem milderen Phänotyp führt
• Häufigkeit: ca. 1 : 4000, häufigstes Mikrodeletions-Syndrom
Das 22q11.2-Deletions-Syndrom
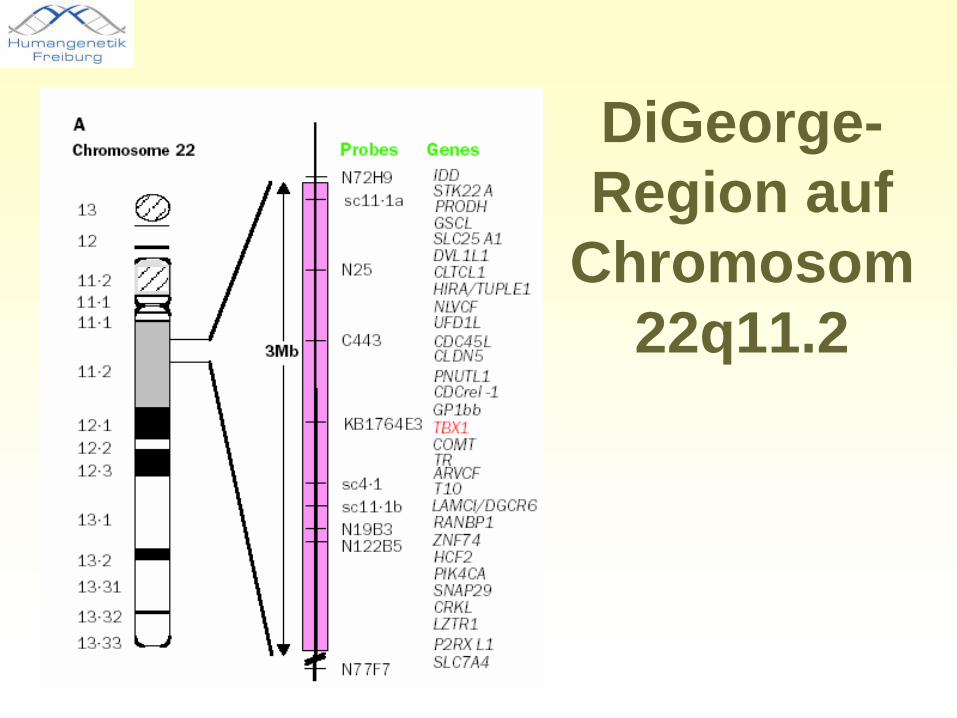
DiGeorge-Region auf
Chromosom 22q11.2
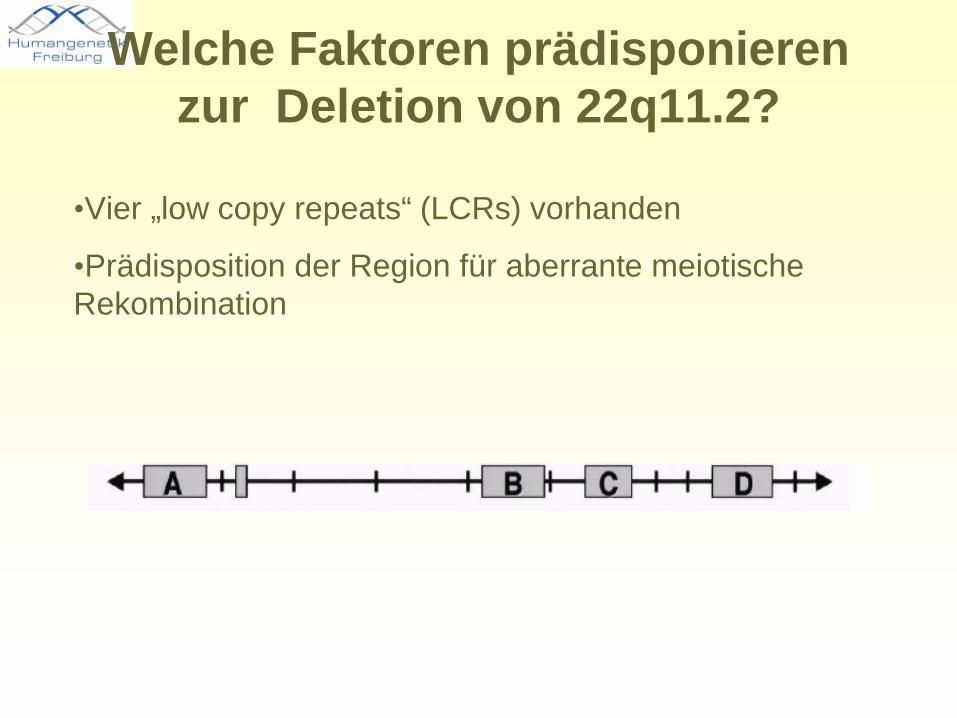
Welche Faktoren prädisponieren zur Deletion von 22q11.2?
•Vier „low copy repeats“ (LCRs) vorhanden
•Prädisposition der Region für aberrante meiotische Rekombination
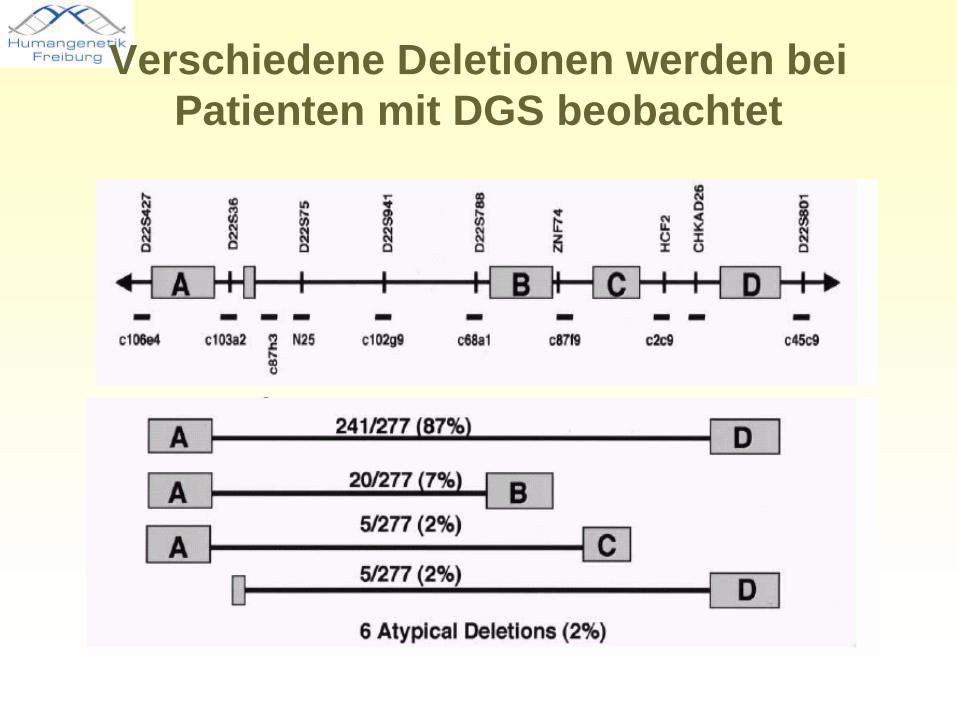
Verschiedene Deletionen werden bei Patienten mit DGS beobachtet
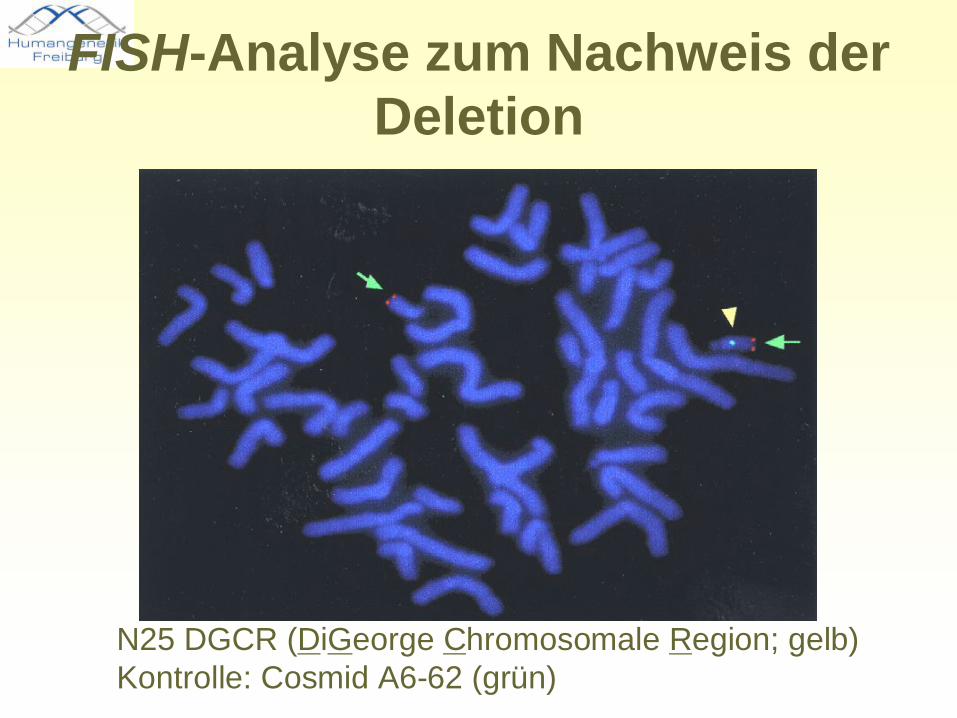
FISH-Analyse zum Nachweis der Deletion
N25 DGCR (DiGeorge Chromosomale Region; gelb)Kontrolle: Cosmid A6-62 (grün)
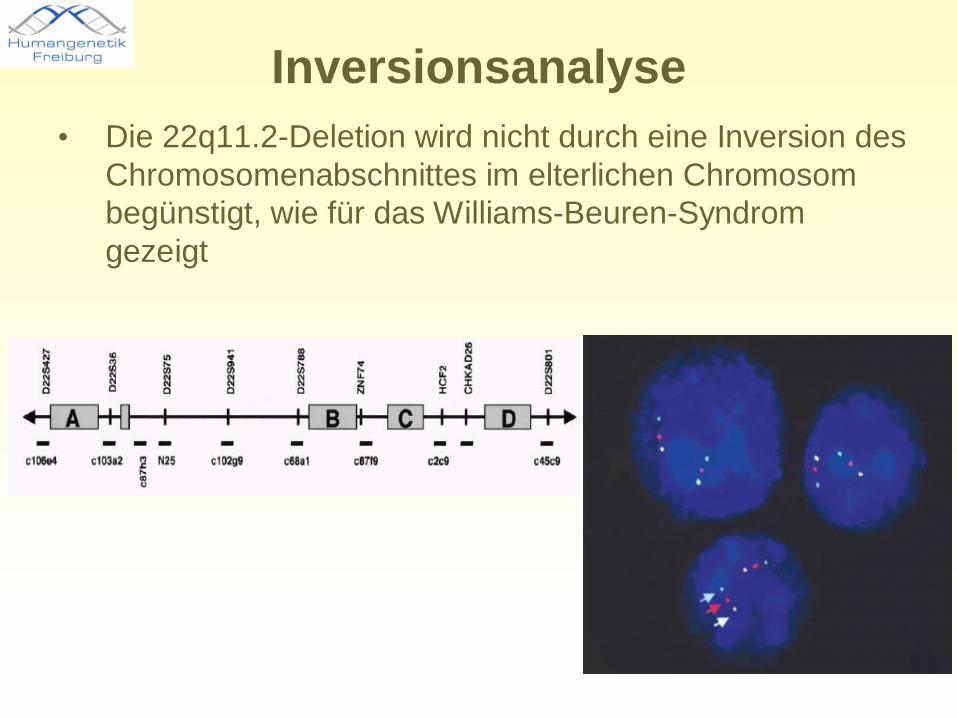
Inversionsanalyse• Die 22q11.2-Deletion wird nicht durch eine Inversion des
Chromosomenabschnittes im elterlichen Chromosom begünstigt, wie für das Williams-Beuren-Syndrom gezeigt
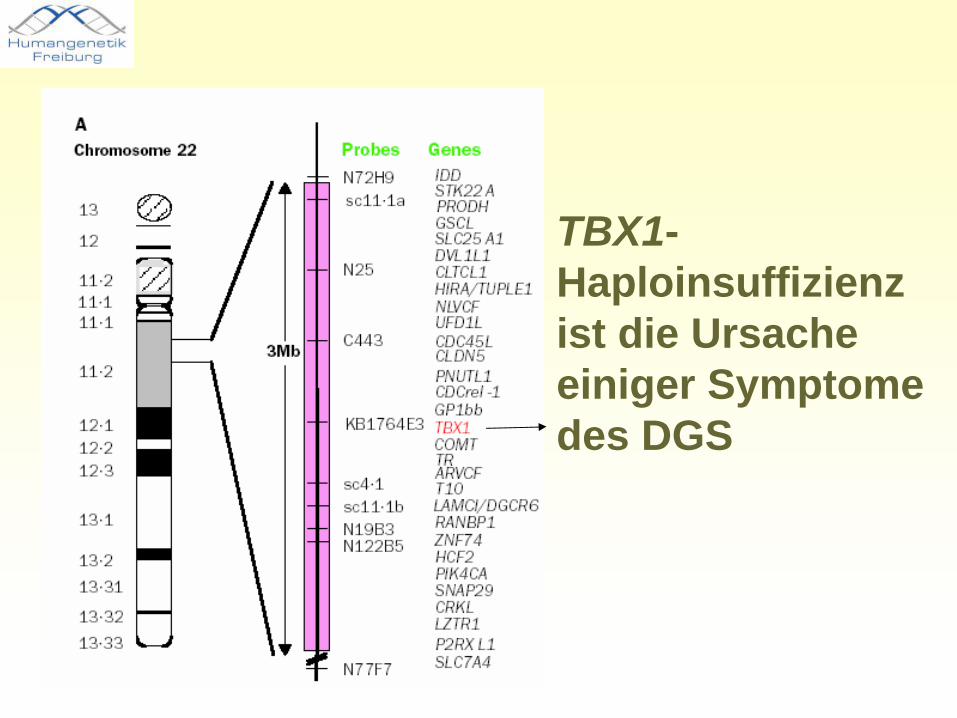
TBX1-Haploinsuffizienz ist die Ursache einiger Symptome des DGS

TBX1 Mutationen sind verantwortlich für 5 Partialphänotypen:
•„Conotruncal anomaly face“- typische Gesichtsdysmorphien•Herzfehler•Hypoplasie des Thymus•Velopharyngeale Insuffizienz mit Gaumenspalte•Nebenschilddrüsendysfunktion mit Hypokalzämie
TBX1 Mutationen sind nicht verantwortlich für die mentale Retardierung
Die TBX1-Haploinsuffizienz ist eine genetische Hauptdeterminante des 22q11.2 Deletions-Syndroms
Rolle von TBX1 bei del22q11.2

•Großwuchs-Syndrom, Länge >97. Perzentile•Makrozephalie >97. Perzentile•Grosse Hände und Füße•Mentale Retardierung•Entwicklungsverzögerung•Neonatale Hypotonie •Epileptische Anfälle•Verursacht durch NSD1-Mutationen•Aber: in Japan 50% größere Deletionen, in anderen Populationen seltener als 10 %
Sotos-Syndrom
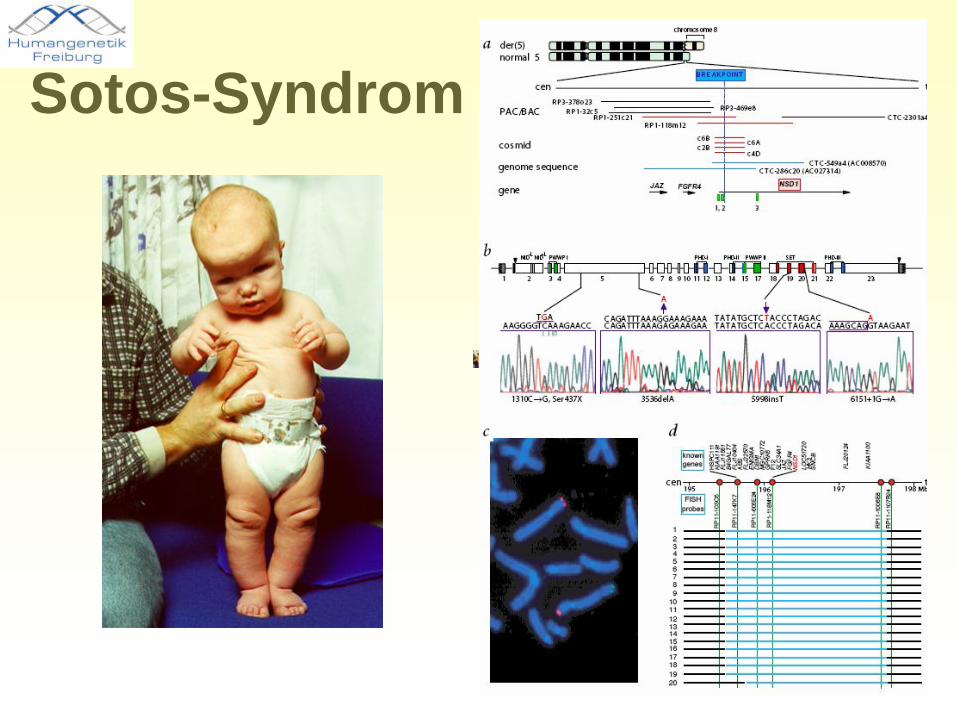
Sotos-Syndrom
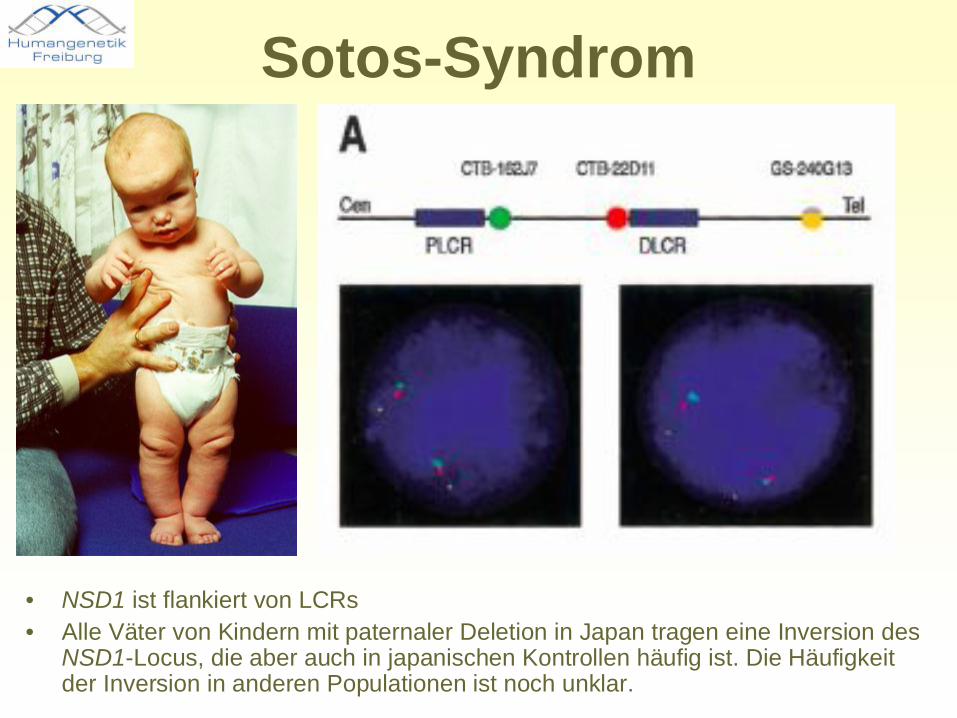
Sotos-Syndrom
• NSD1 ist flankiert von LCRs• Alle Väter von Kindern mit paternaler Deletion in Japan tragen eine Inversion des
NSD1-Locus, die aber auch in japanischen Kontrollen häufig ist. Die Häufigkeit der Inversion in anderen Populationen ist noch unklar.

Moderne Diagnostische MethodikArray-CGH
• Auflösung von Chromosomenanalysen: maximal 5 Mb.• Bei vielen Fehlbildungs-Retardierungssyndromen ist eine
eindeutige klinische Diagnosestellung und damit eine klare Abschätzung des Wiederholungsrisikos nicht möglich.
• Genomprojekt: die Sequenz des humanen Genoms ist nahezu vollständig verfügbar. Damit können Hybridisierungssonden hergestellt werden, um jede Region des Genoms zu untersuchen. Dies kann inzwischen für das gesamte Genom gleichzeitig auf einem Objektträger (Chip, Microarray) geschehen.
• Auflösung: zur Zeit maximal ca. 12 - 50 kb, also 100-mal genauer als die Chromosomenanalyse.
• Bei 12,7% unklarer geistiger Behinderungen wurden mit dieser Methode ursächliche Deletionen/ Duplikationen gefunden, davon 31,7% kleiner als 1 Mb.
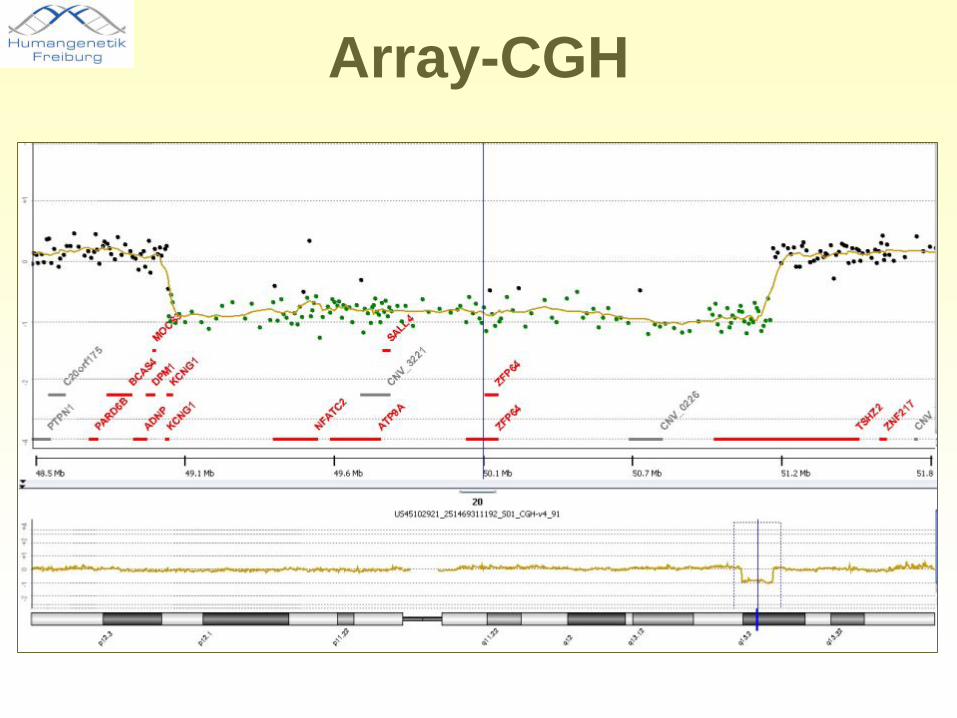
Array-CGH
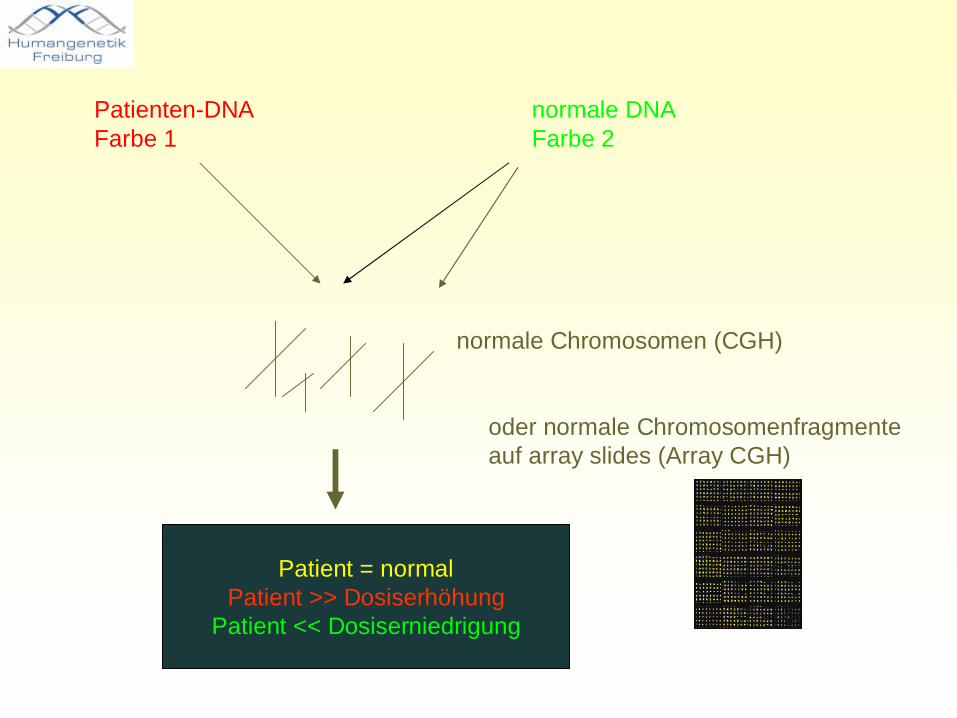
Patienten-DNAFarbe 1
normale DNAFarbe 2
normale Chromosomen (CGH)
Patient = normalPatient >> Dosiserhöhung
Patient << Dosiserniedrigung
oder normale Chromosomenfragmente auf array slides (Array CGH)
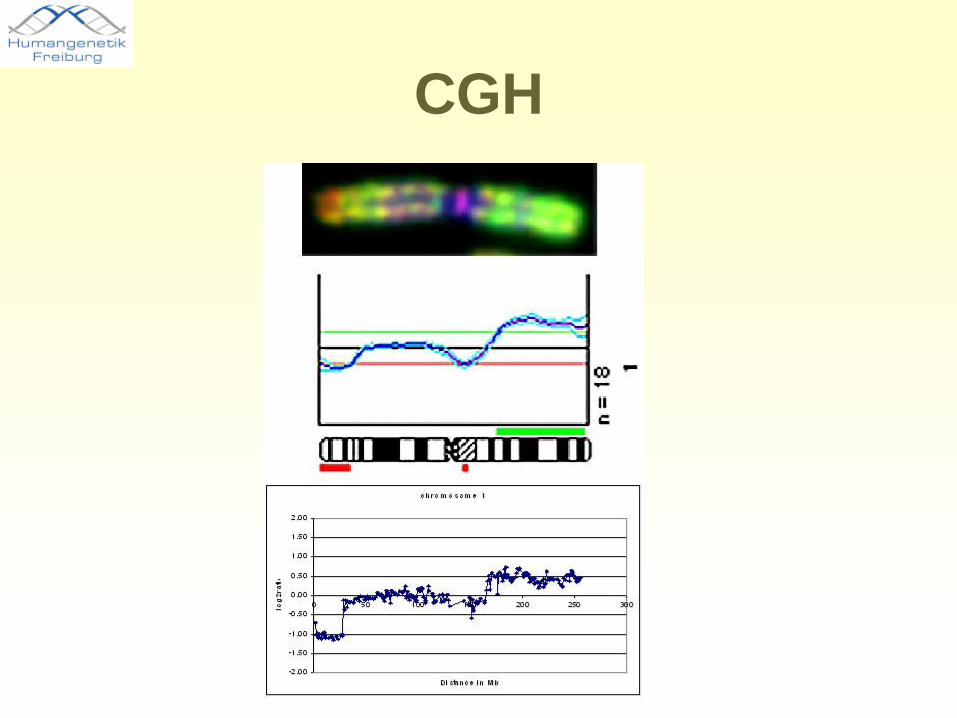
CGH

Sonden in 1 Mb Intervallen
Array CGH - Methode
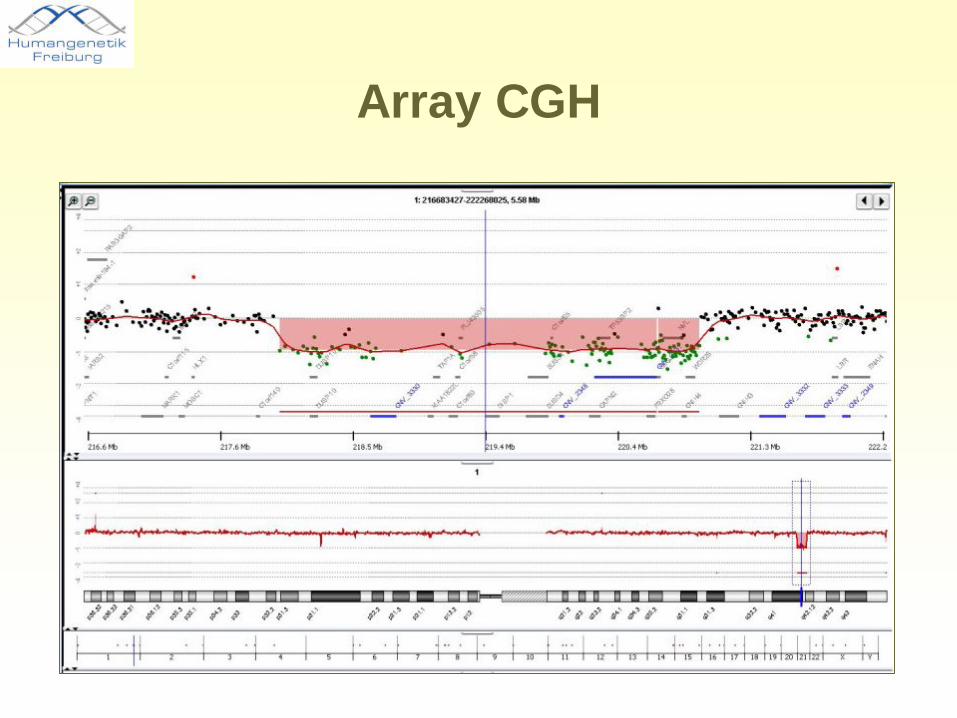
Array CGH
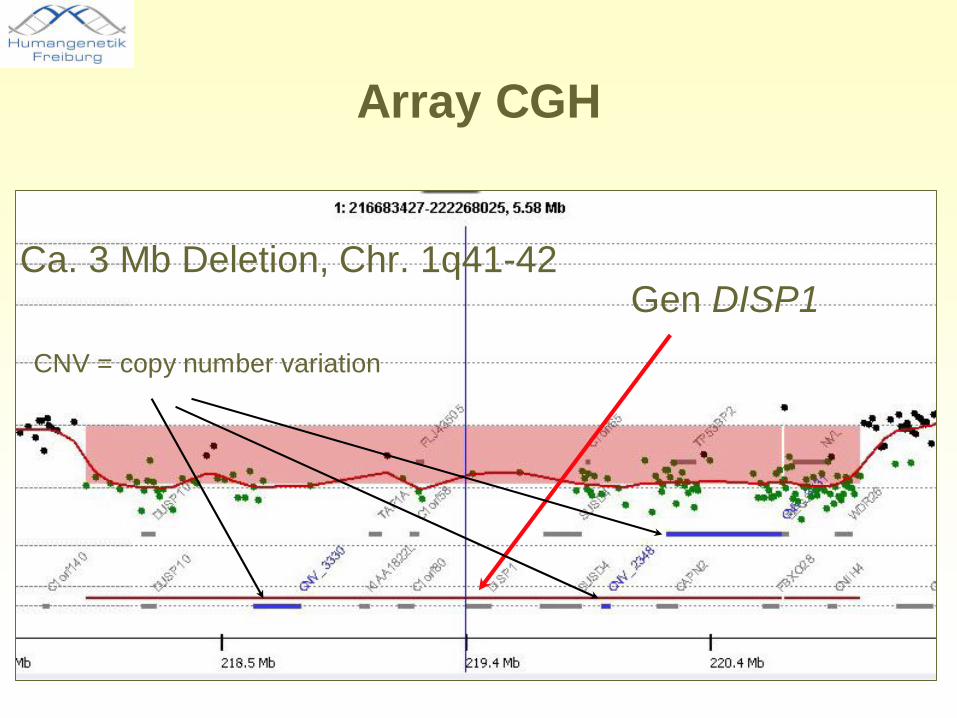
Array CGH
Gen DISP1CNV = copy number variation
Ca. 3 Mb Deletion, Chr. 1q41-42

Abklärung eines unklaren Krankheitsbildes mittels aCGH
• Array-CGH-Ergebnis: heterozygote Deletion auf Chromosom 1q41-q42 (ca. 3 Mb)
• Untersuchung der Eltern (qPCR): unauffällig
• Beurteilung: Deletion 1q41-42 höchstwahrscheinlich ursächlich

Submikroskopische 1q41-q42-Deletionen: ein neues Krankheitsbild
• Geistige Behinderung• Wachstumsretardierung• Schwere
Sprachentwicklungsverzögerung• Anfallsleiden• Distinkte Dysmorphien wie dicke
Lippen, breite Nasenspitze, grobe Gesichtszüge, vorstehende Stirn
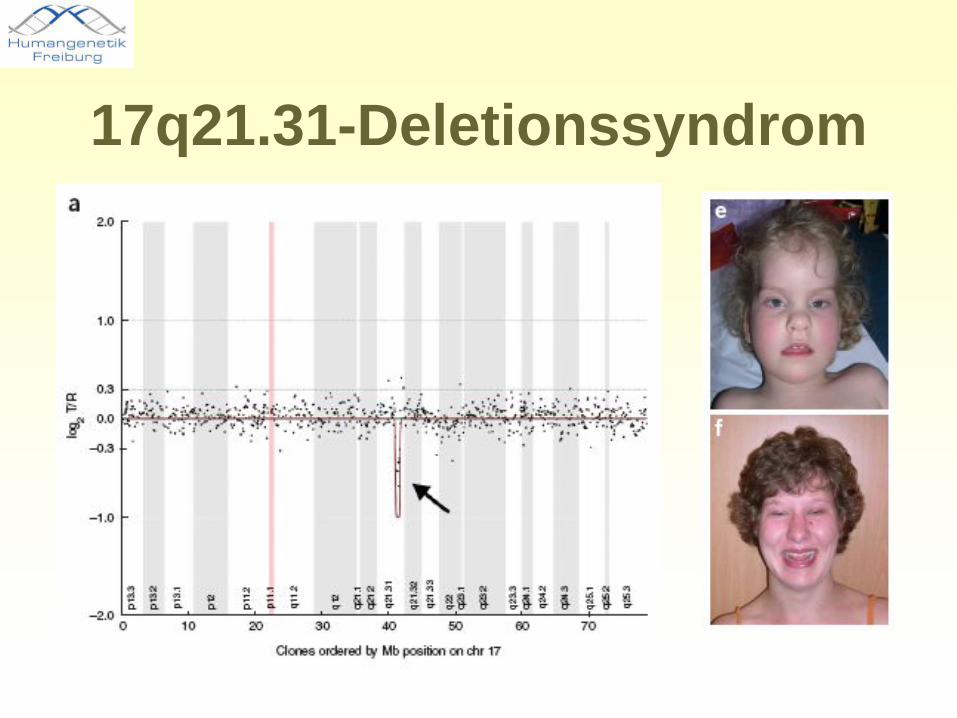
17q21.31-Deletionssyndrom
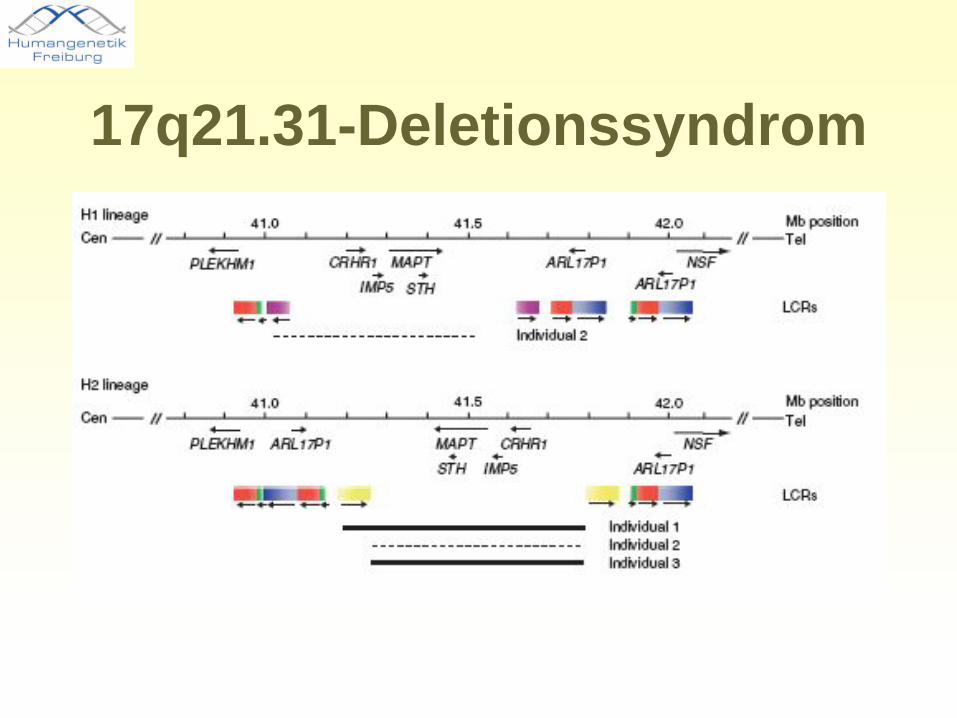
17q21.31-Deletionssyndrom

Lernziele 1
o Welche Mutationstypen- und -wirkungen gibt es?o Was ist autosomal-dominant?o Wie kann ich einen solchen Erbgang erkennen?o Wie berate ich solche Erkrankungen?o Wie ist mit „sporadischen Fällen“ zu verfahren?o Was versteht man unter „Repeaterkrankungen“?o Was ist Penetranz, was Expressivität?o Was ist eine Keimbahnmutation?

Lernziele 2
o Was sind genomische Erkrankungen?o Wie entstehen diese Erkrankungen?o Welche sind die häufigsten Vertreter
dieser Krankheitsgruppe?

Literatur• „Online Mendelian Inheritance In Man“:
(http://www.ncbi.nlm.nih.gov/OMIM)• Strachan/ Read: Human Molecular Genetics 3.
Ed. (Garland Science)• Jorde, Carey, Bamshad, White: Medical Genetics,
3. Ed. (Mosby)• Passarge: Taschenatlas der Genetik, 2. Aufl.
(Thieme)• Buselmaier, Tariverdian: Humangenetik, 4. Aufl.
(Springer)• Murken, Grimm, Holinski-Feder: Taschenlehrbuch
Humangenetik, 7. Aufl. (Thieme)

![TECHNISCHE UNIVERSITÄT MÜNCHEN Klinik und Poliklinik für ... · autosomal-dominat vererbte Neurofibromatose Typ Recklinghausen [34]. Einteilung der Weichteilsarkome Gemäß der](https://img.dokumen.tips/doc/110x75/5dd0985cd6be591ccb61c0e3/technische-universitt-moenchen-klinik-und-poliklinik-fr-autosomal-dominat.jpg)




![Autosomal recessive ichthyosis with limb reduction defect ... · including autosomal dominant, autosomal recessive and X-linked inheritance [1,2]. Associated cutaneous and extracutaneous](https://img.dokumen.tips/doc/110x75/5ec8c9b91adfdf12ab3e663c/autosomal-recessive-ichthyosis-with-limb-reduction-defect-including-autosomal.jpg)






















