Embed Size (px)
Citation preview

Chimica Analitica II
[APPUNTI DEL CORSO] Vademecum del corso del Reisenhofer e alcune definizioni di Adami. Enjoy it

1
Metodi Spettroscopici ................................................................................................................................................. 3
Spettroscopia UV/ vis ............................................................................................................................................. 3
Vantaggi .............................................................................................................................................................. 5
Problematiche ..................................................................................................................................................... 6
Titolazioni spettrofotometriche.............................................................................................................................. 7
Spettroscopia infrarossa ........................................................................................................................................ 7
Svantaggi ............................................................................................................................................................. 8
Assorbiento Atomico ............................................................................................................................................. 8
Spettroscopia ad emissione...................................................................................................................................10
Spettrometria di massa ..............................................................................................................................................11
Metodi Cromatografici ...............................................................................................................................................13
Natura del processo di separazione .......................................................................................................................13
Gas cromatografia ................................................................................................................................................14
Cromatografia Liquida...........................................................................................................................................17
Di adsorbimento .................................................................................................................................................17
Di ripartizione .....................................................................................................................................................17
Problemi .............................................................................................................................................................18
A scambio ionico ...................................................................................................................................................18
Applicazioni ........................................................................................................................................................19
Permeazione su Gel ..............................................................................................................................................19
Tecniche elettroforetiche ......................................................................................................................................20
HPLC(high performance liquid chromatography) ...................................................................................................21
L-L a fasi legate .....................................................................................................................................................21
Tarature ...............................................................................................................................................................22
Confronto fra gas cromatografia e cromatografia liquida .......................................................................................23
Metodi Elettroanalitici................................................................................................................................................24
variazione dell’intensità di corrente con potenziale applicato ..............................................................................24
Elettrogravimetria.................................................................................................................................................25
Sovratensioni (polarizzazioni cinetiche) ...............................................................................................................26
Applicazioni gravimetriche a i = cost ...................................................................................................................27
Applicazioni gravimetriche a Ecat = cost ................................................................................................................27
“Elettrolisi” interne, spontanee ...........................................................................................................................28
Metodi coulombometrici ......................................................................................................................................28
Coulombometria a gas ........................................................................................................................................29
Titolazioni coulombometriche ............................................................................................................................29
Elettrogenerazione esterna di reagenti ...............................................................................................................30
Determinazione end-point ..................................................................................................................................30

2
Metodi polarografici e voltammetrici ....................................................................................................................31
Contributi a ................................................................................................................................................32
Standard addition ...............................................................................................................................................33
Legame fra e la concentrazione di analita .......................................................................................................33
Processi faradici e non faradici ............................................................................................................................34
Titolazioni amperometriche ..................................................................................................................................37
Metodi DEAD-STOP.............................................................................................................................................38
Polarografie derivate e a impulsi ...........................................................................................................................39
Importanza del DROP TIME .................................................................................................................................39
Elettrodi: HDME e MTFE .......................................................................................................................................39
ASV ( anodic stripping voltammetry ) ....................................................................................................................40
Qualità del dato .........................................................................................................................................................41
Criteri per la scelta del metodo analitico .....................................................................................................................42
Tecniche ....................................................................................................................................................................42
Tecnica, metodo, procedura o protocollo? .................................................................................................................42
Procedura analitica totale ..........................................................................................................................................43
Campionamento ........................................................................................................................................................43
Concentrazioni ...........................................................................................................................................................43

3
TEORIA
METODI SPETTROSCOPICI
I metodi spettroscopici si basano sull’interazione dell’onda elettromagnetica con la materia. Queste sono:
1. trasmissione 2. assorbimento 3. rifrazione (deviazione dell’onda alla separazione di fase di due mezzi con diversa densità) 4. riflessione (ritorno di parte dell’onda) 5. scattering (in dispersioni) 6. polarizzazione
Ci interessano in particolare le prime due. Fondamentale è la legge di Beer, che lega l’assorbimento di un dato analita con la sua concentrazione in modo lineare.
Dove è il coefficiente di estinzione molare, che, come A, dipende dalla lunghezza d’onda che
osserviamo, perché ogni specie assorbirà diversamente diverse lunghezze d’onda. b è invece il cammino ottico, e C è la concentrazione. Previa retta di taratura posso dunque conoscere la concentrazione di un analita incognita.
SPETTROSCOPIA UV/ VIS
L’assorbimento è dovuto alla promozione di elettroni della molecola o dell’atomo a livelli energetici più elevati. A seconda dell’energia dei legami fra atomi, si può avere la promozione dell’elettrone a diverse lunghezze d’onda, più o meno energetiche. Doppi legami ed elettroni spaiati daranno probabilmente assorbimento del visibile. Legami più stabili invece daranno picchi nell’UV, come nel caso del benzene, che ha due bande E nell’UV lontano (difficilmente esplorabile perché l’ossigeno dell’aria è opaco a quelle lunghezze d’onda, e si dovrebbe operare in vuoto spinto), e una banda B nel vicino UV, che tuttavia è meno intensa. L’intensità di una banda non è sempre il fattore più importante da considerare. Anche i composti inorganici, a seconda dell’ambiente in cui sono solvatati, assorbono nel visibile. Ad esempio il nichel, verde se solvatato da 6 molecole d’acqua, diventa incolore in ambiente cloridrico, e il rame, in acqua verde, diventa blu in ambiente ammoniacale. I Sali delle terre alcaline, essendo gli orbitali f schermati dagli altri orbitali più interni, come con il cloruro di prosidinio, non cambiano spettro di assorbimento nei diversi ambienti, e quindi sarà più facile una determinazione qualitativa. L’indagine quantitativa invece è più difficile, perché i picchi sono più stretti, e invece si preferiscono picchi più allargati … BANDA PASSANTE!

4
Spettrofotometro a doppio raggio: Sorgenti: visibile: tungsteno, con emissione costante fra 320 e 2500 nm (utilizzata in laboratorio) UV: lampada a deuterio, 180 fino a 375 nm Monocromatori:
- Prisma o vetro nel visibile, opaco però agli UV o quarzo per UV o IR: NaCl, KBr, CsI
- Reticolo di rifrazione o ricorda il CD: il raggio viene scomposto per riflessione. (si avrà raggio leggermente
meno intenso, perché riflesso, ma larga scala di utilizzo). Celle: In vetro o in quarzo. 1 cm (usata per determinare cromo complessato con difenilcarbazone), 1 mm o 2-5 cm. Rilevatori:
cella fotovoltaica, che trasforma il segnale luminoso in elettrico
cella a fotoconduttività, che subisce un cambiamento della resistenza a causa della radiazione.
fotomoltiplicatori, visti in laboratorio. Nel detector ci sono molte superfici di impatto, cosicché il fotone dia un segnale più intenso.
o Tubo fotomoltiplicatore: Il fotomoltiplicatore è costituito da un tubo in vetro al cui interno è stato praticato il vuoto, in cui è presente un anodo e diversi elettrodi dinodi. I fotoni colpiscono attraverso una finestra di ingresso una superficie chiamata fotocatodo, ricoperta di uno strato di materiale che favorisce l'effetto fotoelettrico. A causa di questo effetto vengono emessi degli elettroni, chiamati fotoelettroni che sono focalizzati da un elettrodo verso lo stadio di moltiplicazione. Questo stadio è costituito da una serie di elettrodi ciascuno caricato ad un potenziale superiore al precedente. Il primo elettrone emesso per effetto fotoelettrico subisce una accelerazione a causa del campo elettrico e acquisisce energia cinetica. Quando l'elettrone colpisce il primo elettrodo del dinodo provoca l'emissione secondaria di diversi elettroni di minore energia. La struttura del sistema è progettata in modo che ciascun elettrone emesso da un elettrodo venga accelerato e provochi l'emissione di diversi elettroni dall'elettrodo successivo. Si ha così un fenomeno a cascata per cui un singolo fotone che colpisce
D amp. recorder M S camp.
Blank

5
il tubo provoca il passaggio di moltissimi elettroni. Al termine della sequenza di elettrodi gli elettroni colpiscono un anodo, ed un rapido impulso elettrico indica il rilevamento del fotone. I fotomoltiplicatori devono essere schermati magneticamente, in quanto un campo magnetico esterno (anche quello terrestre) può deviare il percorso degli elettroni al suo interno.
Il segnale uscente deve essere comunque proporzionale all’intensità del raggio entrante. Prima dell’analisi deve essere effettuata una taratura, dopodiché si può usare o l’assorbanza (preferita) o la trasmittanza, che è un valore percentuale tale che:
Questa però non varia proporzionalmente con la concentrazione, ma in modo esponenziale con essa, essendo l’assorbanza il logaritmo di T/100 cambiato di segno. Si imposta il fondo scala, a meno di non voler seguire un metodo di maggiore precisione (extreme precision), con il blank e il limite superiore con qualcosa di opaco, lo shutter. Blank: soluzione in tutto e per tutto uguale ai sample di taratura e al campione, con l’unica differenza di non contenere l’analita che voglio analizzare. Aspetti della legge di Beer:
se ci sono più specie in soluzione, che assorbono alla stessa lunghezza d’onda in modo diverso, l’assorbanza misurata sarà falsata.
la cella stessa darà un minimo di riflessione della radiazione. VANTAGGI
Vasta applicabilità, per analiti organici ed inorganici
Elevata sensibilità: fino a concentrazioni di 10-4 - 10-6 M. (con un attorno ai 10'000-
40'000)
Misure facili e rapide
Selettività, sulla quale si può operare, come vedremo ora. Per migliorare selettività:
Si possono usare reagenti specifici, che reagiscano solo con l’analita, come nel caso di acido sulfanilico e nitriti, che assumono così una colorazione rossa, o con il reagente di Nessler (uno iodiomercurato di potassio) per determinare N ammoniacale (NH3, NH4
+), che diventa rosso mattone. In laboratorio abbiamo visto il caso del difenilcarbazide che complessava ossidandosi con il cromo presente in soluzione, e assumendo colorazione rosa violacea.
Si può operare sull’aggiustamento del numero di ossidazione, come nel caso del ferro (II) a ferro (III), con acqua ossigenata, per poi farlo reagire con tiocianato, e ottenere un complesso rosso sangue.
Controllando il pH, per passare ad esempio da ione cromato a ione bicromato, e spostarne il picco di assorbimento.
Con masking agents, eliminando cioè le specie interferenti
6
PROBLEMATICHE
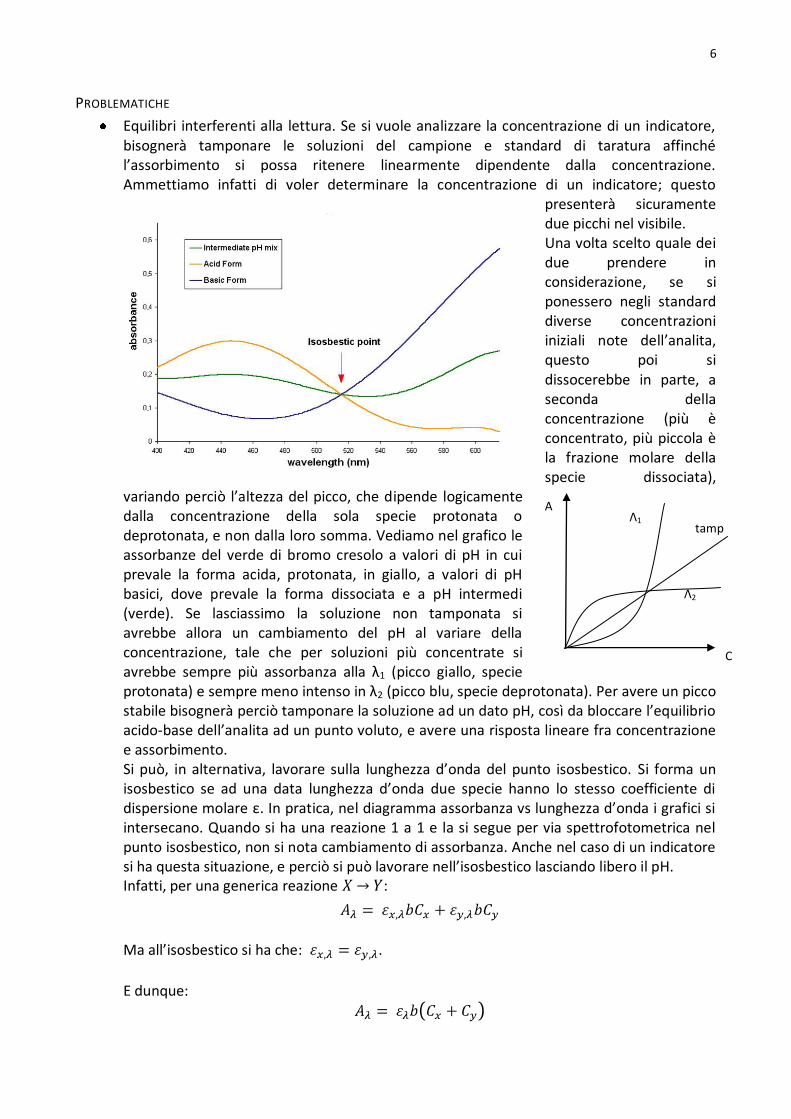
Equilibri interferenti alla lettura. Se si vuole analizzare la concentrazione di un indicatore, bisognerà tamponare le soluzioni del campione e standard di taratura affinché l’assorbimento si possa ritenere linearmente dipendente dalla concentrazione. Ammettiamo infatti di voler determinare la concentrazione di un indicatore; questo
presenterà sicuramente due picchi nel visibile. Una volta scelto quale dei due prendere in considerazione, se si ponessero negli standard diverse concentrazioni iniziali note dell’analita, questo poi si dissocerebbe in parte, a seconda della concentrazione (più è concentrato, più piccola è la frazione molare della specie dissociata),
variando perciò l’altezza del picco, che dipende logicamente dalla concentrazione della sola specie protonata o deprotonata, e non dalla loro somma. Vediamo nel grafico le assorbanze del verde di bromo cresolo a valori di pH in cui prevale la forma acida, protonata, in giallo, a valori di pH basici, dove prevale la forma dissociata e a pH intermedi (verde). Se lasciassimo la soluzione non tamponata si avrebbe allora un cambiamento del pH al variare della concentrazione, tale che per soluzioni più concentrate si avrebbe sempre più assorbanza alla λ1 (picco giallo, specie protonata) e sempre meno intenso in λ2 (picco blu, specie deprotonata). Per avere un picco stabile bisognerà perciò tamponare la soluzione ad un dato pH, così da bloccare l’equilibrio acido-base dell’analita ad un punto voluto, e avere una risposta lineare fra concentrazione e assorbimento. Si può, in alternativa, lavorare sulla lunghezza d’onda del punto isosbestico. Si forma un isosbestico se ad una data lunghezza d’onda due specie hanno lo stesso coefficiente di dispersione molare ε. In pratica, nel diagramma assorbanza vs lunghezza d’onda i grafici si intersecano. Quando si ha una reazione 1 a 1 e la si segue per via spettrofotometrica nel punto isosbestico, non si nota cambiamento di assorbanza. Anche nel caso di un indicatore si ha questa situazione, e perciò si può lavorare nell’isosbestico lasciando libero il pH. Infatti, per una generica reazione :
Ma all’isosbestico si ha che: .
E dunque:
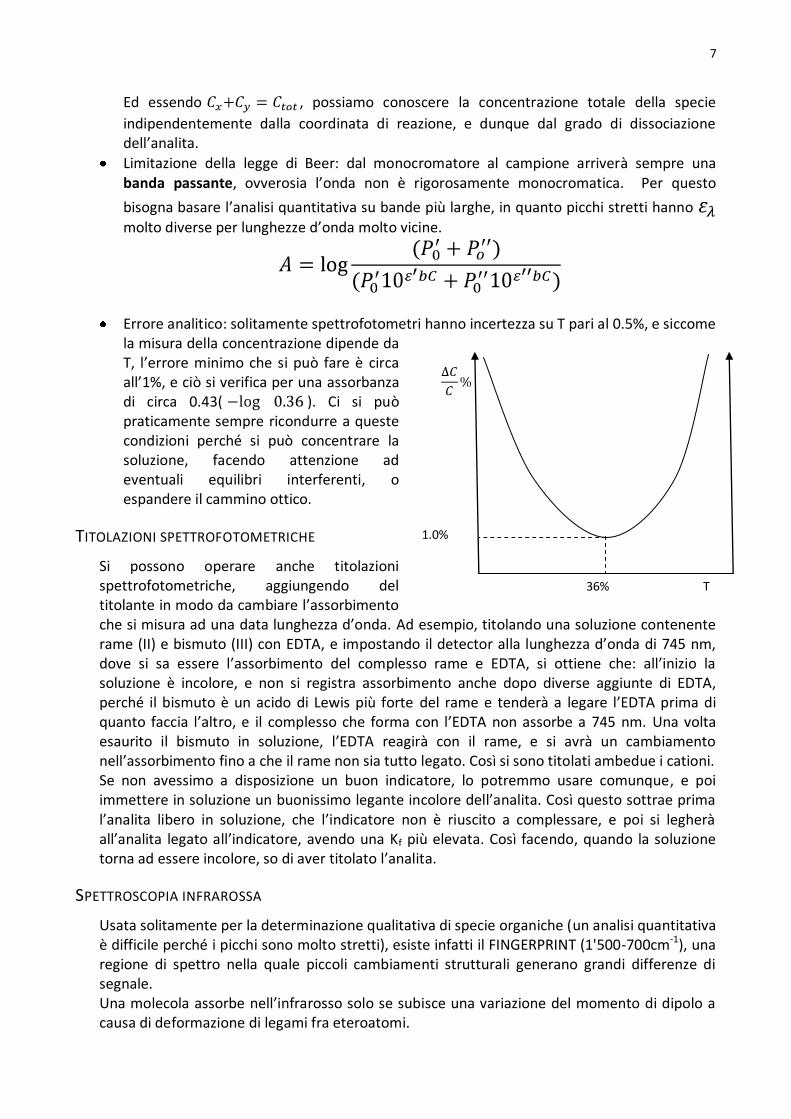
A
C
Λ1
Λ2
tamp
7
Ed essendo , possiamo conoscere la concentrazione totale della specie
indipendentemente dalla coordinata di reazione, e dunque dal grado di dissociazione dell’analita.
Limitazione della legge di Beer: dal monocromatore al campione arriverà sempre una banda passante, ovverosia l’onda non è rigorosamente monocromatica. Per questo
bisogna basare l’analisi quantitativa su bande più larghe, in quanto picchi stretti hanno molto diverse per lunghezze d’onda molto vicine.
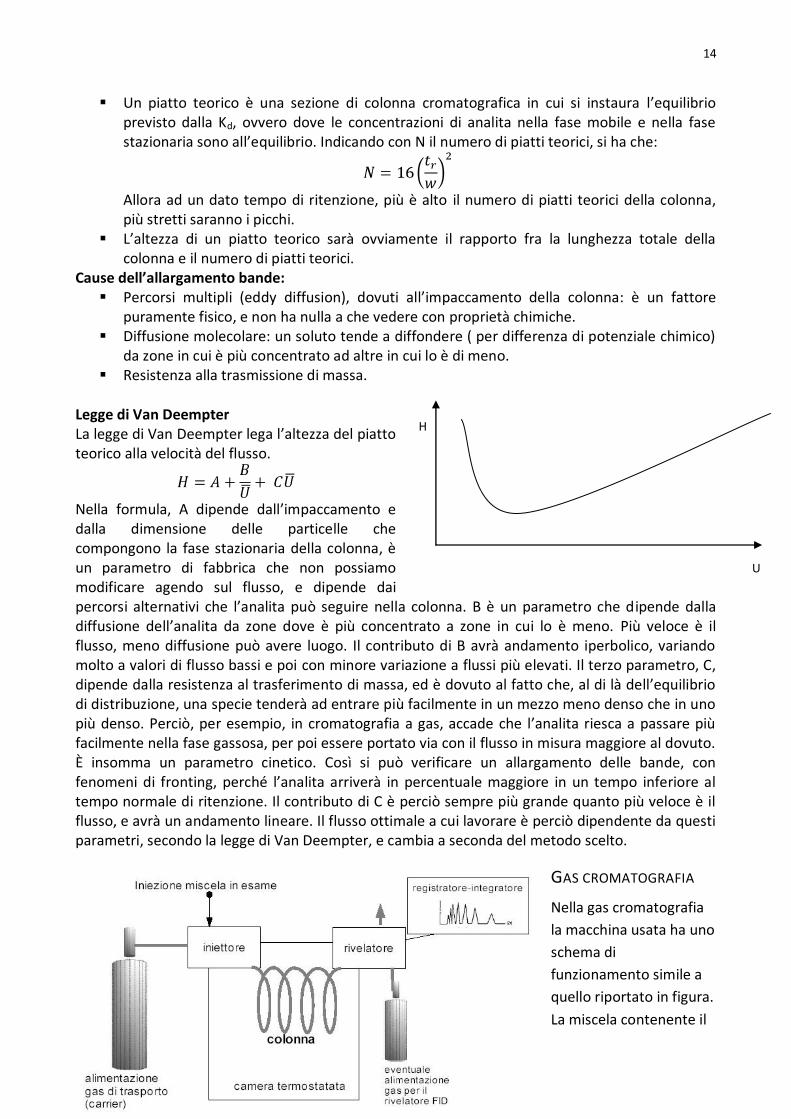
Errore analitico: solitamente spettrofotometri hanno incertezza su T pari al 0.5%, e siccome la misura della concentrazione dipende da T, l’errore minimo che si può fare è circa all’1%, e ciò si verifica per una assorbanza di circa 0.43( ). Ci si può praticamente sempre ricondurre a queste condizioni perché si può concentrare la soluzione, facendo attenzione ad eventuali equilibri interferenti, o espandere il cammino ottico.
TITOLAZIONI SPETTROFOTOMETRICHE
Si possono operare anche titolazioni spettrofotometriche, aggiungendo del titolante in modo da cambiare l’assorbimento che si misura ad una data lunghezza d’onda. Ad esempio, titolando una soluzione contenente rame (II) e bismuto (III) con EDTA, e impostando il detector alla lunghezza d’onda di 745 nm, dove si sa essere l’assorbimento del complesso rame e EDTA, si ottiene che: all’inizio la soluzione è incolore, e non si registra assorbimento anche dopo diverse aggiunte di EDTA, perché il bismuto è un acido di Lewis più forte del rame e tenderà a legare l’EDTA prima di quanto faccia l’altro, e il complesso che forma con l’EDTA non assorbe a 745 nm. Una volta esaurito il bismuto in soluzione, l’EDTA reagirà con il rame, e si avrà un cambiamento nell’assorbimento fino a che il rame non sia tutto legato. Così si sono titolati ambedue i cationi. Se non avessimo a disposizione un buon indicatore, lo potremmo usare comunque, e poi immettere in soluzione un buonissimo legante incolore dell’analita. Così questo sottrae prima l’analita libero in soluzione, che l’indicatore non è riuscito a complessare, e poi si legherà all’analita legato all’indicatore, avendo una Kf più elevata. Così facendo, quando la soluzione torna ad essere incolore, so di aver titolato l’analita.
SPETTROSCOPIA INFRAROSSA
Usata solitamente per la determinazione qualitativa di specie organiche (un analisi quantitativa è difficile perché i picchi sono molto stretti), esiste infatti il FINGERPRINT (1'500-700cm-1), una regione di spettro nella quale piccoli cambiamenti strutturali generano grandi differenze di segnale. Una molecola assorbe nell’infrarosso solo se subisce una variazione del momento di dipolo a causa di deformazione di legami fra eteroatomi.
1.0%
36% T

8
Le tipologie vibrazionali molecolari possono essere: - Stretching (allungamento o accorciamento dei legami) - Bending (in-plane bending: scissoring e rocking; out-of-plane bending: wagging, twisting) Sorgenti: (emettono calore)
Lampade di Nernst (ossidi di terre rare, refrattari)
Globar (lampada in carborundum) Monocromatori: Alogenuri di metalli alcalini: NaCl, KBr, CsBr,LiF (non subisce umidità, al contrario degli altri) Campione:
Se in soluzione: si scioglie preferibilmente in CCl4 (trasparente a 4’000-1'300 cm-1) o solfuro di carbonio (trasparente a 1’300-625 cm-1).
Se come liquido puro: cammino ottico stretto, su pastiglie di NaCl o KBr.
Se solidi (Ø < 2 μ) si disperdono in KBr in rapporto 1 a 100 o si disperdono in Nujol.
Se Ø > 2 μ, la radiazione viene rifratta anziché assorbita e bisogna trattare in qualche modo l’analita, per ricondursi ai casi precedenti.
Detector:
Termocoppie
Balometri (basati su variazione di resistenza)
Celle pneumatiche (variazione della forma)
SVANTAGGI
Scarsissima riproducibilità dell’analisi. È possibile infatti solo un’indagine semi-qualitativa, grazie alla specificità dello spettro. Questo viene registrato in termini di trasmittanza così da poter ricavare subito il rapporto fra l’intensità finale e quella iniziale.
ASSORBIENTO ATOMICO
Una generica molecola può essere sottoposta a:
Dissociazione (formazione di aggregati atomici)
Atomizazione (dissociazione per cui la molecola si scinde nei singoli ioni)
Eccitazione elettronica (promozione elettronica a livelli energetici più alti)
Emissione (ritorno dell’elettrone a stati di eccitazioni inferiori, con emissione di E)
Ionizzazione (formazione di un catione) Un macchinario per spettroscopia ad assorbimento atomico è costituito da: Sorgente: Hollow catode. La sorgente a catodo cavo non emette una radiazione uniforme, bensì uno spettro di emissione specifico dell’elemento di cui il catodo è composto.
Per emettere lo spettro in questione si genera una corrente ionica fra anodo e catodo, che atomizza alcune particelle di quest’ultimo, che a loro volta emettono una radiazione quando

9
tornano nel loro stato fondamentale. Il cilindro è cavo cosicché gli atomi che ricadono sul catodo rimangano lì, e la lampada duri di più. La radiazione così emessa è particolarmente monocromatica, e non si sarebbe potuta ottenere con un monocromatore. La banda passante di un monocromatore infatti sarebbe troppo larga, e la misura dell’assorbimento atomico (differenza fra il segnale emesso e quello finale) sarebbe impossibile, data la sua piccola area, che non influirebbe sulla grande banda policromatica del monocromatore. Invece la banda di emissione atomica ha un’altezza e un area paragonabile a quella dell’assorbimento, e dunque la misura è possibile. Il picco più alto dello spettro di emissione viene detto riga di risonanza, che corrisponde all’emissione più probabile (al primo orbitale libero) dell’elemento. Svantaggi: la lampada può essere usata unicamente per l’analisi dell’elemento di cui il catodo è composto, e dunque bisogna averne una per elemento. Chopper: Siccome anche il campione che viene bruciato, nella sua parte eccitata, emette radiazione, servirà un componente del macchinario che interrompa il cammino della radiazione proveniente dalla lampada in modo da far arrivare al detector solo la radiazione emessa dalla fiamma. In questo modo si può ricavare, per semplice sottrazione del segnale della sola fiamma dal segnale totale, l’effettivo assorbimento della radiazione da parte del campione atomizzato. Infatti la fiamma provoca una lieve emissione atomica indesiderata. Consideriamo l’equazione:
Dove: - è il numero di atomi presenti in un particolare stato eccitato.
- è il numero di atomi presenti allo stato fondamentale. - P sono fattori statistici che fanno riferimento agli stati eccitato e fondamentale, e danno
una misura della probabilità della presenza di atomi eccitati in quel determinato stadio. - E sottosegnato j è l’energia necessaria all’eccitamento. - K è la costante di Boltzman
Alla temperatura della fiamma, di circa 2'500 K, in corrispondenza dell’eccitazione più probabile del sodio, si avranno solamente due atomi eccitati su 10'000 allo stato fondamentale. Per questo nell’ICP (che vedremo più avanti) si arriva a temperature molto più elevate, così da aumentare la percentuale di atomi eccitati. (Questa emissione atomica in realtà sarebbe già sufficiente per un’analisi quantitativa del sodio, ma per altri elementi, in cui le energie di eccitamento diventano più alte, serviranno temperature più elevate per avere emissioni accettabili) Bruciatore:
A fiamma: In questo caso il campione viene nebulizzato e mescolato ad aria e acetilene, per poi essere bruciato. I flussi di aria, acetilene e soluzione campione devono essere il più possibili regolari per evitare fluttuazioni nella rilevazione del segnale. Delle gocce formate dalla nebulizzazione del campione passeranno attraverso l’abbattimento goccioline solo le più minute, mentre le altre verranno drenate e allontanate. Il campione viene bruciato in modo che il campione vada incontro ad atomizzazione, e sia in grado, nella sua percentuale allo stato fondamentale (molto elevata, perché con il bruciatore a fiamma solo 2 atomi su 10'000 saranno eccitati), di assorbire lo spettro emesso dal catodo.
A fornelletto di grafite (flameless)

10
Essendo la grafite un semiconduttore, si può scaldare il cilindretto applicando ad esso una differenza di potenziale. Questa operazione viene eseguita in ambiente inerte (N2 o Ar) di modo che il fornelletto non si ossidi. Il campione viene iniettato nel cilindretto con una minisiringa (di solito automatizzata) e posto, se solido, su una piattaforma detta l’vov. Vantaggi:
o i volumi di soluzione che occorrono sono minimi, mentre nel caso della fiamma si doveva avere un flusso costante, e un volume non inferiore ai 5 mL. Ci sono casi, come con i fluidi corporei, in cui prelevare grandi volumi è problematico.
o Il campione permane nel cilindretto, e quindi nel cammino ottico, più a lungo di quanto non faccia nel caso della fiamma, dove la corrente ascensionale tende a portar via l’analita dopo qualche frazione di secondo. Ne deriva un ordine di grandezza di sensibilità in più.
o Consente di operare con solidi o Si può rilevare la presenza di un elemento in qualsiasi stato di ossidazione dello
stesso, perché il campione viene atomizzato. Monocromatore: perché usarlo? Qual è l’utilità del monocromatore posto solamente dopo il campione? Serve per isolare la riga di spettro che abbiamo deciso di considerare nell’analisi, che potrebbe essere o meno quella di risonanza, a seconda delle interferenze presenti.
SPETTROSCOPIA AD EMISSIONE
Le tecniche di emissione sono diverse… 1. A fiamma: per determinazione analitica di metalli alcalini e alcalino-terrosi si può
direttamente misurare la loro emissione settando il monocromatore sulla riga di risonanza del metallo. Questo perché l’elettrone o gli elettroni dell’ultimo orbitale sono facilmente eccitabili, e dunque una percentuale maggiore dell’analita si ecciterà nel bruciatore, fornendo un’informazione quantitativa.
2. Ad arco: meno utilizzata per la scarsa riproducibilità della scarica, consiste nel porre una piccola quantità di campione fra due elettrodi di grafite ai quali viene applicata una differenza di potenziale di ~ 200 V. Ad un certo punto si ottiene una scarica che eccita il campione, che emette dunque radiazione.
3. A scintilla: anche in questo caso la scintilla e la quantità di campione utilizzata sono difficilmente riproducibili. L’informazione che ne deriva è inoltre solo semi-quantitativa. Il sistema è composto da due circuiti, uno primario, con un generatore di corrente alternata, e uno secondario, al quale viene applicata, attraverso un trasformatore che opera fra le due bobine dei due circuiti, una differenza di potenziale che va da 10'000 a 40'000 V, e che permette di arrivare a temperature più elevate, e quindi riuscire ad eccitare elettroni più legati al loro nucleo. Il circuito secondario riesce ad accumulare carica grazie ad un condensatore, che una volta saturato provocherà due scariche fra le due coppie di

11
elettrodi, delle quali una serve per riferimento, e l’altra è simile a quella descritta per l’arco.
4. I.C.P. (Inductively Coupled Plasma)
Ultima ma non meno importante, è la più usata delle tecniche ad emissione, perchè molto accurata e precisa, e perchè permette di determinare in un’unica analisi le concentrazioni di fino a trenta diversi analiti. Il campione, aspirato tramite una pompa peristaltica per mantenere il flusso il più costante possibile, viene iniettato nello strumento e nebulizzato assieme ad argon, entrando in dei tubi concentrici chiamati Tubi di Crookes. In questi tubi, il gas rarefatto viene bombardato da un flusso di elettroni (una corrente elettrica), che colpendo gli ioni del gas li ionizza. Questa scarica è generata dalla bobina Tesla, che genera un campo magnetico oscillante nel tempo con una frequenza di 27MHz, così da provocare lo sfregamento, molto intenso e regolare, ed estremamente riproducibile, degli ioni di argon, che innescano il plasma. La temperatura sale in alcuni istanti a 6’000-10'000 K, e per non fondere lo strumento è necessario un flusso di Ar refrigerante, e che tenga il gas lontano dalle pareti di quarzo. Le emissioni danno risposte lineari in un grande intervallo di ordini di grandezza (addirittura sei ordini), e dunque serviranno detector molto avanzati, che riescano a rispondere a segnali molto deboli e molto intensi. In laboratorio abbiamo analizzato ferro, zinco, calcio e magnesio in lettura a scansione, mentre sodio e potassio con lettura simultanea.
SPETTROMETRIA DI MASSA
Una generica molecola gassosa bombardata da elettroni può ionizzarsi (come appena visto). Siccome lo ione sarà meno stabile (più energetico) della molecola di partenza, è probabile che lo ione molecolare si scinda e si frammenti. Ciò che può accadere è:
Frammentazione (formazione di specie radicaliche e specie cationiche)
Riarrangiamento
Dimerizzazione (contrariamente alle altre reazioni, la formazione di complessi dimeri è di secondo ordine, e dunque dipende dalla pressione in modo diverso rispetto alle altre. Variando la pressione si può dunque capire che frammenti siano dimeri e quali no)
Comunque sia, ciò che conta è che in seguito a bombardamento elettronico si formano diverse specie cariche, con diverse masse. Strumento per la spettrometria di massa:

12
il campione viene gassificato e introdotto nel macchinario, passando attraverso una camera di ionizzazione dove avviene il bombardamento elettronico. Una volta ionizzate, le diverse particelle generate attraverso i processi di scissione visti sopra vengono accelerate dal campo elettrico presente fra le due piastre di un condensatore, e passano attraverso un foro dell’armatura negativa entrando in un corridoio di accelerazione. Infatti si applica al fascio ionico un campo magnetico perpendicolare al moto degli ioni, che vengono deviati con traiettoria circolare caratterizzata da un raggio che dipende dalla massa e dalla carica dello ione ( che però in ogni caso sarà unitaria). Regolando il campo magnetico posso perciò scegliere quale particella far attraversare tutto il macchinario e arrivare nel collettore, per poi registrare un segnale come corrente ionica. Vediamo come la massa del campione uscente dipende da H, intensità del campo magnetico, e dalla velocità iniziale, v.
ATTENZIONE! Si considerano RISOLTI, in generale, due picchi che non si sovrappongano per più del 10% della loro altezza. Questo anche per le altre tecniche. Esempio Ammettiamo di aver isolato un analita da una matrice, e di volerlo analizzare con questa tecnica. Lo spettro che si ricava variando l’intensità del campo magnetico nel tempo avrà tanti picchi quanti i frammenti carichi formati. Il picco più a destra sarà il segnale del frammento più pesante, in un grafico intensità segnale vs massa del frammento, ma avrà altezza molto bassa, tanto che a volte per essere rilevato deve essere amplificato. Questo perché lo ione molecolare è generalmente instabile, e si frammenta in più pezzi. Fra un alcano lineare e un PAH poliaromatico con pesi molecolari confrontabili, il secondo tenderà ad avere un picco molecolare più elevato, per la maggiore stabilità del catione dovuta alla maggiore distribuzione di carica nello ione molecolare. Si nota inoltre la presenza di più picchi, essendo gli elementi che compongono l’analita composti da una miscela isotopica. Nel caso di Cl e di Br, aventi miscele isotopiche con percentuali simili dei due isotopi che li compongono, l’analisi sarà più complicata. Per conoscere l’intensità del picco a (M+1), dove M è il peso della molecola, non si fa altro che moltiplicare gli elementi per la percentuale dell’isotopo 1 uma più massivo, e farne la somma, per poi paragonarla a M e avere, ad esempio, per il dodecene:
Questa è l’intensità relativa del segnale corrispondente allo ione molecolare con al suo interno un solo isotopo più massivo. Per miscele di analiti, l’altezza totale di un picco sarà la somma delle altezze causate dai vari frammenti aventi lo stesso peso, che dipenderanno ovviamente dalle pressioni a cui i diversi analiti, in fase gassosa si ricorda, vengono immessi nello strumento. Si capisce dunque come l’analisi sia tutt’altro che semplice, sebbene su un composto isolato ( tramite cromatografia, per esempio) dia una risposta qualitativa certa, per cui non si può confondere l’analita con un'altra specie ( per questo è importante avere detector non distruttivi in cromatografia, così da usare in serie uno spettrofotometro di massa).

13
METODI CROMATOGRAFICI
Usati anche come metodi preparativi per separare composti da miscele a più componenti. Una cromatografia si conduce su una colonna cromatografica, nella quale è presente una fase stazionaria, solida o liquida, e si immette una fase mobile, liquida o gassosa. La fase mobile trasporta i componenti lungo la colonna cromatografica, che a seconda della loro affinità per l’una o l’altra fase, usciranno dalla colonna con tempi di ritenzione differenti.
NATURA DEL PROCESSO DI SEPARAZIONE
I componenti della miscela si ripartiranno nelle due fasi a seconda della loro affinità per l’una o per l’altra fase. Il comportamento all’equilibrio di un qualsiasi analita può essere descritto dalla
costante di distribuzione Kd = .
Si può tracciare così una retta isoterma, di pendenza Kd, che descriva la distribuzione dell’analita nelle due fasi a ciascuna concentrazione.
Il grafico dovrebbe essere una retta, ma spesso si riscontrano delle deviazioni dall’idealità, perché all’aumentare della concentrazione totale di analita presente nella colonna si potrebbe registrare una crescente affinità nei confronti di una delle due fasi, e dunque la perdita di linearità. In questo caso i segnali registrati non avranno forma gaussiana, bensì presenteranno una deformazione, detta fronting quando il massimo del picco è spostato verso sinistra e tailing quando è spostato a destra.
Dal coefficiente di distribuzione dipende il tempo di ritenzione di un’analita nella colonna, che è sempre maggiore quanto più questo è affine alla fase stazionaria. Il tempo di ritenzione non va però calcolato dall’inizio dell’analisi, ovvero dall’iniezione del campione, bensì dal cosiddetto tempo morto.
Il tempo morto è il tempo che trascorre dall’iniezione del campione fino all’arrivo della prima goccia di eluente alla fine della colonna, che viene rilevato dal detector a causa di bolle di gas eventualmente presenti, o altre impurità, o tutto ciò che non viene adeguatamente cromatografato dalla colonna. In corrispondenza del tempo morto nel grafico registrato si noterà una leggera oscillazione.
Ovviamente durante l’analisi è bene che il flusso di fase mobile venga controllato, e tenuto costante ( allora il volume di ritenzione sarà proporzionale al tempo di ritenzione) o variato secondo un gradiente. Solitamente si preferisce variare la composizione della fase mobile (in cromatografia liquida, mentre in gas cromatografia si agisce sulla temperatura), così da avere un potere eluente controllato ma che permetta un’analisi più effettiva e veloce.
I picchi sono di forma gaussiana, perché varie particelle di uno stesso analita arriveranno con diversi tempi, a seconda del tragitto seguito e della loro velocità. La distribuzione è perciò statistica.
La risoluzione di due picchi (accettabile dal punto di vista quantitativo solo se sovrapposti per meno del 10% della loro altezza) è data dalla formula:
E quindi più i tempi di ritenzione sono diversi e più sono stretti i picchi (la media delle larghezze delle basi), più due picchi saranno risolti.
L’ampiezza di un picco è determinata dall’efficienza della colonna, a sua volta basata sul numero dei PIATTI TEORICI
14
Un piatto teorico è una sezione di colonna cromatografica in cui si instaura l’equilibrio previsto dalla Kd, ovvero dove le concentrazioni di analita nella fase mobile e nella fase stazionaria sono all’equilibrio. Indicando con N il numero di piatti teorici, si ha che:
Allora ad un dato tempo di ritenzione, più è alto il numero di piatti teorici della colonna, più stretti saranno i picchi.
L’altezza di un piatto teorico sarà ovviamente il rapporto fra la lunghezza totale della colonna e il numero di piatti teorici.
Cause dell’allargamento bande: Percorsi multipli (eddy diffusion), dovuti all’impaccamento della colonna: è un fattore
puramente fisico, e non ha nulla a che vedere con proprietà chimiche. Diffusione molecolare: un soluto tende a diffondere ( per differenza di potenziale chimico)
da zone in cui è più concentrato ad altre in cui lo è di meno. Resistenza alla trasmissione di massa.
Legge di Van Deempter La legge di Van Deempter lega l’altezza del piatto teorico alla velocità del flusso.
Nella formula, A dipende dall’impaccamento e dalla dimensione delle particelle che compongono la fase stazionaria della colonna, è un parametro di fabbrica che non possiamo modificare agendo sul flusso, e dipende dai percorsi alternativi che l’analita può seguire nella colonna. B è un parametro che dipende dalla diffusione dell’analita da zone dove è più concentrato a zone in cui lo è meno. Più veloce è il flusso, meno diffusione può avere luogo. Il contributo di B avrà andamento iperbolico, variando molto a valori di flusso bassi e poi con minore variazione a flussi più elevati. Il terzo parametro, C, dipende dalla resistenza al trasferimento di massa, ed è dovuto al fatto che, al di là dell’equilibrio di distribuzione, una specie tenderà ad entrare più facilmente in un mezzo meno denso che in uno più denso. Perciò, per esempio, in cromatografia a gas, accade che l’analita riesca a passare più facilmente nella fase gassosa, per poi essere portato via con il flusso in misura maggiore al dovuto. È insomma un parametro cinetico. Così si può verificare un allargamento delle bande, con fenomeni di fronting, perché l’analita arriverà in percentuale maggiore in un tempo inferiore al tempo normale di ritenzione. Il contributo di C è perciò sempre più grande quanto più veloce è il flusso, e avrà un andamento lineare. Il flusso ottimale a cui lavorare è perciò dipendente da questi parametri, secondo la legge di Van Deempter, e cambia a seconda del metodo scelto.
GAS CROMATOGRAFIA
Nella gas cromatografia
la macchina usata ha uno
schema di
funzionamento simile a
quello riportato in figura.
La miscela contenente il
H
U

15
campione è immessa nella colonna da un iniettore, che deve provocarne la vaporizzazione e perciò
è riscaldato. Se ci fosse il rischio di decomposizione termica dell’analita, occorrerebbe farlo reagire
in qualche modo per ottenere un prodotto ( in quantità note e proporzionali a quelle di campione
iniziale) più volatile. Ad esempio, se volessimo analizzare un alcol, potremmo trasformarlo in un
etere.
Se la fase stazionaria è liquida si preferisce eseguire una gas cromatografia capillare, di modo che il
liquido si disponga sulle pareti del capillare e permetta un’analisi con picchi più stretti, e dunque
più risolta.
In coda al gas cromatografo si può porre un flussimetro, in modo da valutare eventuali anomalie
del flusso.
Detector:
TCD (thermo conducibility detector): utilizza un ponte di resistenze, sul quale viene fatto scorrere un gas ad alta conducibilità termica, come l’elio. Quando arriva il campione, si avrà un cambiamento di composizione del gas refrigerante in un'unica parte del ponte, causando uno sbilanciamento di resistenze dovuto al cambiamento di temperatura, che verrà poi tradotto in segnale. Vantaggi: universale, NON DISTRUTTIVO Svantaggi: poco sensibile, risposta non sempre lineare e dipendente dalla temperatura di analisi e dal flusso del carrier.
A ionizzazione FID (flame ionization detector): il detector si basa sulla tendenza, soprattutto di sostanze organiche, a dare ioni positivi quando bruciano. Si può ottenere perciò, per combustione, una corrente ionica che può essere utilizzata come segnale. Il campione trasportato dal carrier viene bruciato all’aria fra due piastre di un condensatore, in cui l’anodo è l’ugello stesso, e il catodo è un collettore cilindrico. Quando arriva il campione, la corrente ionica prima generata solo dalla combustione di aria e idrogeno, subisce una grande variazione, che viene dunque registrata. Vantaggi: detector molto sensibile, infatti si arriva ad un LOD di concentrazioni di nanogrammi litro, e la risposta è anche lineare in un intervallo di 6-7 ordini di grandezza. Svantaggi: il detector è distruttivo, e si limita a sostanze organiche. (sarebbe un bene perché se ci fosse stata contaminazione ambientale del campione da parte di acqua o altri gas presenti nell’atmosfera, che non verrebbero ionizzati e dunque non interferiscono con la misurazione)
Termoionico: variante del FID in cui vengono aggiunti sali di Cs, Rb o K, in modo da determinare specie organiche azotate o fosforate.
A cattura di elettroni (CED): il detector registra cambiamenti nell’intensità di corrente elettrica causati dall’assorbimento, da parte dell’analita, di elettroni lenti. È necessario un gas carrier come He, Ar o N2, che venga irradiato con raggi β, emessi a loro volta
dall’isotopo radioattivo depositato su una lamina d’oro. Esso emette elettroni ad elevata energia, lenti, e radiazioni γ e χ, oltre che atomi di elio. Questa corrente elettrica viene letta dal detector, e quando l’analita assorbe gli elettroni, l’intensità cala, fornendo un segnale. Vantaggi: la sensibilità del detector è molto elevata, nell’ordine dei femtogrammi (10 -
15g/s), con composti contenenti atomi elettronegativi, come nitro composti, alogenuri organici, organometalli, mentre con alcoli, chetoni e idrocarburi in generale l’apparecchio perde in sensibilità.
Fasi stazionarie e supporti:

16
Esempi di fasi stazionarie sono - Squalano: miscela di idrocarburi alifatici, con la quale si può operare fino a 150°C, efficace
con idrocarburi saturi, essendo affine ad essi. - “Carbowax 20M”: glicole polipropilenico, col quale si può arrivare fino a 250°C, usato per
alcoli, ammine, composti solfonati, o comunque polari. Queste fasi stazionarie sono liquide, e vanno perciò assorbite su supporti, a meno di non optare per cromatografie capillari. Vediamone alcuni:
- Carboni attivi/ grafitati: supporto apolare, con elevata superficie specifica(800-1’000m2/g), con un grande potere assorbente, essendo finemente suddivisi.
- Gel di silice, o allumina: supporti polari, tendono ad adsorbire molecole d’acqua, e infatti il loro funzionamento si basa su siti attivi che formano legami a idrogeno (nel gel di silice, SiO2, i siti sono Si-OH, per l’allumina, Al2O3, i siti sono Al-OH)
- Setacci molecolari (molecular sieves): sono supporti impaccati in modo da avere cavità di 0.3-1mm.
- Polimeri porosi: hanno un aspetto granulare, ed anch’essi elevata superficie attiva (300-400m2/g).

17
CROMATOGRAFIA LIQUIDA
DI ADSORBIMENTO
È di tipo preparativo, più che analitico. La colonna cromatografica è composta da una fase stazionaria solida e una fase mobile liquida, fra le quali l’analita si ripartisce a seconda dell’affinità per l’una o per l’altra fase. Si parla in questo caso di adsorbimento dell’analita sulla superficie della fase stazionaria, che si lega a dei siti attivi presenti su di essa. Questo si riflette sull’isoterma di adsorbimento, che non è più lineare oltre una certa concentrazione di analita. Infatti ad un certo punto si avrà la saturazione dei siti attivi della fase stazionaria, e l’analita verrà dilavato dalla fase mobile senza essere ritenuto. L’efficienza perciò crolla.
DI RIPARTIZIONE
La cromatografia di ripartizione prevede l’utilizzo di due fasi liquide, l’una adsorbita su di un supporto solido (fase liquida stazionaria) e l’altra usata come eluente. L’eluente e la fase stazionaria devono essere praticamente immiscibili, e dunque avere caratteristiche molto diverse, onde evitare il trascinamento della fase stazionaria stessa lungo la colonna. Questo in realtà avviene comunque, perché due liquidi, per quanto differiscano in proprietà, si disciolgono comunque l’uno nell’altro in piccola parte. Per evitare tutto ciò, si può preventivamente saturare la fase stazionaria con l’eluente e viceversa, così da riscontrare durante la misurazione completa immiscibilità ( è un processo che si può evitare facendo cromatografia L-L a fasi legate). A seconda che la f.s. sia polare o meno si parla di cromatografia di ripartizione diretta o inversa. Per la diretta, si usa:
- F.s.= un solvente polare trattenuto su gel di silice o allumina. - F.m.= solventi più o meno polari, immiscibili con il solvente usato nella fase stazionaria,
esempi sono cloroformio, benzene, CCl4… Per la cromatografia di ripartizione inversa, invece:
- F.s.= oli di silicone, o oli minerali idrofobi assorbiti su carboni attivi - F.m.= acqua, acetone, o comunque solventi polari.
Esempio: TLC La thin layer cromatography, o cromatografia su carta, è una cromatografia di ripartizione diretta, che usa come fase stazionaria gel di silice o allumina distribuiti uniformemente su un foglio di vetro o alluminio, oppure una strisciolina di carta, sul quale è assorbita dell’acqua; come fase mobile viene usato invece un solvente organico. Per normalizzare la cromatografia, è necessario introdurre il fattore di ritenzione, o di ritardo, Rf, definito come il rapporto fra la distanza percorsa sul cromatogramma da parte dell’analita e quella
Cs
Cm
tr
Ctot

18
percorsa dall’eluente, ovvero il fronte della fase mobile. Perciò il fattore di ritenzione sarà sempre minore di 1, ed è caratteristico della specie, come lo è un punto di fusione. Fattori interferenti: l’analisi è influenzata dalla qualità della carta, che non deve variare in spessore di più di 0.2 mm, perché altrimenti il fattore di ritardo potrebbe cambiare. Inoltre, i campioni immessi devono essere piccoli, 0.01-50ng , e vanno inseriti con un capillare, onde evitare una deviazione dalla linearità dell’isoterma di ripartizione e una risoluzione dei picchi insufficiente. Esecuzione del cromatogramma:
- Ascendente, eluizione per capillarità (camera di sviluppo, saturata dai vapori dell’eluente per evitare evaporazione dalla colonna)
- Disendente, eluizione per capillarità e gravità, più veloce della precedente. - Radiale, utile per campioni più grandi. (capsula di Petri, si ritaglia un cerchio di carta e ne si
lascia una strisciolina a pescare nell’eluente) PROBLEMI
- Può capitare che due macchie siano troppo vicine, o addirittura sovrapposte, ma in quel caso si può semplicemente riutilizzare la striscia girandola ed immergendola in un altro eluente, che separi meglio i due analiti.
- Un analita potrebbe essere incolore, e dunque di impossibile individuazione diretta. In questo caso si può operare in diversi modi:
o Nel caso di analita organico e supporto in vetro o alluminio, si può porre il cromatogramma in stufa, e notare annerimenti
o Altrimenti, in una situazione analoga alla precedente, si può annerire l’analita spruzzando dell’acido solforico, notando poi degli annerimenti, per disidratazione.
o Si può porre la piastra su una corrente di I2, nel caso che l’analita sia un composto organico insaturo, perché in quel caso lo iodio reagirebbe con esso a formare iododerivati, colorati.
o Si può usare fluoresceina, sempre per composti organici o Lampade UV/vis
A SCAMBIO IONICO
La cromatografia a scambio ionico viene condotta su colonne impaccate con resine naturali (come argille o zeoliti) o di sintesi, capaci di legare a dei loro siti attivi ioni di carica positiva (resine cationiche) o negativa (resine anioniche, usata in lab!) Gli scambiatori cationici avranno al loro interno funzioni acide, forti o deboli, così che queste, deprotonandosi, assumano carica negativa e dunque attraggano cationi presenti nell’eluente. Gli scambiatori anionici possiedono al contrario funzioni basiche o forti o deboli, capaci dunque di legare, una volta protonate, i diversi anioni presenti nella colonna. Gli scambiatori cationici presentano come siti attivi gruppi solfonici (forti, R-SO3
- H
+) o funzioni carbossiliche (deboli, R-COO- H+), mentre le resine anioniche presentano sali di azoto quaternari (forti, R-N(CH3)3
+OH- ) o ammine (deboli, R-NR2). La separazione di diversi anioni o cationi dipende allora dall’affinità che questi hanno per i siti attivi, che influenzerà la velocità di dilavazione. Infatti si instaura una continua competizione al sito fra ioni dell’eluente, solitamente un tampone, e ioni del campione. Consideriamo la generica reazione A+BR = B+ AR, dove A è il catione o l’anione da analizzare, e B è lo ione idronio o l’anione ossidrile rispettivamente, mentre R è la resina. Allora l’equilibrio sarà descritto da:

19
Ponendo i coefficienti di attività di AR e BR uguali, si ritrova:
Detto coefficiente di equilibrio pratico. Siccome però la misura si conduce su campioni di concentrazione ionica molto minore rispetto allo ione liberato dal sito attivo, e siccome i siti non saranno che in parte occupati dagli ioni dell’analita, la frazione molare di BR sarà molto più grande di AR e dunque costante durante la misura, si ottiene:
Dove Kd è la costante di ripartizione dello ione fra la fase mobile e la resina. Si possono perciò separare diversi ioni, a seconda del loro coefficiente di ripartizione così definito. APPLICAZIONI
- Se volessi determinare la concentrazioni di solfuro per precipitazione come sale di bario, dovrei prima liberare lo ione solfuro legato agli ioni Fe2+ e Ag3+, perché i solfuri di questi anioni sono molto stabili. Si può perciò operare una cromatografia a scambio cationico così da rimuovere i due cationi, avendo essi grandissima affinità per la resina.
- Per concentrare una soluzione acquosa di sali, si può condurre un primo scambio in modo da legare i cationi dei sali presenti alla resina, per poi lavare la resina con una soluzione acida molto concentrata, così da operare un ripristino dei siti acidi, e far fuoriuscire gli ioni, in un più piccolo volume.
- Potrei preparare soluzioni acide o basiche a titolo noto, immettendo in colonna un sale con un catione o un anione molto affine alla resina, così facendo verranno liberati ioni idronio o ossidrile nella stessa quantità, a me nota, di ione immesso nella colonna.
- Separazioni di amminoacidi! Attraverso un gradiente di pH, su resina a scambio cationico con gruppi solfonici, partendo da pH inferiore a 2, di modo che tutti gli amminoacidi siano carichi positivamente e si leghino alla resina. Aumentando la forza ionica e il pH, si distaccheranno i diversi amminoacidi a seconda della loro acidità. Prima si staccheranno gli amminoacidi acidi, poi quelli neutri e infine quelli basici. Un secondo metodo è a concentrazione ISOCRATICA, con resine con siti leganti, composti da ioni metallici di transizione, del tipo:
Dove L è il legante e A è l’analita da separare. Il metallo è solitamente il rame (II), che è un acido di Lewis capace di accettare elettroni, e perciò si lega con i diversi amminoacidi in base alla loro basicità. Si usa come eluente isocratico un tampone ammoniacale, che competerà con gli amminoacidi ai siti.
PERMEAZIONE SU GEL
Questa tecnica cromatografica è volta all’analisi di macromolecole, ed è impostata su colonne il cui riempimento è una specie porosa, che permette la separazione di diversi componenti per esclusione sterica. Il riempimento può essere un polimero idrofilo o idrofobico.

20
Polimeri idrofili, SEPHADEX: Sono usati polimeri di destrano (C6H10O5)n + epicloridrina (1,2-ossi-3-cloropropanolo), commercializzati in due tipi di polimeri, a diversa porosità: uno per molecole di peso ~700 uma, l’altro per macromolecole di 800'000 uma, ovviamente polari. Questi polimeri vengono ovviamente attaccati da ossidanti forti come il cromato, il bicromato e il permanganato, e inoltre hanno un loro campo di stabilità al pH (che va da 2 a 11), ovvero vengono attaccati da acidi e basi forti, siccome contengono gruppi epossidici, che vengono idrolizzati a glicoli con catalisi acida o basica. Polimeri idrofobici, STYRAGEL: Utilizzati con solventi organici e per determinare concentrazioni di analiti apolari, si formano facendo reagire polistirene e divinilbenzene. I polimeri più utilizzati sono: a porosità di Ø6nm, per molecole 800-1'600 uma, e uno a porosità di Ø105nm, per molecole di 20-40 x 106 uma. Importanti concetti:
- Esclusione totale, ovvero quando una molecola è troppo ingombrata per poter permeare nel gel, e dunque viene dilavata dall’eluente nel tempo morto.
- Permeazione totale, che si verifica quando una molecola è così piccola da trovarsi nelle stesse condizioni dentro e fuori dal gel, arrivando al detector assieme a tutte le altre molecole di dimensione ridotta.
- Il detector registrerà picchi alquanto stretti, con un primo segnale al tempo morto ed un ultimo al tempo di permeazione totale.
Applicazioni: - Frazionamento di peptidi in base al loro peso molecolare - Determinazione di peso molecolare di polimeri di sintesi o naturali, usando ovviamente
una molecola di confronto, con un peso molecolare noto. - Dissalatura di soluzioni acquose, con una dialisi, immergendo la soluzione target in acqua
distillata, separata da questa dal polimero, che permetterà il passaggio del sale ma non di molecole più grosse.
TECNICHE ELETTROFORETICHE
L’elettroforesi si utilizza per la separazione di specie cariche in soluzioni elettrolitiche, a cui viene applicato un potenziale elettrico. Si possono analizzare:
- Ioni semplici - Macromolecole - Colloidi - Micro gocce di emulsioni - Specie particellari e batteri - DNA
Si possono distinguere due tipi di elettroforesi: - In fase libera (in soluzione, senza supporto) - Di zona, ovvero su supporto di carta o gel agarosio
La migrazione delle cariche sarà comunque determinata dall’intensità del campo elettrico, dalla carica e dal raggio dell’analita e dalla viscosità del mezzo, secondo l’equazione:

21
L’equazione si può semplificare introducendo la mobilità, u, della specie carica, che lega l’intensità del campo alla velocità della particella.
Fattori che influenzano la migrazione:
- Variazione del pH, che può portare a formazione di complessi e a cambiamenti di carica, come nel caso di aminoacidi.
- Forza ionica, che se diminuita aumenta la velocità - Temperatura, che se aumenta, accelera la diffusione, e dunque il detector rileverà bande
più larghe. Il sistema deve essere dunque termostatato, facendo attenzione a non denaturare eventuali proteine che si vogliono analizzare (t< 42°C)
- Elettrosmosi, fenomeno per cui si crea un movimento di particelle non cariche a causa della migrazione di ioni coordinati a molecole d’acqua.
HPLC(HIGH PERFORMANCE LIQUID CHROMATOGRAPHY)
Questa tecnica permette l’uso di volume di campione molto ridotti , ed ha una buona accuratezza. Il macchinario per HPLC è così composto:
- Serbatoio : contenitore dell’eluente, a volte se ne usano due, così da poterli miscelare in una cromatografia a gradiente.
- Dispositivo (se necessario): serve per miscelare i due eluenti, per ottenere un gradiente di eluizione nel tempo.
- Pompa: immette il campione, le pressioni generate possono arrivare anche a 500 atm. - Colonna: in vetro o in acciaio, Ø 1-6 mm, e altezza 3cm-1m (colonne molto corte), il flusso
dell’eluente è inoltre molto ridotto, 60ml/h. - Riempimento: porosità 3-10 μm, sono sostanzialmente di due tipi:
o a particelle pellicolari: sono impiegate quasi esclusivamente per le colonne di protezione. Sono granuli sferici e non porosi di vetro o materiale polimerico, di dimensione compresa tra i 30 e i 40 μm. Sulla superficie dei granuli viene depositato uno strato poroso di silice, allumina o resina a scambio ionico. Se si necessita di una fase stazionaria liquida, può essere applicata per adsorbimento.
o a particelle porose : hanno diametri compresi tra 5 e 15 μm, il materiale più usato è la silice microporosa, ma possono essere costituite anche di allumina o resina a scambio ionico. Anche in questo caso vengono applicati rivestimenti specifici, legati o per adsorbimento o attraverso legami chimici alla superficie delle particelle.
L’equazione di Van Deempter è leggermente diversa, perché in questo caso compaiono due contributi direttamente proporzionali al flusso. Perciò si lavora a flussi ridotti, di velocità 2-5mm/s. I contributi dipendono dalle dimensioni delle particelle e dall’impaccamento, e dunque non sono modificabili. Il minimo di H si ha per valori di flusso molto piccoli, ma non si possono effettuare cromatografie eterne. Dunque si opera ai valori di flusso riportati.
L-L A FASI LEGATE
Per evitare il pre-miscelamento delle fasi liquide nella cromatografia di ripartizione liquido-liquido, si possono usare polimeri, applicati a supporti solidi inerti, che possono essere polari o apolari. Nel primo caso si parla di cromatografia diretta, nel secondo caso di cromatografia inversa. I polimeri polari usati sono: PEG (polietilenglicole), ovvero , oppure ODPN

22
(ossidiproprionitrile). La fase legata al polimero, ovvero la stazionaria, dovrà essere polare, contenere gruppi -NH2, -CN o –OH. Nella cromatografia inversa invece i polimeri che si usano sono silani, ovvero derivati organici del silicio. I supporti solidi sui quali i polimeri sono applicati possono essere:
- particelle porose: Ø 5-15μm, che hanno una grande superficie attiva e capacità assorbente. Dunque vanno bene per campioni più concentrati. Le bande però si allargano, essendo possibili molti percorsi alternativi.
- Particelle a nucleo solido e rivestimento pellicolare: Ø 37-42μm, che sono molto utilizzati per miscele complesse, permettendo una maggiore risoluzione, perché hanno maggiore efficienza.
TARATURE
Taratura diretta, che consiste nell’analisi, ripetuta più volte per ogni standard, di diverse soluzioni contenenti l’analita in concentrazione nota, così da estrapolare una retta di taratura sulla base della quale si riesca a determinare la concentrazione del campione ignoto. È importante che le concentrazioni del campione e degli standard siano abbastanza simili. Bisogna inoltre accertarsi che le condizioni ambientali e strumentali non varino nel corso della misurazione, così questa deve essere conclusa in un lasso di tempo non troppo lungo.
Standardizzazione interna. Per questo metodo si prepara una serie di soluzioni standard, con l’analita a concentrazione nota, e si aggiunge ad esse un componente, in quantità nota, di modo che si riesca a determinare se il rapporto fra la quantità di analita e standard interno e delle aree dei loro segnali (o delle altezze) varino in modo lineare. Lo standard interno deve:
o essere assente nel campione di partenza o avere un picco risolto rispetto agli altri della miscela o avere concentrazione simile all’analita o Non reagire con esso o Non contenere impurità (o avere una purezza nota)
Se questo è verificato, allora una volta noto il rapporto , sappiamo il valore del
rapporto di masse fra analita e standard interno, e perciò, introducendo una quantità nota e definita di ISD (internal standard) nel campione incognito e registrando il segnale della miscela, si sa che:
Infatti il fattore di risposta relativo dell’analita nei confronti dello standard interno non sarà altro che il coefficiente della retta trovata nel grafico rapporto aree vs rapporto masse.
23
CONFRONTO FRA GAS CROMATOGRAFIA E CROMATOGRAFIA LIQUIDA
L.C. G.C. Segnale descrizione
Eluente a composizione isocratica
Solvente debole (scarsa attitudine a trascinare analiti)
bassa T Tempi di ritenzione molto lunghi, con picchi via via più
larghi. Non tutti i componenti arrivano al detector nel
tempo di analisi.
Eluente a composizione isocratica
Solvente forte
alta T
Picchi non risolti, sovrapposti e stretti
Eluente a composizione isocratica, forza media
T intermedia
Problemi di risoluzione fra i picchi
gradiente programma di temperatura
Picchi risolti e tr non troppo lunghi.
PROCEDURA MIGLIORE

24
METODI ELETTROANALITICI
I metodi elettroanalitici sfruttano reazioni chimiche di ossidoriduzione e misure di grandezze elettriche o gravimetriche che variano con la concentrazione di analita. Introduzione: Si definisce cella elettrochimica un sistema costituito da due semicelle, che contengono due soluzioni elettrolitiche nelle quali pescano due elettrodi, collegati a loro volta da un filo di materiale conduttore, che permetta il passaggio di corrente. Un componente essenziale per il funzionamento della cella è il ponte salino, che può essere sostituito da una membrana semipermeabile, che permetta diffusione di ioni da una semicella all’altra, per evitare che la reazioni si fermi a causa di accumulo di carica netta nei due comparti. Si definisce galvanica la cella elettrochimica nella quale avviene una reazione di ossidoriduzione spontanea, e capace dunque di produrre lavoro elettrico, di generare cioè una differenza di potenziale, calcolabile secondo la legge di Nernst:
Una cella elettrolitica invece è una cella nella quale si fanno avvenire reazioni di ossidoriduzione non spontanee applicando una differenza di potenziale agli elettrodi, e quindi compiendo lavoro esterno. In entrambi i casi il catodo è l’elettrodo al quale la o le specie elettroattive si riducono, mentre l’anodo è l’elettrodo al quale si ossidano. I segni dei due elettrodi invece cambiano a seconda che si prenda in considerazione una cella galvanica o una elettrolitica. Infatti nella cella galvanica il catodo avrà segno positivo, in quanto le specie che si riducono spontaneamente strappano elettroni dall’elettrodo e lo lasciano con una piccola carica positiva; all’anodo avviene invece il processo inverso, perciò l’elettrodo sarà negativo. Nel caso di una cella elettrolitica invece, il catodo è negativo, e si può immaginare che, per far avvenire la riduzione non spontanea di una specie, noi dobbiamo caricare negativamente l’elettrodo, che potrà poi rilasciare elettroni alle specie elettroattive; l’anodo invece dovrà essere carico positivamente, cosicchè le specie elettroattive liberino gli elettroni sulla sua superficie, e si ossidino. VARIAZIONE DELL’INTENSITÀ DI CORRENTE CON POTENZIALE APPLICATO
Nel grafico sono rappresentati i grafici di intensità di
corrente vs differenza di potenziale in diversi sistemi.
Vediamo quali:
1. Nel primo caso abbiamo due elettrodi che
pescano nella stessa soluzione e dunque non
generano alcuna corrente. Se applichiamo un
potenziale alla cella, registreremo nel circuito una
corrente crescente e dipendente dal potenziale
secondo la prima legge di Ohm: .
2. Nel secondo caso si tiene conto della deviazione
dall’idealità dovuta alla polarizzazione degli elettrodi. Infatti in una cella dove avvengono
reazioni elettrodiche di elettrolisi, si generano dei gradienti di concentrazione delle specie
1 2
3
4
5

25
elettroattive: le specie che si ossidano si troveranno in concentrazione maggiore vicino alla
superficie anodica che in soluzione, e saranno invece meno concentrati sulla superficie
catodica che in soluzione gli analiti che si riducono al catodo. Questi gradienti possono
essere diminuiti mescolando la soluzione.
3. La polarizzazione degli elettrodi in questo caso è voluta, e si utilizzano microelettrodi per
aumentare la densità di corrente elettrodica, tanto che, ad un certo punto, non si registra
variazione di corrente all’aumentare del potenziale applicato.
4. Questo è il caso della cella composta da un elettrodo di zinco immerso in una soluzione di
zinco cloruro e un elettrodo ad argento-argento cloruro immerso in una soluzione di
cloruro di sodio. In questo caso la cella genera una differenza di potenziale e un intensità di
corrente con una reazione galvanica. La corrente è convenzionalmente negativa, e
diminuisce man mano che applichiamo una differenza di potenziale contraria a quella della
cella. Si arriva ad una corrente nulla quando il potenziale applicato è in equilibrio con
quello della cella (in realtà ci sono altri contributi che deve eguagliare, ma li vedremo in
seguito). Dopodiché la cella funzionerà in modo elettrolitico, e la corrente assumerà valori
positivi.
5. In questo caso l’elettrodo di zinco è sostituito da un elettrodo a platino, cosicché la cella
non può funzionare come una pila, non essendoci zinco capace di ossidarsi (e platino in
soluzione). Quando applichiamo una differenza di potenziale minore a quella della cella
così composta (che sarà leggermente diverso da quella precedente per questioni di
sovratensione, ma si può trascurare questo fatto perché lo zinco si deposita come solido,
non si ha sviluppo di gas), non si avrà passaggio di corrente all’interno del circuito. A dire la
verità si registrerà una piccola corrente parassita, che aumenta all’aumentare del
potenziale applicato. Applicando invece un potenziale tale da cominciare l’elettrolisi si
registrerà passaggio di corrente. Se interrompessimo l’applicazione di potenziale esterno,
noteremmo che la cella segue il comportamento del grafico 4. Infatti, durante l’elettrolisi,
si è depositato dello zinco, e la cella può funzionare anche spontaneamente.
ELETTROGRAVIMETRIA
Il metodo elettrogravimetrico consiste nell’utilizzare una reazione catodica ( o anodica, meno frequentemente) per depositare un analita elettroattivo all’elettrodo e farne la pesata, per ricavarne la concentrazione. Affinchè ciò sia possibile, la reazione che avviene all’elettrodo deve essere specifica, ovvero deve depositarsi solo la specie che ci interessa, e si deve depositare tutta (fino a raggiungere 10-5C0). Si usa per l’analisi il catodo Winkler, ovvero una reticella di platino, solitamente di geometria cilindrica, così da poter essere posta attorno al contro elettrodo ed essere così interessata da un campo elettrico costante. Si usa il platino perché è un metallo nobile, che non viene facilmente ossidato, e resiste all’attacco degli acidi, a meno che non sia in ambiente ossidante, acido e in presenza di cloro, perché in quel caso tenderebbe a formare sali cloro platinati. Si può operare anche in quelle condizioni a patto di introdurre in soluzione dell’idrazina, che si ossidi liberando azoto, al posto del platino. Per condurre l’analisi bisogna applicare una differenza di potenziale, tale da eguagliare il potenziale di cella nernstiano e superarlo. La differenza di potenziale applicata è però inversa a quella della cella, e in realtà il potenziale di cella non è l’unico contributo che si oppone all’elettrolisi:

26
Gli ultimi contributi sono dovuti alla caduta di potenziale del circuito e alle sovratensioni anodiche e catodiche.
SOVRATENSIONI (POLARIZZAZIONI CINETICHE)
- Sono legate a fenomeni non nernstiani di polarizzazione dell’elettrodo, che sono legate alla cinetica del processo, e per questo hanno questo nome.
- Aumentano con la densità di carica dell’elettrodo, e dunque per intensità di corrente più elevate o per elettrodi più piccoli.
- Dipende dalla natura degli elettrodi. - è notevole se si formano specie gassose
In tabella sono riportati i valori di sovratensione catodica per l’idrogeno e anodica per l’ossigeno, su diversi elettrodi e a diverse densità elettriche.
0.001 amp2/cm2 0.01 amp2/cm2
H2 O2 H2 O2
Pt platinato 0.024 0.721 0.068 0.85
Pt liscio 0.015 0.348 0.030 0.521
Hg 0.9 - 1.1 -
Per platinare l’elettrodo si compie un elettrolisi, ponendo l’elettrodo di platino in una soluzione di acido nitrico e acido cloridrico, cosicché venga ossidato dal primo e formi un sale di cloro (PtCl4), che poi si deposita sull’elettrodo per elettrolisi (Il potenziale di riduzione del platino sarà molto basso). Questo vale anche per l’oro, che posto in acqua regia forma un sale di cloro, AuCl3. Questa operazione è condotta proprio nell’ambiente che si vuole evitare nell’analisi.
Durante l’elettrolisi, tuttavia, il potenziale della cella non rimane costante. Infatti, se ad esempio vogliamo far depositare del rame al catodo, la sua concentrazione diminuirà costantemente in soluzione, facendo variare il potenziale catodico secondo la legge di Nernst, verso valori sempre minori. Il potenziale anodico in questo caso rimarrebbe invece quasi costante, dipendendo solo dal pH e dalla pressione di ossigeno nell’aria (ossidazione dell’ossigeno dell’acqua). Qualora tenessimo il potenziale applicato costante, si riscontrerebbe una diminuzione di intensità elettrica dovuta al fatto che la specie che si riduce si trova a concentrazioni sempre minori in soluzione, e dunque il potenziale catodico tende a cadere. Dovremmo dunque applicare una differenza di potenziale sempre maggiore per avere la stessa intensità di corrente iniziale. Se invece lasciamo il potenziale applicato fisso, diminuisce la velocità dell’elettrolisi, e diminuiscono anche le sovratensioni. Per arrivare alla completezza della reazione bisognerebbe proseguire in eterno, ma ci accontentiamo del limite stabilito in precedenza, 10-5Co. Questo fatto può essere un problema se, volendo determinare la concentrazione di cobalto, in soluzione fossero presenti altre specie capaci di ridursi ai potenziali catodici a cui si arriva ad operare, ad esempio, se in soluzione fosse presente del cadmio (in concentrazione 1M), questo comincerebbe a depositarsi al catodo Winkler una volta giunti a -0.5V (da E0= –0.28V del cobalto). Procedura sperimentale: Prima di cominciare l’analisi bisogna pesare il catodo Winkler con una precisione al decimo di milligrammo. Dopodiché si immerge il catodo nella cella e si conduce l’operazione elettrolitica sotto agitazione, per impedire la polarizzazione dell’elettrodo, con un’ancoretta magnetica o un

27
motore coassiale al working electrode. Quando l’elettrolisi è terminata, si estrae l’elettrodo Winkler dalla soluzione tenendolo sotto tensione, per evitare che la reazione galvanica, che fino a quel momento si è contrastata, disciolga nuovamente l’analita nella cella. Si mette l’elettrodo ad asciugare in stufa, e poi lo si pesa. Dall’incremento in peso si conosce poi la quantità di analita presente. Si ripulisce il catodo usando una soluzione di acido nitrico. Un alternativa all’elettrodo Winkler è l’elettrodo a mercurio, che scioglie i metalli più teneri in leghe chiamate amalgame (il Fe non viene disciolto). APPLICAZIONI GRAVIMETRICHE A I = COST
Questo metodo analitico è adatto a soluzioni semplici, e permette di concludere alquanto rapidamente l’analisi, applicando al sistema elettrolitico un potenziale sempre maggiore per mantenere l’intensità di corrente costante. Alcuni metalli che si possono analizzare con questo metodo sono: Co, Cu, Ni, Sn, Zn e Pb, che però si deposita all’anodo come ossido di piombo, non da reazione catodica come gli altri. Solitamente si fa uso di complessanti, perché il deposito elettrodico diventa molto più compatto e facile da trattare per la pesata. Ad esempio, con argento e cadmio si può usare del cianuro, a patto di non lavorare in ambiente acido, per evitare la formazione dell’acido cianidrico, mortale se inspirato, e dunque è necessario lavorare sotto cappa. Per l’argento, ad esempio, si forma il complesso:
Nulla avrebbe vietato di usare nitrato di argento, ma il deposito sarebbe stato molto più poroso, e avrebbe trattenuto più impurità, oltre ad essere più difficile da pesare. Come si nota dall’equazione, il segno di una specie non conta nello stabilire se la reazione è anodica o catodica, quello che conta è il potenziale di riduzione della specie stessa. Ovviamente specie cariche negativamente si ridurranno tendenzialmente a potenziali applicati più elevati rispetto a specie cariche positivamente, dove l’elettrone riducente riuscirà a reagire più facilmente per questioni elettrostatiche. All’uso di complessanti si ricorre ad esempio nella galvanoplastica, ovvero il processo con cui si ricopre un metallo con un altro metallo più nobile, così si hanno le cromature, orature, platinature, zincature, argentature… APPLICAZIONI GRAVIMETRICHE A ECAT = COST
Il metodo polarografico a potenziale costante si usa per soluzioni complesse, ovvero quando più metalli siano presenti nella stessa soluzione, onde evitare reazioni interferenti all’elettrodo di lavoro. Per fare ciò dobbiamo inserire nel circuito esterno alla cella un potenziostato, e utilizzare un terzo elettrodo, oltre all’elettrodo di lavoro e al contro elettrodo, ovvero un elettrodo di riferimento. Questo perché dobbiamo essere in grado di conoscere ad ogni istante il potenziale catodico, e
variare di conseguenza la E applicata, grazie al potenziostato ed a un cursore posto su una resistenza. Siccome consideriamo completa la reazione quando C=10-5C0, sappiamo che, a seconda della quantità di elettroni che servono a ridurre l’analita, il potenziale applicato finale sarà: 0.30V, 0.15V, 0.10V
Infatti, per la legge di Nernst:
E quindi quando la concentrazione si riduce a quella considerata finale, si ha semplicemente:

28
Alcune applicazioni di questa tecnica sono: - per depositare solo argento, e non rame, da una soluzione ottenuta sciogliendo
galena(PbS) in ambiente acido. Infatti i due metalli si trovano spesso presenti nella roccia. - Per depositare cadmio in presenza di zinco, dalla blenda, solfuro di zinco. - Per separare tramite elettrolisi i singoli metalli da una miscela del tipo:
Cu2+, Bi2+, Pb2+, Sn2+, Cd2+, Zn2+. Questa matrice complessa di metalli sarebbe altrimenti impossibile da analizzare per via gravimetrica. Operativamente, si aggiunge acido tartarico (pKa~7) così da complessare il rame, il piombo, il bismuto e lo stagno. Dopodiché si setta l’elettrodo a :
, , ,
Dopodiché si aggiunge ammoniaca, così da complessare i restanti cadmio e zinco, e li si precipita a:
,
“ELETTROLISI” INTERNE, SPONTANEE
Un esempio è la pila Daniell, nella quale la reazione spontanea porta al deposito di rame sul catodo Winkler. Si usano spesso in questo caso membrane semipermeabili di allumina, che permettano il passaggio degli ioni ma impediscano il diretto rimescolamento delle soluzioni anodiche e catodiche. Questo metodo permette di sostituire il ponte salino, ma si presenta un inconveniente, infatti si genera sulla membrana una piccola divisione di carica, che da luogo ad una differenza di potenziale, Ej, di giunto (junction), che non è eliminabile ed è presente in tutte le celle a giunto liquido (~0.02V) Una diretta conseguenza di questo fatto è che, siccome il pH di una soluzione viene generalmente misurato usando un elettrodo a vetro, che presenta un giunto liquido, non si può determinare il pH alla terza cifra decimale, perché il potenziale di giunto falsa la misura. Un applicazione potrebbe essere quella di determinare la quantità di Ag e Pb nella galena (sciolta in ambiente acido). In questo caso si possono preparare due soluzioni uguali, e usare in entrambe il catodo Winkler, ma in una un contro elettrodo a zinco, mentre nell’altra uno a piombo. Infatti, essendo E0
Ag=+0.80, E0Pb=-0.126 e E0
Zn=-0.76, nella prima cella si depositeranno spontaneamente sia l’argento che il piombo, mentre nella seconda cella si depositerà solo l’argento. Per differenza fra le due pesate si riesce così a determinare la quantità di piombo.
METODI COULOMBOMETRICI
Consistono nel calcolare la concentrazione di analita, in base alla carica passata nel sistema nell’intervallo di tempo dell’analisi, facilmente ricavabile dalla formula:
Dove F è il Faraday, n il numero di elettroni necessari alla reazione, Q è la carica, M.W. è il peso molecolare del composto. Perciò si possono utilizzare i metodi coulombometrici nel caso non si potesse pesare la specie formata, o per confronto con il dato ottenuto da un’analisi elettrogravimetrica. Questo metodo inoltre offre una precisione maggiore, essendo solitamente la quantità di carica molto grande, e dunque meno soggetta ad errore rispetto ad una quantità di deposito di peso molto piccolo. Ad esempio, nella determinazione del trinitrofenolo si ha una

29
produzione di 18F per ogni mole che si riduce, e dunque ad un deposito non molto massivo corrisponde una quantità di carica grandissima. Vediamo i casi:
- Se , allora semplicemente , e si riesce ad ottenere la quantità di analita evitando la pesata. Questo procedimento viene anche detto titolazione coulombometrica, perché in qualche modo analoga alle titolazioni con buretta. Infatti, con un circuito in cui un interruttore provochi interruzione o passaggio di corrente in modo simultaneo in due maglie, una con un cronometro e l’altra con il sistema cella-potenziometro, si riesce a registrare l’intensità di corrente passata semplicemente dal tempo trascorso e dalla caduta di potenziale letta dal potenziometro agli estremi di una resistenza. L’interruttore quindi ha la stessa funzione del rubinetto di una buretta contenente titolante, e il cronometro sostituisce la graduatura della buretta. In realtà l’interruttore fa si che la corrente prodotta dal generatore fluisca o attraverso il sistema della cella (e chiuda il circuito del cronometro) oppure in un’altra maglia, dove è presente una resistenza molto alta, cosicché il generatore continui a lavorare, per evitare sbalzi di corrente.
- Se varia nel tempo, allora si può procedere in due modi: si può mettere nel circuito un coulombometro, che integri in continuo l’intensità di corrente e misuri la quantità di carica. In questo caso però si potrebbero avere delle oscillazioni nel segnale al coulombometro perché il sistema deve essere mescolato, onde evitare polarizzazione. Altrimenti, per evitare errori connessi a queste oscillazioni, si può operare con un coulombometro a gas.
COULOMBOMETRIA A GAS
In serie alla cella dove avviene l’elettrolisi dell’analita a cui si è interessati si pone una cella costituita da una soluzione elettrolitica nella quale è immersa una micro buretta (2-5mL), con al suo interno due elettrodi in platino collegati al circuito. La buretta è riempita di una soluzione elettrolitica acida di un sale di idrazina, che reagisce secondo la reazione:
Dove l’azoto si ossida e l’idrogeno si riduce. In base al volume di gas generato, a determinate condizioni di temperatura e pressione (secondo la legge dei gas), si può conoscere quanto la reazione sia proseguita e dunque la quantità di carica passata nella seconda cella. Essendo la cella in questione in serie alla cella primaria, la quantità di carica passata nelle due celle sarà identica. TITOLAZIONI COULOMBOMETRICHE
Alcune applicazioni: - Se volessimo titolare una soluzione di alogenuri, potremmo operare un’elettrolisi in una
cella con elettrodi ad argento e zinco (o meglio piombo, o un metallo meno nobile dell’Ag), in modo che l’argento si ossidi per elettrolisi e passi in soluzione, precipitando come sale di alogeno.
Vale lo stesso per il mercurio, che precipita come sale mercuroso, dimero di due mercuri (I).
- Si possono effettuare titolazioni di ioni calcio, zinco, piombo e rame con EDTA per via coulombometrica, immettendo l’EDTA complessato con il mercurio (II), che si riduce all’elettrodo a potenziali applicati molto bassi, liberando l’EDTA che si trova dunque nelle

30
condizioni di complessare gli altri ioni. Bisogna allontanare O2, essendo l’analisi effettuata a pH basico per aumentare l’efficienza dell’EDTA. Quindi bisogna insufflare N2.
- Si deve insufflare N2 anche nel caso di titolazioni acido-base, essendo l’anidride carbonica disciolta nell’acqua interferente con la titolazione.
- Si possono effettuare titolazioni previa generazione di specie instabili agli elettrodi, ad esempio argento (II) o manganese (III), così come rame (I).
ELETTROGENERAZIONE ESTERNA DI REAGENTI
Si possono preparare reagenti per via coulombometrica. Vediamo un esempio: si immette in una buretta bifida (a due punte) una soluzione di Na2SO4, e si conduce un elettrolisi con due elettrodi di platino e un batuffolo di lana di vetro che funge da setto poroso. Gli ioni dei due sali sono però più stabili di quanto non sia l’acqua stessa, il cui idrogeno si ridurrà al catodo, generando H2 e ione idrossido, e il cui ossigeno si ossiderà all’anodo, generando ossigeno gassoso e ione idronio. Così si possono preparare acido solforico e idrossido di sodio per via elettrolitica, e possiamo conoscere la quantità di reagente generato in base alla carica passata.
DETERMINAZIONE END-POINT
Si possono usare: - indicatori visuali (esempio 2. e 3.) - misure basate su proprietà elettriche:
o Amperometriche (esempio 1.) o Potenziometriche (nel caso della misura del pH con elettrodo a vetro, si può
applicare nell’esempio 3.). o Conduttimetriche: in soluzioni elettrolitiche, che avendo una buona concentrazione
ionica al loro interno conducono bene la corrente. In titolazioni di precipitazione infatti si può avere un cambiamento di conducibilità per allontanamento di ioni. Si pongono perciò in soluzione due elettrodi di platino e si misura la resistenza del circuito. Più questa aumenta, più la reazione procede. (applicabile all’esempio 3.)
o Spettrofotometriche: basate sul fatto che molti metalli cambiano colore a seconda del complessante a cui sono legati.
Vediamo degli esempi:
1. Determinazione concentrazione del cicloesene. Per l’analisi si fa uso di una cella elettrolitica con due elettrodi destinati all’elettrolisi per generare reagenti e due microelettrodi ai quali si applica una piccola differenza di potenziale, ai quali reagiranno solo eventuali coppie redox reversibili presenti in soluzione. Sono perciò usati come indicatori di raggiungimento dell’end-point. Si procede in questo modo: Si immette il campione in una soluzione acida per acido acetico, con metanolo e un sale di bromo. Il mercurio catalizza la reazione, e fra i due elettrodi della cella viene applicata una
V costante, così da fare avvenire la reazione:

31
Come si vede, il prodotto della reazione catodica si allontana perché gassoso, e dunque non è necessario dividere i due elettrodi in comparti separati. Una volta generato bromo dalla reazione elettrodica, esso reagirà con il cicloesene presente in soluzione formando un 1,2-dibromocicloesano. Si nota che, fino al punto di equivalenza, il bromo prodotto è in difetto, e reagisce tutto con il cicloesene, mentre dopo il punto di equivalenza il bromo prodotto e il bromuro ancora presente in soluzione formeranno una coppia redox reversibile, che sarà in grado di reagire ai microelettrodi presenti nella cella. Solo dopo il punto di equivalenza si avrà passaggio di corrente nel circuito a cui sono collegati i microelettrodi, e dunque il segnale funge da indicatore di end-point.
2. Potremmo in alternativa usare l’indicatore metilarancio, che viene bromurato solo una volta che il cicloesene si è esaurito, colorandosi. Questo è possibile perché il metilarancio reagisce con il bromo meno di quanto non faccia il cicloesene. (costanti di stabilità)
3. Standardizzazione HCl
Si usa in questo caso un indicatore acido-base, essendo il pH al punto di equivalenza neutro. In soluzione si può mettere un elettrodo ad argento e uno a platino, che reagiscano così:
METODI POLAROGRAFICI E VOLTAMMETRICI
Questi metodi si basano sul fenomeno della polarizzazione elettrodica, e pongono in relazione il potenziale applicato con l’intensità di corrente passante nel circuito per ottenere informazioni quantitative. Infatti, si è detto che, lavorando con soluzioni statiche e microelettrodi, si arriva ad un valore limite dell’intensità di corrente, che non varia al variare del potenziale applicato. Si può fare in modo, attraverso alcuni accorgimenti, che questa corrente sia proporzionale alla concentrazione di specie elettroattiva in soluzione. Vediamo un esempio di cella per polarografia. Può essere usato un elettrodo a goccia di mercurio (DME), che consiste di un serbatoio di mercurio e un capillare che pesca nella soluzione elettrolitica, dal quale stilla la goccia di mercurio, che funge da microelettrodo (Ø ~ 1mm), e che andrà da volume nullo a volume massimo ripetutamente, ad ogni ciclo di vita della goccia. Come contro elettrodo si può usare un elettrodo di platino, o lo stesso mercurio che si accumula sul fondo della cella. In alcuni casi si vuole che il contro elettrodo funga anche da reference, e questo è solitamente possibile, perché per l’analisi vengono usate correnti elettriche di mA o addirittura μA di intensità, e dunque la quantità di analita in soluzione non varia apprezzabilmente. Questo comporta due cose:
- L’analisi può essere ripetuta molte volte sulla stessa soluzione, senza falsare il dato quantitativo
- Il contro elettrodo ha un potenziale che può essere considerato costante, e dunque può essere usato anche come elettrodo di riferimento. Si preferisce tuttavia utilizare un reference electrode a calomelano.
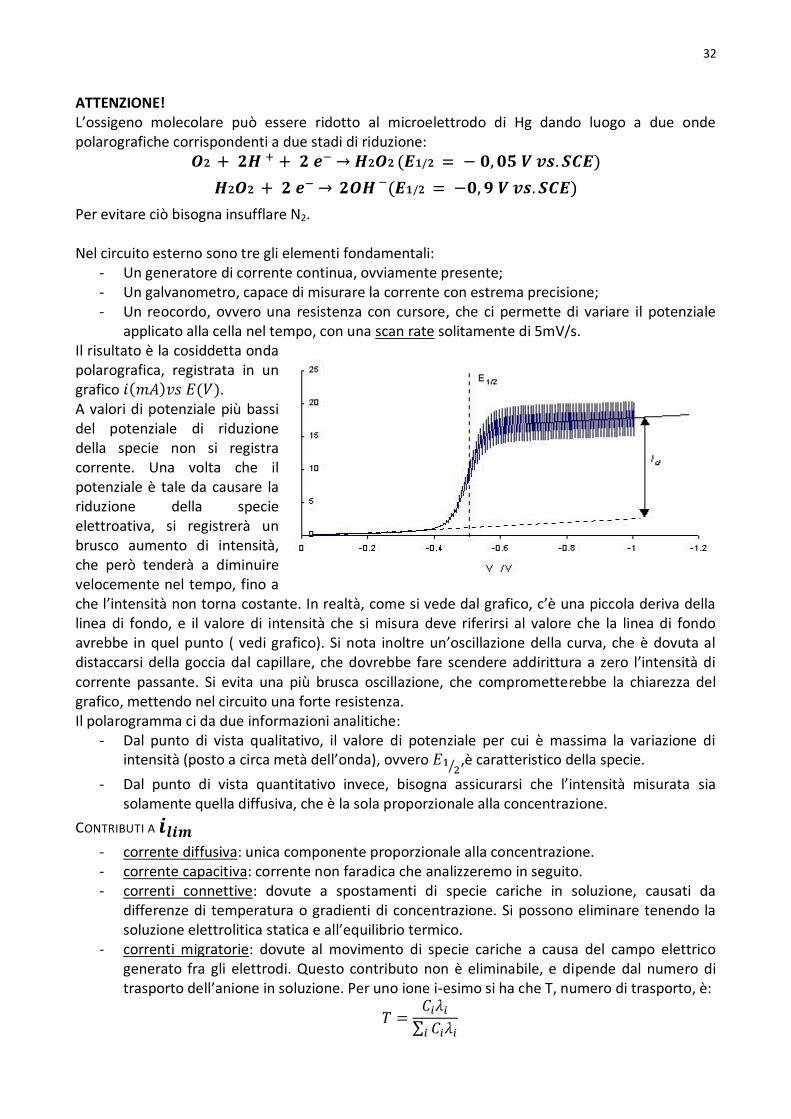
32
ATTENZIONE! L’ossigeno molecolare può essere ridotto al microelettrodo di Hg dando luogo a due onde polarografiche corrispondenti a due stadi di riduzione:
Per evitare ciò bisogna insufflare N2. Nel circuito esterno sono tre gli elementi fondamentali:
- Un generatore di corrente continua, ovviamente presente; - Un galvanometro, capace di misurare la corrente con estrema precisione; - Un reocordo, ovvero una resistenza con cursore, che ci permette di variare il potenziale
applicato alla cella nel tempo, con una scan rate solitamente di 5mV/s. Il risultato è la cosiddetta onda polarografica, registrata in un grafico . A valori di potenziale più bassi del potenziale di riduzione della specie non si registra corrente. Una volta che il potenziale è tale da causare la riduzione della specie elettroativa, si registrerà un brusco aumento di intensità, che però tenderà a diminuire velocemente nel tempo, fino a che l’intensità non torna costante. In realtà, come si vede dal grafico, c’è una piccola deriva della linea di fondo, e il valore di intensità che si misura deve riferirsi al valore che la linea di fondo avrebbe in quel punto ( vedi grafico). Si nota inoltre un’oscillazione della curva, che è dovuta al distaccarsi della goccia dal capillare, che dovrebbe fare scendere addirittura a zero l’intensità di corrente passante. Si evita una più brusca oscillazione, che comprometterebbe la chiarezza del grafico, mettendo nel circuito una forte resistenza. Il polarogramma ci da due informazioni analitiche:
- Dal punto di vista qualitativo, il valore di potenziale per cui è massima la variazione di intensità (posto a circa metà dell’onda), ovvero ,è caratteristico della specie.
- Dal punto di vista quantitativo invece, bisogna assicurarsi che l’intensità misurata sia solamente quella diffusiva, che è la sola proporzionale alla concentrazione.
CONTRIBUTI A
- corrente diffusiva: unica componente proporzionale alla concentrazione. - corrente capacitiva: corrente non faradica che analizzeremo in seguito. - correnti connettive: dovute a spostamenti di specie cariche in soluzione, causati da
differenze di temperatura o gradienti di concentrazione. Si possono eliminare tenendo la soluzione elettrolitica statica e all’equilibrio termico.
- correnti migratorie: dovute al movimento di specie cariche a causa del campo elettrico generato fra gli elettrodi. Questo contributo non è eliminabile, e dipende dal numero di trasporto dell’anione in soluzione. Per uno ione i-esimo si ha che T, numero di trasporto, è:
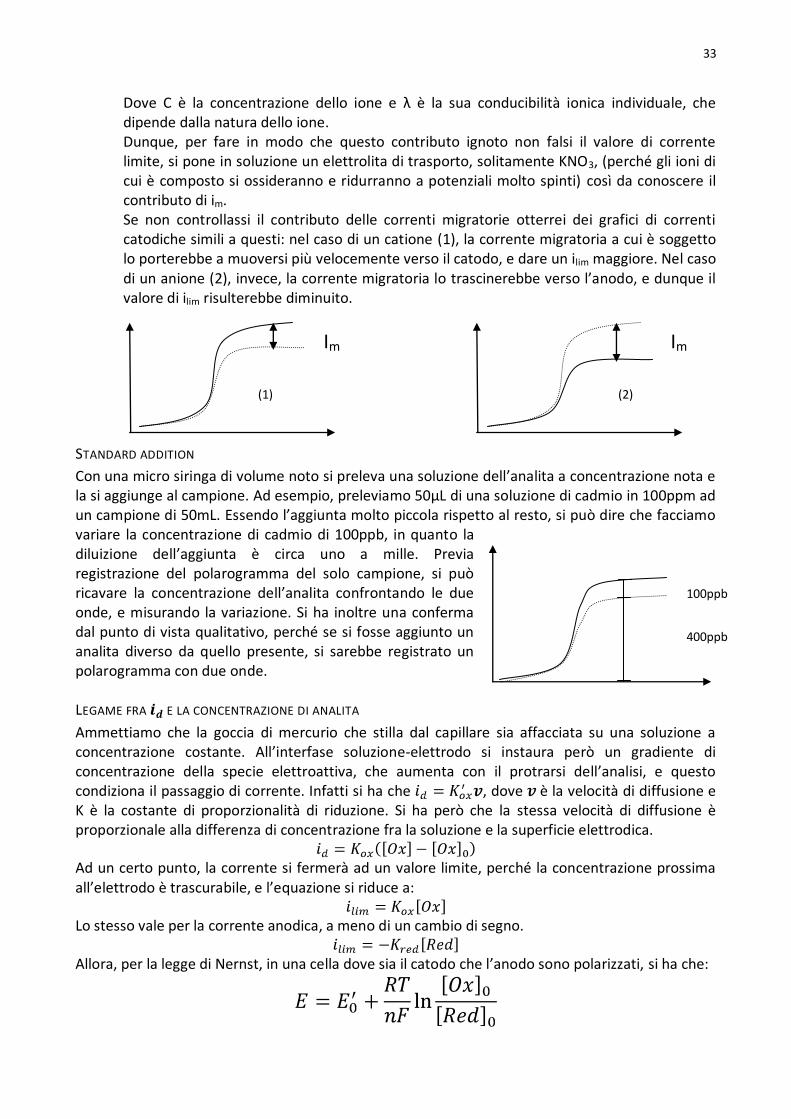
33
Dove C è la concentrazione dello ione e λ è la sua conducibilità ionica individuale, che dipende dalla natura dello ione. Dunque, per fare in modo che questo contributo ignoto non falsi il valore di corrente limite, si pone in soluzione un elettrolita di trasporto, solitamente KNO3, (perché gli ioni di cui è composto si ossideranno e ridurranno a potenziali molto spinti) così da conoscere il contributo di im. Se non controllassi il contributo delle correnti migratorie otterrei dei grafici di correnti catodiche simili a questi: nel caso di un catione (1), la corrente migratoria a cui è soggetto lo porterebbe a muoversi più velocemente verso il catodo, e dare un ilim maggiore. Nel caso di un anione (2), invece, la corrente migratoria lo trascinerebbe verso l’anodo, e dunque il valore di ilim risulterebbe diminuito.
STANDARD ADDITION
Con una micro siringa di volume noto si preleva una soluzione dell’analita a concentrazione nota e la si aggiunge al campione. Ad esempio, preleviamo 50μL di una soluzione di cadmio in 100ppm ad un campione di 50mL. Essendo l’aggiunta molto piccola rispetto al resto, si può dire che facciamo variare la concentrazione di cadmio di 100ppb, in quanto la diluizione dell’aggiunta è circa uno a mille. Previa registrazione del polarogramma del solo campione, si può ricavare la concentrazione dell’analita confrontando le due onde, e misurando la variazione. Si ha inoltre una conferma dal punto di vista qualitativo, perché se si fosse aggiunto un analita diverso da quello presente, si sarebbe registrato un polarogramma con due onde. LEGAME FRA E LA CONCENTRAZIONE DI ANALITA
Ammettiamo che la goccia di mercurio che stilla dal capillare sia affacciata su una soluzione a concentrazione costante. All’interfase soluzione-elettrodo si instaura però un gradiente di concentrazione della specie elettroattiva, che aumenta con il protrarsi dell’analisi, e questo condiziona il passaggio di corrente. Infatti si ha che , dove è la velocità di diffusione e K è la costante di proporzionalità di riduzione. Si ha però che la stessa velocità di diffusione è proporzionale alla differenza di concentrazione fra la soluzione e la superficie elettrodica.
Ad un certo punto, la corrente si fermerà ad un valore limite, perché la concentrazione prossima all’elettrodo è trascurabile, e l’equazione si riduce a:
Lo stesso vale per la corrente anodica, a meno di un cambio di segno.
Allora, per la legge di Nernst, in una cella dove sia il catodo che l’anodo sono polarizzati, si ha che:
100ppb
400ppb
Im
(1)
Im
(2)

34
Dove E0 è soprasegnata perché invece delle attività si sono usate le concentrazioni delle specie, che fanno riferimento ovviamente alla concentrazione sulla superficie dell’elettrodo, che è l’unica che conta per la reazione di ossidoriduzione.
Essendo:
e:
L’equazione diventa:
Sperimentalmente si ottiene che: per ogni goccia di mercurio, la corrente è descritta dalla formula:
Nel grafico i vs t, a potenziale applicato costante si riscontra un andamento analogo a quello dell’onda polarografica in piccola scala. La corrente media passante nel tempo di vita della goccia è
solitamente .
La corrente media può essere anche espressa tramite l’equazione di Ilkovič:
Dove n è il numero di elettroni coinvolti nella reazione, D è il coefficiente di diffusione, che è caratteristico della specie (dipende dall’ingombro sterico dell’analita e dall’ordine di reazione), C è la concentrazione, m è la massa di mercurio uscente al secondo dal capillare, t è il tempo espresso in secondi e 605 è un parametro che è valido con queste unità di misura. PROCESSI FARADICI E NON FARADICI
Come accennato prima, la corrente diffusiva non è l’unica corrente sviluppata all’elettrodo. Infatti la corrente limite è dovuta anche a processi non faradici, ovvero non dovuti a ossidazioni e riduzioni. Il catodo in particolare, richiamerà dalla soluzione specie cariche negativamente, che si disporranno attorno alla superficie della goccia, formando una specie di condensatore. Perciò si sviluppa una corrente detta capacitiva, fra le due armature, dipendente dalla capacità del condensatore, che è data da:
Sappiamo però che la carica sull’elettrodo sarà uguale a quella dell’armatura del condensatore costituita dalla soluzione. Sperimentalmente si trova che la carica q è uguale a:
Dove la C è la capacità per unità di superficie, A è l’area dell’elettrodo e Em è il potenziale dell’elettrodo quando il tempo di vita della goccia finisce (area massima). Inoltre, essendo l’intensità di corrente la derivata prima della carica rispetto al tempo, si ottiene:
Sperimentalmente, si trova che:
tmax

35
E quindi:
È importante tenere a mente che:
Si può notare allora che la corrente capacitiva è sempre presente, non dipende dalla concentrazione dell’analita in soluzione. Inoltre essa diminuisce, anche bruscamente col passare del tempo, e quindi se potessimo misurare le due intensità indipendentemente, avremmo un andamento di questo tipo: Questo dunque non influisce molto sul valore di ilim, essendo il contributo della corrente capacitiva sempre minore col passare del tempo. Vediamo ora come varia ic al variare del potenziale. Se applichiamo all’elettrodo potenziali molto positivi, allora l’elettrodo attirerà cariche negative, mentre se applichiamo ad esso potenziali negativi le cariche che attirerà saranno positive. C’è un valore di potenziale applicato, tuttavia, al quale la corrente capacitiva assume valore nullo. Infatti a quel dato potenziale l’elettrodo e la soluzione non accumulano cariche, e dunque non si comportano come armature di un condensatore. Questo valore è nel caso di soluzioni acquose -0.45 V (vs SCE, standard calomelan electrode). A queste condizioni il tempo di vita della goccia è massimo, e al variare del potenziale verso valori più spinti si osserva una diminuzione sempre più grande del life time. In figura si vede una cella per polarografia. La cella è a forma di tronco di cono per permettere di usare volumi alquanto ridotti (solitamente 20mL). È presente un terzo elettrodo, di riferimento, a calomelano, che a volte è necessario separare dalla soluzione, onde evitare che lo ione Cl- interferisca con le reazioni elettrolitiche. Si nota anche che ci sono due tubicini per l’ingresso dell’azoto nella cella: uno che entra nella soluzione, per far gorgogliare l’azoto e dunque eliminare i gas presenti e interferenti (O2 e CO2); uno fuori dalla soluzione, così da saturare il sistema senza agitare la soluzione quando si compie l’analisi. Infatti si vuole ottenere la polarizzazione dell’elettrodo, che non avviene se la soluzione è tenuta sotto agitazione. Applicazioni:
id
Ic
-2.0 +0.4

36
- Se analizzando una matrice complessa ci si trova di fronte ad un polarogramma con delle onde alquanto sovrapposte, si può ricorrere a dei complessanti, che si leghino con l’analita la cui onda è poco risolta e ne spostino l’E1/2. Ad esempio, se volessimo determinare la concentrazione di ferro (III) e rame (II) in soluzione in ambiente cloridrico, le due specie darebbero un polarogramma con due potenziali di mezz’onda sovrapposti. Basta aggiungere un sale di fluoro affinché si formi l’esafloruro ferrico (FeF6
3-), che ha un potenziale di mezz’onda molto più negativo, essendo la specie carica negativamente e dunque più difficilmente riducibile. Le due onde così si splittano, ed è possibile determinare la concentrazione dei due metalli.
- Dovrebbe stupire il fatto che, sebbene spesso sia necessario condurre le analisi in ambiente acido (onde evitare formazione di idrossidi), lo ione idronio non si riduca al DME. Questo è dovuto al fatto che l’idrogeno ha un’elevata sovratensione catodica sul mercurio, di circa 1V. Per questo è possibile arrivare fino a potenziali tali da ridurre Zn e Ni.
- L’importanza del pH è notevole, quando la concentrazione (attività) dello ione idronio rientra nella reazione elettrodica (nel caso di reazioni anodiche la concentrazione dello ione ossidrile). Ad esempio, per la riduzione di BrO3
-, IO3-, Cr2O7
2-, NO2-. Infatti, nel caso del
bromato, la reazione catodica è:
Il pH deve dunque rimanere costante, affinché l’intensità di corrente generata dalla reazione non vari nel tempo.
- Certi anioni danno reazioni anodiche a valori di potenziale alquanto bassi (poco positivi o addirittura negativi), come ad esempio: Cl-, Br-, I-, CN-,SCN-. Questo fatto riduce sensibilmente il campo di indagine dell’analisi polarografica, infatti le reazioni anodiche danno onde negative, che precludono l’analisi a quei valori di potenziale. Si può ampliare il campo di analisi nel caso di ioni come NO3
- e ClO4-.
- Si possono analizzare anche specie organiche, e i gruppi funzionali elettroattivi sono: o Carbonilico o Carbossilico o Perossidi ed epossidi o Nitrogruppi, azogruppi o Alogenuri organici o Idrochinoni e mercaptani (che danno reazione anodica, ossidando dunque il
mercurio. I mercaptani prendono il loro nome proprio da questo fatto)

37
- Se si volesse operare a voltaggi più positivi di quelli permessi dalla polarografia, si potrebbe sostituire l’elettrodo a Hg con uno ad oro, più nobile e meno facilmente ossidabile. In questo caso si parla di voltammetria.
- Gli elettroliti di supporto che si possono utilizzare dipendono dal solvente. o Con solventi polari e protici, come acqua e alcoli, in genere si usano: KCl, KNO3,
KClO4… o Con solventi organici e aprotici come il benzene, la piridina, l’acetilnitrile (CH3CN),
dimetilformammide (DMF) e dimetilsolfossido (DMSO) si usano: più raramente LiCl, mentre più frequentemente sali di tetralchilammonio. Ad esempio, tetrametilammonio, tetraetilammonio (E1/2=-2.4V) , e tetrabutilammonio(E1/2=-2.7V) Questi sali permettono di raggiungere potenziali estremamente negativi, ma con i dovuti accorgimenti, infatti la vita della goccia dipende dal potenziale, come già visto, e perciò bisogna usare elettrodi con tempi in principio molto lunghi.
TITOLAZIONI AMPEROMETRICHE
È un’alternativa alla polarografia, e consiste nel determinare la concentrazione di un analita grazie ad un titolante, dei quali almeno uno deve essere elettroattivo, e registrare l’intensità di corrente sviluppata nel tempo. La cella usata è solitamente composta da un contro elettrodo, una buretta per le aggiunte di titolante, e un elettrodo molto simile al DME, ma nel quale la goccia di mercurio è sostituita da una pallina di platino, per evitare le fluttuazioni continue del DME. Si nota, se l’analita è elettroattivo, e il prodotto della reazione non lo è, che ad ogni aggiunta di titolante la id si abbassa. Infatti si registrano polarogrammi con corrente limite sempre più bassa. In realtà non serve a nulla registrare ogni volta il polarogramma, ed è molto più pratico portarsi direttamente ad una valore Ex di potenziale tale che la corrente sia limite, e misurare quanto questa vari ad ogni aggiunta di volume. Dopodiché si possono riportare i punti trovati in un diagramma , in modo da trovare per estrapolazione il punto di equivalenza. Si noti che, a differenza delle titolazioni acido-base, dove i punti importanti per l’analisi sono quelli vicini al pt. finale, in questo caso è ininfluente la vicinanza al punto di equivalenza, basta che i valori di intensità non siano sfalsati. Applicazioni:
- Nella titolazione di Pb2+ con ioni cromato o dicromato, per precipitazione, si possono avere due grafici diversi, a seconda del potenziale a cui si opera. Infatti a potenziali catodici più positivi di -0.4 il piombo non si riduce, e quindi non è elettroattivo e non genera nessuna corrente limite. Il grafico sarebbe di questo tipo: Mentre se si opera a potenziali minori di -0.4, il piombo si riduce all’elettrodo, e il grafico avrebbe il primo braccio più inclinato, in questo modo:
- Con il nitrato di piombo, invece, si possono precipitare come sali di piombo solfati, molibdati, floruri e cloruri, e titolarli per via amperometrica a potenziali o più elevati della

38
E1/2 della reazione anodica o più bassi di quella di riduzione del piombo. Altrimenti, a valori di potenziale intermedi, non si registrerebbe corrente in tutta la titolazione.
- Con 8-idrossichinolina (chelante) sono titolabili per complessazione molti metalli di transizione.
METODI DEAD-STOP
- Si basano sulla determinazione del punto finale non per estrapolazione ma per azzeramento d’intensità di corrente passante fra due microelettrodi in soluzione. A questi due microelettrodi è infatti applicata una differenza di potenziale di solo 0.2 V, tale da non essere sufficiente a fare avvenire ossidoriduzione di coppie redox non reversibili. Perciò ammettendo di voler titolare Fe2+ con Ce3+, si può operare in questo modo: Si aggiunge del cerio (IV), in modo che ossidi il ferro a Fe3+. Così si genera la coppa redox ferro (II)-ferro(III), che reagirà ai microelettrodi perché reversibile, dando una corrente che è proporzionale alla specie delle due in difetto, ovvero il ferro (III). Una volta raggiunto il punto di semiequivalenza si otterrà un massimo di intensità di corrente ai microelettrodi, essendo le concentrazioni delle specie della coppia esattamente uguali. Si avrà invece un valore nullo di intensità una volta che tutto il ferro (II) si sia trasformato in ferro(III), ovvero al punto di equivalenza. Dopodichè la corrente agli elettrodi comincerà ad aumentare in relazione alle aggiunte di cerio (IV), che ai microelettrodi formerà una coppia redox con il cerio reagito con il ferro. Attenzione: le coppie redox per dare passaggio di corrente devono essere reversibili, ovvero ossidarsi e ridursi allo stesso potenziale, essere ovvero in equilibrio fra le due specie. Il ferro da una coppia reversibile in ambiente ossalico, ma non in ambiente pirofosforico.
- Metodo di Karl-Fischer, per la determinazione di tracce di acqua e quindi dell’anidricità di un solvente organico. Si usa iodo e diossido di zolfo sciolti in piridinia, cosicché lo iodio si consumi, finché è presente acqua, producendo ioduro. Una volta esaurita l’acqua, aggiungendo iodio si da origine a una corrente generata ai microelettrodi dalla coppia I2/I-. La reazione che avviene è:
- Con l’arsenico (III), titolato con iodio, la situazione è diversa, essendo la coppia dell’acido arsenico ed arsenioso non reversibile. Si ha perciò un grafico dapprima costante, schiacciato sulla linea di fondo, e poi al punto di equivalenza la coppia reversibile iodio-ioduro darà passaggio di corrente e quindi una retta inclinata positivamente.
- Titolando lo iodio con solfiti, si ha il caso inverso: una campana tipica del dead-stop, che dopo lo zero di corrente non presenta una retta inclinata.

39
POLAROGRAFIE DERIVATE E A IMPULSI
Come determinare con la polarografie concentrazioni per cui ? Derivate: se il segnale di un determinato analita non è molto forte, e dunque si vorrebbe amplificare, o se due segnali di due diversi analiti sono molto vicini e sovrapposti, si può operare con il metodo della derivazione. Infatti, essendo il grafico del polarogramma del tipo (1), il grafico della derivata sarà come il (2), dove il massimo è in E1/2, che si distinguerà molto meglio dalla linea di fondo se amplificato. Inoltre, se si avessero due specie, come il cadmio e l’indio, aventi potenziali di mezz’onda talmente ravvicinati da dare un polarogramma con un'unica onda, si può derivare il grafico trovato, riuscendo così a distinguere molto meglio i due picchi. Così si ha una molto più semplice analisi qualitativa. Per un analisi quantitativa tuttavia bisognerebbe ricorrere a complessanti, perché le bande sono ancora molto sovrapposte. A impulsi: operando con questo sistema si riesce ad avere una risposta strumentale molto più sensibile. Il circuito viene programmato in modo da dare un leggero impulso di potenziale (5-100mV) ogni qualvolta la goccia del DME stia per cadere. Questo fa sì che si registri un cambiamento di intensità di corrente registrata, che sarà diverso a seconda del punto dell’onda polarografica. All’inizio l’impulso causerà una piccola differenza di intensità, mentre quando ci si avvicina al potenziale di mezz’onda il diverrà più grande. Così, riportando in un grafico i valori di trovati contro il potenziale, si ottiene una curva con massimo, leggermente dentata, e per il resto praticamente analoga al caso della derivata. Questa tecnica può essere applicata anche ad elettrodi stazionari. Comunque sia, è molto importante che il tempo di vita della goccia sia un parametro conosciuto. IMPORTANZA DEL DROP TIME
Il tempo di vita della goccia è molto importante nella polarografia ad impulsi, dovendo settare il circuito in modo da avere l’impulso di potenziale poco prima del drop-time. Si capisce che il tempo di vita della goccia dipende dall’altezza del capillare, che più sarà lungo più tenderà a stillare velocemente. Si può mantenere costante il livello di mercurio nel serbatoio a cui è collegato il capillare, in modo da non avere variazioni di altezza della colonna durante l’analisi, con un sistema di refill. Un fattore di cui tenere conto è che a voltaggi più negativi la vita della goccia diventa più breve, e dunque il polarogramma tenderebbe a regredire in questo modo: Perciò generalmente si impiegano DME con tempi di goccia lunghi (~10s), e poi si fa cadere la goccia meccanicamente ogni 2 o 3 secondi, così da avere tempi sempre uguali e riproducibili.
ELETTRODI: HDME E MTFE
HDME( hanging drop mercury elecrtode), o di Kemula: è costituito da un capillare e da un serbatoio a tenuta stagna, per cui il mercurio, anziché gocciolare come nel DME, tende a rimanere attaccato al capillare. L’elettrodo si prepara aspirando il mercurio nel serbatoio (~4mL), dopodiché si avvita un tappo per un certo numero di tacche in modo da spingere il mercurio nel capillare, e formare la goccia. Questo metodo si usa nella determinazione di Pb e Cd nelle acque di fiume e di mare, con il metodo dell’ASV. (vedi dopo)
(1)
(2)

40
MTFE(mercury thin film electrode): Può essere usato in alternativa all’HDME, per determinare concentrazioni di metalli in acqua, anche molto diluiti, come zinco, cadmio, piombo e rame. È costituito da un piccolo dischetto di grafite vetrosa (glassy carbon, GC), molto poco porosa e dunque facile da lavare, posta su un supporto di Teflon e collegata al circuito tramite un filo conduttore. Alla soluzione che si vuole analizzare si aggiunge del nitrato di mercurio (II) ,Hg(NO3)2. Il mercurio del sale si ridurrà a potenziali molto ridotti, e dunque si depositerà sull’elettrodo di grafite una sottilissima pellicola (10-20 Å), nel quale poi si amalgameranno gli altri metalli, nell’ASV, che vediamo ora.
ASV ( ANODIC STRIPPING VOLTAMMETRY )
Per soluzioni molto diluite si possono usare gli elettrodi HDME e MTFE per la voltammetria a scansione anodica. La procedura consiste nel applicare all’elettrodo un potenziale abbastanza negativo da provocare la riduzione delle specie metalliche presenti in soluzione di cui si vuole determinare la concentrazione, e lo si applica per un tempo detto di deposizione (pre-elettrolisi). In questo lasso di tempo i diversi metalli si depositeranno sull’elettrodo, sciogliendosi nel mercurio a formare un amalgama, e concentrandosi sempre di più, tanto più lungo è il tempo di pre-elettrolisi. La soluzione dovrà essere agitata, onde evitare la polarizzazione dell’elettrodo, che bloccherebbe il deposito. Una volta che i metalli si siano concentrati, si inizia l’analisi vera e propria, diminuendo gradualmente e costantemente nel tempo il potenziale applicato all’elettrodo (ecco perché stripping). Questo fa sì che le specie che si erano ridotte comincino a ossidarsi e a passare in soluzione, con una reazione anodica (da qui il nome anodic). L’AVS si può condurre in scansione lineare, o a impulsi, ovvero la DP-AVS (differential pulses AVS) generalmente preferita perché la deriva della linea di fondo diminuisce di molto, e la risposta strumentale è dieci volte più sensibile. I parametri da controllare sono:
- L’efficienza dell’elettrolisi - Modalità di stripping (impulsi o lineare) - Tipo di elettrodo da usare:
o Con MTFE si ottengono picchi più stretti di quelli registrati in un AVS con HDME, perché nel primo caso gli ioni passano in soluzione praticamente simultaneamente, data la struttura patinale dell’elettrodo MTFE, mentre per l’hanging drop si avrà un passaggio più graduale dei metalli disciolti in soluzione, perché si distribuiranno fra la superficie della goccia e il corpo della stessa.
- Formazione di complessi intermetallici nell’amalgama, in situazioni di concentrazioni di ppm. Ad esempio, in soluzione di zinco e rame, quando questi si depositano all’elettrodo si legano in un complesso difficilmente ossidabile. Dunque si può aggiungere del gallio, così da formare una lega fra Ga e Cu, e liberare lo zinco di cui si può registrare il segnale. E il rame? Non c’è problema, si può ripetere l’operazione registrando il segnale del rame, liberandolo utilizzando un complessante dello zinco. Questo è possibile perché il calo di concentrazione del metallo in soluzione non influisce sulla misura successiva, essendo molto ridotto.

41
-
LABORATORIO
QUALITÀ DEL DATO
Valore vero: indicato con μ, è il risultato dell’analisi compiuta da un’analista esperto che conduca
un numero ipoteticamente infinito di repliche. In pratica si usano i CRM (Certified Reference
Material), garantiti da organismi di preparazione e di controllo, che sono campioni reali in cui la
concentrazione di ogni specie di interesse analitico o interferente è riportata in un certificato.
Errore assoluto: l’errore assoluto E esprime la differenza fra il valore vero e il valore ottenuto. In
pratica:
Dove D è l’errore sistematico (bias), d è l’errore casuale, che può essere calcolato su base
statistica, G è l’errore grossolano, di calcolo o di procedimento, è l’errore imprevedibile,
associato a parametri esterni da cui il sistema dipende.
Esattezza: (trueness) esprime la vicinanza di un dato al valore vero (o assunto come tale). Il
metodo più comune per valutarla è utilizzare dei CRM, oppure, in mancanza di un materiale di
riferimento certificato per un dato analita, aggiungere ad un blank certificato una concentrazione
di analita nota. Questa matrice prende il nome di campione spiked.
Precisione: la precisione descrive l’accordo fra due o più misure replicate, ed è correlata ad errori
casuali, all’interno di un set di misurazioni. Il termine precisione può essere sostituito nello
specifico da ripetibilità, qualora le repliche fossero effettuate nello stesso laboratorio, dallo stesso
analista, e riproducibilità, quando le misure vengono ripetute in più laboratori, e in tempi più
lunghi. La precisione dipende perciò dal tempo in cui si effettua l’analisi (ricorda l’esperienza 1 di
laboratorio, dove la concentrazione di Cr è scesa nel tempo)
LOD: limit of detection, è la minima concentrazione di analita rilevabile con un segnale, cioè
corrisponde ad un segnale molto simile al bianco ma da esso diverso, in modo da poter essere
ricondotto all’analita in questione secondo qualche criterio specifico. Il limite di rilevabilità è
strettamente correlato al rumore di fondo, noise, e da questo si calcola secondo l’equazione:
Dove h è l’altezza della linea di fondo, calcolata come media delle misure del bianco, e s è la
deviazione standard calcolata su un numero adeguato (>20) di misure del bianco.
MDL: minimum detection limit, è un altro modo per definire il limite di rilevabilità, usato da
produttori di strumentazione scientifica, è definito come un rapporto segnale:rumore di 2:1.

42
LOQ: limit of quantification, minima altezza che il picco può avere per ottenere un’informazione
quantitativa, definito come:
CRITERI PER LA SCELTA DEL METODO ANALITICO
1. Strumentazione disponibile ed equipaggiamento richiesto
2. Confronto fra precisione richiesta e quella della tecnica per cui si vuole optare
3. Confronto del tempo di analisi rispetto a quello di urgenza dei risultati
4. Tempo per la messa a punto del metodo
5. Numero di analiti da determinare
6. Costi
TECNICHE
- Classiche: si utilizzano solo reazioni chimiche, la normale vetreria e strumentazione di
laboratorio e bilance, si misurano dunque solo volumi e masse.
- Strumentali: determinano la quantità di analita in base a una o più propietà fisiche
correlate alla concentrazione. Si basano su scoperte scientifiche e tecnologiche e
prevedono utilizzo di strumentazione specifica, come ad esempio nel caso della
spettrofotometria, della cromatografia o di analisi elettrochimiche.
Possono essere analisi distruttive o non distruttive.
Un’analisi quantitativa presuppone una taratura dello strumento, mentre un’analisi
qualitativa si basa su confronto del campione con sostanze di riferimento.
TECNICA, METODO, PROCEDURA O PROTOCOLLO?
È importante definire questi concetti basilari della chimica analitica.
Tecnica: è l’insieme di principi teorici ed accorgimenti sperimentali che permettono di
determinare la composizione di una matrice in base a uno o più fondamentali fenomeni scientifici.
Metodo: è l’applicazione pratica di una tecnica, volta a risolvere un specifico problema analitico.
Procedura: successione degli stadi operativi principali per utilizzare un metodo analitico, delle
istruzioni pratiche che definiscano gli stadi della misurazione.
Protocollo: l’insieme di direttive dettagliate e da seguire rigidamente affinché i risultati dell’analisi
siano accettati per fini particolari, come in controversie legali o nell’ambito di verifica di
rispondenza ad un certo parametro stabilito per legge.

43
PROCEDURA ANALITICA TOTALE
Basata sul rapporto cliente-analista: le frasi sottolineate prevedono la collaborazione fra operatore
e cliente, perché è necessario stabilire adeguatamente alcuni parametri.
1. Definizione del problema dal punto di vista generale.
2. Definizione del problema analitico.
3. Scelta del metodo, tecnica, procedura o protocollo.
4. Campionamento.(il cliente può conoscere meglio il luogo)
5. Trattamento campione
6. Analisi
7. Valutazione dei dati
8. Conclusioni
9. Relazione.
CAMPIONAMENTO
Il campionamento è il prelievo di una massa di campione da una matrice più estesa tale da essere
sufficiente a permettere le analisi previste e non essere troppo ingombrante o poco maneggevole.
Il campione deve essere tale che la proporzione della proprietà che si vuole accertare rappresenti
la proporzione della stessa propietà nella matrice, entro un dato limite d’errore.
Basi di un qualsiasi sistema di campionamento:
1. Definizione dell’obbiettivo
2. Determinazione dello scopo e dell’accuratezza voluta
3. Identificazione del tipo di campione che si deve raccogliere
4. Scelta dei punti e dei luoghi di campionamento
5. Definizione dei parametri da inserire nel programma di raccolta (temperatura, umidità..)
6. Precauzioni di sicurezza e di igiene
7. Messa a punto di un piano che stabilizzi il numero di campioni da prelevare in base al
tempo richiesto dall’operazione e all’accessibilità e distanza del luogo.
8. Selezione equipaggiamento per il campionamento
9. Individuazione degli adeguati sistemi di manipolazione, stoccaggio e trasporto dei campioni
10. Considerazione di eventuali metodi di analisi su campo
11. Definizione del metodo di documentazione
Il contenitore è molto importante: meglio di vetro nel caso di analiti organici e di plastica nel caso
di analiti inorganici.
CONCENTRAZIONI
Si opta per diverse unità di misura nel caso di:
- Soluzioni acquose:
o mg/L, ma per campioni con meno di 0.1mg/L si esprime il dato in μg/L.

44
o per concentrazioni maggiori di 10’000mg/L si può esprimere la concentrazione, se
la densità è d=1, in %: 10’000mg/L=1% (siccome 1L pesa 1Kg e 10 g ne sono la
centesima parte).
o In ogni caso si può esprimere la concentrazione in M o m.
- Solidi o acque di scarico (d≠1):
o Percentuale in peso
o ppm
o ppb
- Atmosfera:
o In peso: mg/m3 o μg/m3.
o In volume: ml/m3, chiamati anche ppm a 1atm e 25°C
o La concentrazione di particelle si esprime anche in numero di particelle per
centimetro cubo pp/cm3, in base a misure granulometriche.





























