Embed Size (px)
Citation preview

ISSN 0891�4168, Molecular Genetics, Microbiology and Virology, 2011, Vol. 26, No. 4, pp. 159–165. © Allerton Press, Inc., 2011.Original Russian Text © S.V. Seregin, I.V. Babkin, I.D. Petrova, L.N. Yashina, E.M. Malkova, V.S. Petrov, 2011, published in Molekulyarnaya Genetika, Mikrobiologiya i Virusologiya,2011, No. 4, pp. 18–23.
159
INTRODUCTION
The Rubella virus causes acute infectious disease,mainly in children and adolescents, which, in 50% ofcases, is asymptomatic [1]. Rubella poses a major haz�ard for a fetus. When women at an early stage of preg�nancy are infected, abnormal fetus development (con�genital rubella syndrome) may occur in 80% of cases[12]. In adults, the disease occurs in severe complica�tions in the form of arthritis and arthralgias and, rarely,as encephalitis and meningoencephalitis [5]. In Rus�sia, the incidence of rubella is significantly higher thanin European countries and has a wavelike nature withvarying frequency of ups and downs. In particular, it isendemoepidemic in major cities: moderate every 3–5 years (450–800 per 100 thousand population) andmore significant every 10–12 years (1500–2000 per100 thousand population) [4].
The World Health Organization (WHO), in its pro�gram for elimination of congenital rubella syndrome,highlights the importance of molecular�epidemiolog�ical studies, with the help of which we can trace thechannels of rubella and take steps to localize the nidusof infection [16]. Up to the present, research findingson the genetic diversity of the Rubella virus circulatingin Russia is very limited; in western Siberia, with theexception of ours [2, 3, 6, 7], there are none at all.
Isolation of the virus and its genetic characteristicsare the most important stages of rubella observation.In today’s world, two major genetic groups (or clades)have been discovered of Rubella virus (1 and 2), withthe nucleotide differences between them ranging from5.5 to 10.3%. Each of these genotypes divides into sub�groups with the letter designation of the genotype. In arevision of the standard nomenclature for the classifi�cation and labeling of wild�type strains of Rubellavirus, the WHO recognizes the final nine (1B, 1C, 1D,1E, 1F, 1G, 1H, 2A, and 2B) and four provisional gen�otypes (2c, la, li, and lj), which is an extended versionof the nomenclature set up in 2005 [8] that was basedon phylogenetic analysis of nucleotide sequence of agenome section of 739 nucleotides in length (from8731 to 9469), encoding a protein fragment E1—theso�called window 739. The currently recommendedSP�section (or area) of the Rubella virus genome 3261nucleotide length (from 6481 to 9741) encodes allthree structural proteins (C, E2, and E1). As a result,the viral isolates that have been previously applied tothe preliminary genotype lg are now divided into fourgroups: 1G, 1H (final), li, and lj (preliminary).
The WHO recommendations of November 20,2007 [17], on the genotyping of Rubella virus strainsassign the following basic requirements for determina�
Appearance of the Rubella virus Genotype 1H in Western Siberia
S. V. Seregina, I. V. Babkinb, I. D. Petrovaa, L. N. Yashinaa, E. M. Malkovaa, and V. S. Petrova
a Vector State Research Center of Virology and Biotechnology, Federal Service for Surveillance in Consumer Rights Protection and Human Well�Being, Koltsovo, Novosibirsk oblast, 630559 Russia
b Institute of Chemical Biology and Fundamental Medicine, Siberian Branch, Russian Academy of Sciences,Novosibirsk, 630090 Russia
e�mail: [email protected], [email protected], [email protected], [email protected], [email protected], [email protected], [email protected], [email protected]
Received December 30, 2010
Abstract—A molecular epidemiological study of novel strain of Rubella virus isolated during the outbreak inWestern Siberia in 2004 is described. A detailed phylogenetic analysis performed based upon entire SP�sec�tion, which encodes all three Rubella structural proteins (C, E2, and El), is implemented. This analysis pro�vides characterization of this strain and classifies it in the 1H genotype, thereby correcting a previous classi�fication of this strain based upon a shorter nucleotide sequence only encoding El protein. Therefore, thisstudy identified the genotype of the Rubella virus not previously detected in western Siberia (and even theentire Russian Federation), which highlights the importance of more extensive characterization of geneticvariability of the Rubella virus, especially with regard to the potential influence of vaccination on the Rubellavirus mutagenesis.
Keywords: Rubella virus, phylogenetic analysis, nucleotide sequences, genotype.
DOI: 10.3103/S0891416811040070
EXPERIMENTAL WORKS

160
MOLECULAR GENETICS, MICROBIOLOGY AND VIROLOGY Vol. 26 No. 4 2011
SEREGIN et al.
tion of a new genotype: the existence of nucleotidesequences of not less than two viral strains in a data�base; carrying out of phylogenetic analysis by twoindependent methods; a high level of support forbranch nodes of the phylogenetic tree (no less than80%) that separates the members of one genotypefrom the other; similarity of the differences within thenew genogroup and with other groups to the values ofdifferences within (and between) already establishedgroups; and similarity between phylogenetic relation�ships during the construction of phylogenetic trees, asin the SP�area, and on the areas encoding some struc�tural proteins, particularly E1. This work was carriedout in the framework of the described WHO recom�mendations.
The aim of this study was to conduct a detailedphylogenetic analysis for adequately genotypingRubella virus circulating in western Siberia in recentyears. An analysis was carried out in this paper of allthe known nucleotide sequences of Rubella virus ofvirus genome on the SP�section, coding all threestructural proteins (C, E2, and E1), which is moreinformative for accurate reflection of the phylogeneticrelationships than is studying shorter sections of thegenome [18]. The obtained results suggest that, inwestern Siberia, representatives of two groups ofRubella virus, presented as the previously establishedgenotype 1E and the 1H new genotype, are circula�ting.
OBJECTS AND METHODS
Strains of Rubella virus: RVi/Nvk13�23.RUS/04(registration number in the GenBank is EF421977)was isolated from blood of a patient during an outbreakof rubella in Novokuznetsk, Kemerovo oblast, inOctober 2004. RVi/Bar4�108.RUS/06 (registrationnumber in the GenBank is EF421978) was isolatedfrom nasopharyngeal wash of a patient in Barnaul inJanuary 2006. Exploratory work on the virus was car�ried out on a passaged culture of Vero cells by themethod described in [6].
Isolation of Rubella virus RNA, cDNA amplifica�tion, and nucleotide sequence determination were car�ried out in correspondence with the techniques out�lined previously in [6, 7]. Table 1 shows a complete listof the oligonucleotide examples used in the research,indicating the exact location of the virus in the genomeby nucleotide positions.
The nucleotide sequences of Rubella virus wereobtained from the GenBank database (http://www.ncbi.nih.gov). Full list of virus strains and isolates,genomic sequences of which were used in the research,are presented in the Table 2 (studied isolates are inbold).For multiple alignment of nucleotide and aminoacid sequences we used programs BioEdit v.7.0 [9] andClustalX v.1.8 [15].
Phylogenetic analysis and calculation of genetic dis�tances among groups of strains of Rubella virus carriedout by the method of association of nearest neighborsusing the program Mega v.3.1 [14] using models ofnucleotide substitutions of Kimura with two parameters[13] and permutation analysis to 1000 iterations, as wellas in the program MrBayes v.3.1 [11] to 200 thousandgenerations of Markov chains with a record of thechain parameters and the corresponding topologyafter every hundred generations, during which the first250 samples were excluded from the analysis.
RESULTS AND DISCUSSION
For phylogenetic analysis, we used nucleotidesequences of the SP�sections of 46 strain genomesRubella virus of differing geographical origin, includ�ing two sequences, presented in this research for iso�lates from western Siberia (described in Materials andMethods). A list of oligonucleotide primers used toprepare a series of overlapping fragments of DNA andtheir sequencing, allowing reconstruction of thenucleotide sequence of the SP�section of the Rubellavirus genome of 3261 nucleotides’ length, is shown inTable 1.
Multiple alignment of the nucleotide sequences ofthe SP�section of the genome of different strains andRubella virus isolates revealed the presence of15 unique nucleotide positions for representatives of anew 1H genetic group: G�432, C�810, T�1185,T�1190, T�1233, T�1404, T�1464, A�1878, A�1932,T�2127, C�1509, A�1878, A�1932, T�2127, G�2157,A�2349, C�2448, G�2916. The nucleotides that arecharacteristic for all known (more than 20) represen�tatives of the genogroup 1H in the area of the genome(“window 739”), encoding a fragment of the proteinE1 (according to the results of phylogenetic analysiscarried out on the section of the Rubella virusgenome), are in bold type. A numeration of the studiedopen frame of translation, called the SP�section, isgiven. Such a study of amino acid sequences showedthat only a unique nucleotide substitution at position1233 leads to the substitution of amino acid residue atposition 411 to phenylalanine (F) that is characteristiconly for members of genetic group 1H. The represen�tatives of the other genotypes at this position are ala�nine (A), serine (S), or threonine (T).
Analysis of intragroup differences showed that themost similar strain is the RVi/Nvkl3�23.RUS/04 virusfrom Belarus RVi/Minsk. BLR/28.05/2 is differentfrom those selected in western Siberia in primarystructure only because of the 17 nucleotide positions(12 of which are silent, that is, do not lead to thereplacement of an amino acid residue at the corre�sponding position), while the representatives of theclose genogroup 1G RVi/UGA/20.01 and geneticgroup la RVi/WistarRA27/3 (vaccine strain) havemore significant differences from the studied strain,
MOLECULAR GENETICS, MICROBIOLOGY AND VIROLOGY Vol. 26 No. 4 2011
APPEARANCE OF THE Rubella virus GENOTYPE 1H 161
according to 63 and 124 nucleotide positions, respec�tively.
Illustrative material for the above studies is not pre�sented because of its complexity.
For a more rigorous strain genotyping of those iso�lated in western Siberia, we followed the recommen�dations of the WHO and conducted a thorough phylo�genetic analysis using different methods (see Materialsand Methods).
Table 3 shows the mean values of genetic distancesboth within and between different genotypes ofRubella virus. We also want to make a note that themost closely to the genotype 1H, as noted above, is thegenotype 1G (genetic distance between them is 0.041
change to site, that is to nucleotide position).As seenin Table 3, this value is greater than the distancesbetween the genotypes 1E and 1D, as well as betweenthe genotype 1a and most of the other genotypes. Thisallows us to conclude the absolute belonging of strainRVi/Nvkl3�23.RUS/04, selected by us in Novokuz�netsk in 2004, to the new genotype of Rubella virus 1H.At the same time, it should be noted that the strainRVi/Bar4�108.RUS/06, dedicated in Barnaul in 2006,is a representative of the genetic group 1E, as weshowed earlier in an analysis of the genomic fragmentencoding the protein E1 [6, 7]. The most obvious con�firmation of the findings can serve as a graphical rep�
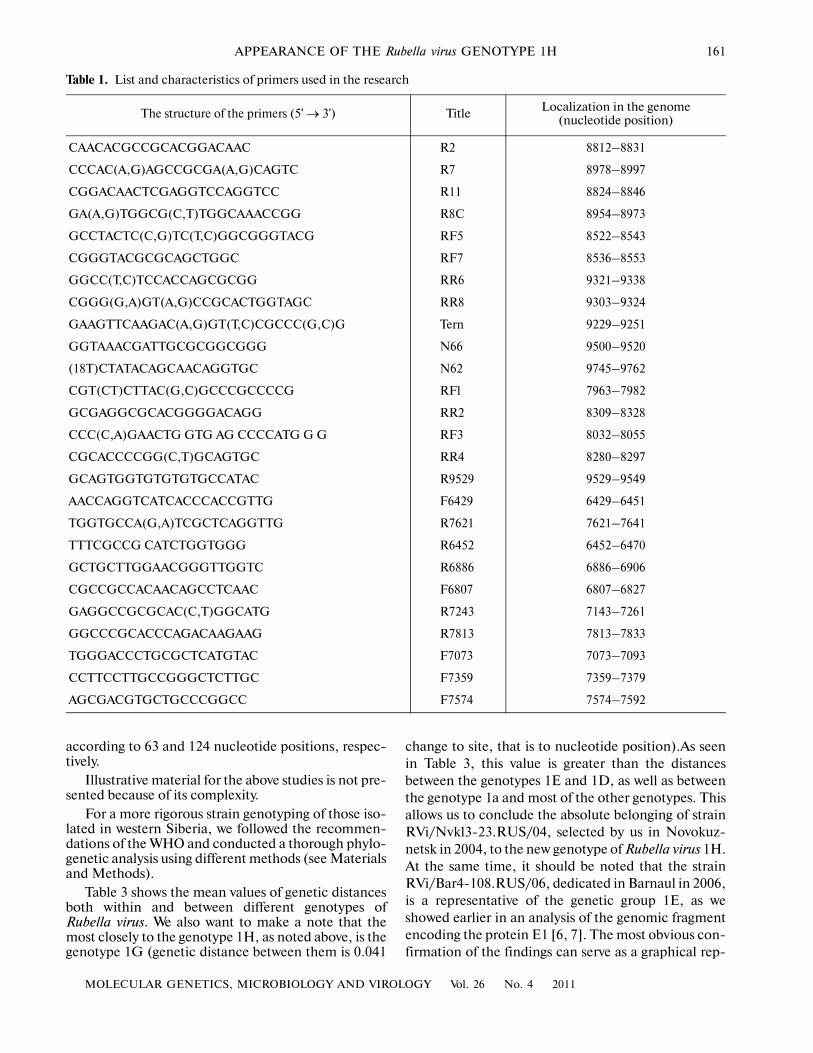
Table 1. List and characteristics of primers used in the research
The structure of the primers (5' → 3') Title Localization in the genome (nucleotide position)
CAACACGCCGCACGGACAAC R2 8812–8831
CCCAC(A,G)AGCCGCGA(A,G)CAGTC R7 8978–8997
CGGACAACTCGAGGTCCAGGTCC R11 8824–8846
GA(A,G)TGGCG(C,T)TGGCAAACCGG R8C 8954–8973
GCCTACTC(C,G)TC(T,C)GGCGGGTACG RF5 8522–8543
CGGGTACGCGCAGCTGGC RF7 8536–8553
GGCC(T,C)TCCACCAGCGCGG RR6 9321–9338
CGGG(G,A)GT(A,G)CCGCACTGGTAGC RR8 9303–9324
GAAGTTCAAGAC(A,G)GT(T,C)CGCCC(G,C)G Tern 9229–9251
GGTAAACGATTGCGCGGCGGG N66 9500–9520
(18T)CTATACAGCAACAGGTGC N62 9745–9762
CGT(CT)CTTAC(G,C)GCCCGCCCCG RFl 7963–7982
GCGAGGCGCACGGGGACAGG RR2 8309–8328
CCC(C,A)GAACTG GTG AG CCCCATG G G RF3 8032–8055
CGCACCCCGG(C,T)GCAGTGC RR4 8280–8297
GCAGTGGTGTGTGTGCCATAC R9529 9529–9549
AACCAGGTCATCACCCACCGTTG F6429 6429–6451
TGGTGCCA(G,A)TCGCTCAGGTTG R7621 7621–7641
TTTCGCCG CATCTGGTGGG R6452 6452–6470
GCTGCTTGGAACGGGTTGGTC R6886 6886–6906
CGCCGCCACAACAGCCTCAAC F6807 6807–6827
GAGGCCGCGCAC(C,T)GGCATG R7243 7143–7261
GGCCCGCACCCAGACAAGAAG R7813 7813–7833
TGGGACCCTGCGCTCATGTAC F7073 7073–7093
CCTTCCTTGCCGGGCTCTTGC F7359 7359–7379
AGCGACGTGCTGCCCGGCC F7574 7574–7592
162
MOLECULAR GENETICS, MICROBIOLOGY AND VIROLOGY Vol. 26 No. 4 2011
SEREGIN et al.
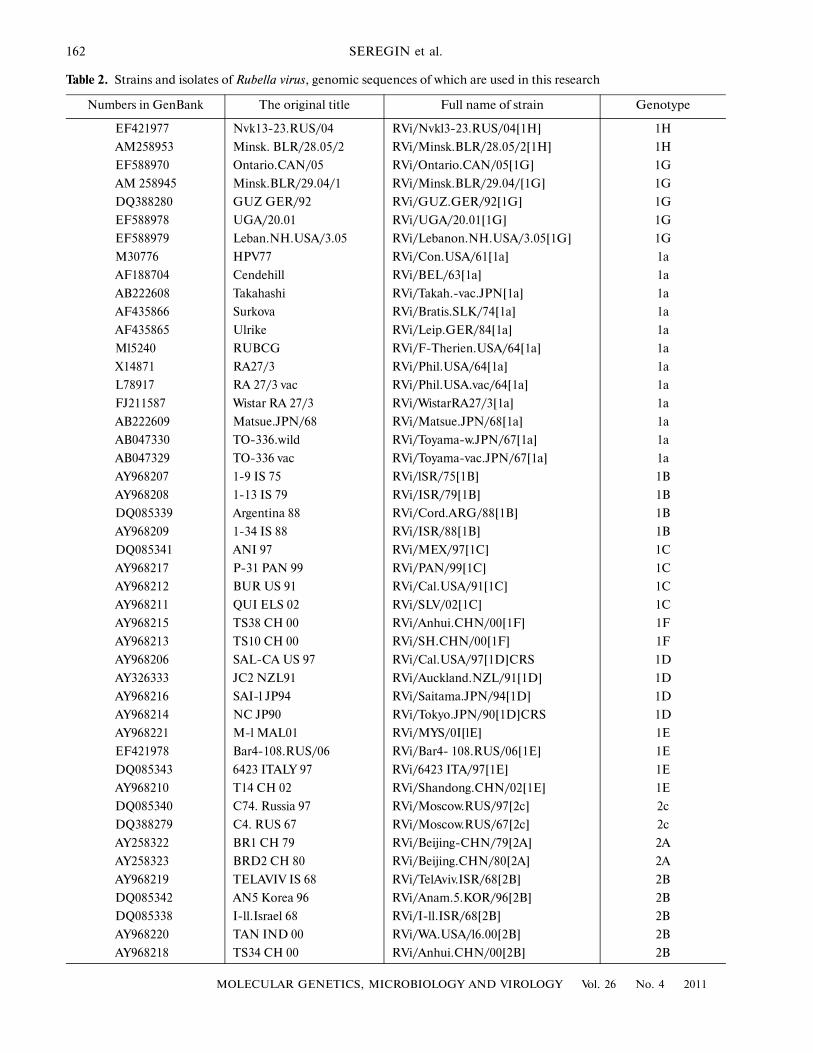
Table 2. Strains and isolates of Rubella virus, genomic sequences of which are used in this research
Numbers in GenBank The original title Full name of strain Genotype
EF421977 Nvk13�23.RUS/04 RVi/Nvkl3�23.RUS/04[1H] 1H
AM258953 Minsk. BLR/28.05/2 RVi/Minsk.BLR/28.05/2[1H] 1H
EF588970 Ontario.CAN/05 RVi/Ontario.CAN/05[1G] 1G
AM 258945 Minsk.BLR/29.04/1 RVi/Minsk.BLR/29.04/[1G] 1G
DQ388280 GUZ GER/92 RVi/GUZ.GER/92[1G] 1G
EF588978 UGA/20.01 RVi/UGA/20.01[1G] 1G
EF588979 Leban.NH.USA/3.05 RVi/Lebanon.NH.USA/3.05[1G] 1G
M30776 HPV77 RVi/Con.USA/61[1a] 1a
AF188704 Cendehill RVi/BEL/63[1a] 1a
AB222608 Takahashi RVi/Takah.�vac.JPN[1a] 1a
AF435866 Surkova RVi/Bratis.SLK/74[1a] 1a
AF435865 Ulrike RVi/Leip.GER/84[1a] 1a
Ml5240 RUBCG RVi/F�Therien.USA/64[1a] 1a
X14871 RA27/3 RVi/Phil.USA/64[1a] 1a
L78917 RA 27/3 vac RVi/Phil.USA.vac/64[1a] 1a
FJ211587 Wistar RA 27/3 RVi/WistarRA27/3[1a] 1a
AB222609 Matsue.JPN/68 RVi/Matsue.JPN/68[1a] 1a
AB047330 TO�336.wild RVi/Toyama�w.JPN/67[1a] 1a
AB047329 TO�336 vac RVi/Toyama�vac.JPN/67[1a] 1a
AY968207 1�9 IS 75 RVi/lSR/75[1B] 1B
AY968208 1�13 IS 79 RVi/ISR/79[1B] 1B
DQ085339 Argentina 88 RVi/Cord.ARG/88[1B] 1B
AY968209 1�34 IS 88 RVi/ISR/88[1B] 1B
DQ085341 ANI 97 RVi/MEX/97[1C] 1C
AY968217 P�31 PAN 99 RVi/PAN/99[1C] 1C
AY968212 BUR US 91 RVi/Cal.USA/91[1C] 1C
AY968211 QUI ELS 02 RVi/SLV/02[1C] 1C
AY968215 TS38 CH 00 RVi/Anhui.CHN/00[1F] 1F
AY968213 TS10 CH 00 RVi/SH.CHN/00[1F] 1F
AY968206 SAL�CA US 97 RVi/Cal.USA/97[1D]CRS 1D
AY326333 JC2 NZL91 RVi/Auckland.NZL/91[1D] 1D
AY968216 SAI�l JP94 RVi/Saitama.JPN/94[1D] 1D
AY968214 NC JP90 RVi/Tokyo.JPN/90[1D]CRS 1D
AY968221 M�l MAL01 RVi/MYS/0I[lE] 1E
EF421978 Bar4�108.RUS/06 RVi/Bar4� 108.RUS/06[1E] 1E
DQ085343 6423 ITALY 97 RVi/6423 ITA/97[1E] 1E
AY968210 T14 CH 02 RVi/Shandong.CHN/02[1E] 1E
DQ085340 C74. Russia 97 RVi/Moscow.RUS/97[2c] 2c
DQ388279 C4. RUS 67 RVi/Moscow.RUS/67[2c] 2c
AY258322 BR1 CH 79 RVi/Beijing�CHN/79[2A] 2A
AY258323 BRD2 CH 80 RVi/Beijing.CHN/80[2A] 2A
AY968219 TELAVIV IS 68 RVi/TelAviv.ISR/68[2B] 2B
DQ085342 AN5 Korea 96 RVi/Anam.5.KOR/96[2B] 2B
DQ085338 I�ll.Israel 68 RVi/I�ll.ISR/68[2B] 2B
AY968220 TAN IND 00 RVi/WA.USA/l6.00[2B] 2B
AY968218 TS34 CH 00 RVi/Anhui.CHN/00[2B] 2B
MOLECULAR GENETICS, MICROBIOLOGY AND VIROLOGY Vol. 26 No. 4 2011
APPEARANCE OF THE Rubella virus GENOTYPE 1H 163
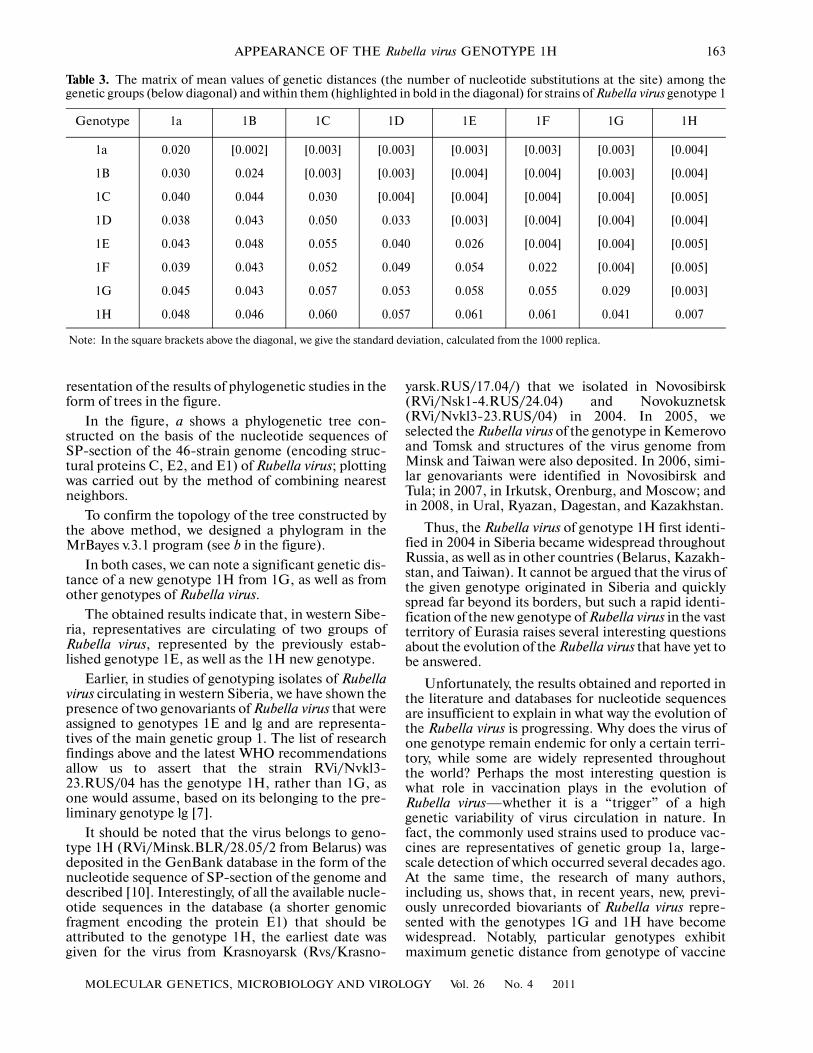
Table 3. The matrix of mean values of genetic distances (the number of nucleotide substitutions at the site) among thegenetic groups (below diagonal) and within them (highlighted in bold in the diagonal) for strains of Rubella virus genotype 1
Genotype 1a 1B 1C 1D 1E 1F 1G 1H
1a 0.020 [0.002] [0.003] [0.003] [0.003] [0.003] [0.003] [0.004]
1B 0.030 0.024 [0.003] [0.003] [0.004] [0.004] [0.003] [0.004]
1C 0.040 0.044 0.030 [0.004] [0.004] [0.004] [0.004] [0.005]
1D 0.038 0.043 0.050 0.033 [0.003] [0.004] [0.004] [0.004]
1E 0.043 0.048 0.055 0.040 0.026 [0.004] [0.004] [0.005]
1F 0.039 0.043 0.052 0.049 0.054 0.022 [0.004] [0.005]
1G 0.045 0.043 0.057 0.053 0.058 0.055 0.029 [0.003]
1H 0.048 0.046 0.060 0.057 0.061 0.061 0.041 0.007
Note: In the square brackets above the diagonal, we give the standard deviation, calculated from the 1000 replica.
resentation of the results of phylogenetic studies in theform of trees in the figure.
In the figure, a shows a phylogenetic tree con�structed on the basis of the nucleotide sequences ofSP�section of the 46�strain genome (encoding struc�tural proteins C, E2, and E1) of Rubella virus; plottingwas carried out by the method of combining nearestneighbors.
To confirm the topology of the tree constructed bythe above method, we designed a phylogram in theMrBayes v.3.1 program (see b in the figure).
In both cases, we can note a significant genetic dis�tance of a new genotype 1H from 1G, as well as fromother genotypes of Rubella virus.
The obtained results indicate that, in western Sibe�ria, representatives are circulating of two groups ofRubella virus, represented by the previously estab�lished genotype 1E, as well as the 1H new genotype.
Earlier, in studies of genotyping isolates of Rubellavirus circulating in western Siberia, we have shown thepresence of two genovariants of Rubella virus that wereassigned to genotypes 1E and lg and are representa�tives of the main genetic group 1. The list of researchfindings above and the latest WHO recommendationsallow us to assert that the strain RVi/Nvkl3�23.RUS/04 has the genotype 1H, rather than 1G, asone would assume, based on its belonging to the pre�liminary genotype lg [7].
It should be noted that the virus belongs to geno�type 1H (RVi/Minsk.BLR/28.05/2 from Belarus) wasdeposited in the GenBank database in the form of thenucleotide sequence of SP�section of the genome anddescribed [10]. Interestingly, of all the available nucle�otide sequences in the database (a shorter genomicfragment encoding the protein E1) that should beattributed to the genotype 1H, the earliest date wasgiven for the virus from Krasnoyarsk (Rvs/Krasno�
yarsk.RUS/17.04/) that we isolated in Novosibirsk(RVi/Nsk1�4.RUS/24.04) and Novokuznetsk(RVi/Nvkl3�23.RUS/04) in 2004. In 2005, weselected the Rubella virus of the genotype in Kemerovoand Tomsk and structures of the virus genome fromMinsk and Taiwan were also deposited. In 2006, simi�lar genovariants were identified in Novosibirsk andTula; in 2007, in Irkutsk, Orenburg, and Moscow; andin 2008, in Ural, Ryazan, Dagestan, and Kazakhstan.
Thus, the Rubella virus of genotype 1H first identi�fied in 2004 in Siberia became widespread throughoutRussia, as well as in other countries (Belarus, Kazakh�stan, and Taiwan). It cannot be argued that the virus ofthe given genotype originated in Siberia and quicklyspread far beyond its borders, but such a rapid identi�fication of the new genotype of Rubella virus in the vastterritory of Eurasia raises several interesting questionsabout the evolution of the Rubella virus that have yet tobe answered.
Unfortunately, the results obtained and reported inthe literature and databases for nucleotide sequencesare insufficient to explain in what way the evolution ofthe Rubella virus is progressing. Why does the virus ofone genotype remain endemic for only a certain terri�tory, while some are widely represented throughoutthe world? Perhaps the most interesting question iswhat role in vaccination plays in the evolution ofRubella virus—whether it is a “trigger” of a highgenetic variability of virus circulation in nature. Infact, the commonly used strains used to produce vac�cines are representatives of genetic group 1a, large�scale detection of which occurred several decades ago.At the same time, the research of many authors,including us, shows that, in recent years, new, previ�ously unrecorded biovariants of Rubella virus repre�sented with the genotypes 1G and 1H have becomewidespread. Notably, particular genotypes exhibitmaximum genetic distance from genotype of vaccine

164
MOLECULAR GENETICS, MICROBIOLOGY AND VIROLOGY Vol. 26 No. 4 2011
SEREGIN et al.
(a)
(b)
RV
i/M
insk
.BL
R/2
805/
2[1H
]R
vi/N
vk13
�23.
RU
S/0
4[1H
]
RV
i/L
eban
on
/NH
/US
A/3
.05[
1G]
RV
I/U
GA
/20.
01[1
G]
RV
i/G
UZ
.GE
R/9
2[1G
]R
Vi/
Min
sk.B
LR
/29.
04/1
[1G
]
RV
i/O
nta
rio
.CA
N/0
5[1G
]R
Vi/
BE
L/6
3[1a
]
RV
i/T
akah
.�va
c.JP
N[1
a]R
Vi/
Co
n.U
SA
/61[
1a]
RV
i/B
rati
s.S
LK
/74[
1a]
RV
i/L
eip.
GE
R/8
4[1a
]
RV
i/F
�Th
erie
n.U
SA
/64[
1a]
RV
i/P
hil
.US
A/6
4[1a
]R
Vi/
Ph
il.U
SA
.vac
/64[
1a]
RV
i/W
ista
rRA
27/3
[1a]
RV
i/M
atsu
e.JP
N/6
8[1a
]R
Vi/
To
yam
a�w
.JP
N/6
7[1a
]R
Vi/
To
yam
a�va
c.JP
N/6
7[1a
]R
Vi/
ISR
/75[
1B]
RV
i/IS
R/7
9[1B
]R
Vi/
Co
rd.A
RG
/88[
1B]
RV
i/IS
R/8
8[1B
]
RV
i/M
EX
/97[
1C]
RV
i/P
AN
/99[
1C]
RV
i/C
al/U
SA
/91[
1C]
RV
i/S
LV
/02[
1C]
RV
i/A
nh
ui.
CH
N/0
0[1F
]R
Vi/
SH
.CH
N/0
0[1F
]
RV
i/C
al.U
SA
/97[
1D]C
RS
RV
i/A
uck
lan
d.N
ZL
/91[
1D]
RV
i/S
aita
ma.
JPN
/94[
1D]
RV
i/T
oky
o.J
PN
/90[
1D]C
RS
RV
i/M
YS
/01[
1E]
RV
i/B
ar4�
108.
RU
S/0
6[1E
]R
Vi/
6423
IT
A/9
7[1E
]R
Vi/
Sh
and
on
g.C
HN
/02[
1E]
RV
i/M
osk
ow
.RU
S/6
7[2c
]
RV
i/B
eijin
g.C
HN
/79[
2A]
RV
i/B
eijin
g.C
HN
/80[
2A]
RV
i/I�
11.I
SR
/68[
2B]
RV
i/T
elA
viv.
ISR
/68[
2B]
RV
i/W
A.U
SA
/16.
00[2
B]
RV
i/A
nam
5.K
OR
/96[
2B]
RV
i/A
nh
ul.
CH
N/0
0[2B
]
RV
i/M
osk
ow
.RU
S/9
7[2c
]
RV
i/S
han
do
ng.
CH
N/0
2[1E
]R
vi/6
43IT
A/9
7[1E
]R
Vi/
Bar
4�10
8.R
US
/06[
1E]
RV
i/M
YS
/01[
1E]
RV
i/L
eban
on
.NH
.US
A/3
.05[
1G]
RV
i/M
insk
.BL
R/2
8.05
/2[1
H]
RV
i/O
nta
rio
.CA
N/0
5[1G
]
RV
i/B
EL
/63[
1a]
RV
i/T
akah
.�va
c.JP
N[1
a]
RV
i/C
on
.US
A/6
1[1a
]
RV
i/B
rati
s.S
LK
/74[
1a]
RV
i/L
eip
.GE
R/8
4[1a
]
RV
i/F
�Th
erie
n.U
SA
/64[
1a]
RV
i/W
ista
rRA
27/3
[1a]
RV
i/M
atsu
e.JP
N/6
8[1a
]R
Vi/
To
yam
a�w
.JP
N/6
7[1a
]
RV
i/T
oya
ma�
vac.
JPN
/67[
1a]
RV
i/IS
R/7
5[1B
]R
Vi/
ISR
/79[
1B]
RV
i/C
ord
.AR
G/8
8[1B
]R
Vi/
ISR
/88[
1B]
RV
i/M
EX
/97[
1C]
RV
i/P
AN
/99[
1C]
RV
i/C
al/U
SA
/91[
1C]
RV
i/S
LV
/02[
1C]
RV
i/A
nh
ui.
CH
N/0
0[1F
]R
Vi/
SH
.CH
N/0
0[1F
]R
Vi/
Cal
.US
A/9
7[1D
]CR
SR
Vi/
Au
ckla
nd
.NZ
L/9
1[1D
]R
Vi/
Sai
tam
a.JP
N/9
4[1D
]R
Vi/
To
kyo
.JP
N/9
0[1D
]CR
S
RV
i/N
vk13
�23.
RU
S/0
4[1H
]
RV
i/U
GA
/20.
01[1
G]
RV
i/G
UZ
.GE
R/9
2[1G
]R
Vi/
Min
sk.B
LR
/29.
04/1
[1G
]
RV
i/P
hil
.US
A.v
ac/6
4[1a
]
RV
i/P
hil
.US
A/6
4[1a
]
1H
1G
1a
1B
1C
1F
1D
1E
0.01
1H
1G
1a
1B
1C
1F
1D
1E
2c
2A 2B
0.00
5
100
94
81
95
99100
100
100
100
100
100
100
100
100
100
100
100
100
100
10010
010010
0
100
100
100
100
99
9993
100
100
99
100
100
8297 10
0 100
95 80
100
100
100 10
0
9570
99
70
100
9910
010
0
100
100
100
100
97
100
Th
e st
rain
s w
e ob
tain
ed a
nd
inve
stig
ated
are
mar
ked
wit
h b
lack
cir
cles
. On
th
e ri
ght
are
the
gen
otyp
es o
f th
e vi
rus.
Mor
e in
form
atio
n o
n t
he
subm
itte
d st
rain
s is
sh
own
in T
able
1.
Bel
ow is
giv
en a
sca
le o
f gen
etic
dis
tan
ces
(sub
stit
utio
ns
or s
ite)
. On
th
e tr
ee, w
e sh
ow t
he
valu
es o
f th
e re
liabi
lity
of n
odes
from
70%
an
d ab
ove.
Sch
eme
opti
ons
of t
he
phyl
ogra
mar
e gi
ven
in M
ater
ials
an
d M
eth
ods.
P
hyl
ogra
m c
onst
ruct
ed b
y co
mbi
nin
g th
e m
eth
od o
f nea
rest
nei
ghbo
rs a
ccor
din
g to
nuc
leo
tide
seq
uen
ces
of t
he
SP
�sec
tion
of t
he
Rub
ella
vir
us g
enom
e in
th
e M
ega
v.3.
1 pr
o�gr
am (
a) a
nd
a si
mil
ar p
hyl
ogra
m (
for
the
maj
or g
enet
ic g
roup
I),
con
stru
cted
in t
he
MrB
ayes
v.3.
1 pr
ogra
m (
b).

MOLECULAR GENETICS, MICROBIOLOGY AND VIROLOGY Vol. 26 No. 4 2011
APPEARANCE OF THE Rubella virus GENOTYPE 1H 165
strains in comparison with other types of genotypes ofgenetic group 1 (see Table 3). It is particularly interest�ing that the new genotype 1H, first identified in theSiberian region of the Russian Federation in 2004,propagated rapidly, at least in the countries of Eurasia.In our opinion, this phenomenon is a result, not somuch of natural virus evolution of Rubella virus innature, as of artificial selection of new genovariantsduring prolonged existence of barrier—that is, vacci�nation—as we mentioned above.
CONCLUSIONS
Thus, in conclusion, it should be mentioned that,at this time, it seems most relevant to study the effectof vaccination on the molecular evolution of Rubellavirus to create fundamentally new and effective vac�cines for reliable prevention of this dangerous disease.
ACKNOWLEDGMENTS
The authors are grateful to G.I. Tyunnikov (aformer employee) for kindly providing viral material.
REFERENCES
1. Zemlyanskii, O.A., Shinkarenko, N.N., and Mezent�seva, A.L., Epid. Infekts. Bol., 2004, no. 1, pp. 36–39.
2. Malkova, E.M., Petrova, I.D., Smerdova, M.A., et al.,Zh. Mikrobiol. Epidemiol. Immunobiol., 2007, no. 2,pp. 44–48.
3. Petrova, I.D., Kazaeva, E.V., Malkova, E.M., et al.,Infekts. Bol., 2007, no. 5, pp. 16–19.
4. Polyakov, V.E., Smirnova, T.N., Kazakova, S.I., et al.,Epid. Infekts. Bol., 2004, no. 1, pp. 59–61.
5. Postovit, V.A., Detskie kapel’nye infektsii u vzroslykh(Infantile Droplet infections in adults), St. Petersburg,1997.
6. Tyunnikov, G.I., Yashina, L.N., Seregin, S.V., et al., Zh.Mikrobiol. Epidemiol. Immunobiol., 2007, no. 6,pp. 26–29.
7. Yashina, L.N., Tyunnikov, G.I., Petrova, I.D., et al.,Vopr. Virusol., 2007, no. 2, pp. 16–19.
8. Caidi, H., Abernathy, E., Benjouad, A., et al., J. Clin.Virol., 2008, vol. 42, pp. 86–90.
9. Hall, T.A., Nucl. Acids Symp. Ser., 1999, vol. 41,pp. 95–98.
10. Hübschen, J.M., Yermalovich, M., Semeiko, G., et al.,J. Gen. Virol., 2007, vol. 88, pp. 1960–1966.
11. Huelsenbeck, J.P. and Ronquist, F., Bioinformatics,2001, vol. 17, pp. 754–755.
12. Katow, S., Pediatr. Int., 2004, vol. 46, pp. 207–213.13. Kimura, M., J. Mol. Evol., 1980, vol. 16, pp. 111–120.14. Kumar, S., Tamura, K., and Nei, M., Brief. Bionform.,
2004, vol. 5, pp. 150–163.15. Thompson, J.D., Gibson, T.J., Plewniak, F., et al.,
Nucleic Acids Res., 1997, vol. 24, pp. 4876–4882.16. World Health Organization. Standartization of the
Nomenclature for Genetic Characterization of Wild�Type Rubella Viruses, Wkly Epidemiol. Rec., 2005,vol. 14, pp. 126–132.
17. World Health Organization. Update of StandardNomenclature for Wild�Type Rubella Viruses, WklyEpidemiol. Rec., 2007, vol. 82, pp. 216–222.
18. Zheng, D.�P., Frey, T.K., Icenogle, J., et al., Emerg.Infect., 2003, vol. 9, pp. 1523–1530.





























