Embed Size (px)
Citation preview

Manual Teórico de Patologia do Sistema Hemolinfopoiético
1
SISTEMA HEMOLINFOPOIÉTICO – CONCEITOS GERAIS: - Elementos envolvidos na hematopoiese ( produtores e/ou destruidores dos
elementos figurados do sangue): • medula óssea (MO) e células dela derivadas - parte mielóide • timo • linfonodos parte linfóide • baço - HEMATOPOIESE:
Na vida embrionária, ilhotas sanguíneas surgem no saco amniótico ao redor da 3a semana gestacional. No 3o mês, ocorre migração destas células para o fígado, que se torna o órgão hematopoiético principal até poucas semanas que antecedem o nascimento. A MO começa a funcionar no 4o mês IU e tem todos os seus sítios funcionantes até a puberdade, com principais áreas nas medulas de osos chatos e longos (vértebras, costelas, esterno, calota craniana,pelve, epífises proximais do úmero e fêmur). Nos outros ossos, aproximadamente metade das MO tornam-se adiposas, amareladas e inativas na vida adulta.
- DIFERENCIAÇÃO DAS CÉLULAS DA LINHAGEM HEMATOPOIÉTICA:
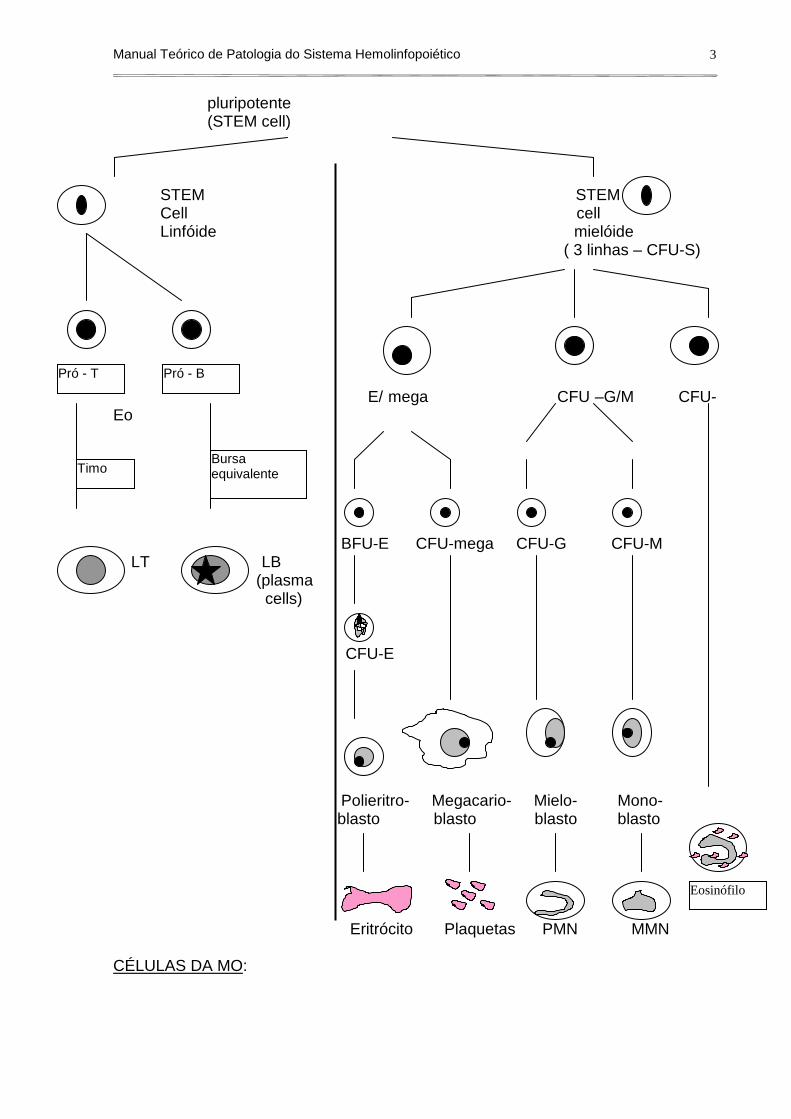
Na MO, há células em todos os estágios de diferenciação e maturação, que no entanto se originaram de um mesmo tipo celular: células pluripotentes ou STEM cell, que origina uma STEM cell de linhagem linfóide e uma STEM cell de linhagem mielóide (trilinhagem mielóide – CFU –S), ambas multipotentes. A partir daí, cada uma delas origina diversos precursores que maturam, perdem ou ganham estruturas e se tornam células maduras (diagrama 1).
- CONSTITUINTES DO SANGUE : A) Elementos Figurados: eritrócitos - leucócitos - plaquetas. - eritrócitos: glóbulos vermelhos responsáveis pelo transporte do oxigênio aos
tecidos para a produção de energia. Células lábeis com aproximadamente 120 dias de vida média normal.
- Leucócitos : células brancas responsáveis pela defesa imunológica e resposta
inflamatória celular a antígenos. Podem ser subdividios em 3 grandes grupos: granulócitos,linfócitos e monócitos de acordo com a diferenciação e maturação celular. Cada um estes grupos possui diferentes atuações, visando sempre a defesa do hospedeiro. Sendo assim : 1) granulócitos: são os polimorfonucleados (PMN), representados
specialmente pelos mielócitos (neutrófilos e segmentados). Estes executam a resposta inflamatória imediata, fazendo fagocitose de antígenos, toxinas, etc... , ativando outras células inflamatórias por quimiotaxia , a cascata do complemento e a liberação de substâncias vasoativas que complementarão a resposta de defesa humoral.

Manual Teórico de Patologia do Sistema Hemolinfopoiético
2
2) Linfócitos : são divididos em linfócitos B (bursa dependentes ) responsáveis em parte por produção de globulinas auxiliando a resposta humoral e linfócitos T (timo dependentes) produtores de linfocinas e geradores da resposta imune celular.
3) Monócitos : são macrófagos mais especializados encontrados com amior frequência na resposta imune celular.
# Toda doença da Medula Óssea implica doenças do sangue periférico. B) Plasma: fatores de coagulação. Origem: fígado. # Toda doença hepática implica deficiência da coagulação. - ANORMALIDADES: anemia - policitemia leucopenia - leucocitose trombocitopenia - trombocitose déficit dos fatores de coagulação
Diagrama 1: hematopoiese. célula
Manual Teórico de Patologia do Sistema Hemolinfopoiético
3
pluripotente (STEM cell) STEM STEM Cell cell Linfóide mielóide ( 3 linhas – CFU-S)
E/ mega CFU –G/M CFU-Eo BFU-E CFU-mega CFU-G CFU-M LT LB (plasma cells) CFU-E Polieritro- Megacario- Mielo- Mono-
blasto blasto blasto blasto Eritrócito Plaquetas PMN MMN CÉLULAS DA MO:
Timo
Pró - T Pró - B
Bursa equivalente
Eosinófilo

Manual Teórico de Patologia do Sistema Hemolinfopoiético
4
1) pronormoblastos, mielobastos e monoblastos: 10 a 20 micra de diâmetro, com citoplasma abundante e marcadamente basófilo e núcleos redondos com cromatina homogênea e nucléolos visíveis
2) Os demais tipos celulares são de difícil separação antes da maturação PROPORÇÕES DOS ELEMENTOS DA MO: • granulócitos : 60% • precurssores eritróides: 20% • linfócitos, monócitos e precurssores: 10% • células não identificáveis ou em desintegração: 10% • Taxa mielóide/eritróide = 3/1 • Predominância mielóide: mielócitos, metamielócitos granulócitos • Predominância eritróide: normoblastos policromatofílicos e ortocromáticos. MO – MORFOLOGIA NORMAL: • sinusóides justapostos separados por camada de tecido endotelial, com média e
adventícia intermitentes • Entre os capilares: células adiposas (1:1 no adulto normal) e hematopioéticas em
diferentes estágios de maturação • Células se movem por migração transcelular
ANEMIAS

Manual Teórico de Patologia do Sistema Hemolinfopoiético
5
- MANIFESTAÇÕES CLÍNICAS: Decorrem da queda do O2 tissular. Pode ser por falha na: 1. Captação de O2: Tensão do O2 atmosférico Função pulmonar Concentração de Hb Afinidade da Hb pelo O2 2. Liberação de O2: Débito cardíaco Afinidade da Hb pelo O2 Concentração de Hb - MECANISMOS DE DEFESA: Somente após a "derrota" destes mecanismos que aparecem os sintomas. Aumento do débito cardíaco 3-4 vezes facilitado pela queda da viscosidade; Aumento do 2-3 difosfoglicerato (2.3 DPG) - faz com que diminua a afinidade da Hb pelo O2, portanto aumenta a liberação de O2 tissular. Hipóxia renal - > aumento da Eritropoetina - > estímulo para medula óssea Diminuição do esforço físico (questiona-se ser um mecanismo voluntário) - MANIFESTAÇÕES DA ANEMIA:. Variam conforme o grau de Hb: A. 9 - 12 g % anemia discreta mecanismos compensadores - > ausência de sintomas exercícios intensos - > sintomas palpitações - dispnéia - sudorese excessiva B. 7 - 10 g % anemia moderada sintomas mais evidentes fadiga fácil sonolência (como mecanismo compensador para poupar esforço) C. 3 - 7 g % anemia severa astenia hipotensão ortostática insônia e cefaléia (por irritação das células nervosas pela hipóxia) anorexia ICC * Tais sintomas dependem também do tipo da anemia e da sua forma de instalação. - CLASSIFICAÇÃO ETIOLÓGICA DAS ANEMIAS: I- Produção deficiente de hemácias: A. Deficiência de nutrientes essenciais

Manual Teórico de Patologia do Sistema Hemolinfopoiético
6
ferro ác. fólico vitamina B12 proteínas B. Hipoplasia eritroblástica 1. Anemias aplásticas hereditárias idiopáticas induzidas por agentes químicos e físicos 2. Hipoplasia eritroblástica pura: timoma agentes químicos antagonistas ác. fólico anticorpos hereditários C. Infiltração Medula Óssea: leucemias e linfomas mieloma múltiplo carcinomas e sarcomas mielofibrose D. Anemia Sideroblástica E. Endocrinopatias F. Insuficiência renal crônica (IRC) G. Doenças Inflamatórias Crônicas H. Hepatopatias II- Destruição excessiva de hemácias: A. Fatores Extracorpusculares anticorpos agentes físicos e químicos traumatismo eritrocitário B. Adquiridos Hemoglobinúria paroxística noturna III - Perda aguda de sangue A. Anemia pós-hemorrágica - HEMOGRAMA x ÍNDICES NORMAIS: VGM ou VCM = (VG x 10)/ E ____90 + - 7 fL HGM ou HCM = (Hb x 10)/ E ____29 + - 2 pg

Manual Teórico de Patologia do Sistema Hemolinfopoiético
7
CHCM = (Hb x 100)/VG ________34 + - 2 g/dL * nunca a HCM passará a 1/3 do VCM, pois é o máximo que um eritrócito comporta de Hb. - CLASSIFICAÇÃO MORFOLÓGICA DAS ANEMIAS: I - Anemia Macrocítica: VCM: 97 - 160 fL HCM: 32 - 50 pg CHCM: 32 - 36 g/dL A. Megaloblásticas: deficiência de B12 - folato síntese deficiente de DNAs (hereditária ou por drogas) B. Não-megaloblásticas: eritropoiese acelerada miscelanias: hepatopatias / hipoplasias / etc... II - Anemia Hipocrômica Microcítica (AHM): VCM: 50 - 80 fL HCM: 12 - 27 pg CHCM: 24 - 32 g/dL depleção de ferro (a grande maioria da AHM) talassemia minor anemias sideroblásticas doenças crônicas III - Anemia Normocrômica Normocítica: VCM: 83 - 97 fL HCM: 27 - 31 pg CHCM: 32 - 36 g/dL pós-hemorrágica hemolíticas hipoplasia medular doenças crônicas renal crônico endocrinopatias excesso de volume plasmático - OUTRAS ALTERAÇÕES NAS HEMÁCIAS: anisocitose poiquilocitose esferócitos - células em alvo eliptócitos acantócitos - estomatócitos fragmentos eritrocitários células em gota policromatofilia

Manual Teórico de Patologia do Sistema Hemolinfopoiético
8
basofilia ponteado basófilo - TRATAMENTO: * Anemia não é diagnóstico, mas manifestação! * Procurar defeito básico. * Transfusão de sangue = RISCOS. ANEMIAS FERROPRIVAS – INTRODUÇÃO
A hemoglobina é f ormada por 3 partes essenciais: 1. Anel Porfirínico (tetrapirrólico) ou Protoporfirina IX 2.Um átomo de Ferro, responsável pelo transporte do O2. 3.Uma cadeia de Globina, formada por várias cadeias de amino-ácidos. Então: Hemoglobina = P IX + Fe + Globina. * O átomo de Ferro é sempre divalente. Quando trivalente chama-se MetaHemoglobina e não tem capacidade de transportar O2. - Distúrbios do Metabolismo do Ferro * Ferro Total Mulheres: + - 35 mg/kg Homens: + - 50 mg/kg De 3 a 5 gramas. * Ferro na Hemoglobina 2/3 do total (60 a 70%) De 1,5 a 3 gramas. * Ferro de Depósito ou Tubulares Estoque disponível: 0,6 a 1,5 g sob a forma de ferritina e hemossiderose. Essencial não - disponível: mioglobina, enzimas (citocromos, penicilases, etc). * Ferro Plasmático 80 a 150 ug/dL. 3 a 4 mg ligado a transferrina ou siderofilina. * Siderofilina 200 a 400 ug/dL. Aproximadamente 8 gramas. * Índice de Saturação (IS) É o índice de saturação da transferrina. Utiliza os valores: Ferro Plasmático: 80 - 150 ug

Manual Teórico de Patologia do Sistema Hemolinfopoiético
9
Transferrina: 200 - 400 ug. Reflete que existe uma grande parte da transferrina livre do Ferro. Portanto, o IS é normal de 25 - 35%. Se IS < 20, Distúrbio no metabolismo do Ferro; Se IS < 10, Anemia Ferropriva. - Absorção do ferro: Duodeno e porções altas do delgado 5 a 10% ou 0,5 a 2,0 mg/dia - Excreção do ferro: 1 mg/dia - suor, urina, descamação pele e intestino. 2 mg/dia - na perda menstrual - Déficit por utilização de ferro: Infecções Doenças Crônicas Anemias Sideroblásticas - Necessidades aumentadas de ferro: Crianças em desenvolvimento Mulheres em período fértil Grávidas ANEMIA FERROPRIVA- ASPECTOS GERAIS : Tipo mais comum de anemia no mundo todo. Patogênese: qualquer anormalidade que leve à rotura do equilíbrio do metabolismo do ferro. Duas fases distintas: Depleção de Ferro e Anemia - Causas fundamentais de anemia ferropriva: Aumento da demanda fisiológica Perda de sangue (no nosso meio - ancilostomose) Ingestão deficiente - Causas mais importantes: Mulheres em período fértil menstruação gestação perdas patológicas dieta defeituosa Homens e Mulheres antes da menarca e após a menopausa Perda crônica de sangue Crianças Dieta pobre

Manual Teórico de Patologia do Sistema Hemolinfopoiético
10
Ferro diminuído ao nascimento * A Anemia erropriva demora de 6 a 8 meses para se instalar e necessita de mais 6 meses para se recuperar com o tratamento. 1. Principais Causas de Perda Crônica de Sangue Hemorróidas Úlcera Péptica Hérnia de Hiato Ca de Estômago Ca de Cólon Varizes de Esôfago Retocolite Ulcerativa Ingestão Crônica de Aspirina Ancilostomose 2. Principais causas de necessidades aumentadas de ferro Crianças em Crescimento Mulher no Período Fértil 3. Ingestão Inadequada de ferro 4. Principais causas de absorção diminuída de ferro Síndrome de Má-absorção Gastrectomia CLÍNICA DA ANEMIA FERROPRIVA: 1. Decorrente da Anemia - Início Insidioso - Palpitações - Lassidão - Angina - Astenia - ICC e hepatomegalia - Dispnéia - Palidez de pele e mucosas Esplenomegalia (apenas em 10% dos pacientes. É uma tentativa do organismo em aumentar órgãos produtores de sangue através de uma metaplasia mielóide do baço) 2. Decorrente de lesões nos tecidos (turn over celular comprometido pela falta de ferro) Unhas finas e quebradiças Glossite e Leucoplasia Estomatite angular Cabelos finos e quebradiços Disfagia LABORATÓRIO NA ANEMIA FERROPRIVA:

Manual Teórico de Patologia do Sistema Hemolinfopoiético
11
Hemograma Anemia HIPOcrômica MICROcítica ou nas fases iniciais NORMO - NORMO. CHbCM < 32 % VGM < 83 fl HCM < 27 pg Plaquetas aumentadas (por estímulo à MO para aumentar a produção) Ferro sérico e Capacidade de transporte IS < 10% Medula Óssea Análise de Ferro em depósitos Depósitos em hemossiderina diminuídos Sideroblastos ausentes ou diminuídos DIAGNÓSTICO DIFERENCIAL DA ANEMIA FERROPRIVA: Anemia das doenças crônicas Talassemia minor Anemia Sideroblástica TRATAMENTO DA ANEMIA FERROPRIVA Eliminar a causa Ferro: reposição oral com Sulfato Ferroso. Há necessidade de tratar com 120 - 200 mg de Ferro elementar. Sulfato Ferroso tem apenas 20% de Ferro. Corrigir com regra de três. Portanto, para 120 - 200 mg de Ferro deve-se ministrar de 600 - 1000mg de Sulfato Ferroso. ANEMIAS MACROCÍTICAS Anemia Megaloblástica
• Anemia Macrocítica: VGM > 97 fL • * Deficiência: B12 e/ou ácido fólico (o ácido fólico só existe na forma de
folatos) B12:
• É essencial como coenzima de reações na síntese de ác. nucléicos.
(principalmente DNA) • O nosso requerimento diário: 1 - 2,5 microgramas • Fator Intrínseco (é necessário para que haja absorção de B12 no estômago) • A B12 é absorvida no íleo terminal, após se ligar aos receptores no
estômago. Ocorre através da transcobalamina II (transportedora). E é depositada nos músculos e fígado principalmente através da transcobalamina I. Que é a forma de depósito
• B12 sérica: 200-900 pg/ml. Sua dosagem não existe pela bioquímica. Somente através de radioimunoensaio ou imunobiolóligo o que encarece muito.

Manual Teórico de Patologia do Sistema Hemolinfopoiético
12
• Estoques: 5000 microgramas (2000 - 10000). Só no fígado: aproximadamente 1700 microgramas. Devido a estes estoques a anemia demora de 4 a 5 anos para surgir. Ainda atrasa mais devido a circulação entero-hepática
Ácido Fólico:
• É importantíssimo para a síntese dos ácidos nucléicos (principalmente DNA) • Necessidade diária: 50 - 100 microgramas. • Absorção: duodeno e jejuno (porções distais) • Sérico: 5 - 20 mg/100ml. A dosagem é tão onerosa quanto a da B12. • Depósitos: 5 a 10 mg (miligramas). Esgotam em 8 meses.
Os deficientes em B12 são os vegetarianos estritos. Existe uma importante interação entre B12 e Ácido F ólico:
• O metabolismo do folato necessita de B12. Isto reflete histologicamente uma hemácia com citoplasma maduro e núcleo imaturo = MEGALOBLASTO.
• A Medula Óssea do anêmico megaloblástico é hipercelular. Só que devido ao defeito as hemácias são destruídas na própria medula, o que acarreta num aumento de bilirrubinas.
Hemácias, Leucócitos e Plaquetas além de todas as outras células de tecidos que tenham grande capacidade de divisão celular estarão afetadas na deficiência de B12. Por isso a Anemia Megaloblástica reflete - ANEMIA, LEUCOPENIA e PLAQUETOPENIA . A deficiência de B12, através da Adenosil colamatase pode acarretar em sintomatologia neurológica, até mesmo à "Loucura Megaloblástica" - que pode ser reversível tratando esta deficiência de B12. Deficiência de B12: A- Aporte diminuído de B12:
• Ingestão deficiente • Absorção alterada: Anemia Perniciosa (ver adiante) • Competição (Síndrome de Alça Cega; Doença Diverticular; Tênia Européia)
B- Necessidades aumentadas de B12: • Neoplasias (grande destruição celular) • Hipertireoidismo (hipermetabolismo)
C- Utilização prejudicada de B12: • Deficiências enzimáticas; • Transporte TC II (falta ou anormal) • Administração de NO2 (óxido nitroso)
Deficiência de Ácido Fólico: A- Aporte deficiente de ácido fólico:
• Dieta • Alcoolismo • Hemodiálise • Absorção alterada
B- Requerimentos Aumentados de ácido fólico: • Gestação (múltiplas principalmente) • Crianças em crescimento • Hipertireoidismo

Manual Teórico de Patologia do Sistema Hemolinfopoiético
13
• Hematopoiese acelerada • Neoplasias • Dermatite expoliativa
C- Utilização prejudicada de ácido fólico: • Drogas anti-neoplásicas (Metrotrexate - é um anti-fólico) • Triamtereno (muito pouco usado) • Trimetropin • Deficiências enzimáticas
Aos pacientes deficientes de folato usa-se ácido fólico por 30 dias. Não respondem ao uso de B12 e Ácido Fólico:
• Inibidores Metabólicos - Drogas anti-neoplásicas
• Erros Inatos - Sd de Lesch-Nyham
- Oroticoaciduria hereditaria - Deficiência enzimática
• Inexplicáveis - Piridina
- Tiamina - Eritroleucemia
ANEMIA PERNICIOSA
• Toda anemia perniciosa é uma anemia megaloblástica. • Hereditária • Fator intrínseco deficiente (ou Anticorpos contra células parietais ou Ac contra
fator intrínseco) • Alterações neurológicas • Diagnóstico: MO // Schilling ( administrar vitamina B12 marcada VO e analisar
urina) • Pelo teste de Schilinng descobrimos que não adianta para A. perniciosa
ministrar apenas vit B12 tem que dar concomitante fator intrínseco. Só que este é muito caro. Portanto ministra-se B12 que não necessite de fator intrínseco para ser absorvida, ou seja, que não por via oral. A via escolhido é parenteral (IM). Inicia com 1 ampola/dia por 1 semana e depois 1 ampola a cada 3 meses ad eternum.
ANEMIAS HEMOLÍTICAS

Manual Teórico de Patologia do Sistema Hemolinfopoiético
14
Definição: são doenças resultantes de um aumento no índice de destruição eritrocitária, que leva à diminuição da vida média das hemácias (normalmente de 120 dias). A parte metabolizada da hemácia corresponde ao anel porfirínico que se torna bilirrubina após passagem pelo fígado, que é excretada em forma de estercobilinogênio � estercobilina nas fezes ou urobilinogênio � urobilina na urina. Os demais componentes destas células (globulina, heme etc) são reaproveitados. - DEFEITOS FUNDAMENTAIS: 1) Ïntrínsecos (ou intracorpusculares): as células são defeituosas; o defeito está na
própria célula. Portanto células normais sobrevivem nos vasos destes pacientes, mas células destes pacientes são destruídas, uma vez que são reconhecidas como defeituosas.
2) Extrínsecos (extracorpuscular): O defeito está no plasmaou nos vasos; as hemácias do paciente são normais.
CLASSIFICAÇÃO : 1) anemia hemolítica por defeito intracorpuscular (todas as doenças congênitas):
a) defeitos na membrana: - esferocitose hereditária ( + comum) - estomatocitose - eliptocitose
a) eritroenzimopatias (qualquer das enzimas do ciclo da glicose):
- defeitos no ciclo de Embden-Meyerhoff ( piruvato quinase (PK ), fosfofrutoquinase (PFK), 6 DGM, etc...)
- defeitos na via hexose-monofosfato ( G6PD)
a) hemoglobinopatias ( hemoglobinas anormais): +- 600 tipos de Hb descritas (normais) - Hb com agregação anormal : S (anemia falciforme) , C, D... - Hb instável com corpúsculos de Heins (deposição de Hb dentro da
hemácia visível com coloração em MO) : Hb Zurich-Köhn - Hb com afinidade anormal pelo O2
a) Talassemias ( Hb normal mas presente em época anormal) e)Hemoglobinúria paroxística norturna (HPN): defeito intracorpuscular com manifestação tardia de urina cor de coca-cola, em surtos e geralmente após o sono. É a única anemia hemolítica intracelular adquirida.
1) anemia hemolítica por defeito extracorpuscular:
a) com Ac: - isoimune (ABO, Rh) - auto-imunes (quente, fria, drogas)

Manual Teórico de Patologia do Sistema Hemolinfopoiético
15
a) sem Ac: - Ação direta de tóxicos (arsina – A3H3) - Toxinas bacterianas (sépsis por G- (Clostridium) ou G+) - Parasitas: malária
a) microangiopáticas: - eclâmpsia - próteses cardíacas (turbilhonamento) - HAS maligna - PTT (púrpura trombocitopênica trombótica)
1) Interação das anormalidades:
a) favismo (células com anormalidades morfológicas –feijão fava - + deficiência de G6PD)
b) enzimas + drogas (cloroquina p/ tto. Malária + defeito enzimático) c) Hb instáveis + drogas ou infecções d) Chumbo
ESFEROCITOSE HEREDITÁRIA • Fisiopatologia: 30 a 60% das vezes a alteração é de proteínas do citoesqueleto
como espectrina actina, anquirina. Em 5 a 25% dos casos a alteração é da proteína 3 de sustentação. A hemácia tem estas proteínas para conferir maleabilidade e permitir a sua passagem para a microcirculação. Quando há defeito nestes sítios, a hemácia fica “presa” na microcirculação esplênica onde é então destruída antes do tempo previsto, gerando o quadro anêmico hemolítico (sequestração esplênica).
• Clínica:
- anemia + icterícia ( 1ºs anos)
ANEMIA HEMOLÍTICA x DOENÇA HEMOLÍTICA:
HEMÓLISE NEM SEMPRE É ANEMIA HEMOLÍTICA!!! Só se pode considerar um
doente com anemia hemolítica a partir do momento que sua capacidedade de regeneração celular medular (6 a 8x) for suplantada pela destruição das células
circulantes. Se há’ crise de hemólise mas a medula consegue regenerar as células perdidas, diz-se que o paciente tem DOENÇA HEMOLÍTICA. No entanto, tanto na
doença como na anemia hemolítica, a MEDULA É HIPERFUNCIONANTE

Manual Teórico de Patologia do Sistema Hemolinfopoiético
16
- esplenomegalia - crises hemolíticas (febre etc...) - infecções sem causa aparente - colecistopatia calculosa (50% dos casos) - úlceras de perna
• sangue e laboratório: - anemia + esferocitose - reticulocitose (10 a 20%) - aumento de bilirrubina indireta - fragilidade osmótica dos eritrócitos (membrana frágil evidenciando
defeito; alteração da turgescência celular, com rompimento da célula em soluções próximas da isotônica)
- dosagem de espectrina, anquirina, proteína 3 • Tratamento:
- esplenectomia, que irá corrigir o efeito deletério (mecanismo hemolítico), mas não a causa da doença ( se fizer hemograma PO, ainda haverá esferócitos, porém paciente é assintomático e vive bem). O paciente contiua doente mas não mais anêmico.
- deve-se aguardar nos casos infantis até 10-12 anos de idade para a realização do procedimento pelo risco de sépsis pós-esplenectomia por Haemoophilus (causa de morte PO esplenectomia comum em 1a e 2a infância)
- graus variáveis : dominante (<) ; recessivo (>); leve ou discreta a moderada.
ANEMIA FALCIFORME • 1ª doença molecular descrita • ocorre pela troca de um ácido glutâmico por valina na cadeia beta, o que
confere menor solubilidade, ciclização e enfoiçamento em baixas tensões de O2, formando hemoglobina S ( HbS).
• Heterozigotos: 9% dos negros americanos; 20% população africana
- HbS = 25 a 40% - HbA = 60 a 75%
- Clínica ( geralmente em vigência de baixas concentrações de O2)
- hematúria / dor lombar - infarto esplênico - resistência relativa ao P. falciparum - aos esforços estenuantes : IAM
• Homozigotos: 0.3 a 1.3% dos negros americanos - HbS > 45%
Obs: enfoiçamento in vivo; anemia hemolítica crônica (após 6 meses); fenômenos tromboembólicos; expectativa de vida reduzida.

Manual Teórico de Patologia do Sistema Hemolinfopoiético
17
- Clínica:
- Desde a infância - Rara nos 1ºs 6 meses - S/S de anemia - Crises de dor óssea ( dolantina) - Crises de dor abdominal ( abdome agudo não cirúrgico) - Febre - Coma - Litíase vesicular - Osteomielite por Samonella - Úlceras de perna
- baço: esplenomegalia na criança e nas associações (talassemias,
Hb C, D, E) � infartos esplênicos de repetição � autoesplenectomia � atrofia progressiva � adulto com baço ausente ou botão esplênico fibroso
- sangue: Hb entre 6 e 9 g% com anisopoiquilocitose intensa e MO
com quadro hiperplásico
- Dx: enfoiçamento das hemácias observado ao hemograma + eletroforese de proteínas (HbS)
- Prognóstico: reservado
- TTO: paliativo : TMO ( só se o homozigoto tiver irmão heterozigoto
p/ a patologia; em estudo)
TALASSEMIAS São doenças hereditárias caracterizadas pela alteração da produção de cadeias de globulinas alfa e beta, partes formadoras da hemoglobina. É uma anemia hipoproliferativa, hemolítica e com deficiência de hemoglobina circulante. Cursa com:
- microcitose não compatível com o grau de anemia - história familiar positiva para talassemias ou HMP com longa
história de anemia microcítica - morfologia celular alterada com: microcitose, acantocitose, células
alvo (dianócitos) - Níveis elevados de HbA2 ( talassemias alfa) ou F(talassemias beta)
Deve-se observar que no adulto normal, aproximadamente 98% da Hb circulante é do tipo A (2 cadeias alfa e 2 cadeias beta), com 1 a 2% de HbF ou A2

Manual Teórico de Patologia do Sistema Hemolinfopoiético
18
(2 cadeias alfa e 2 cadeias gama). Quando há alteração nestas proporções, diagnosticam-se as talassemias, que podem ser alfa ou beta, de acordo com a cadeia alterada. a) talassemias alfa: ocorrem por deleção no cromosomo 16, com consequente
diminuição de produção de cadeias alfa
Epidemiologia: - especialmente em povos do SE da Ásia, China e em negros
Quadro clínico: - o paciente normal ou portador silencioso não exibe clínica ou alterações laboratoriais significativas a) talassemias beta: mutações pontuais no cromossomo 11 que resultam em
diminuição (beta +) ou ausência (beta 0) de expressão dos genes para a formação das cadeias beta (erros na transcrição de DNA). O excesso de cadeias alfa resultante levará estas cadeias a uma ligação isntável, precipitação e diminuição da efeitvidade da MO (hemólise intramedular).
Epidemiologia:
- especialmente povos mediterrâneos (Itália, Grécia), seguidos por asiáticos e negros
Quadro Clínico:
- anemia severa no 1º ano de vida com início de sintomatologia aos 6 meses (transição HbF� HbA). S/S : palidez cutâneomucosa, hepatoesplenomegalia, icterícia, deformidades ósseas e fraturas patológicas (osteopenia por tentativa compensatória da MO que se torna hiperplásica mas inefetiva pois produz células defeituosas = aumento do éritron). Este quadro pode ser amenizado com transfusões sanguíneas, que no entanto resultarão em hemocromatose e hemossiderose e deposição em tecidos nobres como fígado e coração, que levarão a óbito por insuficiência hepática ou ICC ao redor dos 20 ou 30 anos.
- Laboratório:
• anemia microcítica e hipocrômica • Ht de 10% ou menos se não transfundido • poiquilocitose, células alvo, basofilia, hemacias nucleadas • eletroforese de proteínas: HbA <<< ou 0; HbA2 ( quantidade variável); HbF >>> • alteração da função hepática (TGO, TGP, fosfatase alcalina, TAP, albumina
sérica) – fase tardia com infiltração sideroblástica no órgão.
ANEMIAS MEDIADAS POR ANTICORPOS (AC)

Manual Teórico de Patologia do Sistema Hemolinfopoiético
19
Representa a mais importante ramificação dentro das anemias hemolíticas e ocorre em indivíduos previamente hígidos. Tipos de AC:
- aloanticorpos : adquiridos em transfusões ou gestação com contato sangue mãe – feto (são os chamados isoAC).
- Autoanticorpos : podem ser : # quentes : manifestam-se em temperatura >37ºC, com prova de Coombs direta +, IgG não fixando complemento, especificidade anti Rh, resultando em hemólise no baço (portanto hemoglobinemia e hemoglobinúria difíceis de ocorrer). Gera anemia menos severa. # Frios: manifestam-se em temperaturas <20ºC, com anemia variável, fenômenos de Raynaud, IgG fixando complemento (hemólise intravascular = hemoglobinemia e hemoglobinúria), crioaglutininas agudas ou crônicas.
ANEMIA HEMOLÍTICA AUTO-IMUNE: Pode ser:
- primária ou ididopática - secundária a:
• linfomas • Leucemia linfocítica crônica • Lupus eritematoso disseminado • Tumores (pulmão e rim) • Drogas ( alfa metildopa, penicilinas, quinidina) Etiopatogenia:
- modificação dos antígenos (Ag) eritrocitários (não provada no homem)
- Ac com reação cruzada (ex: crioAc da pneumonite por Micoplasma) - Insuficiência de controle de autoimunização - Genética (em estudo)
Quadro Clínico:
- anemia hemolítica - esplenomegalia discreta a moderada - 100% das vezes é doença adquirida
Laboratório: • anemia discreta a moderada • esferócitos antes inexistentes • provas de Coombs direta + • doença de base (marcadores tumorais, FAN, célula LE, VHS etc...) Tto:
- corticóides - aquecimetno (nos casos de AC a frio) - esplenectomia - imunosupressores - TMO (para casos refratários aos ttos. Acima)

Manual Teórico de Patologia do Sistema Hemolinfopoiético
20
HEMOGLOBINÚRIA PAROXÍSTICA NOTURNA É a única das anemias hemolíticas com manifestações clínicas tardias. Decorre de sensibilização alterada ao complemento (fração C3b), que geralmente tem sua atividade aumentada durante as horas de repouso (noite) pela discreta acidose respiratória fisiológica decorrente do sono. A hemólise pode ocorrer continuamente ou em crises, mas sempre após o sono. Quadro clínico + laboratório:
- hemoglobinemia (hemólise intravascular) - hemoglobinúria e hemosidenúria - pancitopenia - teste da sacarose + - teste de HAM (soroácido) +
Patogenia:
- sensibilidade ao C3b do complemento - diminuição da acetilcolinesterase - células jovens (mais sensíveis à lise pelo C)
Tto:
- não há. - Pode-se fazer transfusão com papa de hemácias lavadas (para
diminuir a quantidade de plasma, e consequentemente C,aderida às células)
Prognóstico:
- leucoses agudas - anemias aplásticas
PANCITOPENIAS Definição: Diminuição de todos os elementos figurados Causas: A) DROGAS mecanicamente reversível mecanicamente irreversivel relacionado a dose ou fator imunológico B) DOENÇAS ENVEOLVENDO A MO leucemias tumores mieloma

Manual Teórico de Patologia do Sistema Hemolinfopoiético
21
linfoma aplasia medular mielodisplasias C)DOENÇAS ENVOLVENDO BAÇO linfomas esplenomegalias congestivas infiltração neoplásica calazar tb D) DEFICIÊNCIA DE FATORES ESSENCIAIS anemia megaloblástica E) HEMOGLOBINÚRIA PAROXÍSTICA NOTURNA doença clonal da célula tronco hematopoiética Mecanismo: 1) hematopoiese ineficaz com morte da medula óssea ( anemia megaloblástica ) 2) formação de células defeituosas removidas rapidametne da circulação 3) mecanismo Ag-Ac com destruição celular 4) sequestro de células normais em qeu SREhipertrofiado é ativo ANEMIA APLÁSTICA Ococrre em adultos jovens dos 15 aos 30 anos. Não há destruição total da MO e sim substituição de grande parte por tecido gorduroso. Fisiopatologia: Insuficiência da MO na produição de : leucócitos, eritrócitos, plaquetas devido a : célula tronco incapaz de repopular a medula ambiente medular impróprio para a produção celular - Causas: idiopática: 50 % dos casos hereditária ( Sd de Fanconi- alterações hematopoiéticas, renais, ósseas ) agentes físicos e químicos - Agentes físicos e químicos : A) QUE PRODUZEM APLASIA DEPENDENDO DA DOSE E TEMPO : radiaçao benzeno e derivados citostáticos arsênico - ouro B) QUE OCASIONALMENTE PRODUZEM APLASIA: definitivamente tóxicos ( cloranfenicol, butazona, antitireoideanos ) potencialmente tóxicos

Manual Teórico de Patologia do Sistema Hemolinfopoiético
22
- Clínica: Dça insidiosa na maior parte das vezes Febre, hemorragia, infecções de repetição Se ao Exame físico: baço palpável, até que se prove o contráio, não é anemia aplástica. Sangue: pancitopenia , ferro elevado ( não há produção e o indivíduo continua ingerindo ferro ) Biópsia de MO : Não usar só aspirado - Tratamento: medicamentoso , transplante ( TMO ). Politransfundidos ( mais de 15 transfusões ): TMO não tem bons resultados. AGRANULOCITOSE: - Causas: mesmos agentes da pancitopenia asiniopirina, fenilbutazona, tiouracil, tapazol. - Patogenia: Imunológico - Sintomatologia : início brusco febre, calafrios, angionecrotizante palidez de pele ( não anemia ) lesões ulceradas de mucosas duração de 3 a 9 dias fatal nos 1os. dias - Diagnóstico: hemograma M.O - Tratamento: tentar manter vivo para recuperação isolametno reverso antibiótico.
POLICITEMIA:
Também chamada eritrocitose, corresponde a um aumento no número de eritrócitos, com aumento corresponente de Hb. Este aumento pode ser:

Manual Teórico de Patologia do Sistema Hemolinfopoiético
23
- relativo: decorrente de hemoconcentração (desidratação, vômitos contínuos, diarréias, uso excessivo de diuréticos, stress (Sd. De Gaisböck)).
- absoluto: aumento na massa total de eritrócitos, que pode ser primário (defeito intrínseco da stem cell – policitemia vera, considerada um distúrbio mieloproliferativo) ou secundário a níveis aumentados de eritropoetina (altitudes, cardiopatias cianóticas, doenças pulmonares, tumores (secreção inapropriada) –renais, hepáticos, cerebelares (hemangioblastoma)).
Tto: eliminar a causa base.
COAGULOPATIAS Geralmente resultam de: 1) maior fragilidade da parede dos vasos; 2)alterações plaquetárias; 3) alteração dos fatores e/ou mecanismos de coagulação (maioria produzida no fígado, portanto associar com hepatopatias) ou 4) combinação destes. 1) ALTERAÇÃO DAS PAREDES VASCULARES Também chamadas de púrpuras não trombocitopênicas, são geralmente comuns.Podem levar à formação de petéquias na pele ou mucosas ou sangramentos gengivais sem maiores repercussões hemodinâmicas. Sangramentos musculares, articulares, subperiostais, nasais ou menorragias ocorrem com menor frequência. Laboratório: • contagem de plaquetas • tempo de sangramento geralmente normais • TAP Etiologias:
- infecções � especialmente meningococcemia, sarampo, ricketsiose. Desencadeiam dano microvascular ou coagulação intravascular disseminada (CIVD)
- reações a drogas � pela formação de complexos Ag-Ac que se depositam nas paredes dos vasos com reação leucocitoclástica de hipersensibilidade.
- Sd. De Scurvy e Ehlers-Danlos, idosos e Sd. De Cushing � formações anormais ou desgaste do colágeno que suporta as paredes vasculares
- Púrpura de Henoch-Schölein � deposições de complexos imunes nas camadas mesangiais de causa desconhecida resultando em rash purpúrico, cólicas abdominais (hemorragias focais TGI), poliartralgias e glomerulonefrite aguda (GNA)
- Telangectasia hemorrágica hereditária � distúrbio autosômico dominante com vasos tortuosos, dilatados com paredes finas. No geral o sangramento ocorre em mucosas nasal (epistaxe), oral, língua e olhos.

Manual Teórico de Patologia do Sistema Hemolinfopoiético
24
2)TROMBOCITOPENIAS (Menor número de plaquetas)
- Causa importante de sangramento generalizado (vasos pequenos) , com contagem plaquetária <100.000/mm3 (valores normais entre 150.000 e 300.000/mm3.).
- Abaixo de 20.000/mm3, ocorrem sangramentos espontâneos - Entre 20.000 e 50.000/mm3, pensar em etiologia de sangramento
pós-traumático. - Sítios comuns nestas desordens: pele, mucosas trato
gastrintestinal (TGI) e trato genitourinário (TGU) - Etiologias ( menos comuns):
- doenças da MO = anemia aplástica, leucemia (distúrbio nos megacariócitos)
- megacariocitopoiese inefetiva � anemias megaloblásticas (deficiência de folatos ou vitamina B12).
- Maior destruição plaquetária � anemias hemolíticas (ingestão de drogas, infecções, auto-imune)
- Trombocitopenia X AIDS � é uma das manifestações mais comuns da doença e ocorre nos primeiros estágios (reações Ag-Ac, supressão megacariocítica pelo vírus)
- Trombocitopenia pós-viral aguda ( púrpura trombocitopênica idiopática) � em crianças após quadros exantemáticos ou infecção de vias aéreas superiores (IVAS) de etiologia viral. Ocorre formação de Ac anti plaquetários com resolução espontânea entre 4 e 6 semanas.
- Destruição mecânica ( hemólise por próteses valvares cardíacas), HAS ou aterosclerose.
- Esplenomegalia ou hiperesplenismo (sequestro de aproximadamente 90% das plaquetas circulantes nestas situações). Tto = esplenenctomia
- Tranfusões sanguíneas maciças � plaquetas são viáveis em sangue estocado por até 24 horas. Após isto, seu número se reduz e o paciente recebe menos do que o esperado, resultando em quadro de trombocitopenia.
-Etiologias (mais comuns): TROMBOCITOPENIA ISOIMUNE NEONATAL E PÓS-TRANSFUSIONAL
Resultam do desenvolvimento de Ac diretamente contra isoantígenos plaquetários (Duzo, PL, Bak), além dos Ag ABO e Rh. O mecanismo pelo qual ocorre a trombocitopenia é semelhante ao que ocorre na doença hemolítica do recém-nato (DHRN), pois a mãe não possui o Ag e o feto sim. Aquela passa a produzir Ac específicos que cruzam a barreira placentária e destroem as plaquetas em formação. Fisiopatologia parecida é atribuída à sensibilização pelas transfusões sanguíneas com fatores positivos (como o PLA1).

Manual Teórico de Patologia do Sistema Hemolinfopoiético
25
PÚRPURA TROMBOCITOPÊNICA IDIOPÁTICA (PTI) Doença autoimune crônica que cursa com destruição plaquetária por Ac
imunologicamente mediados, à semelhança do que ocorre na AIDS, lúpus eritematoso sistêmico (LES), infecções virais, complicação de terapias medicamentosas diversas (PURPURAS TROMBOCITOPÊNICAS SECUNDÁRIAS) , que devem ser devidamente investigadas e descartadas antes de se diagnosticar esta desordem. - Patogênese:
- complexos glicoprotéicos plaquetários IIb/IIIa e Ib/IX estão no plasma e /ou aderidos às plaquetas de 80% dos pacientes com PTI;
- geralmente Ac IgG com coexistência de aproximadamente 40% de IgM.
- Semelhante ao que ocorre nas anemias hemolíticas: Plaquetas opsonizadas e com complexos glicoprotéicos � fagocitadas por monomorfonucleados (MMN) � destruição plaquetária � menor número � fenômenos hemorrágicos Obs1: baço é o principal produtor de Ac, recolhedor e destruidor (sistema retículo-endotelial – S R E) das plaquetas sensibilizadas, o que faz da esplenectomia um tratamento a ser pensado nestes casos. Obs 2: cogita-se a possibilidade de sensibilidade e destruição das precurssoras plaquetárias (megacariócitos). De fato isto ocorre mas não se pode atribuir isso à repercussão clínica presente nestes pacientes. - Epidemiologia:
- pico de incidência entre 20 e 50 anos, com proporção de 2 mulheres para cada homem acometido
- Quadro clínico e laboratório:
- sangramentos em virtualmente todos os tecidos do organismo, com petéquias e pequenas hemorragias em mucosa oral. Epicárdio e endocárdio, trato genito-urinário (TGU), sistema neroso central (SNC), nasofaringe e trato gastrintestinal (TGI) também estão mais sujeitos a sangramentos (melena, hematúria, alteração de nível de consciência, epistaxes, sangramentos menstruais excessivos, etc)
- pacientes em bom estado geral e afebris - ausência de esplenomegalia ( BAÇO NORMAL, mas com
formação aumentada de centros germinativos) - contagem plaquetária < 10.000/microlitro com megatrombócitos - discreta anemia (pelos episódios de sangramento) - outras linhagens celulares normal - 10% dos pacientes têm anemia hemolítica autoimune associada
(Sd. De Evan) = anemia, reticulocitose, esferócitos - testes de coagulaçào normais - MO: linhagens celulares normais, com aumento no número de
megacariócitos, muitos imaturos e com um núcleo grande e não-lobulado, demonstrando eritropoiese acelerada
- Tratamento:

Manual Teórico de Patologia do Sistema Hemolinfopoiético
26
- remissão espontânea em poucos casos - corticóide - esplenectomia - imunoglobulinas EV - imunosupressores
- Prognóstico: Bom. PÚRPURA TROMBOCITOPÊNICA TROMBÓTICA (PTT) Síndrome incomum que ocorre provavelmente por alterações plaquetárias, dentre elas a presença de um fator de aglutinação destas células, com papel controverso na patogenia até agora. A doença cursa com:
- Anemia hemolítica microangiopática - Trombocitopenia, anormalidades neurológicas e renais, febre - Testes de coagulação normais - LDH sérica elevada
- Etiologia: desconhecida ( fator de aglutinação plaquetária ? perturbação endotelial?) - Patogenia:
- fator de aglutinação plaquetária � controverso - perturbação endotelial � diminuição da síntese de prostaciclinas
e aumento nos índices de óxido nítrico, o que acarreta vasodilatação e diminui a agregação plaquetária. Este dano endotelial ainda seria reasponsável pela liberação de grandes quantidades de fator VIII, que causariam agregações e trombose microvascular
- Epidemiologia:
- adultos jovens (entre 20 e 50 anos), com discreta preponderância feminina. Pode ser precipitada ocasionalmente por estrogênio ou gravidez
- grande associação com indivíduos HIV + - Quadro Clínico e laboratório: - geralmente em estado geral regular ou ruim, febris
- anemia, sangramentos (palidez, púrpuras, petéquias) - anormalidades neurológicas (cefaléia, confusão, alteração do nível
de consciência – letargia a coma - , afasia, hemiparesias, convulsões) - falência renal progressiva (Obs: NA SÍNDROME HEMOLÍTICO-
URÊMICA o quadro é semelhante com poreponderância para sinais e sintomas renais; mecanismo patogênico semelhante)
- hemoglobinemia e hemoglobinúria - dor abdominal (de origem renal ou pancreática – pancreatite) - anemia moderada a severa

Manual Teórico de Patologia do Sistema Hemolinfopoiético
27
- reticulocitose, hemácias nucleadas, esquistócitos, células em capacete e triangulares
- trombocitopenia + aumento na contagem de neutrófilos - aumento na bilirrubina indireta (hemólise intravascular) - LDH aumentada proporcionalmente à hemólise subjacente
- Tratamento:
- plasmafereses com plasma fresco congelado, dextrans - antiinflamatórios não esteróides (AINES), corticóides - Esplenectomia
- Prognóstico: 20% dos casos são crônicos e recidivantes; 80-90% bom se realizadas as plasmafereses necessárias. 3)ALTERAÇÃO NOS MECANISMOS OU FATORES DE COAGULAÇÃO ALTERAÇÃO DOS MECANISMOS DE COAGULAÇÃO
Defeitos qualitativos nas plaquetas são congênitos ou adquiridos. - Defeitos Congênitos:
• de adesão (Sd de Bernard- Soulier � deficiência no complexo glicoprotéico GpIb/IX, que resulta em menor número de receptores para o fator von Willerbrand)
• de agregração (trombastenia de Gianzmann � defeito autossômico recessivo
que resulta em falha de agregação plaquetária com colágeno, ADP, epinefrina ou trombina, devido a um defeito no receptor de fibrinogênio (GpIIb/IIa)
• de secreção (reações de liberação) � respostas anromais na produção de
prostaglandinas (PG) ou liberação de ADPs aderidos.
- Defeitos adquiridos: • ingestão excessiva de anti-inflamatórios não esteróides � em especial AAS, que
se liga de forma irreversível às membranas plaquetárias e desta forma aumenta o tempo de sangramento. Os antiinflamatórios não esteróides inibem a ciclooxigenase 2 (COX2) enzima responsável pela produção de PG e tromboxane A2, que favorece a agregação plaquetária .
• uremia � mecanismo desconhecido para a alteração da função plaquetária. Geralmente sangramentos em mucosa oral e trato gastrintestinal , que podem ser severos. Sempre associar à severidade da Insuficiência renal concomitante.
ALTERAÇÃO NOS FATORES DE COAGULAÇÃO:
Manifestam-se quase sempre como grandes equimoses ou hematomas após traumas menores, assim como tempo de sangramento prolongado após lacerações ou cirurgias. Sangramentos do trato gastrintestinal e articular também é comum.
Quase todos os fatores de coagulação possuem descrições de alterações, que podem ser adquiridas ou hereditárias e as mais comuns são destacadas a seguir:

Manual Teórico de Patologia do Sistema Hemolinfopoiético
28
- adquiridas: Geralmente são múltiplas. Por exemplo, deficiências de vitamina K = menor síntese de fatores II, VII, IX , X e proteína C. Doenças hepáticas = diátese hemorrágica e deficiência de quase todos os fatores de coagulação.
- Hereditárias: No geral evolvem apenas um fator de coagulação deficiente. Por exemplo, fator VIII (hemofilia A), fator IX (hemofilia B ou doença de Christmans) são desordens com transmissão ligada ao sexo.
a) Hemofilia A:
- caráter recessivo ligado ao X somente afetando homens - atividade diminuída do fator VIII - antígeno do fator VIII normais
- Quadro clínico e laboratório:
- hemartroses espontâneas (joelhos, tornozelos, cotovelos), sangramentos musculares e de trato gastrintestinal
- trombocitopenia � associar à contaminação por HIV - tempo parcial de tromboplastina (PTT) é prolongado - tempo de sangramento, tempo de protrombina e outras provas de
coagulação = normais - níveis de fator VIII:C diminuídos - níveis do fator Von Willebrand ( proteína grande que se liga ao
fator VIII, mas também a outras estruturas como colágeno, heparina e membranas plaquetárias) são normais.
- Tratamento : infusões com fator VIII concentrado (crioprecipitado) - Prognóstico: depende da disponibilidade de fator VIII para a reposição. b)Doença de von Willebrand:
- história familiar de distúrbio autossômico dominante (tipo I) - tempo de sangramento prolongado basal ou após desafio com
aspirina - níveis reduzidos de antígeno de fator VIII ou cofator ristocetina - desordem de fator de coagulação congênita mais comum - sistema de agregação plaquetária normal - fator von Willebrand � sintetizado em megacariócitos e células
endoteliais e circulante no organismo como multímeros de tamanhos variados
- Aproximadamente 20 variantes, sendo o tipo I responsável por 80% dos casos
- Quadro Clínico e laboratório:
- sangramento de mucosas (oral, nasal = epistaxe, menorragia), após manipulação cirúrgica ou dentária, por exemplo

Manual Teórico de Patologia do Sistema Hemolinfopoiético
29
- sangramento diminui na vigência de estrogênios ou gravidez - contagem e morfologia plaquetária = normais - tempo de sangramento quase sempre prolongado (basal ou com
teste da aspirina) - níveis de fator vW diminuídos / antígeno de fator VIII diminuído
- Tratamento: evitar antiinflamatórios não esteróides. Desmopressina. Concentrados de fator VIII (crioprecipitados) - Prognóstico: excelente. c)Hemofilia B ( doença de Christmas)
- herança recessiva ligada ao X, afetando somente homens - níveis baixos de fator IX (Christmas) - 1/3 dos casos com valores normais de fator IX mas disfunção
molecular define clínica - Quadro clínico e laboratório: - semelhante à hemofilia A
- hemastroses espontâneas - PTT prolongado - Tempo de sangramento normal - Níveis de fator IX diminuídos
- Tratamento: concentrados de fator IX (crioprecipitado) - Prognóstico: idem hemofilia A. COMBINAÇÃO DOS FATORES ACIMA
Representado especialmente pela coagulação intravascular disseminada (CIVD), que gera tanto trombose quanto hemorragias (diátese hemorrágica)
COAGULAÇÃO INTRAVASCULAR DISSEMINADA (CIVD)
- ocorre geralmente quando organismo encontra-se sob stress de
grande intensidade ou contínuo por determinado período de tempo. Este stress pode ser uma infecção grave ou sepse, resposta metabólica a um trauma de grande intensidade, síndrome consuptiva ou outra doença de base grave, dentre elas:
• complicações obstétricas: abortamento incompleto, retido ou séptico; embolia de líquido amniótico; toxemia
• infecções: sepse por Gram -, meningococcemia, histoplasmose, aspergilose,malária, ricketsioses
• neoplasias: CA de pâncreas, próstata, pulmão e estômago; leucemia aguda pró-mielocítica (LpmA)

Manual Teórico de Patologia do Sistema Hemolinfopoiético
30
• miscelânea: hemólise intravascular aguda, picadas de animais peçonhentos, hemangiomas gigantes, choques, vasculites, aneurisma de aorta, doenças hepáticas
- anemia hemolítica microangiopática - hipofibrinogenemia, trombocitopenia, produtos de degradação da
fibrina, PT prolongado. Estas alterações são consequência do consumo de plaquetas, fibrina e fatores de coagulação e secundariamente da ativação dos mecanismos fibrinolíticos.
- Etiologia: depende da doença base que levou o paciente a esta condição. - Patogenia:
- não é doença primária; é consequência de quadros graves subjacentes;
- lembrar que coagulação pode ser iniciada por duas vias : extrínseca � ativada pela tromboplastina liberada dos tecidos acometidos; intrínseca � ativação do fator XII pelo contato com colágeno ou outras substâncias negativamente carregadas. Este processo é regulado or fatores de inibição da coagulação, como o sistema fibrinolítico com a geração de plasmina, a fagocitose dos fatores de coagulação pelos monomorfonucleados ou fígado e ativação de fatores anticoagulantes endógenos como a proteína C.
- mecanismos que desencadeiam a CIVD: • liberação exagerada de tromboplastina tecidual: 1) pela placenta – nas complicações obstétricas 2) pelos grânulos das células pró-mielocíticas da LpmA 3) pelo muco liberado pelos CA descritos acima, que ativam fator X diretamente,
independentemente do fato VII. 4) Na sepse por G-, bactérias produzem toxinas, expõem membranas e aumentam
a liberação de tromboplastina dos monócitos. Estes, por sua vez, liberam interleucina 1 (IL-1) e fator de necrose tumoral alfa ( alfa –TNF), ambos aumetando a expressão de fatores endoteliais e diminuindo a expressão de trombomodulina, responsável pela ativação da proteína C (anticoagulante intrínseco). O alfa – TNF, além destas ações, regula a expressão de moléculas de adesão nas células endoteliais, o que favorece a marginação e adesão leucocitária, o que ocasiona dano endotelial pela liberação de radicais livres de oxigênio e proteases pré-formadas.
• Lesão endotelial: liberação de fatores pró-agregação plaquetária, ativando a via
intrínseca da coagulação . Lesão endotelial generalizada pode ocorrer por deposição de complexos Ag-Ac (LES), extremos de temperatura (queimados), ou microrganismos (meningococos, ricketsias). Hipóxia, acidose e choque também levam à lesão endotelial.
- Consequências:
1º ) deposição de fibrina em toda a microcirculação � anemia hemolítica microangiopática � isquemia dos órgãos afetados � microtrombose disseminada e progressiva

Manual Teórico de Patologia do Sistema Hemolinfopoiético
31
2º) diátese hemorrágica resultante do consumo das plaquetas e fatores de coagulação e ativação do plasminogênio (quebra fibrina mas digere fatores V e VIII). Além disso, a fibrinólise leva à formação de produtos de degradação da fibrina, que inibem a agregação plaquetária e a polimerização da fibrina, além de possuir atividade antitrombínica.
Estas 2 consequências levam ao desequilíbrio hemodinâmico, ao colapso circulatório e à falência de múltiplos órgãos (FMO) que levarão o paciente a óbito se não manejado rápida e precisamente. - Epidemiologia: - Aproximadamente 50% dos pacientes com CIVD são obstétricas; 33% têm carcinomatoses. Os demais associam-se aos outros fatores. - Quadro Clínico e laboratório:
- Trombos são encontrados no (em ordem decrescente): cérebro (sintomatologia de sistema nervoso central (SNC) – alteração do nível de consciência, sinais de HIC), coração, pulmões (SARA) , rins (IRA), adrenais (sd. De Waterhouse-Friderischen na meningococcemia), baço e fígado. Na hipófise, causam sd. De Shehan (necrose de hipófise anterior puerperal).
- Sintomas respiratórios: dispnéia, distress respiratório (SARA), cianose
- Convulsões, alterações psicomotoras e confusão mental, letargia e coma
- IRA com oligo/anúria - Diátese hemorrágica predomina em politraumatizados e pacientes
obstétricas - Trombose microvascular predomina em pacientes com neoplasias - anemia hemolítica microangiopática - hipofibrinogenemia, trombocitopenia, produtos de degradação da
fibrina, PT prolongado. Estas alterações são consequência do consumo de plaquetas, fibrina e fatores de coagulação e secundariamente da ativação dos mecanismos fibrinolíticos.
- Tratamento: tratar a causa base; oxigenioterapia; fluídos; drogas vasoativas; etc. - Prognóstico : depende da causa base, da rapidez no diagnóstico e da pronta instituição de terapêutica adequada em UTI de preferência.

Manual Teórico de Patologia do Sistema Hemolinfopoiético
32

Manual Teórico de Patologia do Sistema Hemolinfopoiético
33
DOENÇAS DOS LEUCÓCITOS E LINFONODOS Normal: • Linfócitos e Monócitos estão difundidos pelo corpo e têm alta concentração em
aglomerados organizados de células: linfonodos, timo, baço, amigdalas, adenóides e placas de Peyer.
• Linfonodos Normais: estruturas encapsuladas permeadas por vasos linfáticos
aferentes e preenchidas por camadas de células distintas: - cápsula - córtex � linfócitos B - zona paracortical � linfócitos T - cordões medulares � vasos linfáticos, plasmócitos e poucos linfócitos. • Não são estruturas estáveis: alteram-se de acordo com a doença de base. Patologia:
I. LEUCOPENIA: • representada por neutropenia ou, então, quando a diminuição de células é
drástica, é chamada de agranulocitose. Pode ter 2 causas: (1) queda na produção ou (2) aumento da destruição celular.
• baixa produção: (1) supressão das células mielóides primitivas (neutropenia + anemia + trombocitopenia); (2) supressão das células precursoras de granulócitos e (3) anemia megaloblástica (células precursoras aormais que morrem ainda na medula óssea).
• alta remoção celular: (1) lesão idiopática dos neutrófilos ( Síndrome de Felty); (2) seqüestro esplênico (destruição de neutrófilos + hemácias + plaquetas) ou aumento da utilização de neutrófilos na periferia (infecções graves).
• As maiores neutropenia (agranulocitoses) são as causadas por medicamentos, p.ex. quimioterápicos supressores da medula óssea, que geram agranulocitose + anemia aplástica. Outros medicamentos que geram queda de neutrófilos: amonipirina, cloranfenicol, sulfonamida, clorpromazina, tiouracil e fenilbutazona.
• Morfologia: alterações na medula óssea: (1) destruição excessiva ou granulopoiese ineficaz geram hipercelularidade compensatória na medula óssea; (2) precursores granulocíticos afetados geram hipocelularidade.
• Clínica: agranulocitose manifesta-se com infecções como: lesões necrosantes ulcerativas em gengiva, assoalho de boca, mucosa bucal, faringe e, menos freqüentemente, pele, vagina, ânus e intestino. Evolução: sintomas e sinais são os das infecções bacterianas: quadro semelhante ao quadro gripal; se a neutropenia for extremamente grave, pode levar a óbito.

Manual Teórico de Patologia do Sistema Hemolinfopoiético
34
II. PROLIFERAÇÕES INFLAMATÓRIAS 1. Leucocitose É o aumento do número de células brancas do sangue: • leucocitose PMN � inflamação aguda • leucocitose neutrofílica � infecções piogênicas, necrose tecidual (queimaduras,
infarto do miocárdio) • neutrófilos com grânulos tóxicos � sepse • leucocitose eosinofílica � alergias • monocitose � infecções crônicas (tb, endocardite bacteriana, etc.) • linfocitose � típica de mononucleose infecciosa, mas pode aparecer em outras
infecções virais agudas. 2. Linfadenite aguda inespecífica • Inflamação aguda dos linfonodos. • Drenagem microbiológica direta: infecções dentárias e amigdalianas, apendicite
aguda. • Linfadenopatia generalizada � infecções virais e bacteremia em crianças. • Morfologia: aumento de volume e ingurgitação dos linfonodos; folículos linfóides
proeminentes. Se o agente etiológico for piogênico � necrose dos centros dos folículos � massa supurada. Infiltrado inflamatório (neutrófilos + histiócitos) presentes.
• Clínica: dor local + aumento de volume. Abscesso importante � linfonodos flutuantes. Mediante necrose podem fistulizar para a pele.
3. Linfadenite crônica inespecífica Presença de 3 padrões distintos: (A) hiperplasia folicular, (B) hiperplasia linfóide paracortical e (C) histiocitose sinusal. • Hiperplasia folicular: morfologia: (1) preservação da arquitetura do linfonodo, (2)
variação na forma e tamanho dos tecidos linfóides, (3) pleomorfismo e (4) aumento da atividade fagocitária.
• Hiperplasia linfóide paracortical: afeta células T que se transformam em imunoblastos. Causada por reações imunológicas induzidas por medicamentos e vacinação contra varicela.
• Histiocitose sinusal: ingurgitação dos linfonodos que drenam cânceres, p.ex. mama.
• Clínica: linfadenites crônicas são indolores e afetam mais linfonodos axilares e inguinais.
III. PROLIFERAÇÕES NEOPLÁSICAS Compreendem os linfomas malignos, as leucemias, as discrasia plasmocitárias e as histiocitoses. A) Linfomas malignos Representados pelos linfomas não-Hodgkin e pela doença de Hodgkin.

Manual Teórico de Patologia do Sistema Hemolinfopoiético
35
1. Linfomas não-Hodgkin (LNH) • É diagnóstico diferencial de linfadenopatia infecciosa. • Características gerais: - 80-85% células B - Têm 2 padrões de crescimento celular: nodular e difuso. - Em ambos há destruição da arquitetura folicular. - Linfomas nodulares afetam células B e têm melhor prognóstico. - Células B tornam-se imunoblastos ativados na seguinte sequência: Pequena célula clivada � grande célula clivada � pequena célula ñ-clivada � grande célula ñ-clivada →→→→ imunoblastos Working Formulation para uso clínico nos linfomas não-Hodgkin: Baixo Grau � sobrevida de 45% em 10 anos Linfomas linfocíticos de pequenas células, difuso (tipo LLC) Folicular, com predomínio de pequenas células clivadas Folicular, misto de pequenas células clivadas e grandes células Grau Intermediário � sobrevida de 26% em 10 anos Folicular, predominantemente de grandes células Difuso, de pequenas células clivadas Difuso, misto de pequenas e grandes células Difuso, de grandes células Alto Grau � sobrevida de 23% em 10 anos Imunoblástico de grandes células Linfoblástico Pequenas células não-clivadas (Burkitt ) Outros Baixo grau Linfoma Linfocítico de Pequenas Células (Padrão LLC ) • 4% dos LNH • Componente de medula óssea � células neoplásicas no sangue � quadro de
leucemia linfocítica.crônica (LLC) • Clínica: acomete 6a. e 7a. décadas de vida; linfadenopatia generalizada, discreta
hepatoesplenomegalia. Prognóstico de 45% de sobrevida em 10 anos. Linfomas foliculares • Células B neoplásicas: são maiores, com citoplasma escasso, contorno nuclear
clivado. • Pode haver grandes células clivadas ou não, até 20% (linfoma folicular misto). • 40% dos LNH. • Acomete mais homens do que mulheres, por volta de 50 a 60 anos de idade. • Linfadenopatia indolor + comprometimento da medula óssea. • Longa história natural. Sobrevida de 7 a 10 anos.

Manual Teórico de Patologia do Sistema Hemolinfopoiético
36
Grau intermediário Linfoma folicular de grandes células • 15% dos LNH; células neoplásicas grandes. • Evoluem para linfomas de grandes células; mau prognóstico. Linfoma difuso de pequenas células • Semelhante ao folicular de pequenas células. • Relação homem:mulher mais alta. • Sobrevida menor: 2 a 4 anos Linfoma difuso misto de pequenas e grandes células � raro. • Grandes células clivadas X grandes células não-clivadas Linfoma difuso de grandes células • Grau intermediário mais comum. • Clínica: semelhante à do linfoma imunoblástico de grandes células (alto grau). Alto grau Linfoma imunoblástico de grandes células • Células tumorais plasmocitóides. • 80% provêm de células B. • Representam metade dos LNH em adultos. • Média de idade: 60 anos. • Doença localizada X manifestações extra-ganglionares. Linfoma linfoblástico • Relacionado à LLA-T. • Homem : mulher = 2:1 • Média de idade: menor que 20 anos. • Massa mediastinal proemintente em 50 a 70% dos casos (origem tímica?). • Doença progressiva: medula óssea � sangue � meninges. Linfomas de pequenas células não-clivadas (Burkitt) • São tumores de célula B. • Casos africanos (Burkitt) e não-africanos. • Linfoma de Burkitt � criança de etnia africana. Comprometimento da maxila e
mandíbula. • Boa resposta à QT: aumenta a sobrevida. Outros • Clínica peculiar. • Originários de células T. • Ex.: micose fungóide (S. De Sézany / Leucemia); linfoma de células T no adulto. Obs.: o diagnóstico é firmado por exame histológico do linfonodo.’

Manual Teórico de Patologia do Sistema Hemolinfopoiético
37
Estadiamento: pode-se usar o estadiamento dos LH, mas o padrão dos LNH é pouco previsível. 2. Doença de Hodgkin (LH) Difere dos linfomas não-Hodgkin por: • células gigantes neoplásicas de Reed-Steinberg (células RS); • associado a manifestações sistêmicas � febre • doença maligna do adulto jovem Classificação: 4 tipos. (1) predominância linfocitária; (2) celularidade mista; (3) depleção linfocítica e (4) esclerose nodular. ESTÁGIOS CLÍNICOS DOS LINFOMAS DE HODGKIN E NÃO-HODGKIN (CLASSIFICAÇÃO DE ANN ARBOR)* Estágio Distribuição da Doença I Comprometimento de uma única região ganglionar (I) ou
comprometimento de um único órgão ou sistema linfático (IE). II Comprometimento de 2 ou mais regiões ganglionares do mesmo lado do
diafragma isoladamente (II) ou ou com comprometimento localizado de órgão ou tecido extralinfático contíguo (IIE).
III Comprometimento de regiões ganglionares em ambos os lados do diafragma (III), o que pode incluir o baço (IIIS) e/ou comprometimento limitado de órgão ou sítio extralinfático contíguo (IIIE, IIIES).
IV Focos múltiplos ou disseminados de comprometimento de um ou mais órgãos, tecidos extralinfáticos com ou sem comprometimento linfático.
* Todos os estágios são ainda subdivididos com base na ausência (A) ou presença (B) dos seguintes sintomas sistêmicos: febre significativa, suores noturnos e/ou perda de peso inexplicada superior a 10% do peso corporal normal. 1. Doença de Hodgkin de predominância linfocítica • RS raras. • Células características: “células pipoca”. • Acomete mais homens, < 35 anos, bom prognöstico. • Predomínio linfocítico X tumores de células B 2. Doença de Hodgkin de celularidade mista • Muitas células RS, poucos linfócitos e muitos eosinófios. • Comprometimento difuso dos linfonodos. • Padrão celular heterogêneo. • Acomete mais homens que mulheres. 3. Doença de Hodgkin com depleção linfocítica • Linfócitos escassos e RS não predominantes, porém não raros. • Dois tipos morfológicos: fibrose difusa e variantes reticulares. • Acomete mais homens, idosos; manifesta-se sob forma de doença sistêmica. 4. Doença de Hodgkin tipo Esclerose Nodular

Manual Teórico de Patologia do Sistema Hemolinfopoiético
38
• É a forma mais comum de doença de Hodgkin. • Tem 2 peculiaridades: - célula lacunar (variante da RS) - faixas de colágeno � nódulos. • Único tipo que predomina em mulheres; adolescentes ou adultos jovens. • Acomete linfonodos cervicais inferiores, supraclaviculares e mediastinais. • Bom prognóstico em estágio I ou II. • Evolução clínica da doença de Hodgkin: - jovens � estágios I e II � sem manifestações sistêmicas � sobrevida de 90% / 5a. - doença disseminada � estágios III e IV � manifestações sistêmicas � 60-70%/5a. DIFERENÇAS CLÍNICAS ENTRE OS LINFOMAS HODGKIN E NÃO-HODGKIN Doença de Hodgkin Linfoma não-Hodgkin Restrito a um único grupo axial de linfonodos (cervical, mediastinal, para-aórtico).
Mais freqüente o comprometimento de múltiplos linfonodos periféricos.
Propagação ordenada por contigüidade. Propagação não-contígua. Linfonodos mesentéricos e anel de Waldeyer raramente comprometidos.
Anel de Waldeyer e linfonodos mesentéricos comumente comprometidos.
Comprometimento extra-ganglionar incomum.
Comprometimento extra-ganglionar comum.
B) Leucemias e doenças mieloproliferativas Leucemias são neoplasias malignas das células primitivas hematopoiéticas, caracterizadas pela substituição difusa da medula óssea (MO). São classificadas em: - agudas X crônicas - linfocíticas X mielocíticas 1. Leucemias Agudas * Fisiopatologia: • Bloqueio na diferenciação das células primitivas hematopoiéticas normais por
substituição. • Implicações clínicas: - manifestações resultam da queda do número de hemácias, leucócitos e plaquetas normais. - o tratamento visa a diminuição do clone leucêmico para restabelecimento de linhagem normal. *Manifestações clínicas: São decorrentes da anemia, granulocitopenia e trombocitopenia. • Início súbito � 3 meses após o início da proliferação celular. • Tríade clássica: fadiga (anemia) + febre (granulocitopenia) + hemorragia
(trombocitopenia). • Dor e sensibilidade óssea � expansão da medula óssea.

Manual Teórico de Patologia do Sistema Hemolinfopoiético
39
• Infiltração de órgãos � linfadenopatia generalizada, esplenomegalia, hepatomegalia.
• Manifestações SNC: cefaléia, vômitos, paralisia nervosa resultante de propagação meníngea. Mais comuns em LLA que em LMA; mais comum em crianças que em adultos.
• Achados laboratoriais: - anemia - leucocitose - reticulocitose - plaquetas < 100.000/µL - medula óssea hipercelular (descarta anemia aplástica) Leucemia Linfoblástica Aguda (LLA) • Mais comuns em adultos jovens e crianças (80% das leucemias agudas da
infância). • Pico = 4 anos de idade. Caucasianos. Acomete mais o sexo masculino.
ASPECTOS CITOLÓGICOS
L1
L2
L3
Tamanho da
célula
Predomínio de pequenas células
Grande, heterogêneo em tamanho.
Grande e homogêneo
Quantidade de
Citoplasma
Escasso
Variável, amiúde moderadamente
abundante.
Moderadamente abundante.
Nucléolos
Não visíveis ou pequenos e
inconspícuos
1 ou + presentes, amiúde grandes
1 ou + presentes, amiúde proeminentes.
Cromatina Nuclear
Homogênea, em qualquer caso determinado.
Variável, heterogênea em qualquer caso
determinado.
Finalmente pontilhada e homogênea.
Forma do Núcleo Regular, fendas ou identações ocasionais.
Irregular, fendas ou identações
Regular, oval ou redondo.
Basofilia do Citoplasma
Variável Variável Basofilia intensa
Vacuolização citoplasmática
Variável Variável Proeminenente
Leucemia Mieloblástica Aguda • Adultos de 15 a 39 anos. Representa 20% das leucemias da infância. • Extraordinariamente heterogênea. • Morfologia: - mieloblastos diferem de linfoblastos por coloração Wright-Giemsa - delicada cromatina nuclear, 3 a 5 nucléolos - grânulos azurofílicos mieloperoxidase positiva no citoplasma - bastonetes de Auer (mais comum na promielocítica) - MO com > 30% de mieloblásticos (fecha diagnóstico) - TdT presente em < 5% dos casos. Classificação (Segundo FAB)
Classe Incidência Morfologia medular / comentários

Manual Teórico de Patologia do Sistema Hemolinfopoiético
40
(% de LMA) M10 - LMA minimamente
diferenciada 2-3% Blastos desprovidos de marcadores citológicos e
citoquímicos definitivos de mieloblastos, mas expressam antígenos de linhagem mielóide e
assemelham-se a mieloblastos do ponto de vista ultraestrutural.
M1 - LMA sem diferenciação ~20% Células muito imaturas ≥3% são peroxidadase +. Poucos grãnulosou bastonetes de Auer e pouca
maturação além do estágio de mieloblasto M2 - LMA com maturação 30-40% Série total de maturação mielóide até granulócitos;
bastonetes de Auer presentes na maioria dos casos; a presença de t(8;21) define um subgrupo
prognosticamente favorável. M3 - Leucemia
promielocítica aguda 5-10% A maioria das células são promielócitos
hipergranulares amiúde com muitos bastonetes de Auer por célula; os pacientes são masi jovens (35-40
anos) e muitas vezes desenvolvem CID; a translocação t(15;17) é característica
M4 - Leucemia mielomonocítica aguda
15-20% A diferenciação mielocítica e monocítica é evidente; os elementos mielóides assenelham-se a M2; as células monocíticas são positivas para esterases inespecíficas; a presença de anormalidades no
cromossomo 16 define um subgrupo com eosinófilos na MO e excelente prognóstico.
M5 - Leucemia monocítica ag.
~10% Os monoblastos (peroxidase-negativos, esterase-inespecíficos positivos e pró-monócitos predominam;
essa condição tende a osorrer em pacientes mais velhos, sendo caracterizada por uma incidência muito
alta de organomegalia, linfadenopatia e infiltração tecidual; a hipertrofia de gengivas e a infiltração de
pele são comuns. M6 - Eritroleucemia aguda ~5% Eritroblastos anormais (alguns megaloblastóides,
outros com núcleos gigantes ou múltiplos) predominam como alguns mieloblastos presentes; os
afetados têm idade avançada. M7 - Leucemia
megacariocítica aguda ~1% Os blastos de linhagem megacariocïtica predominam;
reagem com anticorpos plaqueta-específicos contra GPIIb/IIa ou vWF; mielofibrose ou reticulina medular
aumentada na maioria dos casos. • Tratamento: - QT - TMO • Prognóstico: - 60% = remissão completa com QT. - Destes, 70-85% recidivam em 5 anos. 2. Síndromes mielodisplásicas • Definição: grupo de afecções de células primitivas clonais caracterizado por
defeitos de maturação que resultam em hematopoiese ineficaz e maior risco de transformação em LMA.
• Idosos entre 60 e 70 anos. • MO parcial ou totalmente substituída por células clonais primitivas pluripotenciais
mutantes e que retêm a capacidade de diferenciação. • MO mono ou hipercelular. • Esfregaço periférico = pancitopenia.

Manual Teórico de Patologia do Sistema Hemolinfopoiético
41
• Morfologia: - diferenciação displásica afetando as 3 linhagens celulares. - Precursores mielóides hipogranulares com maior proporção de células blásticas (neutrófilos uni ou bilobulados). - micromegacariócitos, plaquetas agranulares. • Classificação: - 5 categorias; - anormalidades cromossômicas em até 2/3 dos pacientes. • Quadro clínico: - 50% assintomáticos - fraqueza + infecções + hemorragias • Prognóstico: - 10-40% evoluem para LMA - Sobrevida de 9 a 29 meses, influenciada pela forma específica de mielodisplasia e por anormalidades cariotípicas. 3. Leucemias crônicas Leucemia Mielocítica Crônica (LMC) • 15 a 20% de todos os casos de leucemia. • Doença de adultos: 25 a 60 anos. • Pico = 4a. e 5a. décadas de vida. • Fisiopatologia: - transformação neoplásica e expansão clonal de célula primitiva pluripotencial - 3 linhagens envolvidas com predomin&ancia de precursores granulocíticos - anormalidades citogenética e molecular características - 90% dos casos = cromossomo pH, que representa translocação recíproca t(9;22)(q34;q11) = gene de fusão BCR-C-ABL, codificador da proteína de fusão de 210-kd com potente atividade tirosino-quinase. - pacientes pH negativos (minoria), por análise citogenética, revelam rearranjos BCR-C-ABL. - mais de 10 a 20 vezes na massa dos precursores granulocíticos. • Clínica: - início lento e sintomas inespecíficos - decorrentes da anemia ou hipometabolismo por maior renovação celular - fadiga, fraqueza, perda de peso, anorexia. - “tração no abdome” = esplenomegalia muito volumosa. • Hemograma: - leucocitose > 100.000µL, com predomínio de neutrófilos, metamielócitos, eosinófilos e basófilos; 50% dos pacientes têm trombocitose precoce. • Ausência quase total de fosfatase alcalina nos granulócitos (diagnóstico
diferencial com reação leucemóide). • Diagnóstico definitivo: cromossomo pH e rearranjos BCR-C-ABL. • Evolução lenta com sobrevida média de 3 anos. • 50% dos pacientes entram em “fase acelerada”: falta gradual de resposta ao
tratamento, anemia e trombocitopenia crescentes, outras anormalidades citogenéticas e transformação em quadro de leucemia aguda (crise blástica).
• Outros 50%: crises blásticas súbitas sem fase acelerada intermediária.

Manual Teórico de Patologia do Sistema Hemolinfopoiético
42
• 70% dos pacientes têm blastos com características mielóides; 30% restantes: blastos com enzima TdT e antígeno de linhagem B (CD10 e CD19)
• Tratamento: - QT � sem boa resposta - TMO • Após desenvolvimento da crise blástica, todas as formas de tratamento tornam-se
ineficazes. Leucemia Linfocítica Crônica • A mais indolente de todas as leucemias. • 25% de todos os casos de leucemia. • Geralmente em > 50 anos de idade. Pico = 60 anos. • Homem : mulher = 2 : 1 *Fisiopatologia: • Distúrbio neoplásico de células B, semelhante ao linfoma linfocítico de pequenas
células (os casos envolvendo células T representam apenas 5%). • Células B transformadas: fenótipo diferente de LLA; expressam CD5: antígeno
associado às células T e que se manifesta em raras células B; apresentam Ig de superfície em número bem pequeno, simulando negatividade à imunofluorescência; não se diferenciam em plasmócitos secretores de Ac in vivo.
• Rearrajnos de bcl2 em 10 a 15% � evita apoptose � acúmulo de células B. *Anormalidades Cromossômicas • 50% apresentam cariótipos anormais, precisam de tratamento precoce e têm
sobrevida mais curta. • 1/3 � trissomia do 12 � prognóstico sombrio • 25 a 40% � anormalidades em 13q � sem influência na clínica. *Clínica • Geralmente assintomáticos • Sintomas inespecíficos: fatigabilidade, perda de peso e anorexia. • 50 a 60% � linfadenopatia generalizada e esplenomegalia. • Leucometria aumentada � até 200.000/µL • Linfocitose absoluta de pequenos linfócitos de aspecto maduro. • Esfregaços periféricos � células fantasmas (núcleos esmagados de linfócitos). • Hipogamaglobulinemia e > susceptibilidade a infecções bacterianas. • 10 a 15% � auto-anticorpos contra hemácias ou plaquetas � anemia e/ou
trombocitopenia auto-imunes (subgrupo CD5+ de células B). • Sobrevida média de 4 a 6 anos. • Raramente se transforma em leucemia aguda com crise blástica. Tricoleucemia • Células apresentam projeções em forma de fios de cabelo. • Células B neoplásicas apresentam fosfatase ácida tartaratorresistente (TRAP). • Possuem agrupamento antigênico peculiar: CD19, CD20, CD11c e PCA-1. • Acomete mais homens, de idade avançada.

Manual Teórico de Patologia do Sistema Hemolinfopoiético
43
• Manifestações decorrem da infiltração da MO, fígado e baço: esplenomegalia e linfadenopatia são as mais comuns.
• Leucocitose em 25% dos casos. • Esplenectomia: benéfica em 2/3 dos pacientes. • Tratamento: QT (interferon-alfa) � bom prognóstico.

Manual Teórico de Patologia do Sistema Hemolinfopoiético
44
Literatura Complementar 1. COTRAN; KUMAR; COLLINS. Robbins Pathologic Basis of Disease Sixth
Edition. W.B. Saunders Company, 1999, USA. 2. BRASILEIRO Fo.; PITELLA; PEREIRA; BAMBIRRA; BARBOSA. Bogliolo
Patologia 5a Edição. Guanabara Koogan, 1994, Rio de Janeiro.





























