Embed Size (px)
Citation preview

Medical and Pediatric Oncology 7:205-218 (1979)
An Unusual Case of T-cell Lymphoproliferative Disorder George Spanos, MD, Robert J. Kanter, MS, Fred Rosner,MD, and Hans W. Grunwald, MD
Division of Hematology (G.S., H.W.G., R.J.K), Department of Medicine (F. R.), Queens Hospital Center Affiliation of the Long Island Jewish-Hillside Medical Center; and the Health Sciences Center, State University of New York School of Medicine at Stony Brook (F.R., H.W.G.)
A 54-year-old woman with epigastric pain had leukocytosis of 73,0OO/p1 con- sisting mainly of atypical lymphoid cells with convoluted and cleaved nuclei resembling Sizary cells; the bone marrow aspirate was nondiagnostic. Skin biopsy was unremarkable. The patient also had hypercalcemia and hemolysis with a positive direct Coombs’ test, both of short duration.
The arterial oxygen tension was decreased, but there was no demonstrable lung pathology. The patient subsequently developed rapidly enlarging lympha- denopathy. Lymph node biopsy was interpreted as “undifferentiated pleo- morphic lymphoma.” Immunologic functional studies revealed that the majority of the peripheral blood atypical lymphoid cells from involved lymph nodes formed rosettes with sheep erythrocytes.
The lymphadenopathy regressed transiently after the administration of chemotherapy and the white blood cell count decreased from a maximum of 385,00O/pl to 3,5OO/p1, at which point the arterial oxygen tension returned to normal. The unusual features of this patient are discussed in light of the known characteristics of the various types of T-cell lymphoproliferative disorders.
Key words: Tcell lymphoma, atypical SCzary syndrome
INTRODUCTION
Recent advances in immunology are now commonly applied to the characterization and classification of lymphoproliferative disorders. Careful study of cell surface markers as well as morphology and function of the abnormal lymphoid cells are now routinely used in the assessment of patients with such disorders. For many years, the non-Hodgkin lym- phomas were classified according to the histopathologic criteria of Rappaport [l] as either nodular or diffuse, and then further characterized according to the state of differentiation of the neoplastic cells.
0098-1532/79/0703-0205$02.60 0 1979 Alan R. Liss, Inc.

206 Spanos et a1
The newer classifications of Lukes and Collins [2] and Lennert et al [3] are based on immunologic cell surface markers and employ newer morphologic and cytochemical criteria. These functional classifications of lymphomas have increased our understanding of the natural history of these disorders and have provided potentially useful therapeutic information. For example, treatment of certain T-cell malignancies with antithymocyte globulin may be an effective therapeutic approach [4].
The patient described here had an unusual type of convoluted T-cell neoplasm with unique features of Coombs’ positive hemolysis and hypercalcemia with progression to an undifferentiated T-cell lymphoma.
CASE REPORT
A 54-year-old black woman came to the emergency room because of mid-epigastric abdominal pain of several hours’ duration, which radiated diffusely throughout the abdomen. One week earlier, the patient had had a similar episode which lasted for several hours and abated spontaneously. There was no associated diarrhea, constipation, nausea, or vomiting.
Past medical history was unremarkable except for hypertension many years earlier treated for a short time with an antihypertensive agent.
Physical examination revealed an obese black woman in no acute distress. Tempera- ture was 98.2’F; pulse 85 and regular; respirations 19 and regular; blood pressure 170/105 mm Hg. There were no cervical, axillary, or inguinal lymph nodes palpable. Spleen and liver were not palpable. Heart and lungs were normal. Examination of the skin revealed no abnormalities. The hemoglobin was 14.7 gm%, hematocrit 45%. Platelet count was 185,000/ pl. There were 73,000 leukocytes/pl, with 85% atypical lymphocytoid cells, 5% segmented neutrophils, 3% monocytes, 5% normal lymphocytes, 1% eosinophils, and 1% basophils. The atypical lymphocytoid cells had large and irregularly shaped nuclei, many of which were cleaved, convoluted, and occupying about 80% of the cell and surrounded by a pale blue ring of cytoplasm. These cells had negative staining reactions to myeloperoxidase, Sudan black, chloracetate esterase, and a-naphthyl acetate esterase, but did stain positively in the periodic acid-Schiff reaction. The acid phosphatase stain was also positive but became negative after treatment with tartrate.
Leukocyte alkaline phosphatase was 48 Kaplow units. Serum glucose was 112 mg%, blood urea nitrogen 16 mg%, sodium 139 mEq/liter, potassium 4.1 mEq/liter, carbon dioxide 31 mEq/liter, and chloride 101 mEq/liter. Serum glutamic oxalacetic transaminase and pyruvate transaminases were 57 IU/ml and 25 IU/ml, respectively; lactic dehydrogenase was 775 IU/ml, and alkaline phosphatase 98 IU/ml. Total serum bilirubin was 0.7 mg%, creatinine 1.5 mg%, uric acid 8.3 mg%, cholesterol 25 1 mg%, calcium 1 1 mg%, and inorganic phosphorus 4 mg%.
Urinalysis was normal. Hemoglobin electrophoresis revealed an AS pattern. The hemoglobin F was within normal limits.
Total serum protein was 7.8 gm% with 4 gm% of albumin. Serum protein electro- phoresis revealed 20.5% of gamma globulin, 13.2% beta globulin, 4.4% alpha-2globulin, 2.9% alpha-1-globulin, and 59% albumin. Serum immunodiffusion showed an IgG of 1,480 mg%; IgA of 324 mg%; IgM of 120 mg%; and IgD of 0.25 mg%o (normal 0.3-40); IgE was nondetectable.
Serum lysozyme was 10 pg/ml (normal, 2.8-8). Serum lactic dehydrogenase isoenzymes were as follows: fraction 1 was 26% (normal,
18-33), fraction 2 was 33% (normal 28-40), fraction 3 was 27% (normal 18-30), fraction

An Unusual Lymphoproliferative Disorder 207
4 was 11% (normal 6-1 6), fraction 5 was 3% (normal 2-1 3). Parathormone assay was within normal limits. Prostaglandin production by the patient’s abnormal lymphoid cells was measured but was not detectable (kindly performed by Dr. Richard Bockman, Memorial Sloan-Kettering Cancer Center, New York, NY).
barium enema were within normal limits. Liver and spleen scan, bone scan, and skeletal sur- vey and abdominal sonograms were all within normal limits.
Bone marrow aspirate and biopsy revealed a normocellular marrow with a preponder- ance of myeloid elements and a depression of the erythroid series (mye1oid:erythroid ratio of 5 : 1). The number of lymphocytes was normal. A slight increase in plasma cells and eosinophils was present. Aspiration of the bone marrow performed on three different occasions showed essentially similar histologic findings.
Arterial blood gases revealed persistent hypoxia, @O, between 55 and 62), the other parameters being normal. Pulmonary function tests revealed a moderate restrictive ventila- tory impairment with a disproportionate reduction in expiratory reserve volume and a normal diffusion capacity. In addition, there was a right-to-left shunt fraction of 0.2449 (normal 0.05-0.08). Ventilation perfusion scan was normal.
Skin biopsy revealed a dermal infdtrate of round cells scattered in the upper dermis. These cells were mainly mature lymphocytes, with occasional larger hyperchromatic cells that were periodic acid-Schiff negative. Occasional plasma cells and a few melanophores were also present. In one area a few giant cells were seen around birefringent material. A slight hyperkeratosis and a few inflammatory cells were noted within foci of parakeratosis. The above findings were consistent with the presence of a foreign body granuloma (Fig. 1).
Repeated attempts at chromosome analysis of peripheral blood cells with stimulation by a variety of agents were unsuccessful. In the bone marrow, 16 of 26 metaphases were normal (46,XX chromosome complement), one was pseudodiploid, one had 45 chromo- somes, two had 47, one had 48, and one had 49 chromosomes. In addition, four metaphases had fewer than 43 chromosomes. In two metaphases a chromosome was noted which had the size of a G group chromosome, but the banding pattern did not correspond with the pattern characteristic of either chromosome 21 or 22.
Hospital Course
Over a two-week period the serum lactic dehydrogenase increased to over 1,800 IU/ml and remained stable at this level. The leukocyte count reached a maximum of 385,00O/j~l and stabilized between 200,000 and 250,0OO/pl. The serum glutamic oxalacetic transaminase and alkaline phosphatase levels increased to 150 IU/ml and 191 IU/ml, respectively. The serum calcium increased to 13 mg%, necessitating the use of diuretics and oral phosphates. The serum uric acid increased to 9.1 mg%, for which allopurinol therapy was instituted.
Four weeks from initial presentation, two lymph nodes became palpably enlarged approximately 2 X 2 cm in diameter, confined to the right cervical area. The nodes were of rubbery consistency, painless and freely movable.
pleomorphic malignant cellular infiltrate. Some of the cells had hyperchromatic bizarre nuclei, whereas others had vesicular nuclei with one or more prominent nucleoli. Some bizarre multinucleated giant cell forms and numerous mitoses were present. There was no periodic acid-Schiff-positive material in the cytoplasm of the cells. The methyl green pyronin stain was weakly positive in only a few cells. Lymph node imprints revealed many cells with convoluted nuclei.
Upper gastrointestinal radiographs, chest x-ray, tomograms of the mediastium, and
Lymph node biopsy disclosed total replacement of the normal architecture by a

208 Spanos et a1
Fig. 1. Skin biopsy showing a dermal inatrate of round cells in the upper dermis with birefrhgent material within two foreign body giant cells (arrow) (hernatoxylin and eosin, polarized light; magnification, X 250).

An Unusual Lymphoproliferative Disorder 209
The patient was treated with repeated leukapheresis. Because of several hypotensive episodes, this therapy had to be abandoned. One month later the patient developed auto- immune (Coombs’-positive) hemolysis, and the presence of IgG was detected on the erythrocyte surface by specific anti-IgG antibody. The red blood cell eluate reacted as a panagglutinin. The absolute reticulocyte count increased from 90,000 to 600,0OO/p1. Serum haptoglobin was absent and the serum bilirubin increased to 2.5 mg%, half of which was indirect. The blood hemoglobin concentration remained normal. The serum calcium and uric acid levels were now within the normal range. The patient was receiving diuretics and allopurinol. The abdominal pain with which the patient originally presented had dis- appeared and never recurred.
Three months from initial presentation, the patient was again hospitalized because of progressive lymph node enlargement in the cervical, axillary, and inguinal areas. The spleen was now palpable, and a spleen scan confirmed splenomegaly. The liver scan showed a suspicion of parenchymal involvement. Inferior venacavagram and intravenous pyelogram were normal; repeat chest radiographs and tomograms of the mediastinum were within normal limits; bone scan was normal; abdominal sonogram was negative. Treatment with cyclophosphamide, prednisone, and vincristine (CVP) was initiated with a good initial response as evidenced by almost total regression of palpable lymphadenopathy and reduc- tion of the number of atypical lymphoid cells in the peripheral blood. After three courses of CVP the patient was clinically stable with minimal lymphadenopathy. The peripheral blood leukocyte count varied between 3,500 p1 and 10,000 pl. The serum lactic dehydro- genase decreased to 270 IU/ml, and repeated arterial pOz determinations were now within the normal range. Repeat pulmonary function tests were unchanged from the previously performed tests, except for an increase in the resting pOz and a slight increase in diffusion capacity. One month later, the patient was hospitalized because of pulmonary thrombo- embolism and died within a few days. Autopsy confirmed the widespread presence of malignant lymphoma.
SPECIAL STUDIES
Materials and Methods
tion of the density gradient technique of Boyum [5] . Heparinized peripheral blood (10 units of heparinlml of peripheral blood) was collected aseptically. The heparinized peripheral blood was diluted with an equal volume of Hank’s balanced salt solution (calcium and magnesium free) (GIBCO). Four ml of diluted peripheral blood was layered over 3 ml of Ficoll-Hypaque and then centrifuged at 400g for 20 minutes at room temperature. The resultant layer of mononuclear cells was aspirated from the interface and washed three times with Hank’s balanced salt solution. The final cell button was resuspended to a con- centration of 2 X lo6 mononuclear cells/ml. Cell viability was assessed by the trypan blue dye exclusion technique, and monocyte contamination was determined by latex particle ingestion. This final suspension was used for all surface marker assays, enzyme studies, and stimulation studies.
Lymphocyte cell surface markers. Lymphocyte subpopulations were determined by the following cell surface marker techniques:
1) T cells: Aliquots of lymphocytes prepared above were used to determine E-rosette- forming cells according to the technique of Wybran and Fudenberg [ 6 ] . Cells with three or more adherent sheep red cells were scored as positive. At least 200 cells were counted and the results expressed as a percentage of the total cells counted.
Preud’ Homme et al [7] . Aliquots of lymphocytes were stained with either polyvalent or
Lymphocyte preparation. Peripheral blood lymphocytes were isolated by modifica-
2) B cells: Surface immunoglobulin-bearing cells were determined by the method of

210 Spanos et a1
monospecific (for heavy chain class and light chain type) fluorescein isothiocyanate (FITC) conjugated anti-immunoglobin serum (Meloy Labs). A minimum of 200 cells were counted using a Leitz Orthoplan microscope equipped for epi-illumination. Results were expressed as a percentage of tagged cells.
3) C3 receptor-bearing cells: Lymphocytes bearing a membrane receptor for C3 were assayed according to the method of Bianco et a1 [8] . Sheep red blood cells were treated with an optimal dilution of an IgM antibody to sheep erythrocytes and reacted with a 1 : 10 dilution of mouse complement. Equal volumes of lymphocyte suspension and the EAC indicator cells were incubated at 37'C for 30 minutes. An aliquot of this mixture was placed in a hemocytometer and a minimum of 200 cells counted. Lymphocytes with three or more adherent EAC indicator cells were scored as positive. Results were expressed as a percentage of total cells counted.
4) Mouse erythrocyte rosettes: Mouse erythrocyte rosettes were determined accord- ing to the method of Gupta et a1 [lo]. In this assay 50 111 of a lymphocyte suspension (5 X lo6 cells/ml) was incubated at room temperature with 100 pl of a 1% suspension of mouse erythrocytes at room temperature for one hour. The cell pellet was gently resuspended, and at least 200 cells were counted. Results are expressed as a percentage of mouse erythro- cyte-rosette-forming cells.
their ability to respond to stimulation with phytohemagglutinin (PHA), pokeweed mitogen (PWM), and concanavalin A (CON A). These studies were done using a modification of the method of Strong et a1 [9], which has been standardized in our laboratory.
5'nucleotidase. Lymphocyte 5 ' nucleotidase was determined histochemically with a modification of Silber's method [ 11 1. Cells at least 80% covered by a black (lead sulfide) precipitate were recorded as positive. Slides were also incubated with 2'3'adenosine mono- phosphate as a control; these slides were uniformly nonreactive. A minimum of 200 cells was counted on each of duplicate slides and the results expressed as a percentage of positive cells.
-70°C at a concentration of between 5 X lo7 and 2 X lo8 cells/ml. Adenosine deaminase was assayed as described by Tung et a1 [12] . Adenosine deaminase activity was determined spectrophotometrically by measuring the conversion of adenosine to uric acid in the presence of exogenous nucleoside phosphorylase and xanthine oxidase. Results were ex- pressed as nmoles of adenosine deaminase activity / mg protein/hour at 37°C. Protein concentration was determined according to Lowry et a1 [13].
fmed at 0-4'C for three hours in 2.5% gluteraldehyde in 0.1 M cacodylate buffer with added sucrose. After buffer wash and postosmication in 1% osmium tetroxide for two hours, the tissues were dehydrated in graded alcohols and embedded in Epon 812. Sections of 0.5-1 .O I.( diameter were examined by light microscopy after staining with gentian violet. Thin sections from selected areas were cut with glass knives and mounted unsupported on 200 mesh copper grids. These were stained with uranyl acetate and lead acetate and photo- graphed with a Hitachi HS 8-1 electron microscope at 50 kV.
Blastogenic studies. Aliquots of the total lymphocyte population were studied for
Adenosine deaminase. Aliquots of the lymphocytes isolated above were frozen at
Electron microscopy. The buffy coat and portions of lymph nodes were immediately
RESULTS
Immunological Studies
Seventy-five percent of the patient's peripheral blood mononuclear cells formed rosettes with sheep erythrocytes (Fig. 2), and 10% had detectable surface immunoglobulin

An Unusual Lymphoproliferative Disorder 21 1
Fig. 2. Peripheral blood atypical lymphocytoid cell forming a rosette with sheep erythrocytes Wright- Giemsa; magnification, X 1,000).
by the use of fluorescein conjugated polyvalent antisera. There was no rosette formation with mouse erythrocytes. With EACcoated red blood cells, 7.5% of the lymphocytes formed rosettes. Of the T-cells, 5% had receptors for the Fc fragment of IgM (Tp cells), and 5% had receptors for the Fc fragment of IgG (Tr cells), as detected by a rosette assay using erythrocytes coated with rabbit IgG or IgM antibodies.
derivative, varidase, mumps antigen, and dermatophytin. There was no blastogenic lympho- cyte transformation in vitro following stimulation with phytohemagglutinin, concanavalin A, pokeweed mitogen, Candida albicans, Escherischia coli, and Staphylococcus aureus.
The phagocytic ability of the patient's peripheral blood leukocytes was tested by incubating them with staphylococcus aureus organisms. The abnormal lymphoid cells did not exhibit any phagocytosis, whereas the monocytes and granulocytes were actively phagocyt ic.
Enzyme Studies
protein/hour (normal mean 1,452 nmoles). Erythrocyte adenosine deaminase was normal. Lymphocyte 5'nucleotidase was increased in that it was present in 80% of the cells (normally detectable in 15% to 43% of lymphoid cells). Terminal deoxynucleotidyl trans- ferase assay was negative.
There was no skin reaction following the intradermal injection of purified protein
Peripheral blood lymphocyte adenosine deaminase activity was 400 nmoles/mg of

21 2 Spanos et a]
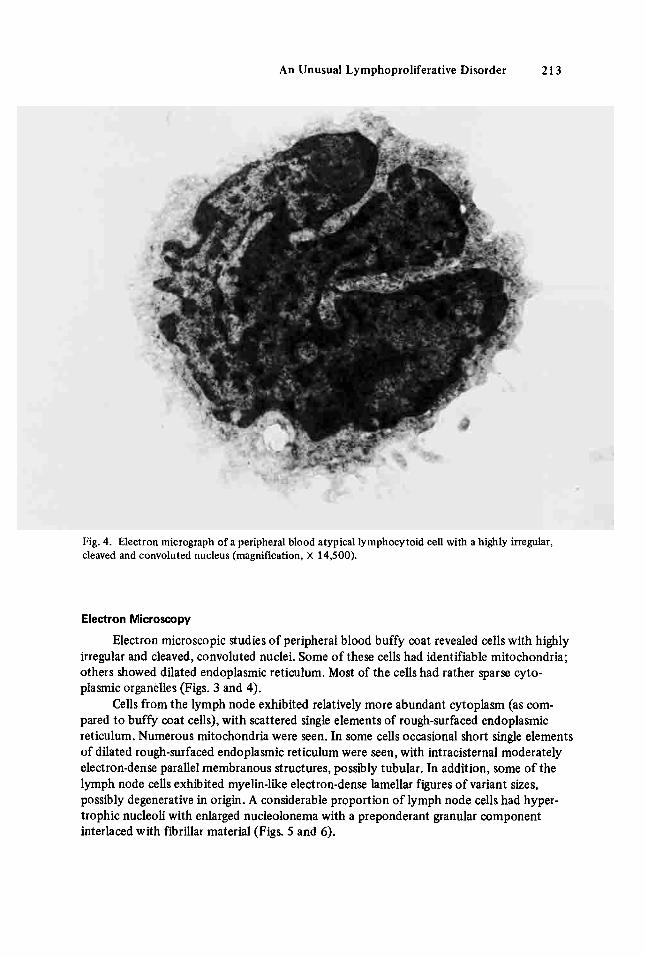
An Unusual Lymphoproliferative Disorder 213
Fig. 4. Electron micrograph of a peripheral blood atypical lymphocytoid cell with a highly irregular, cleaved and convoluted nucleus (magnification, X 14,500).
Electron Microscopy
Electron microscopic studies of peripheral blood buffy coat revealed cells with highly irregular and cleaved, convoluted nuclei. Some of these cells had identifiable mitochondria; others showed dilated endoplasmic reticulum. Most of the cells had rather sparse cyto- plasmic organelles (Figs. 3 and 4).
pared to buffy coat cells), with scattered single elements of rough-surfaced endoplasmic reticulum. Numerous mitochondria were seen. In some cells occasional short single elements of dilated rough-surfaced endoplasmic reticulum were seen, with intracisternal moderately electron-dense parallel membranous structures, possibly tubular. In addition, some of the lymph node cells exhibited myelin-like electron-dense lamellar figures of variant sizes, possibly degenerative in origin. A considerable proportion of lymph node cells had hyper- trophic nucleoli with enlarged nucleolonema with a preponderant granular component interlaced with fibrillar material (Figs. 5 and 6).
Cells from the lymph node exhibited relatively more abundant cytoplasm (as com-

214 Spanos et a1

An Unusual Lymphoproliferative Disorder 215
Fig. 6. Electron micrograph of a lymph node cell showing a myelin-like electron dense lamellar figure, possibly degenerative in origin (arrow) (magnification, X 9,200).
DISCUSSION
The classification of lymphoproliferative disorders has dramatically changed in the last few years as our understanding of immunology has increased. The new classification is based on various membrane properties and functional attributes of the lymphocytes. The known T-cell disorders described to date include mycosis fungoides, Sdzary syndrome, mediastinal lymphoma of children (Sternberg sarcoma), chronic lymphocytic leukemia (about 5 % of cases), acute lymphoblastic leukemia (20-25% of cases), immunoblastic sarcoma, and Lennert lymphoma [2].
We now recognize subpopulations of T-cells, apparently genetically programmed to perform specific functions such as the augmentation and/or supression of the transforma- tion of B-cells into immunoglobulin-producing plasma cells. These T-cells are known as helper cells and suppressor cells, respectively.
This knowledge of lymphocyte differentiation has led not only to a new classification of lymphoproliferative disorders but also to the recognition of diseases that represent homo- geneous expansion of T-cell subpopulations. For example, the SBzary syndrome has been shown, in certain instances, to represent a clonal proliferation of T-helper cells [14].
Our patient’s illness can readily be classified as a T-cell lymphoproliferative disorder based on the morphology of the cells, rosette formation with sheep erythrocytes, absence

216 Spanos et al
of surface immunoglobulin, and absence of mouse erythrocyte rosetting. However, this patient’s illness does not completely conform to known T-cell disorders.
Approximately 5% of chronic lymphocytic leukemias are of the T-cell variety and are characterized by massive splenomegaly, moderate blood and bone marrow infiltration, and frequent skin involvement and neutropenia [15] . In some of these cases, there is a poor mitogenic response t o the plant lectins phytohemagglutinin and concanavalin A. Immunoglobulin levels are usually normal or increased [ 151 . The diagnosis of chronic lymphocytic leukemia could not be sustained in our patient primarily because of the lack of marrow involvement.
Convoluted T-cell lymphoma (Sternberg sarcoma) occurs primarily in childhood and adolescence, and is characterized by a proliferation of immature cells whose nuclei have a characteristic convoluted appearance. Such patients present with infiltrating masses or enlarged lymph nodes, especially of the mediastinum. These lymphoblastic lymphomas have a tendency to transform to leukemia, with peripheral blood and bone marrow invasion. Other than the convoluted cellular appearance under electron and light microscopy, no other similarities exist between these disorders and the present case.
that occur in individuals with abnormal immune systems such as Sjogrens syndrome, rheumatoid arthritis, systemic lupus erythematosus, and in patients receiving immuno- suppressive drugs. These lymphomas may also develop in patients with preexisting immuno- blastic lymphadenopathy. The clinical course of such patients is similar to that of patients with diffuse histiocytic types of lymphomas. The latter part of our patient’s clinical course, characterized by the rapid progression of massive lymphadenopathy, is similar t o an aggres- sive type of diffuse histiocytic lymphoma, and the histopathology of our patient’s lymph node biopsy is consistent with such a diagnosis. However, our patient’s disease. with its initial leukemic phase predating lymph node and organ enlargement and without any bone marrow invasion, differs radically from diffuse histiocytic lymphoma. Furthermore, most histiocytic lymphomas are B-cell disorders.
Lennert lymphoma has been described recently as being a T-cell disorder, and seems t o combine histologic features found in Hodgkin disease with those found in angio- immunoblastic lymphadenopathy. Evaluation of a greater number of patients must f i s t take place t o firmly determine whether this disorder warrants recognition as a separate disease entity or merely represents a variant of an established disease [ 161 .
Although our patient has some features suggestive of the Sezary syndrome, there are major differences. The hallmark of this cutaneous lymphoma is the presence of characteris- tic skin lesions; this patient has no such skin lesions. Although a few cases of Sezary syndrome have been reported with no identifiable skin lesions initially, this was always a transitory phenomenon, with the characteristic lesions appearing during the course of the disease and certainly by the time an overt leukemic phase appears. Our patient had 385,000 circulating lymphocytes per pl and visceral and nodal involvement but no characteristic skin lesions. Furthermore, the rapidity of clinical progression of the disease in our patient is dissimilar to the more indolent course that one usually observes in the SCzary syndrome.
To our knowledge, there is only one report of the SCzary syndrome occurring with increased serum calcium levels [17] . In that case, osteoclast activating factor secretion by the leukemic cells and osteoblastic bone lesions were demonstrated. In the present case, the etiology o f the transient hypercalcemia remains obscure.
A recently reported patient with a T-cell lymphoproliferative disorder [ 181 charac- terized by abnormal T-cells in peripheral blood and lymph nodes but who had prominent
Immunoblastic sarcomas are thought to be lymphomas of transformed B- or T-cells

An Unusual Lymphoproliferative Disorder 217
marrow and visceral involvement at diagnosis, also had a significant unexplained hyper- calcemia, which ultimately led to the patient’s death.
once in the Szary syndrome [ 191 .
absence of anemia and thrombocytopenia despite extreme lymphocytosis. Furthermore, the histochemistry and morphology of these cells under light and electron microscopy were similar to those seen in the SBzary syndrome.
Chromosomal analysis in the Sdzary syndrome has not been very helpful. The chromosome numbers range from diploid to tetraploid, and marker chromosomes have been found in some patients [20] . Our patient had an abnormal but nonspecific karyotype.
synthesis. The absence of adenosine deaminase is associated with certain cases of severe combined immunodeficiency. The level of this enzyme has been found to be somewhat higher in T-cells than in B-cells and is increased in the SCzary syndrome [ 123. In untreated chronic lymphocytic leukemia, however, the lymphocyte adenosine deaminase level is lower than in normal controls but tends to normalize following treatment [21].
S’Nucleotidase in the lymphocytes of patients with chronic lymphocytic leukemia is usually decreased but rarely may be markedly increased [2 11 . 5‘Nucleotidase-negative and 5’nucleotidase-positive subpopulations of lymphocytes may exist. The common 5’nucleotidase-negative and rare 5’nucleotidase-positive types of chronic lymphocytic leukemia may arise from these subpopulations, accounting for the observed differences in enzyme-positive cells in this disorder [21,22] . The significance of these findings in terms of prognosis and treatment remains uncertain [23] .
early in the evolutionary pathway of lymphocytes [24] ; its absence in our patient indicates the mature nature of the cells analyzed.
us to the conclusion that the patient suffered from a Tcell malignancy that presented with a brief initial leukemic phase. The disease evolved into an undifferentiated T-cell lymphoma characterized by generalized lymph node enlargement with replacement of normal nodes by cells having the same surface markers as the abnormal circulating cells. There are signi- ficant similarities between our case and cases of lymphoblastic lymphoma in that both are T-cell diseases, have supradiaphragmatic involvement, and have a tendency to develop into leukemia. In addition, our patient’s syndrome most closely resembles SCzary Syndrome or mycosis fungocides. Skin involvement is the only aspect missing in our patient. Finally, the occurrence of hypercalcemia and hemolysis with a positive direct Coombs’ test in our patient are most unusual in SCzary syndrome. ACKNOWLEDGMENTS
commented on the electron micrographs; to Dr. R. McCaffrey, who performed the terminal deoxynucleotidyl transferase assay; to Dr. Jack Goldstein for the phagocytosis studies; to Mr. Stanley Shapiro’for preparing the electron micrographs; to Dr. Zelma Wessely for interpreting the various pathology specimens; to Ida Freiberger for performing the lymphocyte enzyme studies; to Susana Fort for performing the chromosomal studies; and to Mrs. Miriam Regenworm and Mrs. Sophie Falk for their secretarial skills.
National Leukemia Association, Inc., and the Helena Rubenstein Foundation, Inc.
Our patient also had autoimmune hemolysis, a rare finding previously reported only
As in the Sdzary syndrome, our patient’s illness spared the bone marrow; hence, the
The enzymes adenosine deaminase and S’nucleotidase are essential elements in purine
Lymphocyte terminal deoxynucleotidyl transferase seems to be restricted to cells
The clinical course and various immunological studies performed in our patient lead
The authors are indebted to Dr. Dorothea Zucker-Franklin, who reviewed and
This study was supported in part by grants from the United Leukemia Fund, Inc.,

218 Spanos e t a1
REFERENCES
1. Rappaport H: Tumors of the hematopoietic system. “Atlas of Tumor Pathology,” Sect 111:
2. Lukes RJ, Collins RD: Lukes-Collins classification and its significance. Cancer Treat Rep 61:971-
3. Lennert U, Stein H, Kaiserling E: Cytological and functional criteria for the classification of
4. Fisher RI, Kubato TT, Mandell CL, Broder S, Young RC: Regression of a T cell lymphoma after
5. Boyum A: Separation of leukocytes from blood and bone marrow. Introduction. S a n d J Clin
6. Wybran J, Fudenberg H: Thymusderived rosette forming cells in various human disease states:
Fasc 8. Washington, DC: Armed Forces Institute of Pathology, 1966,91-206.
919,1977.
malignant lymphomata. Br J Cancer 31(Suppl2):29-43,1975.
administration of antithymocyte globulin. Ann Intern Med 88:799-800, 1978.
Lab Invest 2I(Supp1)97:7,1968.
Cancer lymphoma, bacterial and viral infection and other diseases. J Clin Invest 52:1026-1032, 1973.
7. Preud’Homme JL, Seligmann M: Surface bound immunoglobulin, a cell marker in human lympho- proliferative disease, Blood 40:777-794,1972.
8. Bianco C, Patrick R, Nussenzweig V: A population of lymphocytes bearing a membrane receptor for antigen-antibody complement complexes. J Exp Med 132:702-720,1970.
9. Strong DM, Ahmed AA, Thurman GB, Sell KW: In vitro stimulation of murine spleen cells using a microculture system and a multiple automated sample harvester. J Immunol Meth 2:279-291.1973.
lymphocytes. Int Arch Allergy Appl Immunol49:734-742, 1975.
and negative subpopulations. J Clin Invest 56:1325-1327. 1975.
in chronic lymphocytic leukemia. Relationship to B and T cell subpopulations. J Clin Invest
13. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ: Protein measurement with the Folin-phenol
14. Broder S, Edelson RL, Lutzner MA, Nelson DL, MacDermott RP, Durm ME, Goldman CK, Meade
15. Brouet JC, Flandrin G, Sasportes M, Preud’Homme JL, Seligmann M: Chronic lymphocytic
10. Gupta S, Grieco MJ: Rosette formation with mouse erythrocytes: Probable marker for human B
11. Silber R, Conklyn M, Grusky G, Zucker-Franklin D: Human lymphocytes: 5’nucleotidase positive
12. Tung R, Silber R, Quagliata F,Conklyn M , Gottesman J. Huschhorn R: Adenosine deaminase activitc
57 ~756-761.
reagent. J Biol Chem 193:265-275, 1951.
BD, Waldman TA: The Szary syndrome. J Clin Invest 58:1297,1306, 1976.
leukaemia of T cell origin. Immunological and clinical evaluation in 11 patients. Lancet 82390-893, 1975.
lymphoma). Am J Clin Pathol66:l-9, 1976. 16. Burke JS, Butler JJ: Malignant lymphoma with a high content of epithelioid Histiocytes (Lennert’s
17. Broder S: Personal communication, May 1978. 18. Stryckmans PA, Debusscher L, Heyder-Briickner C, Heimann R, Mandelbaum IM, Wybran J: Clonal
19. Amrein P, Ellman L: s z a r y syndrome with warm and cold erythrocyte autoantibodies. Blood
20. Lutzner M, Edelson R, Schein P, Green I, Kirkpatrick C, Ahmed A: Cutaneous T cell lymphomas:
21. LaMantia K, Conklyn M, Quagliata F, Silber R: Immunologic studies of 5’-nucleotidase in normal
22. Kanter RJ, Freiberger IA, Rai KR, Sawitsky A: Lymphocyte populations with S’nucleotidase in
23. Quagliata F, Faig D, Conklyn M, Silber R: Studies on the lymphocyte 5’ nucleotidase in chronic
origin of a T cell lymphoproliferative malignancy. Blood 52:69-76, 1978.
51 ~1229-1230, 1978.
The Szary syndrome, mycosis fungoides, and related disorders. Ann Intern Med 83534-552, 1975.
and chronic lymphocytic leukemia lymphocytes. Blood 46:1042. 1975 (abstr 125).
chronic lymphocyticleukemia. Clin lmmunol lmmunopathol 12:351-357. 1979.
lymphocytic leukemia, infectious mononucleosis, normal subpopulations and phytohemagglutinin- stimulated cells. Qncer Res 34:3197-3202, 1974.
24. Siegal FP, Good RA: Human lymphocyte differentiation markers and their application to immune deficiency and lymphoproliferative diseases. Clin Hematol6:355-423, 1977.





























