Embed Size (px)
DESCRIPTION
neurofibromatosis
Citation preview

E - 4-092-C-10
Neurofibromatosis
Resumen. - Las neurofibromatosis (NF) incluyen al menos dos enfermedades bien identificadas de transmisiónautosómica dominante, la NF1 y la NF2. El gen NF1 ha sido localizado en el cromosoma 17 y el gen NF2 en elcromosoma 22. La NF1 representa el 95 % del conjunto de las NF. Su incidencia es aproximadamente de uncaso por cada 3 500 nacimientos y su prevalencia de una persona por cada 4 000 aproximadamente. La NF1se caracteriza por sus manifestaciones cutáneas, manchas de color café con leche, lentigos y neurofibromas. LaNF2 tiene una incidencia de 1 por cada 33 000 a 40 000 nacimientos. Se caracteriza por schwannomas vesti-bulares bilaterales (antiguamente denominados neurinomas del acústico), schwannomas de otros nervios cra-neales y espinales y meningiomas. Las schwannomatosis o neurilemomatosis son excepcionales y se caracteri-zan por schwannomas múltiples cutáneos y subcutáneos sin schwannomas vestibulares. Según el tipo de NF, elpronóstico, las complicaciones, el consejo genético y el tratamiento son diferentes. Los centros multidisciplina-rios, con sus grupos de expertos, son las estructuras de elección para la asistencia de estos enfermos.
Palabras clave: neurofibromatosis 1, neurofibromatosis 2, manchas de color café con leche, lentigos, schwan-nomatosis, neurilemomatosis, schwannoma, neurofibroma, tumores malignos de las vainas nerviosas.
C 2002, Editions Scíentifiques et Médicales Elsevier SAS, París. Todos los derechos reservados.
P Wolkenstein1 ZellerN Ismaili
Introducción
Bajo el término de neurofibromatosis (NF) seagrupan distintas enfermedades que la
mayor parte de las veces sólo tienen encomún ciertos signos cutáneos ~’3.’z, m: man-chas pigmentarias y tumores. Todas estasafecciones han sido designadas durantemucho tiempo con el nombre de enfermedadde Von Recklinghausen. Actualmente, el tér-mino de enfermedad de Von Recklinghausensólo designa a la más frecuente de las neuro-fibromatosis, la NFl. Forman parte de estegrupo heterogéneo al menos dos enfermeda-des bien identificadas, de transmisión auto-sómica dominante: la NFl y la NF2.
NosologíaHISTORIA NOSOLÓGICA ~’z1
En el siglo XIII, el monje Henricus describióen un manuscrito húngaro al primer enfermoafectado por NF. En el siglo XVI, Aldrovandide Bolonia, en su obra Monstorum historia,
Pierre Wolkenstein : Praticien hospitalier.lacques Zeller : Attaché consultant.Nadia Ismaili : Dermatologue.~rMC~ dp dprmofo/og/p ~ rpx~ou MF, hóp/fo/ ~f7r/ Mondos 3 ~ o~-Service de dermatologie et réseau NF, hópital Henri Mondor, 51, ave-nue du Maréchal-de-Lattre-de-Tassigny, 94010 Créteil cedex, France.
describió un tumor específico de la NF1, elneurofibroma plexiforme. En el siglo XVIII,Buffon, en su Historia natural, refiere el casode un niño que tenía tumores cutáneos ymanchas pigmentadas. Cruveilhier, en suAnatomía patológica del cuerpo, da la primeradescripción precisa de la NFl. Más tarde,Akenside, Ludwig y Tilesius describen susdiferentes aspectos y reconocen la posibilidadde transmisión familiar. Smith en 1849,Hitchcock en 1862 y Von Recklinghausen en1882 precisan su descripción. Wishart (1822)y Knoblauch (1843) describen los «neurino-mas acústicos», actualmente denominadosschwannomas vestibulares, la principalmanifestación de la NF2. Smith y Von
Recklinghausen y, por último, Henneberg yKoch en 1903 describen posteriormente losneurinomas. En 1953, Schull y Neel precisa-ron la importancia diagnóstica de los trastor-nos pigmentarios en lo que siempre se consi-deró que era una afección única denominadaenfermedad de Von Recklinghausen y queagrupa, de hecho, al menos dos enfermeda-des, la NF1 y la NF2. Waardenbourg y mástarde Lich señalaron la frecuencia de losnódulos del iris. La confusión nosológica pro-siguió hasta que en 1970 los trabajos deYoung, Eldridge y Gardner y, posteriormente,los de Riccardi y Mulvihill, establecieron defi-nitivamente la separación de las dos entida-des, la NF1 y la NF2.
A finales de la década de 1980, el caso deJohn Merrick, el hombre elefante, consideradodurante mucho tiempo como una NF1, seasoció al síndrome de Proteo.
NOSOLOGÍA MODERNA
A partir de la conferencia de consenso delNational Institute of Health (NIH) celebrada en1988, se precisaron los criterios diagnósticosde la NF1 y la NF2 (cuadros 1, ll) 1271.
La clasificación de Riccardi (cuadro lll) pre-tendía distinguir otros cuadros clínicos maladaptados a los cuadros precedentes.La validez de los criterios diagnósticos de laconferencia de consenso del NIH ha sido con-firmada por la biología molecular que permi-tió identificar dos genes distintos para la NF1
y la NF2: el gen NF1 está situado en el cromo-soma 17 y el gen NF2 en el cromosoma 22.
Neurofibromatosis 1 oenfermedad de VonRecklinghausen 115,33,38,46]
EPIDEMIOLOGÍA
La NF1 es la enfermedad autosómica domi-nante más frecuente, con una incidencia deaproximadamente 1 de cada 3 000 a 3 500 na-

2
Cuadro I. - Criterios diagnósticos de la neu-rofibromatosis 1 (NF1) (conferencia de con-senso del National Institute of Health, 1988).
Cuadro 11. - Criterios diagnósticos de neuro-fibromatosis 2 (NF2) (conferencia de consen-so del National Institute of Health, 1988).
Cuadro III. - Clasificación de las neurofibro-matosis (NF) según Riccardi.
1
cimientos. Representa el 95 % del conjuntode las NF. Su prevalencia es aproximada-mente de 1 de cada 4 000 personas. Se trans-mite de forma autosómica dominante, sinpredilección étnica. La mitad de los casos sonesporádicos, lo que refleja la frecuencia de lasmutaciones espontáneas. La variación fenotí-pica es importante, incluso en el seno de unamisma familia.
Su morbimortalidad se relaciona con la apa-rición de complicaciones multisistémicas,tumores cerebrales, tumores malignos de lasvainas nerviosas (TMVN) y vasculopatías.
GEN NF1’znu,39~
El gen NFl se identificó mediante clonación
posicional en 1990. Abarca más de 350 kilo-bases en la región pericentromérica del cro-mosoma 17 (17qll.2) y está compuesto por59 exones " ID="I192.28.3">[19]. Se trata de un gen de gran tama-ño cuya exploración es difícil, tanto más por-que existen secuencias homólogas localiza-das en los cromosomas 15, 21 y 22.
La penetrancia del gen se acerca al 100 % alos 5 años.El producto del gen NF1, la neurofibromina,es una proteína citoplasmática compuestapor 2 818 aminoácidos, con una región de 360aminoácidos (dominio GRD) similar al domi-nio catalítico del tipo de la GTPasa de lasproteínas p120GAP de los mamíferos, IRAI,IRA2 y sarl de levaduras (Saccharomyces cere-visiae y S. pombe) y GAP1 de Drosophila. Lafunción común de estas moléculas es estimu-lar la conversión de la forma activa de la pro-teína p2lras unida al GTP en una forma inac-tiva unida al GDP. Por consiguiente, la neu-rofibromina interviene con mucha probabili-dad en el control de la diferenciación y la
proliferación celular. No obstante, los datosactuales no permiten definir de manera for-mal si este control es previo o posterior a laactivación de p2lras. Ciertos resultados per-miten pensar que la neurofibromina podríaser, según el tipo celular, un regulador nega-tivo o un efector directo de p2lras. Existenvarias isoformas de la neurofibromina rela-cionadas con fenómenos de corte y empalmealternativo. La isoforma II, caracterizada porla inserción de 21 aminoácidos en el dominio
GRD, posee una distribución hística seme-jante a la de la isoforma 1. Por el contrario, laisoforma III, caracterizada por la inserción de18 aminoácidos en la parte carboxiterminalde la proteína, se expresa preferentemente enel músculo cardíaco, los músculos esqueléti-cos y lisos. Existe una cuarta isoforma queprocede de la inserción de 10 aminoácidosentre los residuos 420 y 421 de la neurofibro-mina y cuya expresión parece restringirse alsistema nervioso central.
Aproximadamente el 5 % de los pacientesafectados por la NF1 desarrollan tumores
malignos, lo que sugiere que el gen NF1 es ungen supresor tumoral, hecho recientementedemostrado. Por otro lado y, de acuerdo conla hipótesis de Knudson, se han encontradopérdidas de la heterocigosis del gen NF1 en lamayor parte de los tipos de tumores desarro-llados por los pacientes que presentan unaNF1: TMVN (antiguos neurofibrosarcomas),schwannomas, síndromes mielodisplásicos,feocromocitomas e incluso, muy reciente-mente, neurofibromas benignos.Existen grandes variaciones fenotípicas intere intrafamiliares en la NFl. Los criterios
diagnósticos mayores de la NF1, por ejem-plo, las manchas de color café con leche
(MCL), los neurofibromas cutáneos y losnódulos de Lisch están presentes en más del90 % de los casos en la pubertad, aunque elnúmero de lesiones varía enormemente deun individuo a otro. Por el contrario, ciertasmanifestaciones de la NF1 sólo se producenen una minoría de los enfermos; es el caso delas dificultades de aprendizaje, los gliomas delas vías ópticas, las crisis epilépticas, las seu-doartrosis y las escoliosis, las MCL y los feo-cromocitomas. Una minoría significativa deenfermos, aproximadamente del 15 al 20 %,sufre una morbilidad importante relacionadacon la NF1 mientras que la mayoría sólo estáafectada por formas moderadas.
Algunas de estas variaciones de expresiónfenotípica podrían reflejar diferentes muta-ciones en el gen NFl aunque hasta la fecha
hayan sido infructuosos los intentos de esta-blecer una correlación genotipo-fenotipo.Sólo las deleciones importantes del gen NF1han podido asociarse a un fenotipo particu-lar " ID="I192.109.2">(161. Por otra parte, varios grupos intentanactualmente caracterizar las bases molecularesde las asociaciones NFl-síndrome de Noonan.Los datos actuales no permiten definir si estaasociación es una forma alélica de NFl, comoparece ser el caso en el síndrome de Watson, osi está relacionada con la existencia de dosmutaciones distintas situadas en genes genéti-camente ligados (genes contiguos).No obstante, se ha establecido que estas
variaciones en la mutación NF1 no son laúnica fuente de variaciones fenotípicas.Existe también una diferencia de expresiónconsiderable en una misma familia. La varia-ción fenotípica interindividual podría estarrelacionada con genes modificadores, facto-res ambientales o ambos. Los genes modifi-cadores no están situados en el locus NF1
pero su actividad o su represión llevaría a lamodificación de la expresión fenotípica. Estahipótesis parece haber sido confirmadaactualmente. El número de MCL, de NF, laaparición o no de gliomas ópticos, escoliosis,epilepsia o dificultades de aprendizaje pare-cen depender de genes modificadores.
DIAGNÓSTICO CLÍNICO ’?1
Signos cardinales dermatológicosManchas de color café con leche
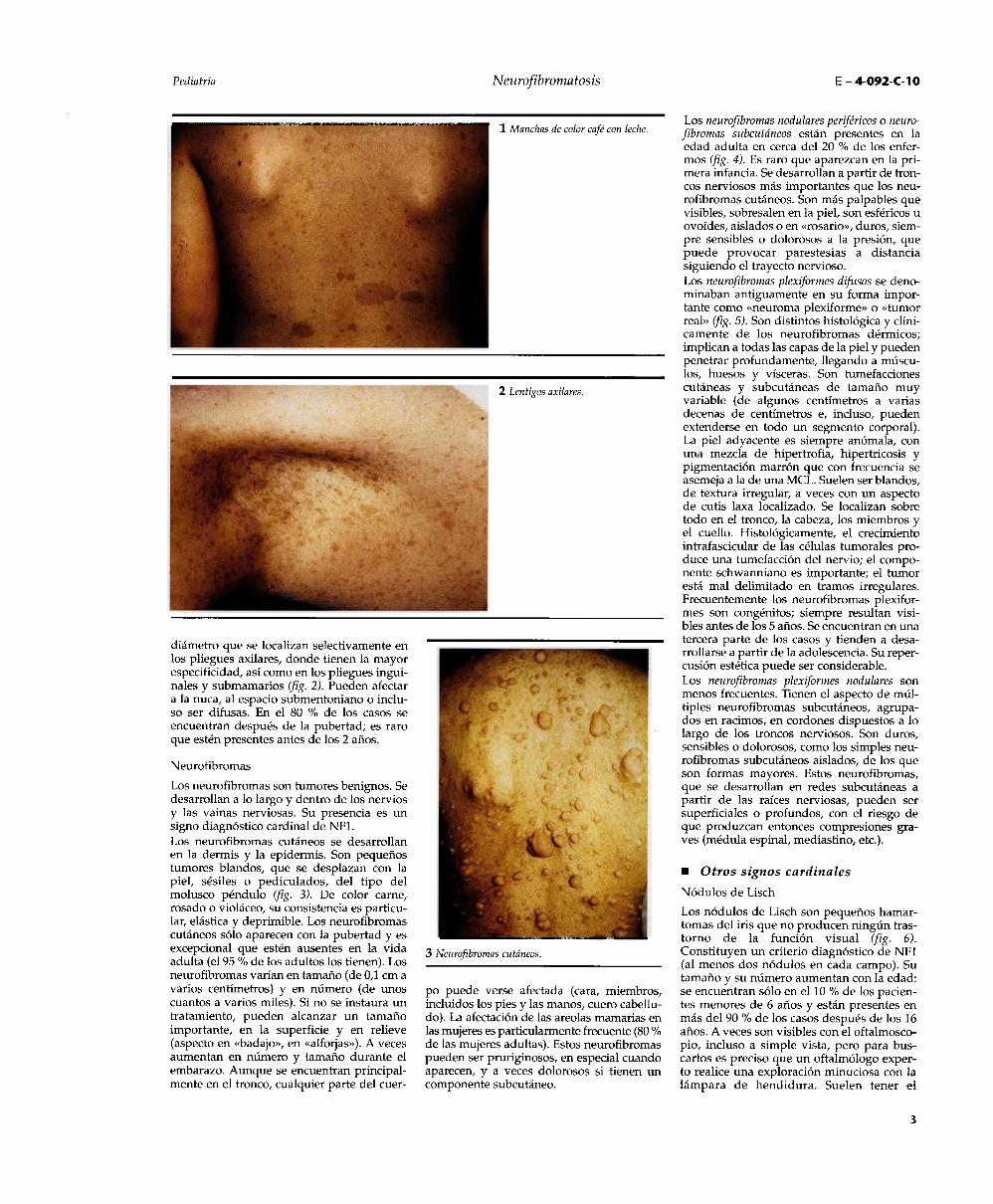
Las MCL son las primeras manifestacionesde la NFI (fig. 1). A menudo son congénitas yaparecen raramente después de los 2 años.Su distribución es aleatoria, sus contornosestán bien delimitados y su tono es marrónmás o menos oscuro, a veces difícilmente
perceptible. El diámetro de las MCL varíaentre 0,5 y 50 cm pero la mayoría midemenos de 10 cm. Las anomalías histológicasse resumen en una gran hiperpigmentaciónen foco de la membrana basal. En el micros-
copio electrónico, los melanosomas gigantesson muy frecuentes pero en la actualidad seconsidera que son poco específicos. Igual-mente, el aumento de las terminaciones ner-viosas que se observó más recientemente enlas MCL no es específico de la NF1 sino quetambién se encuentra en los nevos gigantescongénitos. En la adolescencia, las MCLestán presentes en más del 90 % de los casos.En el transcurso de la vida adulta, las MCLsuelen ser más pálidas, poco visibles y algu-nas desaparecen. Las MCL constituyen losmejores signos diagnósticos de la NF1 y casisiempre están presentes antes de los 5 años deedad. Es importante considerar el tamaño y elnúmero de MCL: las MCL mayores de 0,5 cmen la infancia o de al menos 1,5 cm despuésde la pubertad tienen valor diagnóstico siem-pre que estén en una cantidad igual o supe-rior a seis (más del 25 % de los niños en edadescolar y el 14 % de los adultos tienen de unaa cinco MCL).
LentigosLos lentigos tienen el aspecto de una MCL detamaño pequeño, máculas de 1 a 3 mm de
3
1 Manchas de color café con leche.
2 Lentigos axilares.
diámetro que se localizan selectivamente enlos pliegues axilares, donde tienen la mayorespecificidad, así como en los pliegues ingui-nales y submamarios (fig. 2). Pueden afectara la nuca, al espacio submentoniano o inclu-so ser difusas. En el 80 % de los casos seencuentran después de la pubertad; es raroque estén presentes antes de los 2 años.
Neurofibromas
Los neurofibromas son tumores benignos. Sedesarrollan a lo largo y dentro de los nerviosy las vainas nerviosas. Su presencia es unsigno diagnóstico cardinal de NF1.Los neurofibromas cutáneos se desarrollanen la dermis y la epidermis. Son pequeñostumores blandos, que se desplazan con lapiel, sésiles o pediculados, del tipo delmolusco péndulo (fig. 3). De color carne,rosado o violáceo, su consistencia es particu-lar, elástica y deprimible. Los neurofibromascutáneos sólo aparecen con la pubertad y esexcepcional que estén ausentes en la vidaadulta (el 95 % de los adultos los tienen). Losneurofibromas varían en tamaño (de 0,1 cm avarios centímetros) y en número (de unoscuantos a varios miles). Si no se instaura untratamiento, pueden alcanzar un tamañoimportante, en la superficie y en relieve
(aspecto en «badajo», en «alforjas»). A vecesaumentan en número y tamaño durante elembarazo. Aunque se encuentran principal-mente en el tronco, cualquier parte del cuer-
3 Neurofibromas cutáneos.
po puede verse afectada (cara, miembros,incluidos los pies y las manos, cuero cabellu-do). La afectación de las areolas mamarias enlas mujeres es particularmente frecuente (80 %de las mujeres adultas). Estos neurofibromaspueden ser pruriginosos, en especial cuandoaparecen, y a veces dolorosos si tienen uncomponente subcutáneo.
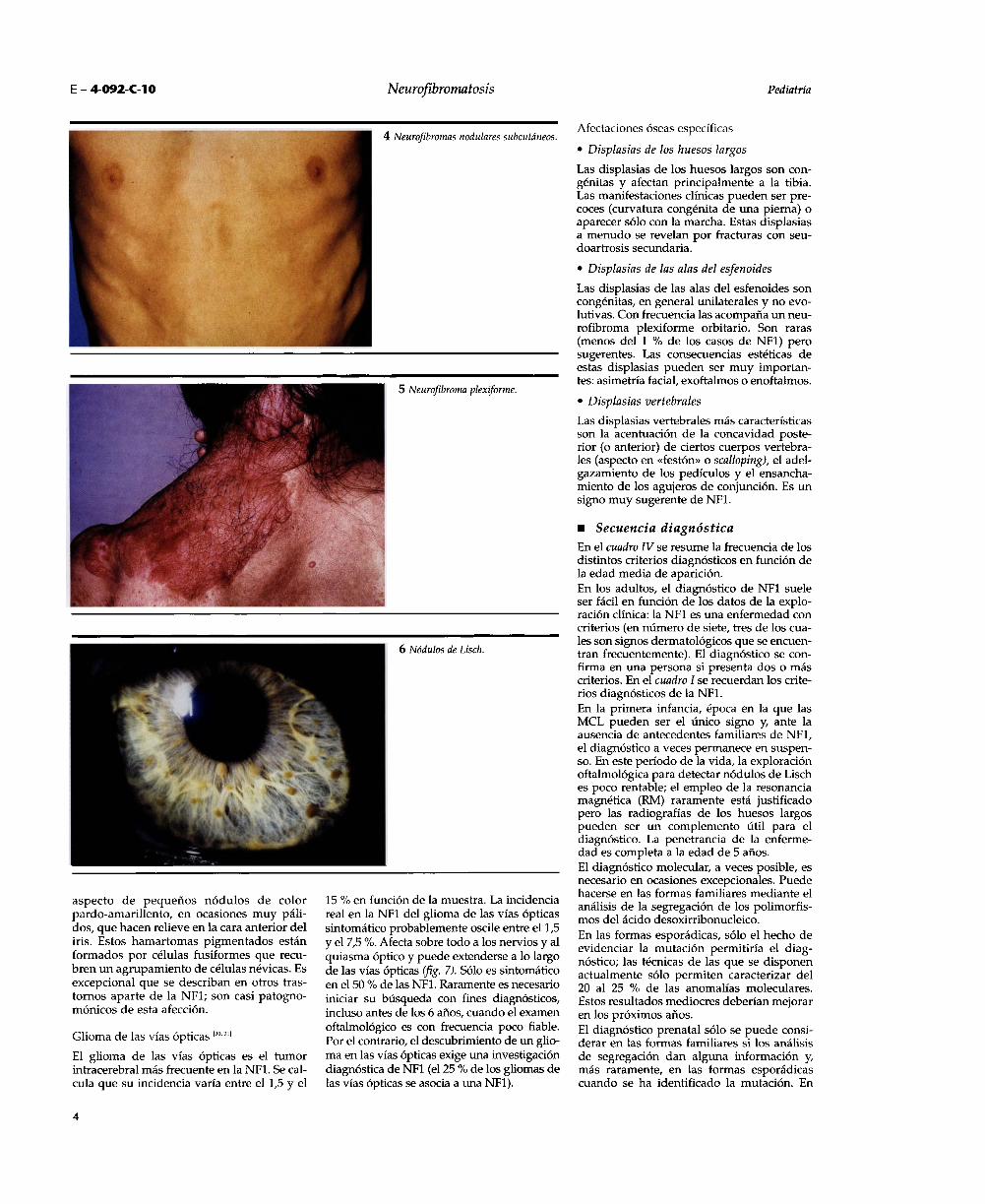
Los neurofibromas nodulares periféricos o neuro-fibromas subcutáneos están presentes en laedad adulta en cerca del 20 % de los enfer-mos (fig. 4). Es raro que aparezcan en la pri-mera infancia. Se desarrollan a partir de tron-cos nerviosos más importantes que los neu-rofibromas cutáneos. Son más palpables quevisibles, sobresalen en la piel, son esféricos uovoides, aislados o en «rosario», duros, siem-pre sensibles o dolorosos a la presión, quepuede provocar parestesias a distancia
siguiendo el trayecto nervioso.Los neurofibromas plexiformes difusos se deno-minaban antiguamente en su forma impor-tante como «neuroma plexiforme» o «tumorreal» (fig. 5). Son distintos histológica y clíni-camente de los neurofibromas dérmicos;implican a todas las capas de la piel y puedenpenetrar profundamente, llegando a múscu-los, huesos y vísceras. Son tumefaccionescutáneas y subcutáneas de tamaño muyvariable (de algunos centímetros a variasdecenas de centímetros e, incluso, puedenextenderse en todo un segmento corporal).La piel adyacente es siempre anómala, conuna mezcla de hipertrofia, hipertricosis ypigmentación marrón que con frecuencia seasemeja a la de una MCL. Suelen ser blandos,de textura irregular, a veces con un aspectode cutis laxa localizado. Se localizan sobretodo en el tronco, la cabeza, los miembros yel cuello. Histológicamente, el crecimientointrafascicular de las células tumorales pro-duce una tumefacción del nervio; el compo-nente schwanniano es importante; el tumorestá mal delimitado en tramos irregulares.Frecuentemente los neurofibromas plexifor-mes son congénitos; siempre resultan visi-bles antes de los 5 años. Se encuentran en unatercera parte de los casos y tienden a desa-rrollarse a partir de la adolescencia. Su reper-cusión estética puede ser considerable.Los neurofibromas plexiformes nodulares sonmenos frecuentes. Tienen el aspecto de múl-tiples neurofibromas subcutáneos, agrupa-dos en racimos, en cordones dispuestos a lolargo de los troncos nerviosos. Son duros,sensibles o dolorosos, como los simples neu-rofibromas subcutáneos aislados, de los queson formas mayores. Estos neurofibromas,que se desarrollan en redes subcutáneas a
partir de las raíces nerviosas, pueden sersuperficiales o profundos, con el riesgo deque produzcan entonces compresiones gra-ves (médula espinal, mediastino, etc.).
Otros signos cardinalesNódulos de Lisch
Los nódulos de Lisch son pequeños hamar-tomas del iris que no producen ningún tras-torno de la función visual (fig. 6).Constituyen un criterio diagnóstico de NF1(al menos dos nódulos en cada campo). Sutamaño y su número aumentan con la edad:se encuentran sólo en el 10 % de los pacien-tes menores de 6 años y están presentes enmás del 90 % de los casos después de los 16años. A veces son visibles con el oftalmosco-
pio, incluso a simple vista, pero para bus-carlos es preciso que un oftalmólogo exper-to realice una exploración minuciosa con lalámpara de hendidura. Suelen tener el
4
4 Neurofibromas nodulares subcutáneos.
5 Neurofibroma plexiforme.
6 Nódulos de Lisch.
aspecto de pequeños nódulos de color
pardo-amarillento, en ocasiones muy páli-dos, que hacen relieve en la cara anterior deliris. Estos hamartomas pigmentados estánformados por células fusiformes que recu-bren un agrupamiento de células névicas. Esexcepcional que se describan en otros tras-tornos aparte de la NFl; son casi patogno-mónicos de esta afección.
Glioma de las vías ópticas " ID="I194.13.6">120,21]
El glioma de las vías ópticas es el tumorintracerebral más frecuente en la NF1. Se cal-cula que su incidencia varía entre el 1,5 y el
15 % en función de la muestra. La incidenciareal en la NF1 del glioma de las vías ópticassintomático probablemente oscile entre el 1,5y el 7,5 %. Afecta sobre todo a los nervios y al
quiasma óptico y puede extenderse a lo largode las vías ópticas (fig. 7). Sólo es sintomáticoen el 50 % de las NFl. Raramente es necesarioiniciar su búsqueda con fines diagnósticos,incluso antes de los 6 años, cuando el examen
oftalmológico es con frecuencia poco fiable.Por el contrario, el descubrimiento de un glio-ma en las vías ópticas exige una investigacióndiagnóstica de NF1 (el 25 % de los gliomas delas vías ópticas se asocia a una NF1). ).
Afectaciones óseas específicaso Displasias de los huesos largosLas displasias de los huesos largos son con-génitas y afectan principalmente a la tibia.Las manifestaciones clínicas pueden ser pre-coces (curvatura congénita de una pierna) oaparecer sólo con la marcha. Estas displasiasa menudo se revelan por fracturas con seu-doartrosis secundaria.
o Displasias de las alas del esfenoidesLas displasias de las alas del esfenoides soncongénitas, en general unilaterales y no evo-lutivas. Con frecuencia las acompaña un neu-rofibroma plexiforme orbitario. Son raras(menos del 1 % de los casos de NF1) perosugerentes. Las consecuencias estéticas deestas displasias pueden ser muy importan-tes : asimetría facial, exoftalmos o enoftalmos.
o Displasias vertebralesLas displasias vertebrales más característicasson la acentuación de la concavidad poste-rior (o anterior) de ciertos cuerpos vertebra-les (aspecto en «festón» o scalloping), el adel-gazamiento de los pedículos y el ensancha-miento de los agujeros de conjunción. Es unsigno muy sugerente de NFl.
a Secuencia diagnósticaEn el cuadro IV se resume la frecuencia de losdistintos criterios diagnósticos en función dela edad media de aparición.En los adultos, el diagnóstico de NFl sueleser fácil en función de los datos de la explo-ración clínica: la NF1 es una enfermedad concriterios (en número de siete, tres de los cua-les son signos dermatológicos que se encuen-tran frecuentemente). El diagnóstico se con-firma en una persona si presenta dos o máscriterios. En el cuadro 1 se recuerdan los crite-rios diagnósticos de la NFl.En la primera infancia, época en la que lasMCL pueden ser el único signo y, ante laausencia de antecedentes familiares de NF1,el diagnóstico a veces permanece en suspen-so. En este período de la vida, la exploraciónoftalmológica para detectar nódulos de Lisches poco rentable; el empleo de la resonanciamagnética (RM) raramente está justificadopero las radiografías de los huesos largospueden ser un complemento útil para el
diagnóstico. La penetrancia de la enferme-dad es completa a la edad de 5 años.El diagnóstico molecular, a veces posible, esnecesario en ocasiones excepcionales. Puedehacerse en las formas familiares mediante elanálisis de la segregación de los polimorfis-mos del ácido desoxirribonucleico.En las formas esporádicas, sólo el hecho deevidenciar la mutación permitiría el diag-nóstico ; las técnicas de las que se disponenactualmente sólo permiten caracterizar del20 al 25 % de las anomalías moleculares.Estos resultados mediocres deberían mejoraren los próximos años.El diagnóstico prenatal sólo se puede consi-derar en las formas familiares si los análisisde segregación dan alguna información y,más raramente, en las formas esporádicascuando se ha identificado la mutación. En

5
7 Glioma de las vías ópticas.
Cuadro IV. -Frecuencia de los diferentes criterios diagnósticos de neurofibromatosis 1 en fun-ción de la edad media de aparición.
estos casos, puede hacerse una biopsia de lasvellosidades coriónicas hacia la décima oundécima semana de gestación, de modo quese pueda autentificar la ausencia o presenciade transmisión del gen NF1 mutado en lascélulas embrionarias. La biopsia de las vello-sidades coriónicas implica un riesgo de abor-to reducido. El diagnóstico prenatal sólo serealiza si los padres pretenden interrumpir elembarazo si el embrión fuera portador delgen NF1 mutado. Además de las eventuales
oposiciones personales de orden moral,sobre todo religioso, se sigue recurriendo deforma muy limitada al diagnóstico prenatal,fundamentalmente debido a la imposibili-dad de predecir la gravedad fenotípica de laforma eventualmente transmitida: del 15 al20 % de los enfermos están gravemente afec-tados por la mutación NFl.No es posible realizar un diagnóstico previoa la implantación.
COMPLICACIONES za
manifestaciones ortopédicasLas cifoescoliosis son frecuentes. La mayoríade las veces se trata de afectaciones discretas,poco evolutivas, inespecíficas, sin distrofiaósea asociada y controlables mediante méto-dos de reeducación funcional o de contención.
Las escoliosis graves son raras (menos del 5 %de los casos), debidas a distrofias vertebralesmás o menos extendidas (deformación de loscuerpos vertebrales, adelgazamiento de lospedículos, ensanchamiento del conductoraquídeo), con o sin distrofia costal. Puedentener una repercusión cardiorrespiratoriaimportante y asociarse a meningoceles o aneurofibromas nodulares paravertebrales ointravertebrales que conllevan el riesgo decompresión medular. Estos trastornos de laestática vertebral aparecen sobre todo en la
primera infancia. Las manifestaciones gravesrequieren tratamiento quirúrgico.Las seudoartrosis afectan del 1 al 3 % de losenfermos de NF1. Su tratamiento, complejo,largo y difícil, conduce a menudo a la ampu-tación, tras años de esfuerzo. La introducciónde técnicas de microcirugía (injertos vascula-rizados) ha mejorado su pronóstico.En el 2 % de los casos se ha comunicado unadeformación del esternón: tórax en embudo
y pectus carinatum. La macrocefalia es fre-cuente en ausencia de anomalías estructura-les o de trastornos funcionales cerebrales, asícomo la talla reducida.
Manifestaciones y complicacionesneurológicas
Pueden desarrollarse astrocitomas de dife-rentes tipos, la mayoría de las veces pilocíti-
cos, casi siempre en la línea media, a veces enla fosa posterior, los hemisferios cerebrales ola médula. Generalmente se consideran pocoo nada evolutivos. Sin embargo, algunospueden producir trastornos graves y evolu-cionar hacia el astrocitoma anaplásico. Lahidrocefalia, presente en el 2 % de los casos,suele ser secundaria a una estenosis del acue-ducto de Silvio, sin tumor identificable. Sehan comunicado casos de vasculopatías,estenosis y aneurismas. La epilepsia parecetener mayor frecuencia. Se producen cefa-leas, a menudo migrañosas, en una gran pro-porción de enfermos.Los neurofibromas nodulares son a veces
múltiples y macizos y producen graves com-presiones medulares (en caso de localizaciónintradural) o nerviosas periféricas (de las raí-ces o los plexos).Las dificultades de aprendizaje constituyenun problema importante de la NFl 16,28]. Sedestacan por su frecuencia, del 30 al 40 % delos casos, y por sus aspectos atípicos. Estostrastornos neurocognitivos en ocasiones al-teran considerablemente la escolarizaciónmientras que los retrasos mentales propia-mente dichos tienen una frecuencia compara-ble a la de la población general. El cuadro seasemeja con frecuencia al de un síndrome dedéficit de atención con o sin hiperactividad:trastornos de la atención, dificultades decoordinación motora, déficit de la memoriareciente, trastornos perceptivos, sobre todovisuales, que producen dificultad para dibu-jar, escribir, leer, calcular, realizar mapas ydiagramas, orientarse en el espacio y desci-frar los gestos; con frecuencia se asocian difi-cultades de la elocución, trastornos disártri-cos y también prosódicos. Desde el punto devista práctico, es importante que los padres ylos profesores sean informados desde la guar-dería sobre las dificultades de aprendizaje,con el fin de proponer precozmente una eva-luación y una ayuda adaptadas a cada caso.Las RM cerebrales de los jóvenes afectadospor la NF1 muestran en el 50 al 70 % de loscasos hiperseñales en T2: zonas bien circuns-critas, hiperintensas sin efecto de masa, obje-tos brillantes no identificados. Estas anoma-lías tienden a desvanecerse en la edad adul-ta. Su significación es incierta; excepto unaasociación posible de estas imágenes contrastornos cognitivos, hasta ahora no se haseñalado ninguna otra anomalía clínica. Enocasiones es difícil interpretar las imágenescerebrales y a veces es delicada la diferencia-ción con un tumor cerebral.
Manifestaciones y complicacionesendocrinas
El feocromocitoma, casi siempre suprarrenal,está presente en menos del 1 % de los casos enla edad adulta y excepcionalmente en la infan-cia. El feocromocitoma suele ser aislado aun-
que, en casos excepcionales, puede formarparte de un síndrome de neoplasias endocri-nas múltiples y afectar a una o ambas glándu-las suprarrenales. En raras ocasiones el feocro-mocitoma se localiza en la bifurcación aórticao en el mediastino. Es muy raro que sea asin-tomático y se asocia la mayor parte de lasveces a signos clínicos intermitentes: crisis
hipertensivas con crisis de sudación, ansie-

6
dad, agitación, cefalea, palpitaciones. La
hipertensión puede ser permanente. La escasafrecuencia del feocromocitoma en la NF1 y sucarácter raramente asintomático hacen que nosea justificable la determinación sistemáticade la concentración de catecolaminas.Los trastornos de la pubertad son raros yasea la pubertad precoz asociada a un gliomadel quiasma o retrasos de la pubertad en el1,5 % de los casos. La pubertad precoz reve-la un tercio de los gliomas de las vías ópti-cas. Es posible observar casos de pubertadprecoz sin afectación de las vías ópticas.
Manifestaciones y complicacionesoftalmológicas
La principal complicación oftalmológica dela NFl es el glioma de las vías ópticas.Aproximadamente el 50 % de los pacientestienen anomalías en la exploración oftalmo-lógica en el momento del diagnóstico delglioma [211. Se trata, la mayor parte de lasveces, de una reducción de la agudeza visualunilateral o bilateral y, más raramente, de unexoftalmos. Estas anomalías pueden ser asin-tomáticas. Se debe insistir especialmente enla dificultad del diagnóstico de reducción dela agudeza visual en niños pequeños 1", 221.Los signos endocrinos son más raros (puber-tad precoz) y están asociados a gliomas delocalización quiasmática. La historia naturalde los gliomas de las vías ópticas en el cursode la NF1 es muy variable: es raro observaruna evolución clínica o radiológica duranteel seguimiento de estos tumores incluso aun-que la lesión sea clínicamente sintomática enel momento del diagnóstico. No obstante, lasevoluciones rápidas que comprometen el
pronóstico visual y vital son posibles y total-mente imprevisibles [221.El período de riesgo de aparición de las com-plicaciones es la primera infancia pero el con-trol clínico (oftalmológico, neurológico yendocrinológico) de estos pacientes debe serprolongado.Sólo se considera una acción terapéutica encaso de un glioma de las vías ópticas que seasintomático y evolutivo.
Se pueden observar muchas anomalías en elcurso de la NF1: lesiones coroideas hamarto-matosas (del 35 al 50 % de los casos); hiper-trofia de los nervios corneales (15 % de loscasos); ptosis palpebral aislada (9 % de loscasos) o asociada a un neurofibroma palpe-bral u orbitario; anomalías de la convergen-cia ; glaucoma congénito (0,5 % de los casos)a menudo asociado a un neurofibroma plexi-forme palpebral.
Trastornos arteriales
Las displasias arteriales parietales fibro-musculares son frecuentes y pueden afectar ala aorta, las arterias mesentéricas, pulmona-res, cerebrales y renales. Esta última localiza-ción a menudo se revela por la presencia dehipertensión. Junto con el feocromocitomasuprarrenal, es la segunda causa de hiperten-sión arterial de la NFI. A veces se puede tra-tar mediante una angioplastia endoluminalpercutánea.
Se han publicado casos de vasculopatías indu-cidas por la radioterapia en niños afectadospor la NF1 y por gliomas de las vías ópticas, loque refleja una susceptibilidad particular ~8l
Trastornos gastrointestinalesAdemás de los tumores carcinoides, se hanreferido casos de neurofibromas y otros
tumores del tubo digestivo en la NFl. Se hacalculado que la frecuencia de los neurofi-bromas digestivos en los enfermos afectadospor la NF1 es aproximadamente del 2 %; sulocalización más frecuente es el yeyuno.También se han publicado casos de leiomio-mas, ganglioneuromas y sarcomas. Los sínto-mas asociados son dolores, dispepsia y estre-ñimiento. La hematemesis y la melena pue-den ser reveladoras.
Complicaciones pulmonares " ID="I196.86.3">1311
Los neurofibromas intrapulmonares son
generalmente asintomáticos. No obstante, encasos excepcionales, se puede revelar la pre-sencia de neurofibromas de gran tamaño porla aparición de tos o de dificultades respira-torias. La existencia de una escoliosis impor-tante implica la mayor parte de las veces unainsuficiencia respiratoria restrictiva porreducción de los volúmenes respiratoriospulmonares y fibrosis pulmonar. Esta insufi-ciencia respiratoria puede comprometer el
pronóstico vital en los casos graves. Las mal-formaciones vasculares pulmonares puedencausar hemoptisis.
Complicaciones del sistemaurinario " ID="I196.102.2">[31 [
Pueden aparecer hidronefrosis o trastornosurinarios. Estos síntomas se deben a la com-
presión de las vías urinarias por neurofibro-mas retroperitoneales o pélvicos.
Complicaciones estéticasy funcionales
Los neurofibromas plexiformes congénitospueden producir complicaciones estéticas
graves (hipertrofia de segmentos corporales);a veces en la cara producen afectación oftal-mológica (ambliopía).Los neurofibromas cutáneos que aparecencon frecuencia en la adolescencia tienen amenudo graves consecuencias psicológicas ysociales debido a su carácter llamativo.
Los hamartomas anémicos son frecuentes enla NFl. A veces también se encuentran placasrosadas atróficas en el tronco, con frecuencia
pretorácicas, deprimidas, planas, con bordesbien delimitados. Los schwannomas cutá-neos o subcutáneos son muy raros en la NFl.
Los xantogranulomas juveniles son excepcio-nales (menos del 1 % de los casos de NF1).Aparecen en los dos primeros años de la viday remiten lentamente. En varias ocasiones seha hecho referencia a su asociación con laleucemia mieloide juvenil crónica. El riesgode leucemia mieloide es muy bajo en la NF1(0,004 % de los casos) pero parece aumentaren caso de asociación con xantogranulomasjuveniles.
El prurito, que con frecuencia se producesimultáneamente con la aparición de los neu-rofibromas, se produce en el 10 % de loscasos aproximadamente.A veces existen zonas hiperpigmentadas queseñalan la presencia de un neurofibroma ple-xiforme y, en situación dorsal media, unaposible afectación subyacente del neuroeje.
Cáncer y neurofibromatosis 1 ~2’~
Tumores malignos de las vainasnerviosas " ID="I196.144.2">íll, 11, 451
Los TMVN, antiguamente denominados neu-rofibrosarcomas, se desarrollan a partir de lascélulas de Schwann o fibroblastos del peri-neuro. Representan aproximadamente el 10 %de los sarcomas de los tejidos blandos peroestán estrechamente relacionados con la NF1.
Así, del 50 al 60 % de los pacientes con TMVNestán afectados por la NFl. La prevalencia delos TMVN en las cohortes de enfermos deNF1 es del orden del 3 al 4 %. Los TMVN se
producen a lo largo del tercer decenio en laNFl y son de alto grado de malignidad en el85 % de los casos. Se desarrollan a partir deneurofibromas plexiformes o de neurofibro-mas nodulares. Los signos de alerta son dolo-res relacionados con la masa tumoral, signosneurológicos como parestesias y aumentorápido del tamaño de un tumor generalmentepreexistente. La aparición de uno de estos sig-nos de alerta debe llevar a la realización deuna biopsia quirúrgica que se repetirá si per-siste la duda.
Otros cánceres
Algunos otros cánceres excepcionales que seproducen en la NF1 tienen indudablementeuna prevalencia acrecentada: glioblastomascerebrales, leucemias, rabdomiosarcoma, ade-nocarcinoma o tumor carcinoide del duodeno,feocromocitoma maligno y tumor de Wilms.
a Alteración de la calidad de vidaen la neurofibromatosis 1 124J
La imprevisibilidad de la evolución de la
NF1, la aparición de complicaciones que dancuenta de la gravedad de la enfermedad perotambién las consecuencias estéticas y la visi-bilidad de la enfermedad tienen un impactosignificativo en la calidad de vida de losenfermos.
VARIANTES SDE LA NEUROFIBROMATOSIS 1 1
Síndrome de Noonan-
neurofibromatosis 1A los signos del síndrome de Noonan (dis-morfia facial con cuello corto, pterygium colli,ptosis palpebral, implantación baja del cabe-llo, orejas con implantación baja y vueltashacia atrás, talla baja, tórax en embudo, linfe-dema, retraso de la pubertad, trastornos cog-nitivos y malformación cardíaca, fundamen-talmente estenosis arterial pulmonar congéni-ta) se asocian en ocasiones a manifestacionesque hacen pensar en la NFl. Existe polémicarespecto a la genética de esta asociación.
7
Síndrome de Watson
Se denomina así a casos familiares de trans-misión autosómica dominante que asocianMCL, estenosis valvular pulmonar e inteli-gencia por debajo de los niveles normales.Parece ser una forma alélica de NFl.
EVOLUCIÓN Y " ID="I197.7.3">GRAVEDAD 1’°,471
La NF1 es una afección evolutiva que, en cadaenfermo afectado, va produciendo mayorestrastornos de año en año. Aunque en la mayorparte de los casos la gravedad de la afectaciónsigue siendo limitada, pueden aparecer com-plicaciones a lo largo de toda la vida, diferen-tes según la edad: neurofibromas plexiformesque siempre se revelan antes de los 5 años,TMVN (neurofibrosarcomas) a partir de laadolescencia (cuadro V). En el 15 al 20 % de lospacientes afectados de NFl, la morbilidad es oserá importante. En los niños, los trastornosdel aprendizaje y el glioma de las vías ópticasconstituyen los principales problemas. En losadultos, la complicación más temible es elTMVN. La esperanza de vida en los pacientesque padecen NF1 se reduce una decena deaños en comparación con la de la poblacióngeneral. Las principales causas de fallecimien-to son las neoplasias, esencialmente TMVN ytumores cerebrales pero también la vasculo-
patía asociada a la NF1 y sobre todo los acci-dentes cerebrovasculares.
La fertilidad de los pacientes que padecenNF1 es normal pero durante el embarazo esfrecuente que se extiendan los neurofibromas
y asimismo que aparezca o se agrave una
hipertensión arterial.
DIAGNÓSTICO DIFERENCIAL- Síndrome de McCune-Albright.Se asocian MCL de bordes irregulares, pu-bertad precoz y displasia fibrosa poliostósica.- Síndrome LEOPARD.Se asocian estenosis arterial pulmonar, lenti-gos múltiples, talla baja y sordera.- Síndrome de Carney.Se asocian lentigos, mixomas (en particularcutáneos y cardíacos) y anomalías endocrinas.- Síndrome de Proteo («el hombre elefante») " ID="I197.46.8">(37].
El hombre elefante no tenía una NF1 sino unsíndrome de Proteo. En este síndrome se aso-cian de forma variable hemihipertrofia cor-poral segmentaria, macrodactilia, hamarto-mas conjuntivos y/o epidérmicos. Es parti-cularmente sugerente el engrosamiento delas palmas y de las plantas en masas cerebri-formes. La histología de las masas subcutá-neas se corresponde con hamartomas lipo-matosos y/o angiomatosos. La mayor partede los casos son esporádicos, probablementerelacionados con una mutación no letal, úni-camente en mosaico.
- Otras afecciones.Se pueden citar el síndrome del hamartomaepidérmico, las lipomatosis y el síndrome deBannayan-Riley-Rulvalcaba, el síndrome deKlippel-Trenaunay (hemangioma, hemihi-pertrofia) y las neoplasias endocrinas múlti-ples (coexistencia de tumores al menos en
dos glándulas endocrinas funcionalmenteindependientes).
SEGUIMIENTO DE LOS ENFERMOSAFECTADOS POR LA
NEUROFIBROMATOSIS 1 14,9,41.43[
En el cuadro V se resume la frecuencia de las
complicaciones que justifican el seguimientoen función de la edad.Además del consejo genético y del trata-miento de las manifestaciones cutáneas queconstituyen la demanda prioritaria de losenfermos adultos, es necesario realizar unseguimiento para la detección precoz de lascomplicaciones de la NF1, muchas de lascuales aparecen en la infancia. La gravedadde la NF1 varía de un paciente a otro y en elseno de una misma familia. La NF1 es unaenfermedad cuya gravedad aumenta gene-ralmente con la edad. No se dispone de sig-nos para predecir la evolución. Incluso anteformas de NF1 que parecen benignas, sedebe proponer el seguimiento; éste debe seresencialmente clínico. Las exploracionesefectuadas con carácter sistemático son
poco rentables para el enfermo [14,411. Con la
exploración clínica se pueden identificarfácilmente complicaciones como la escolio-sis, la seudoartrosis, la hipertensión arterialrelacionada con una estenosis de la arteriarenal o con un feocromocitoma o, incluso,dificultades del aprendizaje escolar cuyadetección debe ser lo más precoz posible.En el cuadro VI se resume la evaluación pro-puesta para un enfermo que padezca NFl.Las pruebas complementarias sólo se realiza-rán basándose en argumentos clínicos. Laúnica excepción polémica puede ser la RMde las vías ópticas para la detección de unglioma potencialmente agresivo, en particu-lar en niños pequeños ya que la exploraciónoftalmológica puede ser difícil.
Algunos neuropediatras y oncopediatras hanpropuesto la siguiente actitud: en los niños,cuando la exploración oftalmológica es difícilpor resultar insuficiente la cooperación yasea debido a la edad (menores de 6 años) obien debido a trastornos cognitivos, la RMcerebral es sistemática. Si no se detecta nin-
guna anomalía en esta prueba, se solicitaráuna nueva RM cerebral al cabo de 2 años si la
exploración oftalmológica anual sistemáticasigue siendo incompleta.En cuanto lo permita la edad del niño, esrecomendable realizar un simple control
oftalmológico anual que incluya una deter-minación de la agudeza visual y un campovisual. Si se detectaran anomalías, se tendríaque realizar una RM.Si en la RM existe una anomalía que hagapensar en un glioma de las vías ópticas, sólolos gliomas de las vías ópticas de evoluciónagresiva justifican medidas terapéuticas.Para evaluar el potencial agresivo del tumor,se propone el siguiente control: exploraciónoftalmológica y RM cada 3 meses durante 6meses, después cada 6 meses durante 1 año ymás tarde todos los años hasta la pubertad.Este protocolo de control permite mejorar elconocimiento sobre la evolución de los glio-mas y validar la asistencia a los niños peque-ños afectados por la NFl.
CONDUCTA TERAPÉUTICA
Tratamiento de las manifestacionescutáneas 1411
Manchas de color café con leche
Se han referido éxitos terapéuticos en las MCLcon los láseres YAG de rubí Q-switched y pul-sados. Sin embargo, parece que los resultadosson temporales. Se han comunicado casos deagravamiento. Por lo tanto, no se puede reco-mendar actualmente este tratamiento.
Cuadro V. - Frecuencia de las complicaciones que justifican el seguimiento de los enfermosque padecen neurofibromatosis 1 en función de la edad.

8
Cuadro VI. - Evaluación de los enfermos afectados por la neurofibromatosis de tipo 1.
Tratamiento de los neurofibromas
0 Neurofibromas cutáneosSe pueden utilizar varios métodos para laexéresis completa o parcial de neurofibromasvoluminosos o que producen molestias.Cuando los neurofibromas cutáneos tienenun tamaño reducido, menor de 2 cm, y sonnumerosos, el método de destrucción prefe-rente es el láser de COZ. Se puede optar porla electrocoagulación cuando los elementosson escasos. La cirugía clásica es necesariacuando las lesiones tienen más de 2 cm.
Según su importancia y su número, estasdestrucciones pueden realizarse bajo aneste-sia local o general. El láser de COZ bajo anes-tesia general permite destruir varias centenasde neurofibromas en una sola sesión.
0 Neurofibromas nodulares periféricosLa cirugía de exéresis de los neurofibromasperiféricos debe hacerse bajo microscopiopara evitar secuelas funcionales neurológicas.
s Neurofibromas plexiformesEl tratamiento sólo puede ser quirúrgico. Laexéresis es casi siempre intralesional. Las
complicaciones hemorrágicas son frecuentes
y se debe proponer la autotransfusión. Losneurofibromas plexiformes de la cara plan-tean problemas de reparación complejos.En la actualidad, está garantizado que lasdestrucciones de los neurofibromas no pro-duzcan ningún riesgo de aceleración del cre-cimiento de los neurofibromas restantescomo tampoco de malignización. El beneficiopsicológico de estas intervenciones es con fre-cuencia considerable y permite esperar que semantenga o se recupere una vida afectiva,social y psicológica normal o casi normal.
Tratamientode las complicaciones
Ciertas escoliosis distróficas requieren untratamiento quirúrgico en la infancia o laadolescencia.El tratamiento de los gliomas ópticos rara-mente está justificado debido a su escasacapacidad evolutiva. La indicación terapéuti-ca (quimioterapia, radioterapia, cirugía) seevalúa según la localización, la evolución, larepercusión y la edad.Para el tratamiento de los TMVN se recurre ala cirugía de exéresis completa asociada aradioterapia con o sin quimioterapia adyu-vante con antraciclinas.
Neurofibromatosisde tipo 2 [51 (antiguaneurofibromatosisacústica o central)
EPIDEMIOLOGÍA Y GENÉTICA " ID="I198.58.4">134.351
La NF2 se denominaba antiguamente neuro-fibromatosis acústica. Es mucho más rara
que la NFl, con una incidencia de 1 de cada33 000 a 40 000 nacimientos. Se caracteriza
por schwannomas vestibulares bilaterales
(antiguamente denominados neurinomas delacústico), schwannomas de otros nervios cra-neales y espinales y meningiomas. El gen dela NF2 ha sido identificado en el cromosoma22, en la región 22q12.2. Éste es un gen supre-sor tumoral. Se ha denominado merlina a la
proteína producida que forma parte de ungrupo de proteínas relacionadas con el cito-esqueleto. La penetrancia del gen NF2 escompleta a los 60 años de edad y las muta-ciones de novo se presentan en el 50 % de loscasos aproximadamente.
CRITERIOS DIAGNÓSTICOS
En el cuadro 11 se recuerdan los criterios diag-nósticos de la NF2.
MANIFESTACIONES CLÍNICAS 125.291
Las manifestaciones cutáneas son inconstan-tes y la mayor parte de las veces discretas.Las MCL están presentes aproximadamenteen la mitad de los casos; son mucho menosnumerosas que en la NF1, generalmente sóloun par.No hay lentigos de los pliegues en la NF2.Los tumores cutáneos, schwannomas y conmenor frecuencia neurofibromas están pre-sentes aproximadamente en el 70 % de lospacientes. Son poco numerosos, menos deuna decena. Estos schwannomas tienen el
aspecto o bien de tumores subcutáneos sensi-bles a la presión, imposibles de distinguir clí-nicamente de los neurofibromas nodularessubcutáneos o bien, lo que es más frecuente,de zonas de engrosamiento cutáneo en«meseta» apenas sobreelevadas, pigmentadasy a menudo pilosas.No se producen neurofibromas plexiformes.Las anomalías oculares son frecuentes. El 70 %de los enfermos tienen una catarata juvenilposterior. Es mucho más raro detectarhamartomas retinianos. Los nódulos deLisch están ausentes.
Los schwannomas vestibulares son práctica-mente constantes (el 90 % de los casos). Sudiagnóstico se hace generalmente en la ter-cera década. Los meningiomas están presen-tes en el 50 % de los casos. Son frecuentes losschwannomas del sistema nervioso central, deotras localizaciones además de la vestibular,los ependimomas o los neurofibromas espina-les. Los gliomas de las vías ópticas no se pre-sentan en la NF2.
PRONÓSTICO ’01
El pronóstico de la NF2 es malo, con unaesperanza de vida del orden de 50 años.

9
Parecen diferenciarse tres fenotipos: leve,moderado o grave. La gravedad del pronós-tico parece relacionarse con la presencia dehamartomas retinianos y de ciertas mutacio-nes. El fenotipo de gravedad se encuentra enlos miembros de una misma familia.
El tratamiento de los schwannomas vesti-bulares depende de la repercusión funcio-nal, del número y el tamaño de los tumoresy de la edad del paciente. Su objetivo prin-cipal es preservar la función auditiva. Si eltumor tiene un crecimiento lento y esdemasiado grande para extirparlo sin cau-sar sordera, están indicadas hacer unaresección parcial o abstenerse de la cirugíay realizar un seguimiento. El tratamientode otros tumores es el mismo, padezca elpaciente una NF2 o no. La asistencia segarantiza más en centros especializadosmultidisciplinarios donde otorrinolaringó-logos, neurólogos y neurocirujanos habi-tuados a este tipo de patología ponen encomún sus conocimientos.
Neurofibromatosissegmentaria (NF5) 1111
Las neurofibromatosis segmentarias son
excepcionales, con una prevalencia inferior al0,001 % [341. Se caracterizan la mayor parte delas veces por la presencia de neurofibromas y,más raramente, de MCL y a veces por lenti-
gos o, excepcionalmente, nódulos de Lisch enun solo segmento corporal, incluso en unhemicuerpo o en más raras ocasiones envarios segmentos de forma bilateral. El diag-nóstico sólo se puede establecer tras haberdescartado el diagnóstico de NF1 o de NF2.Las neurofibromatosis segmentarias po-drían ser una forma mosaico de NFl. El
consejo genético debe ser prudente ya queexisten casos excepcionales de NF1 hereda-da de padres que padecen una forma seg-mentaria (que involucra entonces a las célu-las germinales).
Formas rarasde neurofibromatosis
NEUROFIBROMATOSIS 3 3O NEUROFIBROMATOSIS MIXTA
Algunos casos excepcionales de NF presen-tan a la vez NF1 y NF2.
MANCHAS DE COLOR CAFÉ CONLECHE AISLADAS
O NEUROFIBROMATOSIS 6 6 "J
Las MCL que se mantienen aisladas en laedad adulta al menos dos generaciones, aveces asociadas a otras anomalías de la pig-mentación, definen esta forma particular deNF. A veces se encuentran lentigos de los plie-gues, raramente nódulos de Lisch, pero nin-gún neurofibroma. En ocasiones, existen ano-
malías óseas (tórax en embudo, genu valgum,pies planos), incluso dificultades inespecíficasde la escolarización. La transmisión es autosó-mica dominante. Estas formas excepcionalespueden ser difíciles de distinguir del síndro-me de los lentigos múltiples. Se trataría deuna forma alélica de NF1.
NEUROFIBROMATOSIS SDE COMIENZO TARDÍO
O NEUROFIBROMATOSIS 7
Se trata de una NF con neurofibromas de
aparición tardía, a partir de los 30 años.
SCHWANNOMATOSIS SO NEURILEMOMATOSIS [31
Las schwannomatosis se caracterizan porschwannomas múltiples cutáneos y subcutá-neos sin schwannomas vestibulares. Por con-
siguiente, se trata de una enfermedad próxi-ma a la NF2 pero de mejor pronóstico.
Conclusión
Las NF constituyen un conjunto heterogéneo.La clasificación nosológica de cada caso es
indispensable en el seguimiento. Según las NF,el pronóstico, las complicaciones, el consejogenético y la conducta terapéutica son diferen-tes. Los centros multidisciplinarios de referen-cia, con sus redes de expertos, son las estructu-ras de elección para asistir a estos enfermos.
Cualquier referencia a este artículo debe incluir la mención del artículo original: Wolkenstein P, Zeller 1 et Ismaili N. Neurofibromatoses. Encycl Méd Chir (Editions Scientifiques et Médicales Elsevier SAS, Paris, tous droits réser-vés), Dermatologie, 98-755-A-10, Pédiatrie, 4-092-C-10, 2002, 10 p. p.

10
Bibliografía
[1 ] Arnsmeier SL, Riccardi VM, Palier A5. Familial multiplecafeau iait spots. Arch Dermatoll 994 ;130 :1425-1426
[2] Bahuau M, Vidaud M, Vidaud D. Neurofibromatose. Géné-tique et physiopathologie moléculaire de la NF1. Méd/Thér1997; 3; 623-628
[3] Benchikhi H, Wolkenstein P, Zeller 1, Wechsler 1, Revuz j.Schwannomatosis and its nosologic limitswith neurofibro-matosis type 2. Dermatology 1997; 195 : 228-231 1
[4] Eichenfield LF, Levy ML, Palier AS, Riccardi VM, Farmer ER,Chuang TY et al. Guidelines of care for neurofibromatosistype 1./AmAcadDermatol t 997; 37: 625-630
[5] Evans DG, Huson SM, Donnai D, Neary W, Blair V, Teare Det al. A genetic study of type 2 neurofibromatosis in theUnited Kingdom, 1: prevalence, mutation rate, fitness, andconfirmation of maternal transmission effect on severity.1 Med Genet 1992 ; 29 : 841-846
[6] Ferner RE, Hughes RA, Weinman 1. lntellectual impairmentin neurofibromatosis 1. / Neurol Sci 1996; 138: :125-133
[7] Friedman IM, Birch PH. Type 1 neurofibromatosis: adescriptive analysis of the disorder in 1, 728 patients. Am/ JMed Genet 1997; 70 : 138-143
[8] Grill J, Couanet D, Capelli C, Habrand JL, Rodriguez D,Sainte-Rose C et al. Radiation induced cerebral vasculopa-thy in children with neurofibromatosis and optic patchwayglioma. Ann Ncuro; 1999; 45: 393-396
[9] Gutmann DH, Aykworth A, Carey )C, Korf B, Marks PyeritzRE et al. The diagnostic evaluation and multidisciplinarymanagementof neurofibromatosis 1 and neurofibromato-sis 2. /AMA 1997 ; 278 : 51-57
[10] Gutmann DH, Collins FS. Neurofibromatosis type 1.Beyond positional cioning. Arch Neurol 1993 ; 50 :1185-1193
[11 ] Hager CM, Cohen PR, Tschen JA. Segmental neurofibro-matosis : case report and review. / Am Acad Dermatoll 997 ;37 : 864-869
[12] Huson SM. Neurofibromatosis: historical perspective, clas-sification and diagnostic criteria. In : Huson SM, Hughes RAeds. The neurofibromatoses. A pathogenetic and clinicaloverview. London: Chapman and Hall 1994: 1-22
[7 3] Huson SM. The neurofibromatoses: a pathogenetic andclinical overview. London : Chapman and Halil 994
[14] Huson SM. What leve¡ of care for the neurofibromatoses?Lancet 1999; 353 :1114-1116 6
[7 5] Huson SM, Compston DA, Harper PS. A genetic study ofvon Recklinghausen neurofibromatosis in South EastWales. 11: Guidelines for genetic counselling. 1 Med Genet1989;26:712-721
[16] Kayes LM, Burke W, Riccardi VM, Bennet R, Ehrlich P,Rubenstein A et al. Deletions spanning the neurofibroma-tosis 1 gene: identification and phenotype of five patients.Am/ Hum Genet 1994; 54 : 424-436
[77] KingA,DebaunMR,RiccardiVM,GutmannHD.Malignantperipheral nerve sheath tumors in neurofibromatosis 1. Am¡MedGenet2000; 93: 388-392
[18] Korf BR. Plexfform neurofibromas. Am / Med Genet 1999 ;89 : 31-37
[7 9] Ledbetter DH, Rich DC, O’Connell P, Leppert M, Carey jC.Precise localization of NF1 to 17q11.2 by balanced trans-lation. Am/ Hum Genet 1989; 44 : 20-24
[20] Listernick R, Darling C, Greenwald M, Strauss L, Charrow J.Optic pathwaytumors in children: the effectof neurofibro-matosis type 1 on clinical manifestations and naturalhistory. Peciatrl 995 127: 718-722
[21 Listernick R, Louis DN, Packer RJ, Gutmann DH. Opticpathway gliomas in children with neurofibromatosis 1:consensus statementfrom the NF1 Optic Pathway GliomaTask Force. Ann NfUfoí 1997; 41 : 143-149
[22] Lund AM, Stkovby F. Optic gliomas in children with neu-rofibromatosis type 1 Eurl Pediatr 1991 ; 150: 835-868.
[23] Matsui I, Tanimura M, Kobayashi N, Sawada T, NagaharaN, Akatsuda ]L. Neurofibromatosis type 1 and chiidhoodcancer. Cancer 1993; 72 : 2746-2754
[24] Mauger D, Zellerl, Revuz J, Wolkeinstein P. Retentissementpsychologique de la neurofibromatose de type 1 : analysed’entretiens avec 12 malades en vue d’une évaluation de laqualité de vie. Ann Dermatol Vénéréo11999;126 : 619-620
[25] MautnerVF, Lindenau M, Baser ME, Kluwe L, GottschalkJ. J.Skin abnormalities in neurofibromatosis type 2. Arch Der-matol l 997; 133:1539-1543
[26] NguyenTheT’ichS,MahéJY,CoutantX,GIoanecY,PeuvrelE. Neurofibromatose. Troubles de I’apprentissage: dépis-tage et prise en charge. MédIThér 1997; 3 : 636-639
[27] NIHconsensusdevelopmentconferencestatement.Neu-rofibromatosis. Arch Neurol 1988; 45 : 575-578
[28] North KN, Riccardi V, Samango-Sprouse C, Ferner R,Moore B, Legius E et al. Cognitive function and academicperformance in neurofibromatosis 1 : consensus statementfrom the NF cognitive disorders Task force. Neurology1997;48:1121-1127
[29] Parry DM, Eldridge R, Kaiser-Kupfer MI, Bouzas EA, PikusA,Patronas A. Neurofibromatosis 2 (NF2): clinical character-istics of 63 affected individuals and clinical evidence for he-terogeneity. Am/ Med Genet 1994 ; 52 : 450-461
[30] Parry DM, MacCollin MM, Kaiser-Kupfer MI, Pulaski K,Nicholson HS, Bolesta M et al. Germ-line mutations in theneurofibromatosis 2 gene: correlations with disease seve-rity and retinal abnormalities. Am / Hum Genet 1996 ; 59 :529-539
[31] Pinson S, CréangeA, 8arbarot5,5taider)F, Chaix Y, Rodri-guez D etal. Guidelinesforthe managementof neurofibro-matosis 1. Ann Dermatol Vénéréc12001 128 : 567-575
[32] Riccardi VM. Neurofibromatosis: phenotype, naturalhistory, and pathogenesis. Baltimore : johns Hopkins Uni-versity Pressl 992
[33] Riccardi VM. Von Recklinghausen neurofibromatosis. NEngl/ Med 1981 ; 305 :1617-1627
[34] Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman j,Marineau C etal. Alteration in a newgene encoding a puta-tive membrane-organizing protein causes neurofibroma-tosis type 2. Nature 1993 ; 363 : 51 S-521
[35] Rouleau GA, Wertelecki W, Haines 11, Hobbs Wj, TrofatterjA, Seizinger BR et al. Genetic linkage of bilateral acousticneurofibromatosis to a DNA marker on chromosome 22.Nature 1987; 329 : 246-248
[36] Rubenstein AE, Korf BR. Neurofibromatoses: a handbookfor patients, families, and health-care professionals. NewYork : Thieme Medical Publishers1990
[37] Samiaska CP, Levin SW, lames WD, Benson PM, Walker JC,Perlik PC. Proteus syndrome. Arch Dermatol 1989 ; 125 :1109-1114 4
[38] Samueisson B, Axelsson R. Neurofibromatosis: a clinicaland genetic studyof96 cases in Gothenburg, Sweden. ActaDerm Venereol[suppil 1981 ; 95 : 67-71
[39] Shen MH, Harper PS, Upadhyaya M. Moleculargenetics ofneurofibromatosis type 1 (NF1 ). / Med Genet 1996 ; 33 :2-17 7
[40] Sorensen SA, Mulvihilijj, NieisenA. Long-termfollow-upofvon Recklinghausen neurofibromatosis. Survival andmalignantneoplasms.NEng/jMed1986; 314:1010-1015 5
[41 ] Wolkenstein P, Fréche B, Zeller 1, Revuz J. Usefulness ofscreening investigations in neurofibromatosis type 1: astudy of 152 patients. Arch Dermatol 1996 ; 132 :1333-1336
[42] Wolkenstein P, Mahmoudi A, Zeller, RevuzJ. More on thefrequency of segmental neurofibromatosis. Arch Dermatol1995;131:1465
[43] Wolkenstein P, Zeller mtérét des examens paracliniquesau cours de la neurofibromatose de type 1 et de la sciérosetubéreuse de Bourneville. Nouv Dermatol 1996 ; 15 :497-499
[44] Wolkenstein P, Zeller 1, Mathoret C, Lantieri L. Neurofibro-matose. Traitement des manifestations cutanées de la neu-rofibromatose 1. Méd/Thér1997; 3: 609-613 3
[45] Woodrouff MI. Pathology of tumors ofthe peripheral nervesheath in type 1 neurofibromatosis. Am/ Med Genet 1999 ;89 : 23-30
[46] Zeller j, Hovnanian A. La maladie de von Recklinghausen.Ann Dermatol Vénérécl 1 992 ; 119 : 405-410 0
[47] Zoller M, Rembeck B, Akesson HO, Angervall L. Life expec-tancy, mortality and prognosticfactors in neurofibromato-sis type 1. A twelve-year follow-up of an epidemiologicalstudy in Goteborg, Sweden. Acta Derm Venereol 1995; 75 :136-140







![Cranial MR Imaging in Neurofibromatosis · bromatosis), neurofibromatosis II (bilateral acoustic neurofibromatosis), and other forms [5, 6]. Neuroradiology has traditionally played](https://img.dokumen.tips/doc/110x75/5ed593375be95c6187174771/cranial-mr-imaging-in-bromatosis-neurofibromatosis-ii-bilateral-acoustic-neurofibromatosis.jpg)





















